



This publication is Open Access under the license indicated. Learn More
Integrating Molecular Dynamics Simulations and Single-molecule FRET Spectroscopy: From Computational FRET Estimation to Experimental Data InterpretationClick to copy article linkArticle link copied!
- Stephanie SauveStephanie SauveDepartment of Chemistry and Biochemistry, University of Arkansas, Fayetteville 72701, Arkansas, United StatesMore by Stephanie Sauve
- Ehsaneh KhodadadiEhsaneh KhodadadiDepartment of Chemistry and Biochemistry, University of Arkansas, Fayetteville 72701, Arkansas, United StatesMore by Ehsaneh Khodadadi
- Ahmed ShubbarAhmed ShubbarDepartment of Chemistry and Biochemistry, University of Arkansas, Fayetteville 72701, Arkansas, United StatesMore by Ahmed Shubbar
- Ehsan KhodadadiEhsan KhodadadiDepartment of Chemistry and Biochemistry, University of Arkansas, Fayetteville 72701, Arkansas, United StatesMore by Ehsan Khodadadi
- Mahmoud Moradi*Mahmoud Moradi*Email: [email protected]Department of Chemistry and Biochemistry, University of Arkansas, Fayetteville 72701, Arkansas, United StatesMore by Mahmoud Moradi
The Journal of Physical Chemistry B
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
Molecular dynamics (MD) simulations can characterize biomolecular processes at an exceptional spatiotemporal resolution not able to be accessed experimentally. As the limitations associated with MD simulations lessen and the method advances toward greater capabilities, the simulations are being applied to a wide array of new applications. For example, the integration of MD simulations and single-molecule Förster resonance energy transfer (smFRET) spectroscopy is a newly developing and growing application combining experimental and computational approaches. The integration of these techniques provides valuable insight into the conformational dynamics of biomolecules on an atomic-level, thereby enhancing the understanding of complex biological processes. This review compiles information on simulating FRET dyes and estimating FRET efficiencies from MD simulations and using MD simulations to gain insight into experimental data to shine light on the recent advancements in joining computational and experimental techniques. We discuss notable studies that incorporate the use of both MD simulations and smFRET as well as discuss the challenges that have been faced regarding their integration. The joining of these approaches have provided valuable insights into conformational sampling, binding mechanisms, structural dynamics, and allosteric effects thus far and will continue to advance the understanding of biomolecular dynamics in the future.
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
I. Introduction
Figure 1
Figure 1. A simplified schematic representation showing the overlap (gray) of the donor fluorophore emission spectra shown in green and the acceptor fluorophore absorption spectra shown in red. The overlap of the spectra must be present for FRET to occur. In practice the actual overlap increases with wavelength.
Figure 2
Figure 2. Schematic representation of FRET showing an energy transfer between a donor fluorophore shown in green and an acceptor fluorophore shown in red. The relationship between the distance of the dyes and the efficiency of transfer is shown with the optimal transfer distance being between 1 and 10 nm. The further the dyes move away from each other, the lower the transfer of energy from the donor fluorophore to the acceptor fluorophore.
II. Simulating FRET Dyes and Estimating FRET Efficiencies from MD Simulations
II.A. Simulating Dyes Attached to Proteins
Figure 3
Figure 3. Schematic representation of using coarse-grained systems for MD simulations. In coarse-grained models, groups of atoms are represented as beads, reducing the system’s degrees of freedom. This simplification significantly accelerates simulations, enabling the study of larger systems and longer time scales while reducing computational cost in comparison to to all-atom MD simulations.
II.B. Estimating FRET Efficiency from MD
Figure 4
Figure 4. Schematic representation showing how the three angles that contribute to the orientation factor are defined. θDA is the angle between ûD and ûA (black) where ûD and ûA represent the unit vectors associated with the orientations of the donor and acceptor dyes, respectively. θDR is the angle between ûD and ûR (blue). θAR is the angle between ûA and ûR (green).
Figure 5
Figure 5. (A) Schematic representation showing rotational movement of the dyes on a labeled biomolecule where the dyes on a labeled protein (green) are depicted in blue and red. (B) Schematic representation of the probability distribution of dye angles based on free rotation (green) and hindered rotation (magenta).
III. Using MD to Gain Insight into Experimental Data
III.A. MD and smFRET Use in Parallel
III.B. Parallel Integration of smFRET with MC/MD Simulations
III.C. Use of Enhanced Sampling Methods
Figure 6
Figure 6. Schematic overview of three enhanced sampling techniques commonly used in biomolecular simulations. Left: Schematic representation of metadynamics. A bias potential is constructed by periodically adding repulsive Gaussian hills along selected collective variables, allowing the system to escape local minima and explore rare conformational events. Top right: Go̅-like model, which biases the energy landscape by favoring native contacts. Bottom right: Steered MD, in which an external force is applied along a reaction coordinate to induce transition from outward-facing to inward-facing state.
III.D. MD Based MSM with smFRET Experiments
Figure 7
Figure 7. A schematic showing the integrative approach of combining Markov State Modeling (MSM), smFRET data, and machine learning algorithms to refine and validate MD trajectories.
IV. Summary
Data Availability
No new data is reported in this review article.
Author Information
- Mahmoud Moradi - Department of Chemistry and Biochemistry, University of Arkansas, Fayetteville 72701, Arkansas, United States;
 https://orcid.org/0000-0002-0601-402X;
https://orcid.org/0000-0002-0601-402X;
- Stephanie Sauve - Department of Chemistry and Biochemistry, University of Arkansas, Fayetteville 72701, Arkansas, United States;https://orcid.org/0009-0008-7423-8050
E.K. and A.S. contributed equally.
Biographies
Stephanie Sauve
Stephanie Sauve is a Ph.D. candidate in Cell & Molecular Biology program at the University of Arkansas. She has a B.Sc. in biochemistry from St. Lawrence University, where she gained experience designing stabilized variants of a cyan fluorescent protein through thermal and chemical denaturation studies. Her current Ph.D. research focus includes integration of experimental and computational methodology with emphasis on smFRET and molecular dynamics simulations.
Ehsaneh Khodadadi
Ehsaneh Khodadadi received a B.Sc. and M.Sc. in Plant Breeding and a Ph.D. in Biotechnology. She has also recently received a Ph.D. in Materials Science and Engineering in 2025 from the University of Arkansas. Her research focuses on elucidating the structural and dynamic behavior of membranes and membrane proteins.
Ahmed Shubbar
Ahmed Shubbar has a B.Sc. in pharmacy and a M.Sc. in Pharmacology and is currently pursuing a Ph.D. in Cell & Molecular Biology since 2023 at the University of Arkansas. His main focus is on conformational dynamics of ATP-binding cassette transporters as well as combining experimental and computational techniques with an emphasis on double electron–electron resonance spectroscopy and molecular dynamics simulations.
Ehsan Khodadadi
Ehsan Khodadadi has a B.Sc. and M.Sc. in Plant Breeding and a Ph.D. in Molecular Genetics and is currently pursuing a Ph.D. in Materials Science and Engineering since 2023 at the University of Arkansas. His research is focused on molecular dynamics simulations of drug delivery liposomes.
Mahmoud Moradi
Mahmoud Moradi is a Professor of Chemistry and Biochemistry at the University of Arkansas. He has a B.Sc. and M.Sc. in Physics from the Sharif University of Technology in Iran and a Ph.D. in Physics from the North Carolina State University. His research focuses on theoretical and methodological developments for biomolecular simulations as well as application of these methods to study various proteins.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM147423.
References
This article references 125 other publications.
- 1Sagui, C.; Darden, T. A. MOLECULAR DYNAMICS SIMULATIONS OF BIOMOLECULES: Long-Range Electrostatic Effects. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 155– 179, DOI: 10.1146/annurev.biophys.28.1.155Google ScholarThere is no corresponding record for this reference.
- 2Immadisetty, K.; Moradi, M. Mechanistic Picture for Chemomechanical Coupling in a Bacterial Proton-Coupled Oligopeptide Transporter from Streptococcus Thermophilus. J. Phys. Chem. B 2021, 125, 9738– 9750, DOI: 10.1021/acs.jpcb.1c03982Google ScholarThere is no corresponding record for this reference.
- 3Singharoy, A.; Chipot, C.; Moradi, M.; Schulten, K. Chemomechanical Coupling in Hexameric Protein-Protein Interfaces Harnesses Energy within V-Type ATPases. J. Am. Chem. Soc. 2017, 139, 293– 310, DOI: 10.1021/jacs.6b10744Google ScholarThere is no corresponding record for this reference.
- 4Badiee, S. A.; Kumar, V. G.; Moradi, M. Molecular dynamics investigation of the influenza hemagglutinin conformational changes in acidic pH. J. Phys. Chem. B 2024, 128, 11151– 11163, DOI: 10.1021/acs.jpcb.4c04607Google ScholarThere is no corresponding record for this reference.
- 5Zhang, R.; Jagessar, K. L.; Brownd, M.; Polasa, A.; Stein, R. A.; Moradi, M.; Karakas, E.; Mchaourab, H. S. Conformational cycle of a protease-containing ABC transporter in lipid nanodiscs reveals the mechanism of cargo-protein coupling. Nat. Commun. 2024, 15, 9055 DOI: 10.1038/s41467-024-53420-0Google ScholarThere is no corresponding record for this reference.
- 6Govind Kumar, V.; Ogden, D. S.; Isu, U. H.; Polasa, A.; Losey, J.; Moradi, M. Prefusion spike protein conformational changes are slower in SARS-CoV-2 than in SARS-CoV-1. J. Biol. Chem. 2022, 298, 101814, DOI: 10.1016/j.jbc.2022.101814Google ScholarThere is no corresponding record for this reference.
- 7Polasa, A.; Mosleh, I.; Losey, J.; Abbaspourrad, A.; Beitle, R.; Moradi, M. Developing a rational approach to designing recombinant proteins for peptide-directed nanoparticle synthesis. Nanoscale Adv. 2022, 4, 3161– 3171, DOI: 10.1039/D2NA00212DGoogle ScholarThere is no corresponding record for this reference.
- 8Polasa, A.; Hettige, J.; Immadisetty, K.; Moradi, M. An investigation of the YidC-mediated membrane insertion of Pf3 coat protein using molecular dynamics simulations. Front. Mol. Biosci. 2022, 9, 954262, DOI: 10.3389/fmolb.2022.954262Google ScholarThere is no corresponding record for this reference.
- 9Polasa, A.; Moradi, M. Towards a purely physics-based computational binding affinity estimation. Na. Comput. Sci. 2023, 3, 10– 11, DOI: 10.1038/s43588-023-00396-4Google ScholarThere is no corresponding record for this reference.
- 10Govind Kumar, V.; Polasa, A.; Agrawal, S.; Kumar, T. K. S.; Moradi, M. Binding affinity estimation from restrained umbrella sampling simulations. Nat. Comput. Sci. 2023, 3, 59– 70, DOI: 10.1038/s43588-022-00389-9Google ScholarThere is no corresponding record for this reference.
- 11Isu, U. H.; Polasa, A.; Moradi, M. Differential behavior of conformational dynamics in active and inactive states of cannabinoid receptor 1. J. Phys. Chem. B 2024, 128, 8437– 8447, DOI: 10.1021/acs.jpcb.4c02828Google ScholarThere is no corresponding record for this reference.
- 12Polasa, A.; Badiee, S. A.; Moradi, M. Deciphering the interdomain coupling in a gram-negative bacterial membrane insertase. J. Phys. Chem. B 2024, 128, 9734– 9744, DOI: 10.1021/acs.jpcb.4c02824Google ScholarThere is no corresponding record for this reference.
- 13Agrawal, S.; Kumar, V. G.; Gundampati, R. K.; Moradi, M.; Kumar, T. K. S. Characterization of the structural forces governing the reversibility of the thermal unfolding of the human acidic fibroblast growth factor. Sci. Rep. 2021, 11, 15579 DOI: 10.1038/s41598-021-95050-2Google ScholarThere is no corresponding record for this reference.
- 14Govind Kumar, V.; Agrawal, S.; Kumar, T. K. S.; Moradi, M. Mechanistic Picture for Monomeric Human Fibroblast Growth Factor 1 Stabilization by Heparin Binding. J. Phys. Chem. B 2021, 125, 12690– 12697, DOI: 10.1021/acs.jpcb.1c07772Google ScholarThere is no corresponding record for this reference.
- 15Karplus, M.; Petsko, G. A. Molecular dynamics simulations in biology. Nature 1990, 347, 631– 639, DOI: 10.1038/347631a0Google ScholarThere is no corresponding record for this reference.
- 16Karplus, M.; McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646– 652, DOI: 10.1038/nsb0902-646Google ScholarThere is no corresponding record for this reference.
- 17Hollingsworth, S. A.; Dror, R. O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129– 1143, DOI: 10.1016/j.neuron.2018.08.011Google ScholarThere is no corresponding record for this reference.
- 18Jin, Y.; Johannissen, L. O.; Hay, S. Predicting new protein conformations from molecular dynamics simulation conformational landscapes and machine learning. Proteins 2021, 89, 915– 921, DOI: 10.1002/prot.26068Google ScholarThere is no corresponding record for this reference.
- 19Audagnotto, M.; Czechtizky, W.; De Maria, L.; Käck, H.; Papoian, G.; Tornberg, L.; Tyrchan, C.; Ulander, J. Machine learning/molecular dynamic protein structure prediction approach to investigate the protein conformational ensemble. Sci. Rep. 2022, 12, 10018 DOI: 10.1038/s41598-022-13714-zGoogle ScholarThere is no corresponding record for this reference.
- 20Klyshko, E.; Sung-Ho Kim, J.; McGough, L.; Valeeva, Vi.; Lee, E.; Ranganathan, R.; Rauscher, S. Functional protein dynamics in a crystal. Nat. Commun. 2024, 15, 3244 DOI: 10.1038/s41467-024-47473-4Google ScholarThere is no corresponding record for this reference.
- 21Wang, W. B.; Liang, Y.; Jin, Y. Q.; Zhang, J.; Su, J. G.; Li, Q. M. E484K mutation in SARS-CoV-2 RBD enhances binding affinity with hACE2 but reduces interactions with neutralizing antibodies and nanobodies: Binding free energy calculation studies. J. Mol. Graphics Modell. 2021, 109, 108035, DOI: 10.1016/j.jmgm.2021.108035Google ScholarThere is no corresponding record for this reference.
- 22Chen, J.; Zhang, S.; Wang, W.; Pang, L.; Zhang, Q.; Liu, X. Mutation-Induced Impacts on the Switch Transformations of the GDP- and GTP-Bound K-Ras: Insights from Multiple Replica Gaussian Accelerated Molecular Dynamics and Free Energy Analysis. J. Chem. Inf. Model. 2021, 61, 1954– 1969, DOI: 10.1021/acs.jcim.0c01470Google ScholarThere is no corresponding record for this reference.
- 23Lee, K.-H.; Kuczera, K. Free energy simulations to understand the effect of Met→Ala mutations at positions 205, 206 and 213 on stability of human prion protein. Biophys. Chem. 2021, 275, 106620, DOI: 10.1016/j.bpc.2021.106620Google ScholarThere is no corresponding record for this reference.
- 24Bhadane, R.; Salo-Ahen, O. M. H. High-Throughput Molecular Dynamics-Based Alchemical Free Energy Calculations for Predicting the Binding Free Energy Change Associated with the Selected Omicron Mutations in the Spike Receptor-Binding Domain of SARS-CoV-2. Biomedicines 2022, 10, 2779, DOI: 10.3390/biomedicines10112779Google ScholarThere is no corresponding record for this reference.
- 25Yu, Y.; Wang, Z.; Wang, L.; Tian, S.; Hou, T.; Sun, H. Predicting the mutation effects of protein–ligand interactions via end-point binding free energy calculations: Strategies and analyses. J. Cheminf. 2022, 14, 56 DOI: 10.1186/s13321-022-00639-yGoogle ScholarThere is no corresponding record for this reference.
- 26Zwier, M. C.; Chong, L. T. Reaching biological timescales with all-atom molecular dynamics simulation. Curr. Opin. Pharmacol. 2010, 10, 745– 752, DOI: 10.1016/j.coph.2010.09.008Google ScholarThere is no corresponding record for this reference.
- 27Sahil, M.; Sarkar, S.; Mondal, J. Long-time-step molecular dynamics can retard simulation of protein-ligand recognition process. Biophys. J. 2023, 122, 802– 816, DOI: 10.1016/j.bpj.2023.01.036Google ScholarThere is no corresponding record for this reference.
- 28Mohr, B.; van Heesch, T.; Pérez de Alba Ortíz, A.; Vreede, J. Enhanced sampling strategies for molecular simulation of DNA. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2024, 14, e1712, DOI: 10.1002/wcms.1712Google ScholarThere is no corresponding record for this reference.
- 29Durrant, J. D.; McCammon, J. A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71 DOI: 10.1186/1741-7007-9-71Google ScholarThere is no corresponding record for this reference.
- 30Chodera, J. D.; Mobley, D. L.; Shirts, M. R.; Dixon, R. W.; Branson, Kim.; Pande, V. S. Alchemical free energy methods for drug discovery: Progress and challenges. Curr. Opin. Struct. Biol. 2011, 21, 150– 160, DOI: 10.1016/j.sbi.2011.01.011Google ScholarThere is no corresponding record for this reference.
- 31Odinokov, A.; Yakubovich, A.; Son, W.-J.; Jung, Y.; Choi, H. Exploiting the quantum mechanically derived force field for functional materials simulations. npj Comput. Mater. 2021, 7, 155 DOI: 10.1038/s41524-021-00628-zGoogle ScholarThere is no corresponding record for this reference.
- 32Hu, X.; Amin, K. S.; Schneider, M.; Lim, C.; Salahub, D.; Baldauf, C. System-Specific Parameter Optimization for Nonpolarizable and Polarizable Force Fields. J. Chem. Theory Comput. 2024, 20, 1448– 1464, DOI: 10.1021/acs.jctc.3c01141Google ScholarThere is no corresponding record for this reference.
- 33Grotz, K. K.; Cruz-León, S.; Schwierz, N. Optimized Magnesium Force Field Parameters for Biomolecular Simulations with Accurate Solvation, Ion-Binding, and Water-Exchange Properties. J. Chem. Theory Comput. 2021, 17, 2530– 2540, DOI: 10.1021/acs.jctc.0c01281Google ScholarThere is no corresponding record for this reference.
- 34Lin, F.-Y.; MacKerell, A. D., Jr Force Fields for Small Molecules. In Methods in Molecular Biology; Humana Press, 2019; pp 21– 54 DOI: 10.1007/978-1-4939-9608-7_2 .Google ScholarThere is no corresponding record for this reference.
- 35Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339, DOI: 10.3390/ijms21176339Google ScholarThere is no corresponding record for this reference.
- 36Goolsby, C.; Losey, J.; Fakharzadeh, A.; Xu, Y.; Düker, M.-C.; Getmansky Sherman, M.; Matteson, D. S.; Moradi, M. Addressing the Embeddability Problem in Transition Rate Estimation. J. Phys. Chem. A 2023, 127, 5745– 5759, DOI: 10.1021/acs.jpca.3c01367Google ScholarThere is no corresponding record for this reference.
- 37Ha, T.; Enderle, T.; Ogletree, D. F.; Chemla, D. S.; Selvin, P. R.; Weiss, S. Probing the interaction between two single molecules: Fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6264– 6268, DOI: 10.1073/pnas.93.13.6264Google ScholarThere is no corresponding record for this reference.
- 38Ha, T.; Laurence, T. A.; Chemla, D. S.; Weiss, S. Polarization spectroscopy of single fluorescent molecules. J. Phys. Chem. B 1999, 103, 6839– 6850, DOI: 10.1021/jp990948jGoogle ScholarThere is no corresponding record for this reference.
- 39Weiss, S. Fluorescence spectroscopy of single biomolecules. Science 1999, 283, 1676– 1683, DOI: 10.1126/science.283.5408.1676Google ScholarThere is no corresponding record for this reference.
- 40Weiss, S. Measuring Conformational Dynamics of Biomolecules by Single Molecule Fluorescence Spectroscopy. Nat. Struct. Biol. 2000, 7, 724– 729, DOI: 10.1038/78941Google ScholarThere is no corresponding record for this reference.
- 41Ha, T. Single-Molecule Fluorescence Resonance Energy Transfer. Methods 2001, 25, 78– 86, DOI: 10.1006/meth.2001.1217Google ScholarThere is no corresponding record for this reference.
- 42Hoppe, A.; Christensen, K.; Swanson, J. A. Fluorescence Resonance Energy Transfer-Based Stoichiometry in Living Cells. Biophys. J. 2002, 83, 3652– 3664, DOI: 10.1016/S0006-3495(02)75365-4Google ScholarThere is no corresponding record for this reference.
- 43Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507– 516, DOI: 10.1038/nmeth.1208Google ScholarThere is no corresponding record for this reference.
- 44Felekyan, S.; Sanabria, H.; Kalinin, S.; Kühnemuth, R.; Seidel, C. A. M. Analyzing Förster resonance energy transfer with fluctuation algorithms. In Methods in Enzymology; Elsevier, 2013; Vol. 519, pp 39– 85 DOI: 10.1016/b978-0-12-405539-1.00002-6 .Google ScholarThere is no corresponding record for this reference.
- 45Matsunaga, Y.; Kidera, A.; Sugita, Y. Sequential data assimilation for single-molecule FRET photon-counting data. J. Chem. Phys. 2015, 142, 214115, DOI: 10.1063/1.4921983Google ScholarThere is no corresponding record for this reference.
- 46Margineanu, A.; Chan, J. J.; Kelly, D. J.; Warren, S. C.; Flatters, D.; Kumar, S.; Katan, M.; Dunsby, C. W.; French, P. M. W. Screening for protein-protein interactions using Förster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Sci. Rep. 2016, 6, 28186 DOI: 10.1038/srep28186Google ScholarThere is no corresponding record for this reference.
- 47Wang, J.; Wang, J.; Mu, X. Physical mechanism of concentration-dependent fluorescence resonance energy transfer. Spectrochim. Acta, Part A 2020, 231, 118143, DOI: 10.1016/j.saa.2020.118143Google ScholarThere is no corresponding record for this reference.
- 48Barth, A.; Opanasyuk, O.; Peulen, T.-O.; Felekyan, S.; Kalinin, S.; Sanabria, H.; Seidel, C. A. M. Unraveling multi-state molecular dynamics in single-molecule FRET experiments. I. Theory of FRET-lines. J. Chem. Phys. 2022, 156, 141501, DOI: 10.1063/5.0089134Google ScholarThere is no corresponding record for this reference.
- 49Li, H.; Wang, T.; Han, J.; Xu, Y.; Kang, X.; Li, X.; Zhu, M. Fluorescence resonance energy transfer in atomically precise metal nanoclusters by cocrystallization-induced spatial confinement. Nat. Commun. 2024, 15, 5351 DOI: 10.1038/s41467-024-49735-7Google ScholarThere is no corresponding record for this reference.
- 50Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 1948, 437, 55– 75, DOI: 10.1002/andp.19484370105Google ScholarThere is no corresponding record for this reference.
- 51Brunger, A. T.; Strop, P.; Vrljic, M.; Chu, S.; Weninger, K. R. Three-dimensional molecular modeling with single molecule FRET. J. Struct. Biol. 2011, 173, 497– 505, DOI: 10.1016/j.jsb.2010.09.004Google ScholarThere is no corresponding record for this reference.
- 52Selvin, P. R. Fluorescence resonance energy transfer. In Methods in Enzymology; Academic Press, 1995; Vol. 246, pp 300– 334 DOI: 10.1016/0076-6879(95)46015-2 .Google ScholarThere is no corresponding record for this reference.
- 53Sasmal, D. K.; Pulido, L.; Kasal, S.; Huang, J. Single-Molecule Fluorescence Resonance Energy Transfer in Molecular Biology. Nanoscale 2016, 8, 19928– 19944, DOI: 10.1039/C6NR06794HGoogle ScholarThere is no corresponding record for this reference.
- 54Lakowicz, J. R. Principles of Fluorescence Spectroscopy ; 2006.Google ScholarThere is no corresponding record for this reference.
- 55Khrenova, M.; Topol, I.; Collins, J.; Nemukhin, A. Estimating Orientation Factors in the FRET Theory of Fluorescent Proteins: The TagRFP-KFP Pair and Beyond. Biophys. J. 2015, 108, 126– 132, DOI: 10.1016/j.bpj.2014.11.1859Google ScholarThere is no corresponding record for this reference.
- 56Hoefling, M.; Lima, N.; Haenni, D.; Seidel, C. A. M.; Schuler, B.; Grubmüller, H. Structural Heterogeneity and Quantitative FRET Efficiency Distributions of Polyprolines through a Hybrid Atomistic Simulation and Monte Carlo Approach. PLoS One 2011, 6, e19791 DOI: 10.1371/journal.pone.0019791Google ScholarThere is no corresponding record for this reference.
- 57Förster, T. Modern Quantum Chemistry. Istanbul Lectures. Part III: Action of Light and Organic Crystals; Academic Press: New York and London, 1965; Vol. 3, pp 93– 137.Google ScholarThere is no corresponding record for this reference.
- 58Stryer, L.; Haugland, R. P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. U.S.A. 1967, 58, 719– 726, DOI: 10.1073/pnas.58.2.719Google ScholarThere is no corresponding record for this reference.
- 59Cantor, C. R.; Schimmel, P. R. Biophysical Chemistry; W. H. Freeman, 1980.Google ScholarThere is no corresponding record for this reference.
- 60Clegg, R. M. Fluorescence resonance energy transfer and nucleic acids. In Methods in Enzymology; Elsevier, 1992; Vol. 211, pp 353– 388 DOI: 10.1016/0076-6879(92)11020-j .Google ScholarThere is no corresponding record for this reference.
- 61Haase, T.; Nadine, H.; Robert Kraehling, J.; Behrends, S. Fluorescent Fusion Proteins of Soluble Guanylyl Cyclase Indicate Proximity of the Heme Nitric Oxide Domain and Catalytic Domain. PLoS One 2010, 5, e11617 DOI: 10.1371/journal.pone.0011617Google ScholarThere is no corresponding record for this reference.
- 62Wang, K.; Flaherty, D. P.; Chen, L.; Yang, D. High-Throughput Screening of G-Quadruplex Ligands by FRET Assay. In Methods in Molecular Biology; Springer: New York, 2019; Vol. 2035, pp 323– 331 DOI: 10.1007/978-1-4939-9666-7_19 .Google ScholarThere is no corresponding record for this reference.
- 63Zagotta, W. N.; Sim, B. S.; Nhim, A. K.; Raza, M. M.; Evans, E. G. B.; Vankatesh, Y.; Jones, C. M.; Mehl, R. A.; Petersson, E. J.; Gordon, S. E. An improved fluorescent noncanonical amino acid for measuring conformational distributions using time-resolved transition metal ion FRET. eLife 2021, 10, e70236 DOI: 10.7554/eLife.70236Google ScholarThere is no corresponding record for this reference.
- 64Vimal, T.; Pujar, G. H.; Agraharil, K.; Inamdar, S. R.; Manohar, R. Nanoparticle surface energy transfer (NSET) in ferroelectric liquid crystal-metallic-silver nanoparticle composites: Effect of dopant concentration on NSET parameters. Phys. Rev. E 2021, 103, 022708, DOI: 10.1103/PhysRevE.103.022708Google ScholarThere is no corresponding record for this reference.
- 65Lu, H. P. Probing Single-Molecule Protein Conformational Dynamics. Acc. Chem. Res. 2005, 38, 557– 565, DOI: 10.1021/ar0401451Google ScholarThere is no corresponding record for this reference.
- 66Sobakinskaya, E.; Schmidt Am Busch, M.; Renger, T. Theory of FRET “spectroscopic ruler” for short distances: Application to polyproline. J. Phys. Chem. B 2018, 122, 54– 67, DOI: 10.1021/acs.jpcb.7b09535Google ScholarThere is no corresponding record for this reference.
- 67Kim, S. A.; Heinze, K. G.; Schwille, P. Fluorescence correlation spectroscopy in living cells. Nat. Methods 2007, 4, 963– 973, DOI: 10.1038/nmeth1104Google ScholarThere is no corresponding record for this reference.
- 68Deniz, A. A.; Mukhopadhyay, S.; Lemke, E. A. Single-molecule biophysics: At the interface of biology, physics and chemistry. J. R. Soc., Interface 2008, 5, 15– 45, DOI: 10.1098/rsif.2007.1021Google ScholarThere is no corresponding record for this reference.
- 69Lu, M.; Castillo-Menendez, L. R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D. S.; Chambers, M.; Peng, D.; Zhang, B.; Zhou, T. Associating HIV-1 Envelope Glycoprotein Structures with States on the Virus Observed by smFRET. Nature 2019, 568, 415– 419, DOI: 10.1038/s41586-019-1101-yGoogle ScholarThere is no corresponding record for this reference.
- 70Maslov, I.; Volkov, O.; Khorn, P.; Orekhov, P.; Gusach, A.; Kuzmichev, P.; Gerasimov, A.; Luginina, A.; Coucke, Q.; Andrey, B. Sub-millisecond conformational dynamics of the A2A adenosine receptor revealed by single-molecule FRET. Commun. Biol. 2023, 6, 362 DOI: 10.1038/s42003-023-04727-zGoogle ScholarThere is no corresponding record for this reference.
- 71Ao, Y.; Grover, J. R.; Giffford, L.; Han, Y.; Zhong, G.; Katte, R.; Li, W.; Bhattacharjee, R.; Zhang, B.; Sauve, S. Bioorthogonal click labeling of an amber-free HIV-1 provirus for in-virus single molecule imaging. Cell Chem. Biol. 2024, 31, 487– 501.e7, DOI: 10.1016/j.chembiol.2023.12.017Google ScholarThere is no corresponding record for this reference.
- 72Best, R. B.; Merchant, K. A.; Gopich, I. V.; Schuler, B.; Bax, Ad.; Eaton, W. A. Effect of flexibility and cis residues in single-molecule FRET studies of polyproline. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18964– 18969, DOI: 10.1073/pnas.0709567104Google ScholarThere is no corresponding record for this reference.
- 73Merchant, K. A.; Best, R. B.; Louis, J. M.; Gopich, I. V.; Eaton, W. A. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1528– 1533, DOI: 10.1073/pnas.0607097104Google ScholarThere is no corresponding record for this reference.
- 74Nath, A.; Sammalkorpi, M.; DeWitt, D. C.; Trexler, A. J.; Elbaum-Garfinkle, S.; O’Hern, C. S.; Rhoades, E. The conformational ensembles of α-synuclein and tau: Combining single-molecule FRET and simulations. Biophys. J. 2012, 103, 1940– 1949, DOI: 10.1016/j.bpj.2012.09.032Google ScholarThere is no corresponding record for this reference.
- 75Melo, A. M.; Coraor, J.; Alpha-Cobb, G.; Elbaum-Garfinkle, S.; Nath, A.; Rhoades, E. A functional role for intrinsic disorder in the tau-tubulin complex. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 14336– 14341, DOI: 10.1073/pnas.1610137113Google ScholarThere is no corresponding record for this reference.
- 76Borgia, A.; Borgia, M. B.; Bugge, K.; Kissling, V. M.; Heidarsson, P. O.; Fernandes, C. B.; Sottini, A.; Soranno, A.; Buholzer, K. J.; Nettels, D. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61– 66, DOI: 10.1038/nature25762Google ScholarThere is no corresponding record for this reference.
- 77LeVine, M. V.; Terry, D. S.; Khelashvili, G.; Siegel, Z. S.; Quick, M.; Javitch, J. A.; Blanchard, S. C.; Weinstein, H. The allosteric mechanism of substrate-specific transport in SLC6 is mediated by a volumetric sensor. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 15947– 15956, DOI: 10.1073/pnas.1903020116Google ScholarThere is no corresponding record for this reference.
- 78Meng, F.; Bellaiche, M. M. J.; Kim, J.-Y.; Zerze, G. H.; Best, R. B.; Chung, H. S. Highly disordered amyloid-β monomer probed by single-molecule FRET and MD simulation. Biophys. J. 2018, 114, 870– 884, DOI: 10.1016/j.bpj.2017.12.025Google ScholarThere is no corresponding record for this reference.
- 79Wolf, S.; Sohmen, B.; Hellenkamp, B.; Thurn, J.; Stock, G.; Hugel, T. Hierarchical dynamics in allostery following ATP hydrolysis monitored by single molecule FRET measurements and MD simulations. Chem. Sci. 2021, 12, 3350– 3359, DOI: 10.1039/D0SC06134DGoogle ScholarThere is no corresponding record for this reference.
- 80Spiegel, J. D.; Fulle, S.; Kleinschmidt, M.; Gohlke, H.; Marian, C. M. Failure of the IDA in FRET Systems at Close Inter-Dye Distances Is Moderated by Frequent Low k2 Values. J. Phys. Chem. B 2016, 120 (34), 8845– 8862, DOI: 10.1021/acs.jpcb.6b05754Google ScholarThere is no corresponding record for this reference.
- 81Deplazes, E.; Jayatilaka, D.; Corry, B. Testing the use of molecular dynamics to simulate fluorophore motions and FRET. Phys. Chem. Chem. Phys. 2011, 13, 11045– 11054, DOI: 10.1039/c1cp20447eGoogle ScholarThere is no corresponding record for this reference.
- 82Shoura, M. J.; Ranatunga, R. J. K. U.; Harris, S. A.; Nielsen, S. O.; Levene, S. D. Contribution of Fluorophore Dynamics and Solvation to Resonant Energy Transfer in Protein-DNA Complexes: A Molecular-Dynamics Study. Biophys. J. 2014, 107, 700– 710, DOI: 10.1016/j.bpj.2014.06.023Google ScholarThere is no corresponding record for this reference.
- 83Grotz, K. K.; Nueesch, M. F.; Holmstrom, E. D.; Heinz, M.; Stelzl, L. S.; Schuler, B.; Hummer, G. Dispersion Correction Alleviates Dye Stacking of Single-Stranded DNA and RNA in Simulations of Single-Molecule Fluorescence Experiments. J. Phys. Chem. B 2018, 122, 11626– 11639, DOI: 10.1021/acs.jpcb.8b07537Google ScholarThere is no corresponding record for this reference.
- 84Reinartz, I.; Sinner, C.; Nettels, D.; Stucki-Buchli, B.; Stockmar, F.; Panek, P. T.; Jacob, C. R.; Nienhaus, G. U.; Schuler, B.; Schug, A. Simulation of FRET dyes allows quantitative comparison against experimental data. J. Chem. Phys. 2018, 148, 123321, DOI: 10.1063/1.5010434Google ScholarThere is no corresponding record for this reference.
- 85Best, R. B.; Hofmann, H.; Nettels, D.; Schuler, B. Quantitative interpretation of FRET experiments via molecular simulation: Force field and validation. Biophys. J. 2015, 108, 2721– 2731, DOI: 10.1016/j.bpj.2015.04.038Google ScholarThere is no corresponding record for this reference.
- 86Matsunaga, Y.; Sugita, Y. Linking time-series of single-molecule experiments with molecular dynamics simulations by machine learning. eLife 2018, 7, e32668 DOI: 10.7554/eLife.32668Google ScholarThere is no corresponding record for this reference.
- 87Matsunaga, Y.; Sugita, Y. Refining Markov state models for conformational dynamics using ensemble-averaged data and time-series trajectories. J. Chem. Phys. 2018, 148, 241731, DOI: 10.1063/1.5019750Google ScholarThere is no corresponding record for this reference.
- 88Matsunaga, Y.; Sugita, Y. Use of single-molecule time-series data for refining conformational dynamics in molecular simulations. Curr. Opin. Struct. Biol. 2020, 61, 153– 159, DOI: 10.1016/j.sbi.2019.12.022Google ScholarThere is no corresponding record for this reference.
- 89Reviews in Fluorescence 2005. In Reviews in Fluorescence, 1st ed.; Geddes, C.; Lakowicz, J., Eds.; Springer, 2006.Google ScholarThere is no corresponding record for this reference.
- 90Rothwell, P. J.; Berger, S.; Kensch, O.; Felekyan, S.; Antonik, M.; Wöhrl, B. M.; Restle, T.; Goody, R. S.; Seidel, C. A. M. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1655– 1660, DOI: 10.1073/pnas.0434003100Google ScholarThere is no corresponding record for this reference.
- 91Schröder, G. F.; Alexiev, U.; Grubmüller, H. Simulation of Fluorescence Anisotropy Experiments: Probing Protein Dynamics. Biophys. J. 2005, 89, 3757– 3770, DOI: 10.1529/biophysj.105.069500Google ScholarThere is no corresponding record for this reference.
- 92Henry, E. R.; Hochstrasser, R. M. Molecular dynamics simulations of fluorescence polarization of tryptophans in myoglobin. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 6142– 6146, DOI: 10.1073/pnas.84.17.6142Google ScholarThere is no corresponding record for this reference.
- 93Rindermann, J. J.; Akhtman, Y.; Richardson, J.; Brown, T.; Lagoudakis, P. G. Gauging the Flexibility of Fluorescent Markers for the Interpretation of Fluorescence Resonance Energy Transfer. J. Am. Chem. Soc. 2011, 133, 279– 285, DOI: 10.1021/ja105720jGoogle ScholarThere is no corresponding record for this reference.
- 94VanBeek, D. B.; Zwier, M. C.; Shorb, J. M.; Krueger, B. P. Fretting about FRET: Correlation between κ and R. Biophys. J. 2007, 92, 4168– 4178, DOI: 10.1529/biophysj.106.092650Google ScholarThere is no corresponding record for this reference.
- 95Shoura, M. J.; Vetcher, A. A.; Giovan, S. M.; Bardai, F.; Bharadwaj, A.; Kesinger, M. R.; Levene, S. D. Measurements of DNA-loop formation via Cre-mediated recombination. Nucleic Acids Res. 2012, 40, 7452– 7464, DOI: 10.1093/nar/gks430Google ScholarThere is no corresponding record for this reference.
- 96Piana, S.; Donchev, A. G.; Robustelli, P.; Shaw, D. E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113– 5123, DOI: 10.1021/jp508971mGoogle ScholarThere is no corresponding record for this reference.
- 97Best, R. B.; Zheng, W.; Mittal, J. Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput. 2014, 10, 5113– 5124, DOI: 10.1021/ct500569bGoogle ScholarThere is no corresponding record for this reference.
- 98Abascal, J. L. F.; Vega, C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505, DOI: 10.1063/1.2121687Google ScholarThere is no corresponding record for this reference.
- 99Jorgensen, W. L. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 1981, 103, 335– 340, DOI: 10.1021/ja00392a016Google ScholarThere is no corresponding record for this reference.
- 100Horn, H. W.; Swope, W. C.; Pitera, J. W.; Madura, J. D.; Dick, T. J.; Hura, G. L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665– 9678, DOI: 10.1063/1.1683075Google ScholarThere is no corresponding record for this reference.
- 101Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M.l L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926– 935, DOI: 10.1063/1.445869Google ScholarThere is no corresponding record for this reference.
- 102Nir, E.; Michalet, X.; Hamadani, K. M.; Laurence, T. A.; Neuhauser, D.; Kovchegov, Y.; Weiss, S. Shot-Noise Limited Single-Molecule FRET Histograms: Comparison between Theory and Experiments. J. Phys. Chem. B 2006, 110, 22103– 22124, DOI: 10.1021/jp063483nGoogle ScholarThere is no corresponding record for this reference.
- 103Berger, A.; Kurtz, J.; Katchalski, E. Poly-L-proline. J. Am. Chem. Soc. 1954, 76, 5552– 5554, DOI: 10.1021/ja01650a082Google ScholarThere is no corresponding record for this reference.
- 104Van der Meer, B. W. Kappa-squared: From nuisance to new sense. Rev. Mol. Biotechnol. 2002, 82, 181– 196, DOI: 10.1016/S1389-0352(01)00037-XGoogle ScholarThere is no corresponding record for this reference.
- 105Corry, B.; Jayatilaka, D. Simulation of structure, orientation, and energy transfer between AlexaFluor molecules attached to MscL. Biophys. J. 2008, 95, 2711– 2721, DOI: 10.1529/biophysj.107.126243Google ScholarThere is no corresponding record for this reference.
- 106Loura, L. M. S. Simple estimation of Förster resonance energy transfer (FRET) orientation factor distribution in membranes. Int. J. Mol. Sci. 2012, 13, 15252– 15271, DOI: 10.3390/ijms131115252Google ScholarThere is no corresponding record for this reference.
- 107Wong, C. Y.; Curutchet, C.; Tretiak, S.; Scholes, G. D. Ideal dipole approximation fails to predict electronic coupling and energy transfer between semiconducting single-wall carbon nanotubes. J. Chem. Phys. 2009, 130, 130081104, DOI: 10.1063/1.3088846Google ScholarThere is no corresponding record for this reference.
- 108Li, Y.; Arghittu, S. M.; Dietz, M. S.; Hella, G. J.; Haße, D.; Ferraris, D. M.; Freund, P.; Barth, H.-D.; Lamele, L.; de Jonge, H. Single-molecule imaging and molecular dynamics simulations reveal early activation of the MET receptor in cells. Nat. Commun. 2024, 15, 9486 DOI: 10.1038/s41467-024-53772-7Google ScholarThere is no corresponding record for this reference.
- 109Qin, F. Restoration of Single-Channel Currents Using the Segmental k-Means Method Based on Hidden Markov Modeling. Biophys. J. 2004, 86, 1488– 1501, DOI: 10.1016/S0006-3495(04)74217-4Google ScholarThere is no corresponding record for this reference.
- 110Trexler, A. J.; Rhoades, E. Single Molecule Characterization of α-synuclein in Aggregation-Prone States. Biophys. J. 2010, 99, 3048– 3055, DOI: 10.1016/j.bpj.2010.08.056Google ScholarThere is no corresponding record for this reference.
- 111Trexler, A. J.; Rhoades, E. α-synuclein Binds Large Unilamellar Vesicles as an Extended Helix. Biochemistry 2009, 48, 2304– 2306, DOI: 10.1021/bi900114zGoogle ScholarThere is no corresponding record for this reference.
- 112Brustad, E. M.; Lemke, E. A.; Schultz, P. G.; Deniz, A. A. A General and Efficient Method for the Site-Specific Dual-Labeling of Proteins for Single Molecule Fluorescence Resonance Energy Transfer. J. Am. Chem. Soc. 2008, 130, 17664– 17665, DOI: 10.1021/ja807430hGoogle ScholarThere is no corresponding record for this reference.
- 113Young, T. S.; Ahmad, I.; Yin, J. A.; Schultz, P. G. An Enhanced System for Unnatural Amino Acid Mutagenesis in E. coli. J. Mol. Biol. 2010, 395, 361– 374, DOI: 10.1016/j.jmb.2009.10.030Google ScholarThere is no corresponding record for this reference.
- 114Girodat, D.; Pati, A. K.; Terry, D. S.; Blanchard, S. C.; Sanbonmatsu, K. Y. Quantitative comparison between sub-millisecond time resolution single-molecule FRET measurements and 10-second molecular simulations of a biosensor protein. PLOS Comput. Biol. 2020, 16, e1008293 DOI: 10.1371/journal.pcbi.1008293Google ScholarThere is no corresponding record for this reference.
- 115Best, R. B.; Chen, Y.-G.; Hummer, G. Slow Protein Conformational Dynamics from Multiple Experimental Structures: The Helix/Sheet Transition of Arc Repressor. Structure 2005, 13, 1755– 1763, DOI: 10.1016/j.str.2005.08.009Google ScholarThere is no corresponding record for this reference.
- 116Baucom, D. R.; Furr, M.; Kumar, V. G.; Okoto, P.; Losey, J. L.; Henry, R. L.; Moradi, M.; Kumar, T. K. S.; Heyes, C. D. Transient local secondary structure in the intrinsically disordered C-term of the Albino3 insertase. Biophys. J. 2021, 120, 4992– 5004, DOI: 10.1016/j.bpj.2021.10.013Google ScholarThere is no corresponding record for this reference.
- 117Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603, DOI: 10.1103/PhysRevLett.100.020603Google ScholarThere is no corresponding record for this reference.
- 118Benton, M.; Furr, M.; Kumar, V. G.; Polasa, A.; Gao, F.; Heyes, C. D.; Kumar, T. K. S.; Moradi, M. cpSRP43 is both highly flexible and stable: Structural insights using a combined experimental and computational approach. J. Chem. Inf. Model. 2023, 63, 4125– 4137, DOI: 10.1021/acs.jcim.3c00319Google ScholarThere is no corresponding record for this reference.
- 119Jaru-Ampornpan, P.; Shen, K.; Lam, V. Q.; Ali, M.; Doniach, S.; Jia, T. Z.; Shan, S.-o. ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit. Nat. Struct. Mol. Biol. 2010, 17, 696– 702, DOI: 10.1038/nsmb.1836Google ScholarThere is no corresponding record for this reference.
- 120Gao, F.; Kight, A. D.; Henderson, R.; Jayanthi, S.; Patel, P.; Murchison, M.; Sharma, P.; Goforth, R. L.; Kumar, T. K. S.; Henry, R. L.; Heyes, C. D. Regulation of Structural Dynamics within a Signal Recognition Particle Promotes Binding of Protein Targeting Substrates. J. Biol. Chem. 2015, 290, 15462– 15474, DOI: 10.1074/jbc.M114.624346Google ScholarThere is no corresponding record for this reference.
- 121Durham, R. J.; Latham, D. R.; Sanabria, H.; Jayaraman, V. Structural Dynamics of Glutamate Signaling Systems by smFRET. Biophys. J. 2020, 119, 1929– 1936, DOI: 10.1016/j.bpj.2020.10.009Google ScholarThere is no corresponding record for this reference.
- 122Evensen, G. Data Assimilation: The Ensemble Kalman Filter, 2nd ed.; Springer, 2009.Google ScholarThere is no corresponding record for this reference.
- 123Pande, V. S.; Beauchamp, K.; Bowman, Gr. R. Everything you wanted to know about Markov State Models but were afraid to ask. Methods 2010, 52, 99– 105, DOI: 10.1016/j.ymeth.2010.06.002Google ScholarThere is no corresponding record for this reference.
- 124Bishop, C. M. Pattern Recognition and Machine Learning; Springer, 2006.Google ScholarThere is no corresponding record for this reference.
- 125Schuler, B.; Lipman, E. A.; Steinbach, P. J.; Kumke, M.; Eaton, W. A. Polyproline and the “spectroscopic ruler” revisited with single-molecule fluorescence. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 2754– 2759, DOI: 10.1073/pnas.0408164102Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The Journal of Physical Chemistry B
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
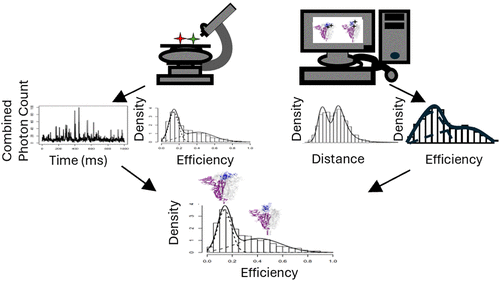
Figure 1

Figure 1. A simplified schematic representation showing the overlap (gray) of the donor fluorophore emission spectra shown in green and the acceptor fluorophore absorption spectra shown in red. The overlap of the spectra must be present for FRET to occur. In practice the actual overlap increases with wavelength.
Figure 2
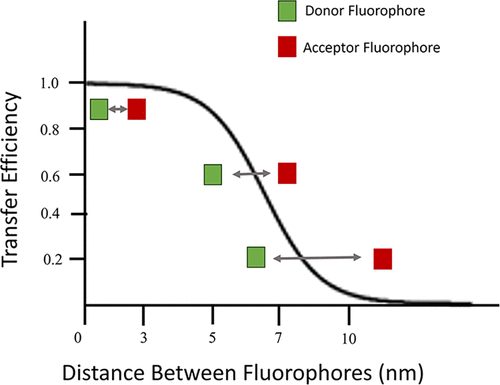
Figure 2. Schematic representation of FRET showing an energy transfer between a donor fluorophore shown in green and an acceptor fluorophore shown in red. The relationship between the distance of the dyes and the efficiency of transfer is shown with the optimal transfer distance being between 1 and 10 nm. The further the dyes move away from each other, the lower the transfer of energy from the donor fluorophore to the acceptor fluorophore.
Figure 3
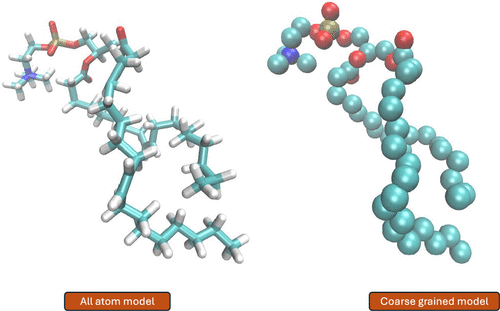
Figure 3. Schematic representation of using coarse-grained systems for MD simulations. In coarse-grained models, groups of atoms are represented as beads, reducing the system’s degrees of freedom. This simplification significantly accelerates simulations, enabling the study of larger systems and longer time scales while reducing computational cost in comparison to to all-atom MD simulations.
Figure 4
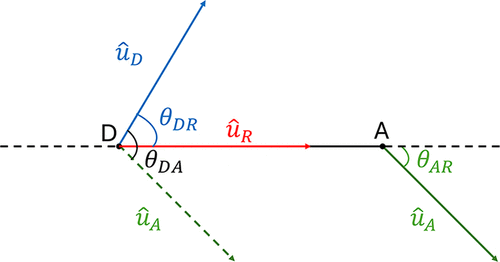
Figure 4. Schematic representation showing how the three angles that contribute to the orientation factor are defined. θDA is the angle between ûD and ûA (black) where ûD and ûA represent the unit vectors associated with the orientations of the donor and acceptor dyes, respectively. θDR is the angle between ûD and ûR (blue). θAR is the angle between ûA and ûR (green).
Figure 5

Figure 5. (A) Schematic representation showing rotational movement of the dyes on a labeled biomolecule where the dyes on a labeled protein (green) are depicted in blue and red. (B) Schematic representation of the probability distribution of dye angles based on free rotation (green) and hindered rotation (magenta).
Figure 6

Figure 6. Schematic overview of three enhanced sampling techniques commonly used in biomolecular simulations. Left: Schematic representation of metadynamics. A bias potential is constructed by periodically adding repulsive Gaussian hills along selected collective variables, allowing the system to escape local minima and explore rare conformational events. Top right: Go̅-like model, which biases the energy landscape by favoring native contacts. Bottom right: Steered MD, in which an external force is applied along a reaction coordinate to induce transition from outward-facing to inward-facing state.
Figure 7

Figure 7. A schematic showing the integrative approach of combining Markov State Modeling (MSM), smFRET data, and machine learning algorithms to refine and validate MD trajectories.
References
This article references 125 other publications.
- 1Sagui, C.; Darden, T. A. MOLECULAR DYNAMICS SIMULATIONS OF BIOMOLECULES: Long-Range Electrostatic Effects. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 155– 179, DOI: 10.1146/annurev.biophys.28.1.155There is no corresponding record for this reference.
- 2Immadisetty, K.; Moradi, M. Mechanistic Picture for Chemomechanical Coupling in a Bacterial Proton-Coupled Oligopeptide Transporter from Streptococcus Thermophilus. J. Phys. Chem. B 2021, 125, 9738– 9750, DOI: 10.1021/acs.jpcb.1c03982There is no corresponding record for this reference.
- 3Singharoy, A.; Chipot, C.; Moradi, M.; Schulten, K. Chemomechanical Coupling in Hexameric Protein-Protein Interfaces Harnesses Energy within V-Type ATPases. J. Am. Chem. Soc. 2017, 139, 293– 310, DOI: 10.1021/jacs.6b10744There is no corresponding record for this reference.
- 4Badiee, S. A.; Kumar, V. G.; Moradi, M. Molecular dynamics investigation of the influenza hemagglutinin conformational changes in acidic pH. J. Phys. Chem. B 2024, 128, 11151– 11163, DOI: 10.1021/acs.jpcb.4c04607There is no corresponding record for this reference.
- 5Zhang, R.; Jagessar, K. L.; Brownd, M.; Polasa, A.; Stein, R. A.; Moradi, M.; Karakas, E.; Mchaourab, H. S. Conformational cycle of a protease-containing ABC transporter in lipid nanodiscs reveals the mechanism of cargo-protein coupling. Nat. Commun. 2024, 15, 9055 DOI: 10.1038/s41467-024-53420-0There is no corresponding record for this reference.
- 6Govind Kumar, V.; Ogden, D. S.; Isu, U. H.; Polasa, A.; Losey, J.; Moradi, M. Prefusion spike protein conformational changes are slower in SARS-CoV-2 than in SARS-CoV-1. J. Biol. Chem. 2022, 298, 101814, DOI: 10.1016/j.jbc.2022.101814There is no corresponding record for this reference.
- 7Polasa, A.; Mosleh, I.; Losey, J.; Abbaspourrad, A.; Beitle, R.; Moradi, M. Developing a rational approach to designing recombinant proteins for peptide-directed nanoparticle synthesis. Nanoscale Adv. 2022, 4, 3161– 3171, DOI: 10.1039/D2NA00212DThere is no corresponding record for this reference.
- 8Polasa, A.; Hettige, J.; Immadisetty, K.; Moradi, M. An investigation of the YidC-mediated membrane insertion of Pf3 coat protein using molecular dynamics simulations. Front. Mol. Biosci. 2022, 9, 954262, DOI: 10.3389/fmolb.2022.954262There is no corresponding record for this reference.
- 9Polasa, A.; Moradi, M. Towards a purely physics-based computational binding affinity estimation. Na. Comput. Sci. 2023, 3, 10– 11, DOI: 10.1038/s43588-023-00396-4There is no corresponding record for this reference.
- 10Govind Kumar, V.; Polasa, A.; Agrawal, S.; Kumar, T. K. S.; Moradi, M. Binding affinity estimation from restrained umbrella sampling simulations. Nat. Comput. Sci. 2023, 3, 59– 70, DOI: 10.1038/s43588-022-00389-9There is no corresponding record for this reference.
- 11Isu, U. H.; Polasa, A.; Moradi, M. Differential behavior of conformational dynamics in active and inactive states of cannabinoid receptor 1. J. Phys. Chem. B 2024, 128, 8437– 8447, DOI: 10.1021/acs.jpcb.4c02828There is no corresponding record for this reference.
- 12Polasa, A.; Badiee, S. A.; Moradi, M. Deciphering the interdomain coupling in a gram-negative bacterial membrane insertase. J. Phys. Chem. B 2024, 128, 9734– 9744, DOI: 10.1021/acs.jpcb.4c02824There is no corresponding record for this reference.
- 13Agrawal, S.; Kumar, V. G.; Gundampati, R. K.; Moradi, M.; Kumar, T. K. S. Characterization of the structural forces governing the reversibility of the thermal unfolding of the human acidic fibroblast growth factor. Sci. Rep. 2021, 11, 15579 DOI: 10.1038/s41598-021-95050-2There is no corresponding record for this reference.
- 14Govind Kumar, V.; Agrawal, S.; Kumar, T. K. S.; Moradi, M. Mechanistic Picture for Monomeric Human Fibroblast Growth Factor 1 Stabilization by Heparin Binding. J. Phys. Chem. B 2021, 125, 12690– 12697, DOI: 10.1021/acs.jpcb.1c07772There is no corresponding record for this reference.
- 15Karplus, M.; Petsko, G. A. Molecular dynamics simulations in biology. Nature 1990, 347, 631– 639, DOI: 10.1038/347631a0There is no corresponding record for this reference.
- 16Karplus, M.; McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646– 652, DOI: 10.1038/nsb0902-646There is no corresponding record for this reference.
- 17Hollingsworth, S. A.; Dror, R. O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129– 1143, DOI: 10.1016/j.neuron.2018.08.011There is no corresponding record for this reference.
- 18Jin, Y.; Johannissen, L. O.; Hay, S. Predicting new protein conformations from molecular dynamics simulation conformational landscapes and machine learning. Proteins 2021, 89, 915– 921, DOI: 10.1002/prot.26068There is no corresponding record for this reference.
- 19Audagnotto, M.; Czechtizky, W.; De Maria, L.; Käck, H.; Papoian, G.; Tornberg, L.; Tyrchan, C.; Ulander, J. Machine learning/molecular dynamic protein structure prediction approach to investigate the protein conformational ensemble. Sci. Rep. 2022, 12, 10018 DOI: 10.1038/s41598-022-13714-zThere is no corresponding record for this reference.
- 20Klyshko, E.; Sung-Ho Kim, J.; McGough, L.; Valeeva, Vi.; Lee, E.; Ranganathan, R.; Rauscher, S. Functional protein dynamics in a crystal. Nat. Commun. 2024, 15, 3244 DOI: 10.1038/s41467-024-47473-4There is no corresponding record for this reference.
- 21Wang, W. B.; Liang, Y.; Jin, Y. Q.; Zhang, J.; Su, J. G.; Li, Q. M. E484K mutation in SARS-CoV-2 RBD enhances binding affinity with hACE2 but reduces interactions with neutralizing antibodies and nanobodies: Binding free energy calculation studies. J. Mol. Graphics Modell. 2021, 109, 108035, DOI: 10.1016/j.jmgm.2021.108035There is no corresponding record for this reference.
- 22Chen, J.; Zhang, S.; Wang, W.; Pang, L.; Zhang, Q.; Liu, X. Mutation-Induced Impacts on the Switch Transformations of the GDP- and GTP-Bound K-Ras: Insights from Multiple Replica Gaussian Accelerated Molecular Dynamics and Free Energy Analysis. J. Chem. Inf. Model. 2021, 61, 1954– 1969, DOI: 10.1021/acs.jcim.0c01470There is no corresponding record for this reference.
- 23Lee, K.-H.; Kuczera, K. Free energy simulations to understand the effect of Met→Ala mutations at positions 205, 206 and 213 on stability of human prion protein. Biophys. Chem. 2021, 275, 106620, DOI: 10.1016/j.bpc.2021.106620There is no corresponding record for this reference.
- 24Bhadane, R.; Salo-Ahen, O. M. H. High-Throughput Molecular Dynamics-Based Alchemical Free Energy Calculations for Predicting the Binding Free Energy Change Associated with the Selected Omicron Mutations in the Spike Receptor-Binding Domain of SARS-CoV-2. Biomedicines 2022, 10, 2779, DOI: 10.3390/biomedicines10112779There is no corresponding record for this reference.
- 25Yu, Y.; Wang, Z.; Wang, L.; Tian, S.; Hou, T.; Sun, H. Predicting the mutation effects of protein–ligand interactions via end-point binding free energy calculations: Strategies and analyses. J. Cheminf. 2022, 14, 56 DOI: 10.1186/s13321-022-00639-yThere is no corresponding record for this reference.
- 26Zwier, M. C.; Chong, L. T. Reaching biological timescales with all-atom molecular dynamics simulation. Curr. Opin. Pharmacol. 2010, 10, 745– 752, DOI: 10.1016/j.coph.2010.09.008There is no corresponding record for this reference.
- 27Sahil, M.; Sarkar, S.; Mondal, J. Long-time-step molecular dynamics can retard simulation of protein-ligand recognition process. Biophys. J. 2023, 122, 802– 816, DOI: 10.1016/j.bpj.2023.01.036There is no corresponding record for this reference.
- 28Mohr, B.; van Heesch, T.; Pérez de Alba Ortíz, A.; Vreede, J. Enhanced sampling strategies for molecular simulation of DNA. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2024, 14, e1712, DOI: 10.1002/wcms.1712There is no corresponding record for this reference.
- 29Durrant, J. D.; McCammon, J. A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71 DOI: 10.1186/1741-7007-9-71There is no corresponding record for this reference.
- 30Chodera, J. D.; Mobley, D. L.; Shirts, M. R.; Dixon, R. W.; Branson, Kim.; Pande, V. S. Alchemical free energy methods for drug discovery: Progress and challenges. Curr. Opin. Struct. Biol. 2011, 21, 150– 160, DOI: 10.1016/j.sbi.2011.01.011There is no corresponding record for this reference.
- 31Odinokov, A.; Yakubovich, A.; Son, W.-J.; Jung, Y.; Choi, H. Exploiting the quantum mechanically derived force field for functional materials simulations. npj Comput. Mater. 2021, 7, 155 DOI: 10.1038/s41524-021-00628-zThere is no corresponding record for this reference.
- 32Hu, X.; Amin, K. S.; Schneider, M.; Lim, C.; Salahub, D.; Baldauf, C. System-Specific Parameter Optimization for Nonpolarizable and Polarizable Force Fields. J. Chem. Theory Comput. 2024, 20, 1448– 1464, DOI: 10.1021/acs.jctc.3c01141There is no corresponding record for this reference.
- 33Grotz, K. K.; Cruz-León, S.; Schwierz, N. Optimized Magnesium Force Field Parameters for Biomolecular Simulations with Accurate Solvation, Ion-Binding, and Water-Exchange Properties. J. Chem. Theory Comput. 2021, 17, 2530– 2540, DOI: 10.1021/acs.jctc.0c01281There is no corresponding record for this reference.
- 34Lin, F.-Y.; MacKerell, A. D., Jr Force Fields for Small Molecules. In Methods in Molecular Biology; Humana Press, 2019; pp 21– 54 DOI: 10.1007/978-1-4939-9608-7_2 .There is no corresponding record for this reference.
- 35Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339, DOI: 10.3390/ijms21176339There is no corresponding record for this reference.
- 36Goolsby, C.; Losey, J.; Fakharzadeh, A.; Xu, Y.; Düker, M.-C.; Getmansky Sherman, M.; Matteson, D. S.; Moradi, M. Addressing the Embeddability Problem in Transition Rate Estimation. J. Phys. Chem. A 2023, 127, 5745– 5759, DOI: 10.1021/acs.jpca.3c01367There is no corresponding record for this reference.
- 37Ha, T.; Enderle, T.; Ogletree, D. F.; Chemla, D. S.; Selvin, P. R.; Weiss, S. Probing the interaction between two single molecules: Fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6264– 6268, DOI: 10.1073/pnas.93.13.6264There is no corresponding record for this reference.
- 38Ha, T.; Laurence, T. A.; Chemla, D. S.; Weiss, S. Polarization spectroscopy of single fluorescent molecules. J. Phys. Chem. B 1999, 103, 6839– 6850, DOI: 10.1021/jp990948jThere is no corresponding record for this reference.
- 39Weiss, S. Fluorescence spectroscopy of single biomolecules. Science 1999, 283, 1676– 1683, DOI: 10.1126/science.283.5408.1676There is no corresponding record for this reference.
- 40Weiss, S. Measuring Conformational Dynamics of Biomolecules by Single Molecule Fluorescence Spectroscopy. Nat. Struct. Biol. 2000, 7, 724– 729, DOI: 10.1038/78941There is no corresponding record for this reference.
- 41Ha, T. Single-Molecule Fluorescence Resonance Energy Transfer. Methods 2001, 25, 78– 86, DOI: 10.1006/meth.2001.1217There is no corresponding record for this reference.
- 42Hoppe, A.; Christensen, K.; Swanson, J. A. Fluorescence Resonance Energy Transfer-Based Stoichiometry in Living Cells. Biophys. J. 2002, 83, 3652– 3664, DOI: 10.1016/S0006-3495(02)75365-4There is no corresponding record for this reference.
- 43Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507– 516, DOI: 10.1038/nmeth.1208There is no corresponding record for this reference.
- 44Felekyan, S.; Sanabria, H.; Kalinin, S.; Kühnemuth, R.; Seidel, C. A. M. Analyzing Förster resonance energy transfer with fluctuation algorithms. In Methods in Enzymology; Elsevier, 2013; Vol. 519, pp 39– 85 DOI: 10.1016/b978-0-12-405539-1.00002-6 .There is no corresponding record for this reference.
- 45Matsunaga, Y.; Kidera, A.; Sugita, Y. Sequential data assimilation for single-molecule FRET photon-counting data. J. Chem. Phys. 2015, 142, 214115, DOI: 10.1063/1.4921983There is no corresponding record for this reference.
- 46Margineanu, A.; Chan, J. J.; Kelly, D. J.; Warren, S. C.; Flatters, D.; Kumar, S.; Katan, M.; Dunsby, C. W.; French, P. M. W. Screening for protein-protein interactions using Förster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Sci. Rep. 2016, 6, 28186 DOI: 10.1038/srep28186There is no corresponding record for this reference.
- 47Wang, J.; Wang, J.; Mu, X. Physical mechanism of concentration-dependent fluorescence resonance energy transfer. Spectrochim. Acta, Part A 2020, 231, 118143, DOI: 10.1016/j.saa.2020.118143There is no corresponding record for this reference.
- 48Barth, A.; Opanasyuk, O.; Peulen, T.-O.; Felekyan, S.; Kalinin, S.; Sanabria, H.; Seidel, C. A. M. Unraveling multi-state molecular dynamics in single-molecule FRET experiments. I. Theory of FRET-lines. J. Chem. Phys. 2022, 156, 141501, DOI: 10.1063/5.0089134There is no corresponding record for this reference.
- 49Li, H.; Wang, T.; Han, J.; Xu, Y.; Kang, X.; Li, X.; Zhu, M. Fluorescence resonance energy transfer in atomically precise metal nanoclusters by cocrystallization-induced spatial confinement. Nat. Commun. 2024, 15, 5351 DOI: 10.1038/s41467-024-49735-7There is no corresponding record for this reference.
- 50Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 1948, 437, 55– 75, DOI: 10.1002/andp.19484370105There is no corresponding record for this reference.
- 51Brunger, A. T.; Strop, P.; Vrljic, M.; Chu, S.; Weninger, K. R. Three-dimensional molecular modeling with single molecule FRET. J. Struct. Biol. 2011, 173, 497– 505, DOI: 10.1016/j.jsb.2010.09.004There is no corresponding record for this reference.
- 52Selvin, P. R. Fluorescence resonance energy transfer. In Methods in Enzymology; Academic Press, 1995; Vol. 246, pp 300– 334 DOI: 10.1016/0076-6879(95)46015-2 .There is no corresponding record for this reference.
- 53Sasmal, D. K.; Pulido, L.; Kasal, S.; Huang, J. Single-Molecule Fluorescence Resonance Energy Transfer in Molecular Biology. Nanoscale 2016, 8, 19928– 19944, DOI: 10.1039/C6NR06794HThere is no corresponding record for this reference.
- 54Lakowicz, J. R. Principles of Fluorescence Spectroscopy ; 2006.There is no corresponding record for this reference.
- 55Khrenova, M.; Topol, I.; Collins, J.; Nemukhin, A. Estimating Orientation Factors in the FRET Theory of Fluorescent Proteins: The TagRFP-KFP Pair and Beyond. Biophys. J. 2015, 108, 126– 132, DOI: 10.1016/j.bpj.2014.11.1859There is no corresponding record for this reference.
- 56Hoefling, M.; Lima, N.; Haenni, D.; Seidel, C. A. M.; Schuler, B.; Grubmüller, H. Structural Heterogeneity and Quantitative FRET Efficiency Distributions of Polyprolines through a Hybrid Atomistic Simulation and Monte Carlo Approach. PLoS One 2011, 6, e19791 DOI: 10.1371/journal.pone.0019791There is no corresponding record for this reference.
- 57Förster, T. Modern Quantum Chemistry. Istanbul Lectures. Part III: Action of Light and Organic Crystals; Academic Press: New York and London, 1965; Vol. 3, pp 93– 137.There is no corresponding record for this reference.
- 58Stryer, L.; Haugland, R. P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. U.S.A. 1967, 58, 719– 726, DOI: 10.1073/pnas.58.2.719There is no corresponding record for this reference.
- 59Cantor, C. R.; Schimmel, P. R. Biophysical Chemistry; W. H. Freeman, 1980.There is no corresponding record for this reference.
- 60Clegg, R. M. Fluorescence resonance energy transfer and nucleic acids. In Methods in Enzymology; Elsevier, 1992; Vol. 211, pp 353– 388 DOI: 10.1016/0076-6879(92)11020-j .There is no corresponding record for this reference.
- 61Haase, T.; Nadine, H.; Robert Kraehling, J.; Behrends, S. Fluorescent Fusion Proteins of Soluble Guanylyl Cyclase Indicate Proximity of the Heme Nitric Oxide Domain and Catalytic Domain. PLoS One 2010, 5, e11617 DOI: 10.1371/journal.pone.0011617There is no corresponding record for this reference.
- 62Wang, K.; Flaherty, D. P.; Chen, L.; Yang, D. High-Throughput Screening of G-Quadruplex Ligands by FRET Assay. In Methods in Molecular Biology; Springer: New York, 2019; Vol. 2035, pp 323– 331 DOI: 10.1007/978-1-4939-9666-7_19 .There is no corresponding record for this reference.
- 63Zagotta, W. N.; Sim, B. S.; Nhim, A. K.; Raza, M. M.; Evans, E. G. B.; Vankatesh, Y.; Jones, C. M.; Mehl, R. A.; Petersson, E. J.; Gordon, S. E. An improved fluorescent noncanonical amino acid for measuring conformational distributions using time-resolved transition metal ion FRET. eLife 2021, 10, e70236 DOI: 10.7554/eLife.70236There is no corresponding record for this reference.
- 64Vimal, T.; Pujar, G. H.; Agraharil, K.; Inamdar, S. R.; Manohar, R. Nanoparticle surface energy transfer (NSET) in ferroelectric liquid crystal-metallic-silver nanoparticle composites: Effect of dopant concentration on NSET parameters. Phys. Rev. E 2021, 103, 022708, DOI: 10.1103/PhysRevE.103.022708There is no corresponding record for this reference.
- 65Lu, H. P. Probing Single-Molecule Protein Conformational Dynamics. Acc. Chem. Res. 2005, 38, 557– 565, DOI: 10.1021/ar0401451There is no corresponding record for this reference.
- 66Sobakinskaya, E.; Schmidt Am Busch, M.; Renger, T. Theory of FRET “spectroscopic ruler” for short distances: Application to polyproline. J. Phys. Chem. B 2018, 122, 54– 67, DOI: 10.1021/acs.jpcb.7b09535There is no corresponding record for this reference.
- 67Kim, S. A.; Heinze, K. G.; Schwille, P. Fluorescence correlation spectroscopy in living cells. Nat. Methods 2007, 4, 963– 973, DOI: 10.1038/nmeth1104There is no corresponding record for this reference.
- 68Deniz, A. A.; Mukhopadhyay, S.; Lemke, E. A. Single-molecule biophysics: At the interface of biology, physics and chemistry. J. R. Soc., Interface 2008, 5, 15– 45, DOI: 10.1098/rsif.2007.1021There is no corresponding record for this reference.
- 69Lu, M.; Castillo-Menendez, L. R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D. S.; Chambers, M.; Peng, D.; Zhang, B.; Zhou, T. Associating HIV-1 Envelope Glycoprotein Structures with States on the Virus Observed by smFRET. Nature 2019, 568, 415– 419, DOI: 10.1038/s41586-019-1101-yThere is no corresponding record for this reference.
- 70Maslov, I.; Volkov, O.; Khorn, P.; Orekhov, P.; Gusach, A.; Kuzmichev, P.; Gerasimov, A.; Luginina, A.; Coucke, Q.; Andrey, B. Sub-millisecond conformational dynamics of the A2A adenosine receptor revealed by single-molecule FRET. Commun. Biol. 2023, 6, 362 DOI: 10.1038/s42003-023-04727-zThere is no corresponding record for this reference.
- 71Ao, Y.; Grover, J. R.; Giffford, L.; Han, Y.; Zhong, G.; Katte, R.; Li, W.; Bhattacharjee, R.; Zhang, B.; Sauve, S. Bioorthogonal click labeling of an amber-free HIV-1 provirus for in-virus single molecule imaging. Cell Chem. Biol. 2024, 31, 487– 501.e7, DOI: 10.1016/j.chembiol.2023.12.017There is no corresponding record for this reference.
- 72Best, R. B.; Merchant, K. A.; Gopich, I. V.; Schuler, B.; Bax, Ad.; Eaton, W. A. Effect of flexibility and cis residues in single-molecule FRET studies of polyproline. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18964– 18969, DOI: 10.1073/pnas.0709567104There is no corresponding record for this reference.
- 73Merchant, K. A.; Best, R. B.; Louis, J. M.; Gopich, I. V.; Eaton, W. A. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1528– 1533, DOI: 10.1073/pnas.0607097104There is no corresponding record for this reference.
- 74Nath, A.; Sammalkorpi, M.; DeWitt, D. C.; Trexler, A. J.; Elbaum-Garfinkle, S.; O’Hern, C. S.; Rhoades, E. The conformational ensembles of α-synuclein and tau: Combining single-molecule FRET and simulations. Biophys. J. 2012, 103, 1940– 1949, DOI: 10.1016/j.bpj.2012.09.032There is no corresponding record for this reference.
- 75Melo, A. M.; Coraor, J.; Alpha-Cobb, G.; Elbaum-Garfinkle, S.; Nath, A.; Rhoades, E. A functional role for intrinsic disorder in the tau-tubulin complex. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 14336– 14341, DOI: 10.1073/pnas.1610137113There is no corresponding record for this reference.
- 76Borgia, A.; Borgia, M. B.; Bugge, K.; Kissling, V. M.; Heidarsson, P. O.; Fernandes, C. B.; Sottini, A.; Soranno, A.; Buholzer, K. J.; Nettels, D. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61– 66, DOI: 10.1038/nature25762There is no corresponding record for this reference.
- 77LeVine, M. V.; Terry, D. S.; Khelashvili, G.; Siegel, Z. S.; Quick, M.; Javitch, J. A.; Blanchard, S. C.; Weinstein, H. The allosteric mechanism of substrate-specific transport in SLC6 is mediated by a volumetric sensor. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 15947– 15956, DOI: 10.1073/pnas.1903020116There is no corresponding record for this reference.
- 78Meng, F.; Bellaiche, M. M. J.; Kim, J.-Y.; Zerze, G. H.; Best, R. B.; Chung, H. S. Highly disordered amyloid-β monomer probed by single-molecule FRET and MD simulation. Biophys. J. 2018, 114, 870– 884, DOI: 10.1016/j.bpj.2017.12.025There is no corresponding record for this reference.
- 79Wolf, S.; Sohmen, B.; Hellenkamp, B.; Thurn, J.; Stock, G.; Hugel, T. Hierarchical dynamics in allostery following ATP hydrolysis monitored by single molecule FRET measurements and MD simulations. Chem. Sci. 2021, 12, 3350– 3359, DOI: 10.1039/D0SC06134DThere is no corresponding record for this reference.
- 80Spiegel, J. D.; Fulle, S.; Kleinschmidt, M.; Gohlke, H.; Marian, C. M. Failure of the IDA in FRET Systems at Close Inter-Dye Distances Is Moderated by Frequent Low k2 Values. J. Phys. Chem. B 2016, 120 (34), 8845– 8862, DOI: 10.1021/acs.jpcb.6b05754There is no corresponding record for this reference.
- 81Deplazes, E.; Jayatilaka, D.; Corry, B. Testing the use of molecular dynamics to simulate fluorophore motions and FRET. Phys. Chem. Chem. Phys. 2011, 13, 11045– 11054, DOI: 10.1039/c1cp20447eThere is no corresponding record for this reference.
- 82Shoura, M. J.; Ranatunga, R. J. K. U.; Harris, S. A.; Nielsen, S. O.; Levene, S. D. Contribution of Fluorophore Dynamics and Solvation to Resonant Energy Transfer in Protein-DNA Complexes: A Molecular-Dynamics Study. Biophys. J. 2014, 107, 700– 710, DOI: 10.1016/j.bpj.2014.06.023There is no corresponding record for this reference.
- 83Grotz, K. K.; Nueesch, M. F.; Holmstrom, E. D.; Heinz, M.; Stelzl, L. S.; Schuler, B.; Hummer, G. Dispersion Correction Alleviates Dye Stacking of Single-Stranded DNA and RNA in Simulations of Single-Molecule Fluorescence Experiments. J. Phys. Chem. B 2018, 122, 11626– 11639, DOI: 10.1021/acs.jpcb.8b07537There is no corresponding record for this reference.
- 84Reinartz, I.; Sinner, C.; Nettels, D.; Stucki-Buchli, B.; Stockmar, F.; Panek, P. T.; Jacob, C. R.; Nienhaus, G. U.; Schuler, B.; Schug, A. Simulation of FRET dyes allows quantitative comparison against experimental data. J. Chem. Phys. 2018, 148, 123321, DOI: 10.1063/1.5010434There is no corresponding record for this reference.
- 85Best, R. B.; Hofmann, H.; Nettels, D.; Schuler, B. Quantitative interpretation of FRET experiments via molecular simulation: Force field and validation. Biophys. J. 2015, 108, 2721– 2731, DOI: 10.1016/j.bpj.2015.04.038There is no corresponding record for this reference.
- 86Matsunaga, Y.; Sugita, Y. Linking time-series of single-molecule experiments with molecular dynamics simulations by machine learning. eLife 2018, 7, e32668 DOI: 10.7554/eLife.32668There is no corresponding record for this reference.
- 87Matsunaga, Y.; Sugita, Y. Refining Markov state models for conformational dynamics using ensemble-averaged data and time-series trajectories. J. Chem. Phys. 2018, 148, 241731, DOI: 10.1063/1.5019750There is no corresponding record for this reference.
- 88Matsunaga, Y.; Sugita, Y. Use of single-molecule time-series data for refining conformational dynamics in molecular simulations. Curr. Opin. Struct. Biol. 2020, 61, 153– 159, DOI: 10.1016/j.sbi.2019.12.022There is no corresponding record for this reference.
- 89Reviews in Fluorescence 2005. In Reviews in Fluorescence, 1st ed.; Geddes, C.; Lakowicz, J., Eds.; Springer, 2006.There is no corresponding record for this reference.
- 90Rothwell, P. J.; Berger, S.; Kensch, O.; Felekyan, S.; Antonik, M.; Wöhrl, B. M.; Restle, T.; Goody, R. S.; Seidel, C. A. M. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1655– 1660, DOI: 10.1073/pnas.0434003100There is no corresponding record for this reference.
- 91Schröder, G. F.; Alexiev, U.; Grubmüller, H. Simulation of Fluorescence Anisotropy Experiments: Probing Protein Dynamics. Biophys. J. 2005, 89, 3757– 3770, DOI: 10.1529/biophysj.105.069500There is no corresponding record for this reference.
- 92Henry, E. R.; Hochstrasser, R. M. Molecular dynamics simulations of fluorescence polarization of tryptophans in myoglobin. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 6142– 6146, DOI: 10.1073/pnas.84.17.6142There is no corresponding record for this reference.
- 93Rindermann, J. J.; Akhtman, Y.; Richardson, J.; Brown, T.; Lagoudakis, P. G. Gauging the Flexibility of Fluorescent Markers for the Interpretation of Fluorescence Resonance Energy Transfer. J. Am. Chem. Soc. 2011, 133, 279– 285, DOI: 10.1021/ja105720jThere is no corresponding record for this reference.
- 94VanBeek, D. B.; Zwier, M. C.; Shorb, J. M.; Krueger, B. P. Fretting about FRET: Correlation between κ and R. Biophys. J. 2007, 92, 4168– 4178, DOI: 10.1529/biophysj.106.092650There is no corresponding record for this reference.
- 95Shoura, M. J.; Vetcher, A. A.; Giovan, S. M.; Bardai, F.; Bharadwaj, A.; Kesinger, M. R.; Levene, S. D. Measurements of DNA-loop formation via Cre-mediated recombination. Nucleic Acids Res. 2012, 40, 7452– 7464, DOI: 10.1093/nar/gks430There is no corresponding record for this reference.
- 96Piana, S.; Donchev, A. G.; Robustelli, P.; Shaw, D. E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113– 5123, DOI: 10.1021/jp508971mThere is no corresponding record for this reference.
- 97Best, R. B.; Zheng, W.; Mittal, J. Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput. 2014, 10, 5113– 5124, DOI: 10.1021/ct500569bThere is no corresponding record for this reference.
- 98Abascal, J. L. F.; Vega, C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505, DOI: 10.1063/1.2121687There is no corresponding record for this reference.
- 99Jorgensen, W. L. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 1981, 103, 335– 340, DOI: 10.1021/ja00392a016There is no corresponding record for this reference.
- 100Horn, H. W.; Swope, W. C.; Pitera, J. W.; Madura, J. D.; Dick, T. J.; Hura, G. L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665– 9678, DOI: 10.1063/1.1683075There is no corresponding record for this reference.
- 101Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M.l L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926– 935, DOI: 10.1063/1.445869There is no corresponding record for this reference.
- 102Nir, E.; Michalet, X.; Hamadani, K. M.; Laurence, T. A.; Neuhauser, D.; Kovchegov, Y.; Weiss, S. Shot-Noise Limited Single-Molecule FRET Histograms: Comparison between Theory and Experiments. J. Phys. Chem. B 2006, 110, 22103– 22124, DOI: 10.1021/jp063483nThere is no corresponding record for this reference.
- 103Berger, A.; Kurtz, J.; Katchalski, E. Poly-L-proline. J. Am. Chem. Soc. 1954, 76, 5552– 5554, DOI: 10.1021/ja01650a082There is no corresponding record for this reference.
- 104Van der Meer, B. W. Kappa-squared: From nuisance to new sense. Rev. Mol. Biotechnol. 2002, 82, 181– 196, DOI: 10.1016/S1389-0352(01)00037-XThere is no corresponding record for this reference.
- 105Corry, B.; Jayatilaka, D. Simulation of structure, orientation, and energy transfer between AlexaFluor molecules attached to MscL. Biophys. J. 2008, 95, 2711– 2721, DOI: 10.1529/biophysj.107.126243There is no corresponding record for this reference.
- 106Loura, L. M. S. Simple estimation of Förster resonance energy transfer (FRET) orientation factor distribution in membranes. Int. J. Mol. Sci. 2012, 13, 15252– 15271, DOI: 10.3390/ijms131115252There is no corresponding record for this reference.
- 107Wong, C. Y.; Curutchet, C.; Tretiak, S.; Scholes, G. D. Ideal dipole approximation fails to predict electronic coupling and energy transfer between semiconducting single-wall carbon nanotubes. J. Chem. Phys. 2009, 130, 130081104, DOI: 10.1063/1.3088846There is no corresponding record for this reference.
- 108Li, Y.; Arghittu, S. M.; Dietz, M. S.; Hella, G. J.; Haße, D.; Ferraris, D. M.; Freund, P.; Barth, H.-D.; Lamele, L.; de Jonge, H. Single-molecule imaging and molecular dynamics simulations reveal early activation of the MET receptor in cells. Nat. Commun. 2024, 15, 9486 DOI: 10.1038/s41467-024-53772-7There is no corresponding record for this reference.
- 109Qin, F. Restoration of Single-Channel Currents Using the Segmental k-Means Method Based on Hidden Markov Modeling. Biophys. J. 2004, 86, 1488– 1501, DOI: 10.1016/S0006-3495(04)74217-4There is no corresponding record for this reference.
- 110Trexler, A. J.; Rhoades, E. Single Molecule Characterization of α-synuclein in Aggregation-Prone States. Biophys. J. 2010, 99, 3048– 3055, DOI: 10.1016/j.bpj.2010.08.056There is no corresponding record for this reference.
- 111Trexler, A. J.; Rhoades, E. α-synuclein Binds Large Unilamellar Vesicles as an Extended Helix. Biochemistry 2009, 48, 2304– 2306, DOI: 10.1021/bi900114zThere is no corresponding record for this reference.
- 112Brustad, E. M.; Lemke, E. A.; Schultz, P. G.; Deniz, A. A. A General and Efficient Method for the Site-Specific Dual-Labeling of Proteins for Single Molecule Fluorescence Resonance Energy Transfer. J. Am. Chem. Soc. 2008, 130, 17664– 17665, DOI: 10.1021/ja807430hThere is no corresponding record for this reference.
- 113Young, T. S.; Ahmad, I.; Yin, J. A.; Schultz, P. G. An Enhanced System for Unnatural Amino Acid Mutagenesis in E. coli. J. Mol. Biol. 2010, 395, 361– 374, DOI: 10.1016/j.jmb.2009.10.030There is no corresponding record for this reference.
- 114Girodat, D.; Pati, A. K.; Terry, D. S.; Blanchard, S. C.; Sanbonmatsu, K. Y. Quantitative comparison between sub-millisecond time resolution single-molecule FRET measurements and 10-second molecular simulations of a biosensor protein. PLOS Comput. Biol. 2020, 16, e1008293 DOI: 10.1371/journal.pcbi.1008293There is no corresponding record for this reference.
- 115Best, R. B.; Chen, Y.-G.; Hummer, G. Slow Protein Conformational Dynamics from Multiple Experimental Structures: The Helix/Sheet Transition of Arc Repressor. Structure 2005, 13, 1755– 1763, DOI: 10.1016/j.str.2005.08.009There is no corresponding record for this reference.
- 116Baucom, D. R.; Furr, M.; Kumar, V. G.; Okoto, P.; Losey, J. L.; Henry, R. L.; Moradi, M.; Kumar, T. K. S.; Heyes, C. D. Transient local secondary structure in the intrinsically disordered C-term of the Albino3 insertase. Biophys. J. 2021, 120, 4992– 5004, DOI: 10.1016/j.bpj.2021.10.013There is no corresponding record for this reference.
- 117Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603, DOI: 10.1103/PhysRevLett.100.020603There is no corresponding record for this reference.
- 118Benton, M.; Furr, M.; Kumar, V. G.; Polasa, A.; Gao, F.; Heyes, C. D.; Kumar, T. K. S.; Moradi, M. cpSRP43 is both highly flexible and stable: Structural insights using a combined experimental and computational approach. J. Chem. Inf. Model. 2023, 63, 4125– 4137, DOI: 10.1021/acs.jcim.3c00319There is no corresponding record for this reference.
- 119Jaru-Ampornpan, P.; Shen, K.; Lam, V. Q.; Ali, M.; Doniach, S.; Jia, T. Z.; Shan, S.-o. ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit. Nat. Struct. Mol. Biol. 2010, 17, 696– 702, DOI: 10.1038/nsmb.1836There is no corresponding record for this reference.
- 120Gao, F.; Kight, A. D.; Henderson, R.; Jayanthi, S.; Patel, P.; Murchison, M.; Sharma, P.; Goforth, R. L.; Kumar, T. K. S.; Henry, R. L.; Heyes, C. D. Regulation of Structural Dynamics within a Signal Recognition Particle Promotes Binding of Protein Targeting Substrates. J. Biol. Chem. 2015, 290, 15462– 15474, DOI: 10.1074/jbc.M114.624346There is no corresponding record for this reference.
- 121Durham, R. J.; Latham, D. R.; Sanabria, H.; Jayaraman, V. Structural Dynamics of Glutamate Signaling Systems by smFRET. Biophys. J. 2020, 119, 1929– 1936, DOI: 10.1016/j.bpj.2020.10.009There is no corresponding record for this reference.
- 122Evensen, G. Data Assimilation: The Ensemble Kalman Filter, 2nd ed.; Springer, 2009.There is no corresponding record for this reference.
- 123Pande, V. S.; Beauchamp, K.; Bowman, Gr. R. Everything you wanted to know about Markov State Models but were afraid to ask. Methods 2010, 52, 99– 105, DOI: 10.1016/j.ymeth.2010.06.002There is no corresponding record for this reference.
- 124Bishop, C. M. Pattern Recognition and Machine Learning; Springer, 2006.There is no corresponding record for this reference.
- 125Schuler, B.; Lipman, E. A.; Steinbach, P. J.; Kumke, M.; Eaton, W. A. Polyproline and the “spectroscopic ruler” revisited with single-molecule fluorescence. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 2754– 2759, DOI: 10.1073/pnas.0408164102There is no corresponding record for this reference.