

About the Cover:
Upconverting nanoparticles (UCNPs) are lanthanide-based phosphors that efficiently convert low-energy light to higher energies, enabling breakthroughs in deep-tissue imaging, nanoscale sensing, single-molecule imaging, optogenetics, vision technologies, optical computing, and more. UCNPs have also proven to be fertile ground for nanocrystal design, automated synthesis, high-throughput characterization, and machine learning approaches.
View the article.Articles

NIR Excitation in Atomically Precise Nanoclusters via Two-Photon and Three-Photon Absorption
Agata Hajda - ,
Patryk Obstarczyk - , and
Joanna Olesiak-Bańska *
Gold nanoclusters exhibit unique optical properties, with absorption similar to that of molecular systems, ranging from the UV to near-infrared (NIR) region with tunable photoluminescence. Nanoclusters also present outstanding multiphoton properties, with the possibility of excitation in the NIR-I and NIR-II ranges via two- or three-photon absorption. Multiphoton microscopy, which relies on multiphoton excitation, offers additional advantages such as the selective excitation of molecules only at the focal point, enabling true 3D optical sectioning and reducing photobleaching and phototoxicity compared with one-photon microscopy. Our review organizes the current knowledge on the multiphoton absorption of noble metal nanoclusters, highlighting key recent advances in the field and addressing current challenges, with particular emphasis on the potential of these nanostructures in biological NIR imaging applications. The Account starts with an introduction to the basis of multiphoton absorption and multiphoton microscopy advantages, followed by examples of applications of nanoclusters as two- and three-photon absorbers and NIR emitters. Thus, current trends and further perspectives clearly show that the application of multiphoton excitation and noble metal nanoclusters provides a highly beneficial approach for efficient NIR bioimaging.
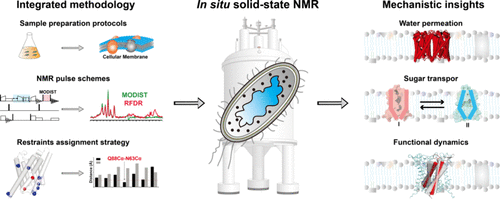
Structure and Dynamics of Membrane Proteins in Native Cellular Membranes Revealed by In Situ Solid-State NMR
Huayong Xie - ,
Weijing Zhao - , and
Jun Yang *
Membrane proteins perform essential functions in the complex and heterogeneous environment of the cellular membrane, where their structure and activity are profoundly influenced by the native lipid milieu. Most membrane protein structures in the Protein Data Bank (PDB) have been determined in vitro in membrane-mimetic environments such as detergents. These membrane-mimetic environments can perturb native protein conformations and may result in a loss of function, which in turn leads to misinterpretations of molecular mechanisms. Direct structural investigations of membrane proteins within native cellular membranes are crucial to understanding the undisturbed molecular mechanism. Solid-state nuclear magnetic resonance (ssNMR) spectroscopy provides a powerful platform for in situ studies of membrane proteins. However, achieving high-resolution structure determination in native membranes faces significant obstacles, including limited spectral sensitivity, background interference, and substantial uncertainties in extracting distance restraints from ssNMR spectra. These issues have long hindered complete chemical shift assignments and the determination of high-resolution structures in native environments. To address these issues, our group has established integrated in situ ssNMR methodologies, enabled by the foundational contributions of many research groups. These advances allow for complete resonance assignments, high-resolution structure determination, and residue-specific dynamics analysis of membrane proteins in native cellular membranes. This Account summarizes our contributions over the past decade to in situ ssNMR studies of membrane proteins organized around two complementary themes: methodological advances in in situ ssNMR and the elucidation of how the membrane environment influences molecular mechanisms. The discussion systematically examines (i) recent methodological progress, including selective membrane sample preparation protocols compatible with both 13C- and 1H-detected ssNMR, signal-enhancing pulse sequences, and structural computation methods and (ii) key structural and dynamic insights from in situ ssNMR, including high-resolution structure determination of diverse membrane proteins such as aquaporin Z (AqpZ), the sugar transporter BjSemiSWEET, and the channel protein MscL, alongside a mechanistic understanding of how the membrane environment regulates their functions. By demonstrating the feasibility of in situ ssNMR for determining membrane protein structures in native settings and providing unprecedented atomic-level insights into their functions, we aim to advance research on membrane-sensitive proteins and promote the broader application of in situ ssNMR methodologies.

The Stability of Organic Field-Effect Transistors: From Materials, Devices to Circuits
Xiaosong Chen - ,
Jialu Xue - ,
Zhongwu Wang - ,
Yinan Huang - , and
Liqiang Li *
Organic field-effect transistors (OFETs) have attracted broad attention in flexible displays, human–machine interaction, and the Internet of Things (IoTs) due to their unique advantages, including flexibility, low cost, large-area fabrication, and biocompatibility. However, their stability remains a key barrier to commercialization. Focusing on this central obstacle toward commercialization, we bridge the stability gap by proposing an integrated “material–device–circuit” stabilization route. Material stability was achieved from both chemical and physical perspectives. Chemically, by removing reactive oxygen species (ROS) and suppressing triplet excitations via vitamin C (VC) treatment, oxidative degradation of n-type organic semiconductor (OSC) films was effectively prevented; Physically, by revealing the aggregation-state evolution governed by interfacial stress, high morphological stability was achieved through oxygen-induced lattice strain (OILS) and quasi-dispersion strengthening with nanoparticles. Device stability encompassing operational stability, environmental stability (thermal stability, photostability, oxygen stability), and storage stability was achieved by microwave annealing, reaching the zero-temperature-coefficient (ZTC) point, constructing an “exciton–polaron quenching” strategy, and strain balance strategy. Circuit stability was further achieved by stabilizing n-type OFETs and adopting an asymmetric dual-gate strategy toward stable CMOS inverters and uniform unipolar logic circuits.
Beyond stability enhancement, we further turned instability into an opportunity. By manipulating the morphological evolution of OSCs, we realized a controllable transformation from polycrystalline to single-crystalline states. By the synthesis of photoresponsive OSCs and the exploiting of the photothermal effect at the electrode/OSC interface of OFETs, the functionalities of UV and IR detection were achieved, respectively. Collectively, our integrated “material–device–circuit” stabilization route establishes a comprehensive and robust framework for the application of OSCs, OFETs, and organic integrated circuits, which expands their potential applications.

From Upconversion Nanoparticles to Proteins: Probing Hydration-Water Density Fluctuations by Luminescence Thermometry
Ramon S. Raposo Filho - ,
Yongwei Guo - ,
Fernando E. Maturi - ,
Carlos D. S. Brites - , and
Luís D. Carlos *

This publication is Open Access under the license indicated. Learn More
Water appears simple, yet its anomalous behavior reveals an unexpected structural complexity. A growing body of evidence indicates that many of water’s anomalies arise from fluctuations between low-density (LD) and high-density (HD) local structural motifs, a form of polymorphism that is well established in the supercooled regime and increasingly supported at ambient conditions. Yet, how these structural motifs manifest within hydration layers, where water interacts with nanoparticles, proteins, and charged interfaces, remains far less understood. This interfacial water governs colloidal stability, biomolecular function, and chemical reactivity, but its microscopic organization is difficult to probe directly with conventional bulk techniques.
In this Account, we describe how luminescence nanothermometry provides a powerful and versatile approach to accessing density fluctuations in the hydration layer. By monitoring temperature-dependent optical and Brownian observables of luminescent probes, structural reorganizations of the surrounding hydration layer can be inferred with nanoscale sensitivity. Over the past several years, our group has shown that lanthanide-doped upconversion nanoparticles (UCNPs) and fluorescent proteins, such as enhanced green fluorescent protein (EGFP), act as local reporters of hydration-water density fluctuations.
A central observation emerging from these studies is the existence of a crossover temperature, Tc, at which hydration-water observables exhibit bilinear temperature dependencies. This Tc correlates with the depletion of LD motifs in the hydration shell and typically falls within the 315–330 K range, close to the minimum of water’s isothermal compressibility. Importantly, Tc depends on the nature of the probe and its interaction with the surrounding water.
By systematically varying nanoparticle size, pH, surface chemistry, and probe type, we show that previously contradictory trends in Tc can be unified by a single parameter: the effective surface charge density of the probe. When Tc is plotted against this quantity, data from UCNPs with different sizes and surface functionalizations, as well as from fluorescent proteins at different concentrations, collapse onto a master curve. This result demonstrates that interfacial electrostatics govern the stability of LD motifs in the hydration layer, providing a physically intuitive framework that links nanoscale charge distributions to local water structure.
We further extend this framework by examining nuclear quantum effects through isotopic substitution. Using EGFP as a model biomolecular probe, we show that replacing H2O with D2O shifts Tc upward by ≈10 K and enhances protein thermal stability, consistent with stronger hydrogen bonding and the displacement of thermodynamic anomalies in heavy water. In contrast, several inorganic and molecular probes fail to resolve a comparable isotopic shift, highlighting that the detectability of LD/HD fluctuations might be probe-dependent. Control experiments in H218O confirm that hydrogen, rather than oxygen, dominates these quantum effects.
Together, these results establish luminescent nanoprobes as sensitive reporters of hydration-water density fluctuations and reveal how interfacial charge, confinement, and quantum effects sculpt water structure at the nanoscale. Beyond resolving long-standing questions about water’s anomalies, this approach opens new avenues for understanding protein stability, designing functional nanomaterials, and exploiting hydration-water density fluctuations in chemical and biological systems.

S-Scheme Shapes Heterojunction Photocatalysis
Mahmoud Sayed - ,
Liuyang Zhang *- ,
Hermenegildo García *- ,
Huogen Yu *- , and
Jiaguo Yu *
A persistent obstacle in heterogeneous photocatalysis is the rapid recombination of photogenerated electrons and holes, a consequence of the strong Coulombic attraction between carriers within conventional semiconductors. This intrinsic limitation significantly constrains the efficiency of solar-to-chemical conversion processes. Heterojunction engineering has therefore become a central strategy for promoting charge separation by coupling semiconductors with complementary electronic structures. Such systems typically outperform their single-component analogues because the interfacial electronic configuration promotes directional charge migration and suppresses bulk recombination losses.
Within this context, S-scheme heterojunctions (SH) offer a mechanistically robust framework that reconciles efficient carrier separation with strong redox capability. An S-scheme couples a reduction photocatalyst (RP) and an oxidation photocatalyst (OP) in a staggered configuration. Under illumination, electrons in the OP selectively recombine with holes in the RP, while the high-energy electrons in the RP and high-energy holes in the OP are spatially retained and directed to catalytic sites. This selective recombination preserves redox power, enhances charge utilization, and accelerates surface reactions.
Since introducing the S-scheme concept in 2019 with the WO3/g-C3N4 system supported by in situ irradiated X-ray photoelectron spectroscopy (ISIXPS)─we have expanded its material scope across multiple dimensional architectures, including perovskite materials, semiconducting quantum dots (QDs), conjugated polymers (CP), metal–organic frameworks (MOFs), and covalent-organic frameworks (COFs). To validate the S-scheme mechanism, elucidate charge transfer dynamics, and resolve reaction mechanisms, we have employed an array of state-of-the-art characterization techniques, such as light-irradiated Kelvin probe force microscopy (KPFM), in situ electron paramagnetic resonance (EPR), in situ X-ray absorption spectroscopy (XAS), and femtosecond-transient absorption spectroscopy (fs-TAS).
Our most recent efforts focus on composition tuning, defect modulation, and interfacial bonding engineering to optimize the separation and lifetime of photogenerated carriers. Through these strategies, we aim to reinforce the internal electric field, regulate band bending, and precisely control charge flow pathways, ultimately maximizing photocatalytic efficiency. This Account provides a concise yet comprehensive overview of the evolution of SH, with emphasis on the design principles and advanced characterization techniques developed and adopted by our group. We summarize key strategies for engineering SH tailored for enhanced charge carrier separation and highlight their applications in major photocatalytic reactions. Finally, we outline promising future directions for the field.
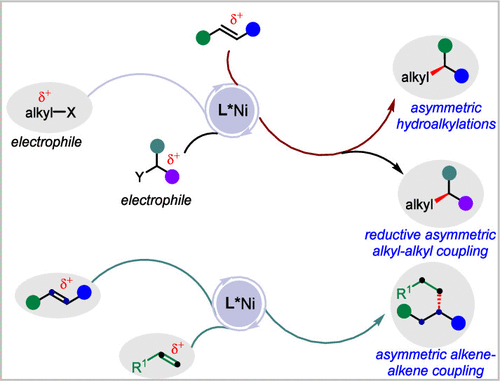
Ni-Catalyzed Asymmetric Alkyl–Alkyl Cross-Coupling: Reaction Mode Development and Applications
Qiong Yu - ,
Muneer-ul-Shafi Bhat - , and
Wei Shu *
Stereogenic carbon centers with an alkyl–alkyl (C(sp3)–C(sp3)) bond constitute a fundamental structural motif in organic molecules, where precise stereocontrol is critical for the development of pharmaceuticals, agrochemicals, and functional materials. Although transition-metal-catalyzed cross-coupling has revolutionized bond formation, enantioselective alkyl–alkyl coupling to access stereogenic carbon centers remains less developed, mainly due to challenges in selectivity control and the inherent instability and side reactions associated with alkyl metallic intermediates. Nickel-catalyzed asymmetric carbon–carbon cross-coupling has emerged as a powerful strategy for building stereogenic carbon centers via asymmetric C(sp2)–C(sp3) cross-coupling. Traditional Ni-catalyzed asymmetric alkyl–alkyl cross-coupling heavily relies on the use of stoichiometric alkyl electrophiles and stoichiometric alkyl metallic reagents as nucleophiles. Recently, Ni-catalyzed asymmetric hydroalkylation of alkenes provides a new solution for the construction of stereogenic carbon centers with an alkyl–alkyl bond, thereby avoiding the use of stoichiometric organometallic species as alkyl nucleophiles. However, significant challenges remain in Ni-catalyzed alkyl–alkyl cross-coupling reactions. To address these challenges, our group has focused on developing asymmetric Ni-catalyzed alkyl–alkyl bond-forming reactions. This Account outlines the development of reaction modes in Ni-catalyzed asymmetric alkyl–alkyl cross-coupling and presents three strategies from our laboratory: Ni-catalyzed asymmetric hydroalkylation of alkenes, Ni-catalyzed reductive asymmetric alkyl–alkyl cross-coupling, and Ni-catalyzed reductive–oxidative asymmetric alkene–alkene cross-coupling. First, we focused on developing Ni-catalyzed asymmetric hydroalkylation of alkenes, which enables direct and enantioselective alkyl–alkyl bond formation from electron-rich, electron-neutral, and electron-deficient alkenes and alkyl electrophiles. Next, we developed a reductive asymmetric alkyl–alkyl cross-coupling that directly couples two alkyl electrophiles. This strategy eliminates the need for preformed organometallic reagents and achieves high chemoselectivity and enantioselectivity in alkyl–alkyl cross-coupling by using alkyl electrophiles as the sole coupling partner. Most recently, we introduced a reductive–oxidative asymmetric alkene–alkene cross-coupling, a distinctive strategy that unites two alkenes through a redox event. Two alkenes act as surrogates for different alkyl metallic nucleophiles in the presence of hydride. This reaction mode enables alkyl–alkyl cross-coupling without the use of stoichiometric alkyl nucleophiles or alkyl electrophiles, allowing for control of the chemoselectivity, regioselectivity, and enantioselectivity. We hope that the strategies and concepts discussed herein will inspire the further development of new nickel-catalyzed methodologies for stereoselective alkyl–alkyl cross-couplings, thereby providing synthetic tools for organic chemistry and related fields.
Photon Avalanching Nanoparticles
Luan N. Passini - ,
Emory M. Chan - , and
Bruce E. Cohen *
Avalanches within nanoparticles seem like science fiction, but if they are avalanches of photons, they open up real-world innovations in imaging, sensing, optical computing, and other unexplored light-driven technologies. Avalanches are outsized events arising from the integration of many smaller inputs, and photon avalanching (PA) was first reported in bulk crystals in 1979 as an unexpectedly large jump in luminescence as excitation intensity was slowly increased. It would be 41 years before PA would be observed at the nanoscale in photon avalanching nanoparticles (ANPs), Tm3+-doped upconverting nanoparticles that show excited-to-ground state absorption inversion greater than 10,000:1 and emission that scales nonlinearly up to the 32nd power of the pump intensity. This extreme nonlinearity enables a real-time 5-fold improvement in the 150-year-old Abbe limit of spatial resolution, achieving 70 nm resolution using only simple scanning confocal microscopy. This extreme nonlinearity also gives rise to a series of highly unusual optical and sensing properties. Tm3+ ANPs show NIR-controlled bidirectional photoswitching, lasting over 1000 cycles in ambient or aqueous conditions with no measurable sign of photodegradation. This enables 2- and 3-dimensional optical nanoscale patterning with full erase and rewrite capabilities. Unlimited photoswitching also underlies the super-resolution technique INPALM, which is capable of sub-Ångstrom localization precision and resolving individual ANPs within tightly packed clusters. Nd3+-based ANPs show the peculiar property of intrinsic optical bistability (IOB), a form of memory in which emission depends on whether the ANPs have previously undergone PA. This stable, history-dependent contrast makes these ANPs analogous to optical transistors and promising materials for optical computing, neuromorphic circuitry, and related photonic technologies. The steep nonlinearity of PA also makes ANPs exceptional sensors of external perturbations, as tiny environmental changes may be amplified into large changes in optical output. As force sensors, Tm3+ ANPs are able to detect forces over a dynamic range of 4 orders of magnitude, from piconewtons to micronewtons, a range that will enable force sensing in complex systems across scales. Application of current ANP designs to imaging and devices, discovery of new PA-associated phenomena, and design of new ANPs with unique properties are all underway as the novelty of this technology cascades toward new fundamental discoveries and applications.
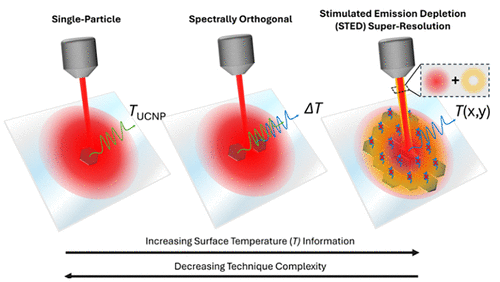
Upconverting Nanoparticle Thermometry beyond the Diffraction Limit
Benjamin Harrington - ,
Ziyang Ye - ,
Laura Signor - , and
Andrea D. Pickel *
This publication is Open Access under the license indicated. Learn More
The growing demand for nanoscale temperature measurement capabilities is motivated by diverse applications such as thermal management of microelectronics and batteries, design of plasmonic systems, mechanistic studies of catalysis, and unraveling intracellular processes. Upconverting nanoparticles (UCNPs) are lanthanide-doped inorganic probes that are popular luminescent thermometers, with advantages including well-understood temperature-dependent behavior, broadly tunable excitation and emission wavelengths, and exceptional thermal and chemical stability. Like other optical thermometry techniques, luminescence thermometry provides the desirable capability of remotely collecting the temperature-dependent signal from the far field. Conventional implementations of luminescence thermometry also share a major limitation of other optical thermometry techniques, namely, their diffraction limited spatial resolution. However, in contrast with other optical thermometry techniques, luminescence thermometry also creates an opportunity to leverage certain unique strategies for circumventing the diffraction limit.
In this Account, we discuss our contributions to initiating or building on three major strategies for achieving UCNP thermometry beyond the diffraction limit. Some of these concepts originate from or have direct parallels in the realm of biological imaging, where optical imaging with spatial resolution below the diffraction limit has been a longstanding goal; conversely, others have no direct bioimaging analogy. Exciting an isolated single UCNP with a diffraction limited laser beam enables thermometry with subdiffraction limited spatial resolution governed by the UCNP size, although this approach is inherently restricted to measurements at a single spatial point. We begin by describing our efforts to extend single-UCNP measurements to smaller UCNP sizes and understand how their temperature-dependent emission can be influenced by external factors such as the excitation laser intensity or the surrounding optical environment, the latter of which is exemplified by an investigation of how single-UCNP emission is altered when the UCNPs are placed on various metallic substrates. Next, we show how the principles underlying single-UCNP thermometry can be expanded to sample multiple temperature points within a subdiffraction region by combining different UCNP compositions with spectrally orthogonal temperature-dependent luminescence. As a practical demonstration, we resolve a nearly 20 K temperature difference over a sub-110 nm distance originating from the steep temperature gradient near a laser-heated Ag nanodisk. Finally, we discuss our adaptation of UCNP-based stimulated emission depletion (STED) super-resolution imaging for super-resolution nanothermometry, combining temperature-dependent STED spectroscopy, self-assembled UCNP monolayer formation, and a detection scheme that enables practical scan times. STED nanothermometry can reveal a temperature gradient on a Joule-heated microstructure that is undetectable with analogous diffraction limited measurements, showcasing the power of this approach. We conclude with our perspective on the outlook for UCNP thermometry methods that circumvent the diffraction limit, highlighting both current research needs to further improve the measurement capabilities and strategies that could facilitate broader adoption of these emerging techniques.

Catalysis over Isolated and Nested Lewis Acid Centers and Noble Metal Centers Anchored by Nested Lewis Acid Centers in Zeolites
Yanfei Zhang - ,
Tingshu Yang - ,
Liang Qi *- , and
Alexis T. Bell *
Highly dispersed transition-metal Lewis acid centers (e.g., Zn, Co, Y, La, Fe, Sn, Hf, and Zr) and Lewis acid-anchored noble metal centers (e.g., Pt–Zn, Pt–Sn, Pt–Fe, Rh–Zn, and Rh–Co) supported on siliceous zeolites are promising catalysts for a number of industrially important reactions, such as alcohol dehydrogenation, aldol condensation, alkane dehydrogenation, and olefin hydroformylation. In this Account, we describe the preparation and characterization of Lewis acid centers grafted onto hydrogen (H)-bonded silanol groups present in zeolites as well as Lewis acid-anchored noble metal centers and discuss the mechanism and kinetics for different reactions occurring over each type of center. We show that isolated and nested Lewis acid centers can be created by the reaction of hydrated cationic species with H-bonded silanol groups on dealuminated beta (DeAlBEA) or Silicalite-1 zeolite. We then demonstrate that isolated and nested Lewis acid centers are effective catalysts for light alkane dehydrogenation. Nested Lewis acid centers can also serve as efficient anchoring sites for dispersing noble metals such as Pt and Rh to generate bimetallic centers that exhibit superior catalytic performance relative to monometallic Pt and Rh for reactions such as alkane dehydrogenation and olefin hydroformylation. Finally, we summarize our recent investigations of isolated and nested Lewis acid centers and the Pt- and Rh-based bimetallic centers as catalysts for ethanol conversion to 1,3-butadiene (ETB), acetone conversion to isobutene (ATI), propane dehydrogenation to propene (PDH), n-butane dehydrogenation to butene and 1,3-butadiene (BDH), and ethene hydroformylation to propanal. We show that the activity of Lewis acid centers for these reactions is affected by their local coordination environments. In particular, we highlight the significance of H-bonding between hydroxyl groups connected to Lewis acid centers in an open configuration (M–OH) and silanol groups on zeolite supports to generate (≡SiO)xMn+–OH···(O(H)–Si≡)y structures, which exhibit aldol condensation activities that are higher than that of (≡SiO)xMn+–OH sites. These studies demonstrate that siliceous zeolites rich in H-bonded silanol groups can be utilized to create highly dispersed Lewis acid centers and can be further employed as an anchoring platform for noble metal atoms to construct atomically dispersed bimetallic centers. Both the chemical structure and the local coordination environment of these centers significantly influence their catalytic performance.

Conjugated Oligoelectrolytes as Optical Probes
Samuel J. W. Chan - ,
Ji-Yu Zhu - , and
Guillermo C. Bazan *
Optical probes are essential tools for interrogating biological and chemical systems invisible to the naked eye, providing insights into molecular interactions, protein activity, and cellular trafficking. Conjugated oligoelectrolytes (COEs), an emerging class of optical probes, are synthetic organic amphiphiles defined by a π-conjugated backbone and charged pendant groups. COEs with a linear conjugated structure and charged groups at the two termini can be designed to mimic the molecular dimensions and arrangements of hydrophobic and hydrophilic groups characteristic of lipid bilayers. This design drives their spontaneous intercalation into and prolonged residence within biological lipid bilayer membranes. By tailoring their molecular building blocks, their electronic and photophysical properties as well as their interactions with cells can be readily tuned, positioning COEs as a versatile platform for developing molecular probes for fundamental research and applied bioimaging across a range of biological systems.
In this Account, we describe the design strategies elaborated by our group for developing COEs as optical probes, with a focus on their applications and uses in elucidation and tracking of cellular membrane properties. We show that COEs can be used to detect and visualize lipid membranes at multiple length scales, ranging from single microbial cells and exogenously isolated small extracellular vesicles and particles to subcellular organelles and whole cells in live animal models. COEs also function as effective nonlinear optical probes that are applicable in advanced imaging modalities such as two-photon microscopy and stimulated emission depletion microscopy to extract spatiotemporal information at high resolution.
We also provide our insights into how COEs can be designed to be functional probes that exhibit predictable photophysical behavior in response to the local molecular and chemical environment. Using fluorescence lifetime imaging microscopy, the time-resolved emission of COEs can be leveraged to provide insight into dynamic processes such as rapid changes in membrane tension and long-term changes in membrane rigidity and composition. We additionally elaborate strategies for modulating interactions with biological membranes, designing membrane-specific probes that respond to specific cellular biophysical parameters, and offer perspectives and opportunities toward developing a new platform for disease detection and diagnosis.
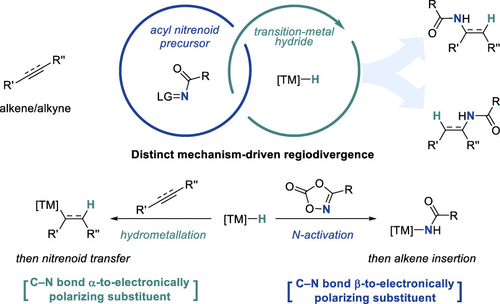
Transition-Metal Hydride Catalysis Meets Nitrenoid Transfer: Design Principles for Precision C–N Bond Formation
Xiang Lyu - ,
Hoonchul Choi - , and
Sukbok Chang *
This publication is Open Access under the license indicated. Learn More
Transition-metal hydride (TMH) catalysis has become a powerful strategy for hydroamination reactions, enabling direct C–N bond formation from simple alkenes and alkynes under mild conditions. In conventional TMH-catalyzed hydroamination, a metal hydride first engages a π system through hydrometalation or related hydrogen-atom-transfer processes to generate organometallic intermediates, wherein stabilization of the incipient carbon center dictates regioselectivity. As a consequence, subsequent coupling with a nitrogen electrophile intrinsically favors C–N bond formation at electronically activated positions. Accordingly, regioselectivity patterns such as α-amination adjacent to electronically polarizing substituents, directing-group-controlled sites, or sterically accessible terminal positions following migration are well-established, whereas complementary β-selective amination remains challenging to achieve.
This Account summarizes our efforts to merge TMH catalysis with nitrenoid transfer chemistry and, in doing so, to uncover two distinct and mechanistically orthogonal hydroamidation regimes governed by the ordering of elementary steps. Using bench-stable dioxazolones as acyl nitrenoid precursors, we first established a canonical TMH manifold in which hydrometalation precedes inner-sphere nitrenoid transfer. In this regime, regioselectivity is programmed at the metal hydride insertion stage and can be predictably controlled across a wide range of TMH catalytic systems, enabling regioselective hydroamidation of alkynes and alkenes with broad scope and high functional-group compatibility.
A conceptual turning point emerged from mechanistic studies of NiH catalysis, where an unexpected β-selective intramolecular hydroamidation exposed a fundamentally different reaction manifold. Rather than initiating with hydrometalation, NiH was found to activate the nitrenoid precursor first, generating a Ni-amido intermediate that subsequently engages the alkene through polarity-matched amidonickelation. This transposed hydroamidation regime inverts the selectivity-determining step, shifting control from alkene hydrometalation to C–N bond formation. As a result, regioselectivity patterns inaccessible under conventional TMH logic, including β-lactam formation, intermolecular β-amidation of conjugated carbonyls, and homobenzylic hydroamidation of vinylarenes, become attainable with high enantioselectivity.
Together, these studies establish step order as a central design parameter in TMH-catalyzed hydroamidation. By deliberately choosing whether hydride delivery or nitrenoid generation occurs first, complementary regio- and stereochemical outcomes can be accessed from the same classes of unsaturated substrates. Beyond nitrenoid chemistry, extension of this transposed logic to carbene transfer processes further underscores its generality. We anticipate that continued mechanistic elucidation and expansion of this framework will transform TMH–nitrenoid synergy from a collection of reactions into a predictive platform for precision C–N bond construction.
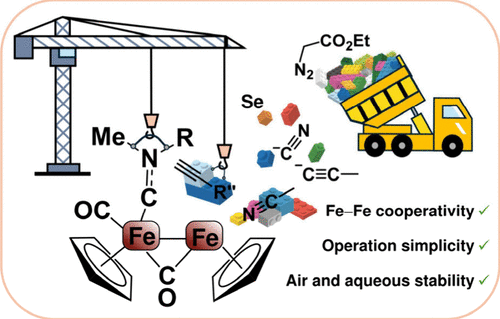
Diiron(I) Bis(cyclopentadienyl) Complexes with Bridging Iminium Ligands: From Foundational Organometallic Chemistry to Unique Reactivity and Biological Potential
Sara Benetti - ,
Alessia Cinci - ,
Chiara Zappelli - , and
Fabio Marchetti *
This publication is Open Access under the license indicated. Learn More
Dimetallic complexes offer a remarkable platform to probe metal–metal cooperativity, enabling ligand reactivity patterns that are inaccessible to mononuclear systems. Starting from [Fe2Cp2(CO)4] (Fp2, Cp = η5-C5H5), diiron μ-aminocarbyne (iminium) complexes are available through a straightforward multigram-scale procedure. Carbonyl removal is key to enabling selective modification of the ligand set and promoting the formation of uncommon hydrocarbyl ligands involving the carbyne center. In this context, the insertion of terminal alkynes into iron–carbynes bond affords a wide diversity of vinyliminium complexes, characterized by a highly versatile and modular reactivity with reasonably broad reaction scopes. Specifically, three representative transformations are discussed in this Account: 1) Cyanide addition, leading to a cyano-aminoallylidene ligand, in which an intramolecular amine–CO interaction dictates the stereochemical outcome and facilitates subsequent thermal CO dissociation, thereby enabling further reaction pathways. 2) Incorporation of a selenium atom through vinyliminium deprotonation, yielding intrinsically stable complexes bearing an almost pure selenolate function. This moiety displays marked nucleophilic reactivity, including facile dimerization to a Fe4 framework via selenide-to-diselenide oxidation, as well as the construction of a selenophene-decorated Fischer alkylidene ligand. Mild hydrolytic cleavage breaks the alkylidene bridge, providing access to a new family of highly functionalized selenophenes. 3) Vinyliminium deprotonation, representing a key entry point to the first family of ferrabenzenes. A multicomponent assembly involving one carbonyl ligand and ethyl diazoacetate generates a six-membered metallacycle, which is ultimately converted into substituted ferrabenzenes through O-alkylation.
Beyond their organometallic reactivity, cationic aminocarbyne and vinyliminium complexes display a combination of properties that are highly attractive for medicinal applications, including straightforward synthesis, air and aqueous stability, broad structural tunability, and amphiphilicity. These features prompted their evaluation as anticancer agents. Their cytotoxicity relies on a molecular “time bomb” behavior, as extensive fragmentation of the diiron scaffold occurs intracellularly, releasing reactive iron(I) species and carbon monoxide. The resulting fragments primarily induce mitochondrial dysfunction, leading to disruption of cellular redox homeostasis. Importantly, both cytotoxicity and mechanism of action can be regulated by the choice of substituents and ligands, and appreciable cancer cell selectivity is generally achieved. Notably, selected complexes confirmed their promise in 3D cellular models and, in one case, in vivo, warranting further development of these diiron-based anticancer agents.
Overall, this Account traces a long-term research journey centered on diiron bis(cyclopentadienyl) complexes. The narrative begins in a historical context where organometallic chemistry was largely confined to inert-atmosphere manipulation and biological or aqueous applications were scarcely envisioned. It then progresses through the discovery of novel organometallic reactivity patterns and motifs enabled by metal–metal cooperativity, with emphasis on the most recent advances, and culminates in the transition toward biological applications. Collectively, these studies illustrate how fundamental organometallic chemistry can naturally evolve into the concepts and principles of modern bioorganometallic chemistry.
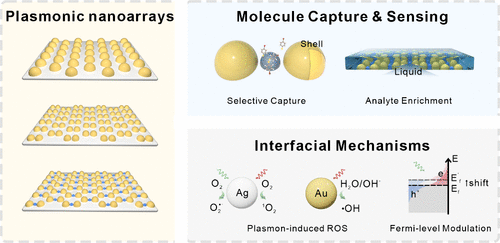
Plasmonic Nanoarrays as SERS Substrates: Advances, Challenges, and Perspectives
Lei Yao - ,
Shuying Chen - ,
Shikuan Yang *- ,
Teng Qiu *- , and
Qi Hao *
Surface-enhanced Raman scattering (SERS) provides a powerful spectroscopic approach for molecular identification and interfacial analysis by combining chemical specificity with ultrahigh sensitivity. While chemically synthesized nanoparticles have enabled broad use of SERS, increasing attention is being paid to how structural uniformity, aggregation behavior, and surface chemistry influence signal reproducibility, reliability, and mechanistic interpretation. In this context, plasmonic nanoarrays fabricated by template-assisted physical deposition offer a complementary and increasingly important SERS platform.
This Account summarizes recent advances in SERS using nanoarrays fabricated by template-assisted evaporation. In these approaches, nanoscale geometry and hotspot distributions are predefined by the template and realized through directional deposition. These template-defined architectures enable reproducible electromagnetic enhancement, polarization-controlled excitation, and stable plasmonic responses. Moreover, physical deposition yields clean, ligand-free metal surfaces, providing a well-defined interface for probing plasmon-molecule interactions and interfacial chemical processes. Using anodic aluminum oxide (AAO) lithography as a representative platform, we illustrate how precise control over template thickness enables angle-resolved deposition and structural programmability, allowing the fabrication of dimers, trimers, and compositionally heterogeneous architectures with nanometer-scale gaps. These capabilities support advanced SERS functionalities, including efficient hotspot activation for enhanced sensitivity, selective molecular trapping, and access to interfacial processes on nonplasmonic or weakly plasmonic materials. Furthermore, integration with transparent substrates and soft supports enables liquid-phase SERS configurations and flexible sensing platforms. These liquid-phase SERS configurations improve signal stability and measurement reliability for real-time, in situ measurements, while mitigating aggregation-related issues commonly encountered in conventional SERS. Beyond molecular detection, nanoarray-based SERS provides a controlled experimental framework for mechanistic studies in plasmonic chemistry. The combination of chemically clean surfaces with nonaggregating and structurally stable architectures enables plasmon-driven interfacial processes to be examined under well-defined and reproducible conditions, and facilitates in situ, real-time tracking of reaction dynamics in liquid-phase SERS measurements. This well-controlled environment serves as a reliable physical model for investigating interfacial reaction mechanisms, allowing direct identification of key reaction intermediates and offering an effective route to resolving long-standing mechanistic debates in plasmonic chemistry.
Overall, this Account underscores the value of template-fabricated plasmonic nanoarrays as a versatile SERS platform that connects sensitive detection with mechanistic insight. Looking ahead, continued advances in template engineering and deposition strategies are expected to further expand their role in well-controlled studies of light-matter interactions and interfacial physics and chemistry.

From Covalent Systems to Bulk Phases: Addressing Structural Complexity with Computational NMR
Giacomo Saielli *
This publication is Open Access under the license indicated. Learn More
The free induction decay (FID) signal acquired in a typical NMR experiment contains information about the chemical shifts, δ, and the spin–spin coupling constants, J, of the system investigated. These two parameters, particularly the chemical shifts, are very sensitive to both intramolecular and intermolecular perturbations. As such, they are also very good probes of the structure of both the molecule and the hosting solvent/matrix. This sensitivity is exploited in natural product studies to deduce the molecular structure of newly isolated compounds from the analysis of their NMR spectra. However, for complex carbon skeletons, the interpretation of the NMR data is far from trivial; the structural information is too deeply buried within the overlapping high-order multiplets and close resonances. In these cases, it is useful to compare the experimental NMR data of the unknown substance with the ones predicted by density functional theory (DFT) based methods for hypothetical molecules. Ideally, one will discard all putative structures resulting in a disagreement with the experiments and will keep the only one exhibiting an agreement within the benchmarked accuracy of the level of theory used. Two other sources of complexity, besides the topological complexity of natural substances, may strongly affect the interpretation of the NMR spectra. One, still related with covalent compounds, is the presence of heavy atoms which brings in relativistic effects in the NMR. Even for a simple molecular structure, they turn the interpretation of the NMR spectrum into a very difficult task since empirical rules often do not allow a full elucidation of the structure; thus, such effects can be accounted for only with relativistic versions of DFT. The other source of complexity is the presence of strong noncovalent interactions of the NMR probe molecule with its environment. In these cases, the full dynamics of the solute and solvent system has to be taken into account and the structure that is responsible for the observed NMR is in fact the average bulk structure of the solute–solvent system. Then, molecular dynamics (MD) simulations have to be coupled with the DFT-NMR calculations in order to predict the NMR properties. In turn, the comparison between the calculated and experimental data can shed light on the force field (FF) parameters used in the MD simulation. Therefore, computational NMR can be used to shed light on both covalent and noncovalent structural problems: in one case, the exploration of a discrete structural space will allow one to select the correct structure of an unknown compound among several hypothesis; in the other one, it will enable the fine-tuning of classical FF parameters over a continuum range of possibilities.

Microheterogeneous Electrolytes: From Chemical Composition to Spatial Architecture─A Paradigm Shift in Electrolyte and Interphase Design
Canfu Zhang - ,
Zhineng Ren - , and
Huilin Pan *
Electrolytes underpin all electrochemical energy-storage devices by mediating ion transport, interfacial reactions, and electrochemical stability. Conventional electrolyte design has long relied on a homogeneous-solution paradigm, wherein ions are uniformly solvated and electrochemical behavior is primarily dictated by composition and bulk thermodynamic properties. Within this framework, tailoring local solvation environments has enabled important incremental improvements. However, ion transport, interfacial chemistry, and electrochemical stability remain intrinsically coupled, rendering the simultaneous optimization of multiple performance metrics fundamentally challenging.
Recent experimental and computational advances have increasingly challenged the assumption of homogeneity in liquid electrolytes. Electrolytes are now recognized as structurally complex soft-matter systems that can spontaneously develop spatial heterogeneity across molecular, nanometric, and mesoscale lengths. Even in macroscopically uniform electrolytes, ions and solvent molecules may self-organize into clusters, micelles or reverse micelles, bicontinuous networks, and microemulsion-like domains. These microscopic heterostructures are not incidental: they play decisive roles in governing ion-transport pathways, interfacial reactivity, and electrochemical stability, yet have largely remained outside traditional electrolyte design frameworks.
Drawing on our work over the past five years, together with related advances from the broader community, this Account introduces the concept of microheterogeneous electrolytes (MHEs), which converts electrolyte design from compositional optimization to microstructural regulation. In MHEs, spatially differentiated domains act as functional units that decouple the otherwise conflicting electrochemical requirements. Ion transport can be accelerated along percolating low-energy barrier pathways; solvent reactivity can be suppressed through confinement and topological control, and interfacial reactions are regulated via selective enrichment of active species.
We first trace the historical development and thermodynamic origins of microheterogeneity (MH) in liquids, elucidating how competition between energetic and entropic contributions stabilizes nanoscale domains without macroscopic phase separation. Building on this foundation, we establish a structure–function paradigm linking solvation topology, mesoscale connectivity, and electrochemical behavior. This framework clarifies how MHEs enable fast ion transport, broaden electrochemical stability windows, and promote adaptive interphase formation under extreme conditions of temperature, voltage, and current density.
To enable the rational design of MHEs, we articulate interaction-level design principles through anion and solvent. Together, these principles transform molecular interactions into programmable electrolyte architectures. We further summarize experimental and computational approaches that render these invisible structures observable and quantifiable. By redefining electrolytes as spatially organized and dynamically adaptive media, this Account establishes MHEs as a general design principle applicable to Li+, Na+, multivalent, and aqueous batteries. More broadly, it builds a conceptual bridge between soft-matter physics and electrochemical engineering, opening new opportunities for designing electrolytes capable of meeting the stringent demands of next-generation energy-storage technologies.

Enhanced Transcytosis and Retention (ETR) of Drug Delivery Nanocarrier in Solid Tumors
Mengmeng Qin - ,
Zhenyu Zhang - ,
Yuliang Zhao - , and
Huan Meng *
Transcytosis, traditionally regarded as biological, constitutes an intrinsic and powerful pathway for macromolecule transport across endothelial and epithelial barriers. The emerging concept of Enhanced Transcytosis and Retention (ETR) is distinct from passive extravasation or tissue leakiness. It recasts nanocarrier delivery as an orchestrated chain of interfacial equilibria in which encoded surface chemistry directs receptor recognition, active barrier crossing, and subsequent accumulation within target tissues. Here, we delineate the chemical framework underpinning ETR-mediated delivery, emphasizing that the chemical identity of a nanocarrier, i.e., its surface functional groups, coordination motifs, hydration shell, reactive ligands, surface free energy, and biocorona, dictates a hierarchical sequence of interactions. To enable ETR access, we propose a triadic interaction model among the nanocarrier, an endogenous or engineered protein-based material, and a specific cellular receptor. This architecture represents a fundamental shift from conventional two-entity protein-adsorption frameworks, converting inherently stochastic protein deposition into a chemically programmable, design-driven active transport process. At the inner interface (between nanocarrier and protein material), surface functional groups, roughness, and topology determine the composition, orientation, and reactivity of a given biomolecule, such as endogenous or engineered proteins. At the outer interface (protein-cell receptor), these nanocarrier–protein complexes engage cell receptors through amino acid sequence-specific molecular recognition, topological complementarity, hydrogen-bond cooperativity, and electrostatic complementarity that collectively trigger ETR active access. Such an ETR framework, first exemplified in solid tumors (e.g., pancreatic cancer and triple negative breast cancer), now extends to diverse pathological contexts including the blood-brain barrier and dystrophic muscle. By viewing ETR drug delivery through a chemical lens, this manuscript integrates structure–reactivity principles with biological transport, providing a molecularly actionable framework for otherwise inaccessible tissues. Noteworthy, artificial intelligence (AI) guided protein engineering, using strategies such as point mutagenesis and noncanonical amino-acid substitution, will enable the creation of highly optimized and artificial ligands that assemble ETR-activating units with molecular-level precision.

Aggregation-Induced Emission: Past, Present, and Future
Pioneer Account
Natalie Y. Baona Tang - ,
Siyuan Wang - ,
Letian Xu - ,
Kun Zhou *- ,
Zheng Zhao *- , and
Ben Zhong Tang *
For centuries, the reductionist view that “the whole equals the sum of its parts” has guided scientific study, particularly materials design. Nature, however, often defies this logic: an aggregate (whole) can display emergent properties that are totally absent in its individual parts. Aggregation-induced emission (AIE) exemplifies this “anomaly”: nonluminescent molecules become emissive upon aggregation, achieving a qualitative “0-to-1” leap that challenges the reductionist tenet and provides a unique lens through which to view the emergence of new properties.
Since it was proposed as a concept in 2001, AIE has been mechanistically understood as arising from the restriction of molecular motion (RMM) in the excited state. In dilute solutions, molecular rotors and vibrators dissipate exciton energy through active motions, leading to nonradiative decay. Upon aggregation, these motions are physically restricted by molecular packing and noncovalent interactions, impeding nonradiative channels and opening radiative pathways. This mechanistic understanding has motivated extensive research into AIE and expanded the field into a diverse platform of aggregation-enabled luminescent systems, including clusteroluminescence (CL), room-temperature phosphorescence (RTP), and circularly polarized luminescence (CPL)─all absent in the isolated molecular constituents and emerging through aggregation.
With accumulated knowledge in AIE, the attention has broadened toward the exploration of aggregation-generated function (AGF). From this perspective, molecular motions─previously viewed as energy “wasted” that reduced emission─can be harnessed to convert excited-state energy into heat through rotations and vibrations. By channeling the same exciton energy that underlies luminescence into nonradiative decay pathways, we can engineer aggregates to exhibit emergent photothermal (PT), photoacoustic (PA), and photocatalytic (PC) activities. These functions open new application avenues, including solar energy conversion, high-resolution deep-tissue imaging, and “intelligent” actuation.
From the serendipitous encounter with AIE to the systematic study of AGF, advances in the field have shifted scientific attention from isolated molecules to complex aggregates. With the elucidation of principles governing emergent properties, it is becoming clear that a paradigm shift is needed─from molecularism to aggregatism or from molecular science to aggregate science (AS). Guided by emergentism, AS studies how molecules, through noncovalent interactions and hierarchical organization, give rise to macroscopic functions absent in their individual constituents. Notably, the materials we use and the life we see around us are all aggregates. This aggregate-level perspective enables the development of new systems with complex functionalities (e.g., advanced multimodal theranostics) and deepens our understanding of life─an archetypal multiary system in which the aggregation of nonliving biomolecular constituents yields a living organism.
In this Account, we detail the intellectual trajectory from AIE to AGF and finally to AS. We distill the guiding principles and outline future directions, including transitions from unary to multiary systems, static structures to dynamic processes, and descriptive aggregate science to prescriptive aggregate engineering. A deeper understanding of AS will enable new scientific discoveries and technological innovations, inviting us to imagine a future designed not merely with matter but with the sophisticated organizational logic that endows it with “life-like” functions.
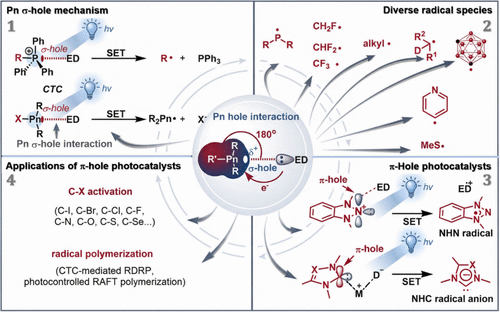
Exploiting Pnictogen σ/π-Hole Interactions for Visible-Light-Induced Radical Transformations
Qiang Liu - ,
Yan Zhang - , and
Xiang-Yu Chen *
The direct and selective activation of chemical bonds under mild, operationally simple conditions remains a longstanding pursuit in organic synthesis. Recently, elemental σ/π-hole interactions have emerged as powerful noncovalent tools, enabling new modes of molecular activation. Despite their promise, the application of pnictogen σ/π-hole interactions in photoinduced radical processes is still at a nascent stage.
In this Account, we describe our recent efforts in leveraging pnictogen σ/π-hole interactions to facilitate the generation of organic radicals under visible-light irradiation. By capitalizing on the properties of pnictogen σ/π-holes─tuned through careful selection of pnictogen elements, electron-withdrawing substituents, and pnictogen hole acceptors─we have developed general strategies for visible-light-induced radical transformations. The key element of this strategy is the use of pnictogen σ/π-hole interactions to assemble charge-transfer complexes (CTCs), which undergo visible-light-induced single-electron transfer (SET) from an electron donor to the pnictogen center. This process generates either pnictogen-centered radicals or substrate-derived radical species, thereby providing a basis for the rational design of new reagents and catalysts. The main advances can be summarized as follows:(1)
We established an efficient, transition-metal-free and photocatalyst-free strategy for the generation of a broad range of radical species─including P(III)-centered, alkyl, carboranyl, fluoromethyl, difluoromethyl, trifluoromethyl, pyridyl, oxyalkyl, dn-alkyl and methylthio radicals─by using pnictogen σ-hole interactions.
(2)On this basis, the scope of pnictogen-hole interaction-enabled photoreactions was further expanded by introducing N-heterocyclic nitrenium (NHN) and N-heterocyclic carbene (NHC) systems. The amphiphilic character and π-hole electron-accepting ability of NHNs promote the formation of photoactive CTCs with suitable electron donors, which, upon single-electron transfer, afford NHN-centered radicals. This approach enables a series of metal-free reductive radical transformations mediated by NHN radicals, including the activation of C–I, C–Br, and activated C–Cl bonds, as well as controlled radical polymerizations. In addition, the combination of NHNs with ligated boryl radicals allows the activation of otherwise inert alkyl chlorides, further broadening the applicability and synthetic utility of this strategy.
(3)The concept of pnictogen interactions was further extended to NHC-based photocatalytic systems. NHCs, which are isoelectronic and isostructural analogues of NHNs and possess vacant pπ orbitals, can accept an electron to generate NHC radical anions. These species can act as strong reductants capable of activating a range of inert bonds, including Caryl–F, Caryl–N, Caryl–S, Caryl–Se, and Caryl–O bonds.
Taken together, these advances underscore the potential of pnictogen σ/π-hole interactions in contemporary radical chemistry.
Mastheads
Issue Editorial Masthead

This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More