



This publication is Open Access under the license indicated. Learn More
Graph Neural Networks for Polymer Characterization and Property Prediction: Opportunities and ChallengesClick to copy article linkArticle link copied!
- Hector Medina*Hector Medina*Email: [email protected]School of Engineering, Liberty University, Lynchburg, Virginia 24515, United StatesMore by Hector Medina
- Rachel DrakeRachel DrakeSchool of Engineering, Liberty University, Lynchburg, Virginia 24515, United StatesMore by Rachel Drake
Journal of Chemical Information and Modeling
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
Using machine learning to accelerate the characterization and prediction of properties of many-molecule systems, such as polymers, is appealing, yet challenging. Polymers are large, complex molecules that have unique properties and potential applications in a wide range of industries. Their potential in advancing fields such as ion-transport polymer for energy storage, lightweighting of structural materials, bioinspired multifunctional materials, etc., provide enough impetus for accelerating the discovery of novel polymeric materials. However, mathematical mapping and the consequent manipulation of polymer structures are still challenging tasks due to their complex configuration and the smorgasbord of motifs encountered naturally and in engineering materials. Traditional methods of polymer structure mapping and property prediction at multiscale domains can include approaches such as Density Functional Theory, Molecular Dynamics, and Finite Element Analysis, which can be time-consuming and computationally expensive. The promise of machine learning to accelerate these tasks is appealing, and currently, researchers are pursuing the development of architectures and composition approaches to accomplish this. Here we discuss the current state of the knowledge on the use of Graph Neural Networks, and related architectures, being developed and/or used for the characterization and prediction of properties of polymers. Many challenges still exist such as the lack of sufficient and comprehensive data sets. To address these issues, efforts are being pursued─such as the so-called CRIPT (Community Resource for Innovation in Polymer Technology) led by a lab consortium that includes representations from private industry, academia, government, and others. We conclude that even though this field is young it has both momentum and promise. The current challenges that must be overcome are also addressed.
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
1. Introduction
2. GNN Architectures
Figure 1
Figure 1. General architecture of a GNN for molecular property prediction. The input molecular graph is processed through multiple message passing layers, where the node embeddings are iteratively updated by propagating information from neighboring atoms. These updated representations are then combined by a readout function (e.g., average or sum) to produce a graph-level embedding, which is fed into a downstream prediction layer.
| model | type | training data | polymer family | target properties |
|---|---|---|---|---|
| PU-gn-exp (106) | MPNN | 697 polymers, collected from literature | conjugated semiconducting polymers | mobility, HOMO, LUMO |
| wD-MPNN (121) | MPNN | 42,966 copolymers, computational (xTB/DFT) | conjugated copolymers | IP, EA |
| GH-GNN (124) | MPNN | 2500–2800 polymer–solvent pairs, experimental | homopolymer–solvent mixtures | activity coefficient at infinite dilution |
| polyGNN (126) | MPNN | 13,388 polymers, computational and experimental | diverse synthetic polymers | multiproperty: electronic, optical, thermal, mechanical, solubility |
| St. John MPNN (127) | MPNN | 91,000 molecules and oligomers, computational (DFT) | OPV candidates | HOMO, LUMO, excitation energy |
| Periodic D-MPNN (123) | MPNN | 15,219 data points, experimental and DFT | homopolymers | atomization energy, bandgap, EA, dielectric, Tg |
| GNN-A/B | MPNN | 372 experimental polyimides (8,205,096 virtual candidates screened) | polyimides | Tg |
| sGNN (108) | MPNN | 20,000 MP2 conformations, computational | PEG, PE fragments | bonding potential energy |
| Multitask GNN (147) | GCN | 876 polymers (5 ns MD), 117 polymers (50 ns MD) | polymer electrolytes | ionic conductivity, diffusivity |
| Chem-DAGNN (148) | GCN | 687 polyimides, experimental | polyimides | Tg |
| Park GCN (149) | GCN | 2687 Organic Polyamides | polyamides | Tg, Tm, density, elastic modulus |
| Hu GCN (133) | GCN | 300–600 polymers, experimental | homopolymers | Tg |
| Hickey GCN (134) | GCN | 7558 polymers, experimental | general polymers | Tg |
| Zeng GCNN (110) | GCN | 1073 polymers, computational (DFT) | organic polymers | dielectric constant, bandgap |
| SweetNet (150) | GCN | 19,775 glycans, experimental | glycans | taxonomy, immunogenicity, pathogenicity, viral glycan bonding |
| Kimmig GCNN (135) | GCN | 2813 nanoparticle measurements, experimental | poly(methacrylates) | nanoparticle size |
| Volgin GCNN (151) | GCN | 6,726,950 synthetic (QSPR) and 214 experimental polyimides | polyimides | Tg |
| GATBoost (144) | GAT | 235 polymers, experimental | acid containing polymers | Tg |
| POLYMERGNN (111) | GAT | 296 polymers, experimental | polyesters | Tg, intrinsic viscosity |
| GNNs (107) | MPNN, GAT, GCN | 1313 glycans, 15,778 peptides, experimental | glycans, peptides | immunogenicity, taxonomy, minimum inhibitory concentration |
2.1. Message Passing Neural Networks
Figure 2
Figure 2. Two graph augmentations in polymer GNN architectures: a virtual edges connecting distant nodes to encode periodic or long-range interactions (left), and a global node connected to all nodes to capture system-level or graph-wide information (right).
2.2. Graph Convolutional Networks (GCN)
2.3. GAT and Attention-Based Architectures
Figure 3
Figure 3. An illustration of GAT operations. The left portion of the diagram shows how attention coefficients αij are computed: the transformed node features Whi and Whj are concatenated and passed through a shared attention mechanism parametrized by the vector a, followed by a LeakyReLU nonlinearity and a softmax normalization over all neighbors j ∈ Ni. The resulting coefficient αij encodes the learned importance of neighbor j when updating node i. The right portion illustrates multihead attention for a specific node (node 1 in the drawing): each neighbor contributes messages through multiple attention heads (depicted by the green, blue and purple lines), producing head-specific weights α1j(k) and transformed messages W(k)hj. The index k refers to the attention head number, where each head has its own learnable weight matrix W(k) and produces its own attention score. These per-head aggregations are combined via concatenation (for intermediate layers) or averaging (for final layers) to yield the updated node embedding hi. The diagram highlights both key components of GATs: attention-based neighborhood weighting and multihead message aggregation.
3. Data Sets
Figure 4
Figure 4. Conceptual depiction of CG in polymer informatics. The top bar represents the (abstract) spectrum of CG strategies from macroscopic, geometry-based top-down strategies to atomistic, energetics-based bottom-up approaches. The molecular structure is overlaid with color-coded regions representing multiple levels of abstraction: atoms (gray), monomer/repeat units (red), substructures, functional groups or motifs (blue), small domains (purple), and large domains (yellow). This hierarchy also illustrates the conceptual transition from atomistic to increasingly coarse-grained graph representations, by grouping chemically or structurally important substructures so that scalable and interpretable ML pipelines are achievable.
| database | types of polymers | size/scope |
|---|---|---|
| Handbook of Polymers (100) | polymers used by plastics, electronics, and medical fields | over 200 different types of polymers |
| Polymer Data Handbook (159) | data on physical properties of polymers | 217 polymers |
| PubChem (98) | wide range of polymers and properties | millions of chemical structures, only a portion are polymers |
| MatWeb (99) | wide range of material properties, including polymers | 185,000 materials |
| RadonPy (160) | 15 properties | over 1000 amorphous polymers |
| PolyInfo (152) | various data for polymeric material design | over 20,000 polymers |
| Polymers: A Property Database (161) | conducting polymers, hydrogels, nanopolymers and biomaterials | over 1000 polymers |
| PI1M (155) | polymers for tasks in density, glass transition temperature, melting temperature, and dielectric constants | 1 million generated polymers |
| Polymer Data set for Property Prediction (162) | for the design of high dielectric constant polymers | 1073 polymers |
| CRIPT (156) | polymer data structures stored as graphs containing metadata on properties | size and scope under development |
| Various small data sets (162−167) | atomization energy, bandgap, dielectric constant, unit cell volume | various sizes |
4. Coarse-Graining in Polymer Informatics
| CG model name | approach type | key features | references |
|---|---|---|---|
| Dissipative Particle Dynamics (DPD) | Top-down | particle-based CG method with soft forces and momentum conservation for mesoscale soft matter | (194−197) |
| Martini Model | Top-down | 4-to-1 (variable) CG model tuned to reproduce experimental thermodynamic data | (198−200) |
| Self-Consistent Field Theory (SCFT) | Top-down | field-theoretic simulation framework for polymer phase behavior | (201,202) |
| TraPPE-CG | Top-down | typically for alkanes, parameters fitted to vapor–liquid coexistence | (203) |
| UNRES | Top-down | united-residue CG force field for protein structure and folding simulations | (204,205) |
| Multiscale CG (MS-CG) | Bottom-up | derives CG potentials by matching atomistic forces (force-matching) | (172) |
| Iterative Boltzmann Inversion (IBI) | Bottom-up | iteratively updates CG potentials to match atomistic pair distributions | (206,207) |
| Single-Chain Boltzmann Inversion | Bottom-up | uses ab initio torsions and intrachain conformational sampling | (208) |
| United Atom/Blobs | Bottom-up | groups atoms into beads; bonded and nonbonded interactions derived from atomistic structure | (209−211) |
| Slip-Spring Model | Bottom-up | represents entanglement effects via slip-link dynamics between chains | (212−214) |
| Bead–Rod Models | Bottom-up | captures chain stiffness using rigid or semiflexible rod-like segments | (215) |
| Kremer-Grest Model | Bottom-up | idealized bead–spring model for simulating entangled polymer melts | (216−219) |
| Adaptive Resolution Scheme (AdResS) | Hybrid | couples atomistic and coarse-grained regions dynamically in one simulation box | (220) |
| SIRAH | Hybrid | CG biomolecular force field combining atomistic mapping with empirical tuning with multiscaling available | (221) |
| Rosetta | Hybrid | fragment-based modeling using statistical potentials and structural bioinformatics data | (175,222,223) |
5. Applications and Implementations
5.1. Electronic and Optical Property Prediction
| study | task | polymer representation | evaluation context | key takeaway/limitation |
|---|---|---|---|---|
| Zhang et al. (106) | mobility, HOMO/LUMO | polymer-unit graph | random split; classification | interpretability; parsing-dependent |
| Aldeghi and Coley (121) | IP/EA | stoichiometry- and architecture-aware monomer graph | random splits | weighted atomic contributions |
| Zeng et al. (110) | bandgap, dielectric | crystal-derived polymer graph | random split; MAE | structure-limited |
| Gurnani et al. (126) | electronic, optical, dielectric | periodic repeat-unit graph | RMSE | multitask; low-data dielectric limits |
| St. John et al. (127) | OPV optoelectronics | 2D connectivity graph | MAE | scalable screening; runtime not quantified |
| Antoniuk et al. (123) | multiproperty electronics | periodic polymer graph | relative error reduction | periodicity dominant |
5.2. Thermal and Mechanical Properties
| study | task | polymer representation | evaluation context | key takeaway/limitation |
|---|---|---|---|---|
| Park et al. (149) | Tg, Tm, ρ, E | monomer-level molecular graph | random split; RMSE/R2 | backbone rigidity captured |
| Gurnani et al. (126) | Tg, Tm, Td, modulus, strength | periodic repeat-unit graph | RMSE | multitask learning |
| Hu et al. (133) | Tg | monomer graph with SMILES augmentation | Random split; RMSE | SMILES augmentation |
| Qiu et al. (109) | High-Tg PI discovery | monomer graph | screening and experimental validation | interpretability |
| Volgin et al. (151) | Tg | monomer graph | transfer vs direct training | synthetic pretraining |
| Hickey et al. (134) | Tg | monomer graph | random split; RMSE/R2 | small-scale interpretability |
| Qiu et al. (148) | high-Tg | chemically aware GNN | random split; RMSE/R2 | data augmentation |
| POLYMERGNN (244) | Tg, intrinsic viscosity | attention-based graph | random split; RMSE | multitask coupling |
| Li et al. (GATBoost) (144) | Tg | SMILES graph | random split; RMSE/R2 | augmentation boosts accuracy; realism unclear |
5.3. Transport and Dynamic Properties
| study | task | polymer representation | evaluation context | key takeaway/limitation |
|---|---|---|---|---|
| Xie et al. (147) | ionic conductivity, diffusivity | polymer graph | MAE | MD–GNN hybrid |
| Sanchez Medina et al. (124) | activity coefficients at infinite dilution | polymer–solvent graphs | MAE/R2 | transfer learning |
| Wang et al. (108) | intramolecular energy (MLIP) | subgraph-based polymer graph | conformational RMSE vs force fields | near MP2 accuracy achieved; limited to intramolecular PES |
| Kimmig et al. (135) | nanoparticle size | polymer graph | train/test MAPE | good generalization across chemistries |
5.4. Biological Properties
6. Discussion and Outlook
6.1. Understanding GNNs: Tools and Insights
6.2. Coarse-Graining in Graph Design
6.3. Outlook: from Prediction to Design
Develop benchmark data sets that integrate property prediction with attribution labels;
Promote modular and hierarchical GNN architectures supporting hybrid input graphs and dynamic behavior predictions;
Enable interpretability with experimental pipelines, e.g., synthesis constraints and descriptors for applied knowledge extractability.
7. Conclusions
Author Information
- Hector Medina - School of Engineering, Liberty University, Lynchburg, Virginia 24515, United States;
 https://orcid.org/0000-0003-1014-2275;
https://orcid.org/0000-0003-1014-2275;
References
This article references 260 other publications.
- 1Liang, Y.; Yu, L. Development of semiconducting polymers for solar energy harvesting. Polym. Rev. 2010, 50 (4), 454– 473, DOI: 10.1080/15583724.2010.515765Google ScholarThere is no corresponding record for this reference.
- 2Baytekin, B.; Baytekin, H. T.; Grzybowski, B. A. Retrieving and converting energy from polymers: deployable technologies and emerging concepts. Energy Environ. Sci. 2013, 6 (12), 3467– 3482, DOI: 10.1039/c3ee41360hGoogle ScholarThere is no corresponding record for this reference.
- 3Abdelhamid, M. E.; O’Mullane, A. P.; Snook, G. A. Storing energy in plastics: a review on conducting polymers & their role in electrochemical energy storage. RSC Adv. 2015, 5 (15), 11611– 11626, DOI: 10.1039/C4RA15947KGoogle ScholarThere is no corresponding record for this reference.
- 4Fan, J.; Njuguna, J. An introduction to lightweight composite materials and their use in transport structures. In Lightweight Composite Structures in Transport; Elsevier, 2016; pp 3– 34.Google ScholarThere is no corresponding record for this reference.
- 5Singh, J.; Srivastawa, K.; Jana, S.; Dixit, C.; Ravichandran, S. Advancements in lightweight materials for aerospace structures: A comprehensive review. Acceleron Aerosp. J. 2024, 2 (3), 173– 183, DOI: 10.61359/11.2106-2409Google ScholarThere is no corresponding record for this reference.
- 6Yeo, S. J.; Oh, M. J.; Yoo, P. J. Structurally controlled cellular architectures for high-performance ultra-lightweight materials. Adv. Mater. 2019, 31 (34), 1803670 DOI: 10.1002/adma.201803670Google ScholarThere is no corresponding record for this reference.
- 7Cui, H.; Zhao, Q.; Zhang, L.; Du, X. Intelligent polymer-based bioinspired actuators: from monofunction to multifunction. Adv. Intell. Syst. 2020, 2 (11), 2000138 DOI: 10.1002/aisy.202000138Google ScholarThere is no corresponding record for this reference.
- 8Weng, M.; Ding, M.; Zhou, P.; Ye, Y.; Luo, Z.; Ye, X.; Guo, Q.; Chen, L. Multi-functional and integrated actuators made with bio-inspired cobweb carbon nanotube-polymer composites. Chem. Eng. J. 2023, 452, 139146 DOI: 10.1016/j.cej.2022.139146Google ScholarThere is no corresponding record for this reference.
- 9Lendlein, A.; Rehahn, M.; Buchmeiser, M. R.; Haag, R. Polymers in biomedicine and electronics. Macromol. Rapid Commun. 2010, 31 (17), 1487– 1491, DOI: 10.1002/marc.201000426Google ScholarThere is no corresponding record for this reference.
- 10Medina, H.; Child, N. A review of developments in carbon-based nanocomposite electrodes for noninvasive electroencephalography. Sensors 2025, 25 (7), 2274 DOI: 10.3390/s25072274Google ScholarThere is no corresponding record for this reference.
- 11Farmer, C.; Medina, H. Effects of electrostriction on the bifurcated electro-mechanical performance of conical dielectric elastomer actuators and sensors. Robotica 2023, 41 (1), 215– 235, DOI: 10.1017/S0263574722001254Google ScholarThere is no corresponding record for this reference.
- 12Medina, H.; Farmer, C.; Liu, I. Dielectric elastomer-based actuators: a modeling and control review for non-experts. In Actuators; MDPI, 2024; Vol. 13, p 151.Google ScholarThere is no corresponding record for this reference.
- 13Korn, D.; Farmer, C.; Medina, H. A detailed solution framework for the out-of-plane displacement of circular dielectric elastomer actuators. Eng. Rep. 2022, 4 (1), e12442 DOI: 10.1002/eng2.12442Google ScholarThere is no corresponding record for this reference.
- 14Peterson, G. I.; Choi, T.-L. Cascade polymerizations: recent developments in the formation of polymer repeat units by cascade reactions. Chem. Sci. 2020, 11 (19), 4843– 4854, DOI: 10.1039/D0SC01475CGoogle ScholarThere is no corresponding record for this reference.
- 15Cao, C.; Lin, Y. Correlation between the glass transition temperatures and repeating unit structure for high molecular weight polymers. J. Chem. Inf. Comput. Sci. 2003, 43 (2), 643– 650, DOI: 10.1021/ci0202990Google ScholarThere is no corresponding record for this reference.
- 16Tezuka, Y.; Oike, H. Topological polymer chemistry: systematic classification of nonlinear polymer topologies. J. Am. Chem. Soc. 2001, 123 (47), 11570– 11576, DOI: 10.1021/ja0114409Google ScholarThere is no corresponding record for this reference.
- 17Gu, Y.; Zhao, J.; Johnson, J. A. A (macro) molecular-level understanding of polymer network topology. Trends Chem. 2019, 1 (3), 318– 334, DOI: 10.1016/j.trechm.2019.02.017Google ScholarThere is no corresponding record for this reference.
- 18Li, Y.; Abberton, B. C.; Kröger, M.; Liu, W. K. Challenges in multiscale modeling of polymer dynamics. Polymers 2013, 5 (2), 751– 832, DOI: 10.3390/polym5020751Google ScholarThere is no corresponding record for this reference.
- 19Schmid, F. Understanding and modeling polymers: The challenge of multiple scales. ACS Polymers Au 2023, 3 (1), 28– 58, DOI: 10.1021/acspolymersau.2c00049Google ScholarThere is no corresponding record for this reference.
- 20Jones, R. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87 (3), 897, DOI: 10.1103/RevModPhys.87.897Google ScholarThere is no corresponding record for this reference.
- 21Parr, R. G. Density-Functional Theory of Atoms and Molecules; Oxford University Press, 1989.Google ScholarThere is no corresponding record for this reference.
- 22Karplus, M.; McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9 (9), 646– 652, DOI: 10.1038/nsb0902-646Google ScholarThere is no corresponding record for this reference.
- 23Hollingsworth, S. A.; Dror, R. O. Molecular dynamics simulation for all. Neuron 2018, 99 (6), 1129– 1143, DOI: 10.1016/j.neuron.2018.08.011Google ScholarThere is no corresponding record for this reference.
- 24Tuckerman, M. E.; Martyna, G. J. Understanding modern molecular dynamics: Techniques and applications. J. Phys. Chem. B 2000, 104, 159– 178, DOI: 10.1021/jp992433yGoogle ScholarThere is no corresponding record for this reference.
- 25Bhavikatti, S. Finite Element Analysis. New Age International, 2005.Google ScholarThere is no corresponding record for this reference.
- 26Kim, N.-H.; Sankar, B. V.; Kumar, A. V. Introduction to Finite Element Analysis and Design; John Wiley & Sons, 2018.Google ScholarThere is no corresponding record for this reference.
- 27Farmer, C.; Medina, H. Hyperelastics. jl: A julia package for hyperelastic material modelling with a large collection of models. J. Open Source Softw. 2024, 9 (96), 6314 DOI: 10.21105/joss.06314Google ScholarThere is no corresponding record for this reference.
- 28Farmer, C.; Medina, H. Impact-4ccs: Integrated modeling and prediction using ab initio and trained potentials for collision cross sections. J. Comput. Chem. 2025, 46 (11), e70106 DOI: 10.1002/jcc.70106Google ScholarThere is no corresponding record for this reference.
- 29Medina, H.; Farmer, C. Current challenges in monitoring low contaminant levels of per-and polyfluoroalkyl substances in water matrices in the field. Toxics 2024, 12 (8), 610 DOI: 10.3390/toxics12080610Google ScholarThere is no corresponding record for this reference.
- 30Farmer, C.; Medina, H. Universal interatomic potentials with dft for understanding orbital localization in polydimethylsiloxane-amorphous silica nanocomposites. ACS Omega 2025, 10 (49), 60812– 60818, DOI: 10.1021/acsomega.5c09273Google ScholarThere is no corresponding record for this reference.
- 31McCormick, T. M.; Bridges, C. R.; Carrera, E. I.; DiCarmine, P. M.; Gibson, G. L.; Hollinger, J.; Kozycz, L. M.; Seferos, D. S. Conjugated polymers: Evaluating dft methods for more accurate orbital energy modeling. Macromolecules 2013, 46 (10), 3879– 3886, DOI: 10.1021/ma4005023Google ScholarThere is no corresponding record for this reference.
- 32Adekoya, O. C.; Adekoya, G. J.; Sadiku, E. R.; Hamam, Y.; Ray, S. S. Application of dft calculations in designing polymer-based drug delivery systems: An overview. Pharmaceutics 2022, 14 (9), 1972, DOI: 10.3390/pharmaceutics14091972Google ScholarThere is no corresponding record for this reference.
- 33Cohen, A. J.; Mori-Sánchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112 (1), 289– 320, DOI: 10.1021/cr200107zGoogle ScholarThere is no corresponding record for this reference.
- 34Kohn, W.; Becke, A. D.; Parr, R. G. Density functional theory of electronic structure. J. Phys. Chem. A 1996, 100 (31), 12974– 12980, DOI: 10.1021/jp960669lGoogle ScholarThere is no corresponding record for this reference.
- 35Dawson, W.; Degomme, A.; Stella, M.; Nakajima, T.; Ratcliff, L. E.; Genovese, L. Density functional theory calculations of large systems: Interplay between fragments, observables, and computational complexity. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2022, 12 (3), e1574 DOI: 10.1002/wcms.1574Google ScholarThere is no corresponding record for this reference.
- 36Bishop, M.; Kalos, M.; Frisch, H. Molecular dynamics of polymeric systems. J. Chem. Phys. 1979, 70 (3), 1299– 1304, DOI: 10.1063/1.437567Google ScholarThere is no corresponding record for this reference.
- 37Han, J.; Gee, R. H.; Boyd, R. H. Glass transition temperatures of polymers from molecular dynamics simulations. Macromolecules 1994, 27 (26), 7781– 7784, DOI: 10.1021/ma00104a036Google ScholarThere is no corresponding record for this reference.
- 38Lau, D.; Jian, W.; Yu, Z.; Hui, D. Nano-engineering of construction materials using molecular dynamics simulations: Prospects and challenges. Compos. Part B 2018, 143, 282– 291, DOI: 10.1016/j.compositesb.2018.01.014Google ScholarThere is no corresponding record for this reference.
- 39Mackerle, J. Finite element analysis and simulation of polymersan addendum: a bibliography (1996–2002). Modell. Simul. Mater. Sci. Eng. 2003, 11 (2), 195 DOI: 10.1088/0965-0393/11/2/307Google ScholarThere is no corresponding record for this reference.
- 40Zeng, X.; Brown, L. P.; Endruweit, A.; Matveev, M.; Long, A. C. Geometrical modelling of 3d woven reinforcements for polymer composites: Prediction of fabric permeability and composite mechanical properties. Compos. Part A: Appl. Sci. Manuf. 2014, 56, 150– 160, DOI: 10.1016/j.compositesa.2013.10.004Google ScholarThere is no corresponding record for this reference.
- 41Alshahrani, H. Characterization and finite element modeling of coupled properties during polymer composites forming processes. Mech. Mater. 2020, 144, 103370 DOI: 10.1016/j.mechmat.2020.103370Google ScholarThere is no corresponding record for this reference.
- 42Thompson, A. P.; Aktulga, H. M.; Berger, R.; Bolintineanu, D. S.; Brown, W. M.; Crozier, P. S.; In’t Veld, P. J.; Kohlmeyer, A.; Moore, S. G.; Nguyen, T. D.; Shan, R.; Stevens, M. J.; Tranchida, J.; Trott, C.; Plimpton, S. J. Lammps-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171 DOI: 10.1016/j.cpc.2021.108171Google ScholarThere is no corresponding record for this reference.
- 43Ratcliff, L. E.; Mohr, S.; Huhs, G.; Deutsch, T.; Masella, M.; Genovese, L. Challenges in large scale quantum mechanical calculations. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2017, 7 (1), e1290 DOI: 10.1002/wcms.1290Google ScholarThere is no corresponding record for this reference.
- 44Fredrickson, G. H. Computational field theory of polymers: opportunities and challenges. Soft Matter 2007, 3 (11), 1329– 1334, DOI: 10.1039/b710604aGoogle ScholarThere is no corresponding record for this reference.
- 45Sindu, B.; Hamaekers, J. Feature-based prediction of properties of cross-linked epoxy polymers by molecular dynamics and machine learning techniques. Modell. Simul. Mater. Sci. Eng. 2025, 33 (6), 065010 DOI: 10.1088/1361-651X/adf56cGoogle ScholarThere is no corresponding record for this reference.
- 46Chew, A. K.; Afzal, M. A. F.; Chandrasekaran, A.; Kamps, J. H.; Ramakrishnan, V. Designing the next generation of polymers with machine learning and physics-based models. Mach. Learn. Sci. Technol. 2024, 5 (4), 045031 DOI: 10.1088/2632-2153/ad88d7Google ScholarThere is no corresponding record for this reference.
- 47He, J.; Rong, F. Prediction of molecularly imprinted polymer binding affinity based on graph neural networks. J. Phys.: Conference Series 2025, 3030, 012009Google ScholarThere is no corresponding record for this reference.
- 48Nagasawa, S.; Al-Naamani, E.; Saeki, A. Computer-aided screening of conjugated polymers for organic solar cell: classification by random forest. J. Phys. Chem. Lett. 2018, 9 (10), 2639– 2646, DOI: 10.1021/acs.jpclett.8b00635Google ScholarThere is no corresponding record for this reference.
- 49Kishino, M.; Matsumoto, K.; Kobayashi, Y.; Taguchi, R.; Akamatsu, N.; Shishido, A. Fatigue life prediction of bending polymer films using random forest. Int. J. Fatigue 2023, 166, 107230 DOI: 10.1016/j.ijfatigue.2022.107230Google ScholarThere is no corresponding record for this reference.
- 50Arora, A.; Lin, T.-S.; Rebello, N. J.; Av-Ron, S. H.; Mochigase, H.; Olsen, B. D. Random forest predictor for diblock copolymer phase behavior. ACS Macro Lett. 2021, 10 (11), 1339– 1345, DOI: 10.1021/acsmacrolett.1c00521Google ScholarThere is no corresponding record for this reference.
- 51Malashin, I.; Tynchenko, V.; Gantimurov, A.; Nelyub, V.; Borodulin, A. Support vector machines in polymer science: a review. Polymers 2025, 17 (4), 491, DOI: 10.3390/polym17040491Google ScholarThere is no corresponding record for this reference.
- 52Ziaee, H.; Hosseini, S. M.; Sharafpoor, A.; Fazavi, M.; Ghiasi, M. M.; Bahadori, A. Prediction of solubility of carbon dioxide in different polymers using support vector machine algorithm. J. Taiwan Inst. Chem. Eng. 2015, 46, 205– 213, DOI: 10.1016/j.jtice.2014.09.015Google ScholarThere is no corresponding record for this reference.
- 53Chen, F.-C. Virtual screening of conjugated polymers for organic photovoltaic devices using support vector machines and ensemble learning. Int. J. Polym. Sci. 2019, 2019 (1), 4538514 DOI: 10.1155/2019/4538514Google ScholarThere is no corresponding record for this reference.
- 54Brereton, R. G.; Lloyd, G. R. Support vector machines for classification and regression. Analyst 2010, 135 (2), 230– 267, DOI: 10.1039/B918972FGoogle ScholarThere is no corresponding record for this reference.
- 55Youshia, J.; Ali, M. E.; Lamprecht, A. Artificial neural network based particle size prediction of polymeric nanoparticles. Eur. J. Pharm. Biopharm. 2017, 119, 333– 342, DOI: 10.1016/j.ejpb.2017.06.030Google ScholarThere is no corresponding record for this reference.
- 56Zhang, Z.; Friedrich, K. Artificial neural networks applied to polymer composites: a review. Compos. Sci. Technol. 2003, 63 (14), 2029– 2044, DOI: 10.1016/S0266-3538(03)00106-4Google ScholarThere is no corresponding record for this reference.
- 57El Kadi, H. Modeling the mechanical behavior of fiber-reinforced polymeric composite materials using artificial neural networksa review. Compos. Struct. 2006, 73 (1), 1– 23, DOI: 10.1016/j.compstruct.2005.01.020Google ScholarThere is no corresponding record for this reference.
- 58Liu, W.; Cao, C. Artificial neural network prediction of glass transition temperature of polymers. Colloid Polym. Sci. 2009, 287 (7), 811– 818, DOI: 10.1007/s00396-009-2035-yGoogle ScholarThere is no corresponding record for this reference.
- 59Chen, L.; Pilania, G.; Batra, R.; Huan, T. D.; Kim, C.; Kuenneth, C.; Ramprasad, R. Polymer informatics: Current status and critical next steps. Mater. Sci. Eng. R 2021, 144, 100595 DOI: 10.1016/j.mser.2020.100595Google ScholarThere is no corresponding record for this reference.
- 60Butler, K. T.; Davies, D. W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547– 555, DOI: 10.1038/s41586-018-0337-2Google ScholarThere is no corresponding record for this reference.
- 61Schleder, G. R.; Padilha, A. C. M.; Acosta, C. M.; Costa, M.; Fazzio, A. From dft to machine learning: Recent approaches to materials science-a review. J. Chem. Inf. Model. 2019, 2 (4), 1347– 1356, DOI: 10.1088/2515-7639/ab084bGoogle ScholarThere is no corresponding record for this reference.
- 62Sha, W.; Li, Y.; Tang, S.; Tian, J.; Zhao, Y.; Guo, Y.; Zhang, W.; Zhang, X.; Lu, S.; Cao, Y.-C.; Cheng, S. Machine learning in polymer informatics. InfoMat 2021, 3 (4), 353– 361, DOI: 10.1002/inf2.12167Google ScholarThere is no corresponding record for this reference.
- 63Martin, T. B.; Audus, D. J. Emerging trends in machine learning: a polymer perspective. ACS Polymers Au 2023, 3 (3), 239– 258, DOI: 10.1021/acspolymersau.2c00053Google ScholarThere is no corresponding record for this reference.
- 64Ramprasad, R.; Batra, R.; Pilania, G.; Mannodi-Kanakkithodi, A.; Kim, C. Machine learning in materials informatics: recent applications and prospects. npj Comput. Mater. 2017, 3 (1), 54, DOI: 10.1038/s41524-017-0056-5Google ScholarThere is no corresponding record for this reference.
- 65Chen, G.; Shen, Z.; Iyer, A.; Ghumman, U. F.; Tang, S.; Bi, J.; Chen, W.; Li, Y. Machine-learning-assisted de novo design of organic molecules and polymers: opportunities and challenges. Polymers 2020, 12 (1), 163, DOI: 10.3390/polym12010163Google ScholarThere is no corresponding record for this reference.
- 66Wu, S.; Yamada, H.; Hayashi, Y.; Zamengo, M.; Yoshida, R. Potentials and challenges of polymer informatics: exploiting machine learning for polymer design, 2020, arXiv:2010.07683. arXiv.org e-Print archive https://arxiv.org/abs/2010.07683.Google ScholarThere is no corresponding record for this reference.
- 67Kumar, J. N.; Li, Q.; Jun, Y. Challenges and opportunities of polymer design with machine learning and high throughput experimentation. MRS Commun. 2019, 9 (2), 537– 544, DOI: 10.1557/mrc.2019.54Google ScholarThere is no corresponding record for this reference.
- 68Wieder, O.; Kohlbacher, S.; Kuenemann, M.; Garon, A.; Ducrot, P.; Seidel, T.; Langer, T. A compact review of molecular property prediction with graph neural networks. Drug Discovery Today: Technol. 2020, 37, 1– 12, DOI: 10.1016/j.ddtec.2020.11.009Google ScholarThere is no corresponding record for this reference.
- 69Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Philip, S. Y. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2021, 32 (1), 4– 24, DOI: 10.1109/TNNLS.2020.2978386Google ScholarThere is no corresponding record for this reference.
- 70Gao, Q.; Dukker, T.; Schweidtmann, A. M.; Weber, J. M. Self-supervised graph neural networks for polymer property prediction. Mol. Syst. Des. Eng. 2024, 9 (11), 1130– 1143, DOI: 10.1039/D4ME00088AGoogle ScholarThere is no corresponding record for this reference.
- 71Inae, E.; Liu, Y.; Zhu, Y.; Xu, J.; Liu, G.; Zhang, R.; Luo, T.; Jiang, M. Modeling Polymers with Neural Networks; American Chemical Society, 2025.Google ScholarThere is no corresponding record for this reference.
- 72Gurney, K. An Introduction to Neural Networks; CRC Press, 2018.Google ScholarThere is no corresponding record for this reference.
- 73Haykin, S.; Network, N. A comprehensive foundation. Neural Netw. 2004, 2, 41Google ScholarThere is no corresponding record for this reference.
- 74LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86 (11), 2278– 2324, DOI: 10.1109/5.726791Google ScholarThere is no corresponding record for this reference.
- 75Li, Z.; Liu, F.; Yang, W.; Peng, S.; Zhou, J. A survey of convolutional neural networks: analysis, applications, and prospects. IEEE Trans. Neural Netw. Learn. Syst. 2022, 33 (12), 6999– 7019, DOI: 10.1109/TNNLS.2021.3084827Google ScholarThere is no corresponding record for this reference.
- 76Gu, J.; Wang, Z.; Kuen, J.; Ma, L.; Shahroudy, A.; Shuai, B.; Liu, T.; Wang, X.; Wang, G.; Cai, J.; Chen, T. Recent advances in convolutional neural networks. Pattern Recognit. 2018, 77, 354– 377, DOI: 10.1016/j.patcog.2017.10.013Google ScholarThere is no corresponding record for this reference.
- 77Ge, W.; De Silva, R.; Fan, Y.; Sisson, S. A.; Stenzel, M. H. Machine learning in polymer research. Adv. Mater. 2025, 37 (11), 2413695 DOI: 10.1002/adma.202413695Google ScholarThere is no corresponding record for this reference.
- 78Platonov, O.; Kuznedelev, D.; Diskin, M.; Babenko, A.; Prokhorenkova, L. A critical look at the evaluation of gnns under heterophily: Are we really making progress? 2023, arXiv:2302.11640. arXiv.org e-Print archive https://arxiv.org/abs/2302.11640.Google ScholarThere is no corresponding record for this reference.
- 79Wang, Y.; Li, Z.; Barati Farimani, A. Graph neural networks for molecules. In Machine Learning in Molecular Sciences; Springer, 2023; pp 21– 66.Google ScholarThere is no corresponding record for this reference.
- 80Hamilton, W.; Ying, Z.; Leskovec, J. Inductive representation learning on large graphs. In NIPS’17: Proceedings of the 31st International Conference on Neural Information Processing Systems; Department of Computer Science: Stanford, CA, 2017; pp 1025– 1035.Google ScholarThere is no corresponding record for this reference.
- 81Xu, K.; Hu, W.; Leskovec, J.; Jegelka, S. How powerful are graph neural networks? 2018, arXiv:1810.00826. arXiv.org e-Print archive https://arxiv.org/abs/1810.00826.Google ScholarThere is no corresponding record for this reference.
- 82Sharma, K.; Lee, Y.-C.; Nambi, S.; Salian, A.; Shah, S.; Kim, S.-W.; Kumar, S. A survey of graph neural networks for social recommender systems. ACM Computing Surveys 2024, 56 (10), 1– 34, DOI: 10.1145/3661821Google ScholarThere is no corresponding record for this reference.
- 83Kumar, S.; Mallik, A.; Khetarpal, A.; Panda, B. S. Influence maximization in social networks using graph embedding and graph neural network. Informat. Sci. 2022, 607, 1617– 1636, DOI: 10.1016/j.ins.2022.06.075Google ScholarThere is no corresponding record for this reference.
- 84Zhang, X.-M.; Liang, L.; Liu, L.; Tang, M.-J. Graph neural networks and their current applications in bioinformatics. Front. Genetics 2021, 12, 690049 DOI: 10.3389/fgene.2021.690049Google ScholarThere is no corresponding record for this reference.
- 85Malla, A. M.; Banka, A. A. A systematic review of deep graph neural networks: Challenges, classification, architectures, applications & potential utility in bioinformatics, 2023, arXiv:2311.02127. arXiv.org e-Print archive https://arxiv.org/abs/2311.02127.Google ScholarThere is no corresponding record for this reference.
- 86Zheng, P.; Wang, S.; Wang, X.; Zeng, X. Artificial intelligence in bioinformatics and drug repurposing: methods and applications. In Frontiers Research Topics , 2022.Google ScholarThere is no corresponding record for this reference.
- 87Reiser, P.; Neubert, M.; Eberhard, A.; Torresi, L.; Zhou, C.; Shao, C.; Metni, H.; van Hoesel, C.; Schopmans, H.; Sommer, T.; Friederich, P. Graph neural networks for materials science and chemistry. Commun. Mater. 2022, 3 (1), 93, DOI: 10.1038/s43246-022-00315-6Google ScholarThere is no corresponding record for this reference.
- 88Fung, V.; Zhang, J.; Juarez, E.; Sumpter, B. G. Benchmarking graph neural networks for materials chemistry. npj Computat. Mater. 2021, 7 (1), 84, DOI: 10.1038/s41524-021-00554-0Google ScholarThere is no corresponding record for this reference.
- 89Maurizi, M.; Gao, C.; Berto, F. Predicting stress, strain and deformation fields in materials and structures with graph neural networks. Sci. Rep. 2022, 12 (1), 21834 DOI: 10.1038/s41598-022-26424-3Google ScholarThere is no corresponding record for this reference.
- 90Kipf, T. N.; Welling, M. Semi-supervised classification with graph convolutional networks, 2016, arXiv:1609.02907. arXiv.org e-Print archive https://arxiv.org/abs/1609.02907.Google ScholarThere is no corresponding record for this reference.
- 91Gilmer, J.; Schoenholz, S. S.; Riley, P. F.; Vinyals, O.; Dahl, G. E. Neural message passing for quantum chemistry. In International Conference on Machine Learning; PMLR, 2017; pp 1263– 1272.Google ScholarThere is no corresponding record for this reference.
- 92Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. “ Graph attention networks, 2017, arXiv:1710.10903. arXiv.org e-Print archive https://arxiv.org/abs/1710.10903.Google ScholarThere is no corresponding record for this reference.
- 93Liu, G.; Inae, E.; Jiang, M. Deep learning for polymer property prediction. In Deep Learning for Polymer Discovery: Foundation and Advances; Springer, 2025; pp 17– 35.Google ScholarThere is no corresponding record for this reference.
- 94Dong, C.; Li, D.; Liu, J. Glass transition temperature prediction of polymers via graph reinforcement learning. Langmuir 2024, 40 (35), 18568– 18580, DOI: 10.1021/acs.langmuir.4c01906Google ScholarThere is no corresponding record for this reference.
- 95Wang, X.; Liao, L.; Huang, C.; Wu, X. Prediction of mechanical properties of cross-linked polymer interface by graph convolution network. Acta Mechanica Sinica 2026, 42 (3), 424627 DOI: 10.1007/s10409-024-24627-xGoogle ScholarThere is no corresponding record for this reference.
- 96Safari, H.; Bavarian, M. Enhancing polymer reaction engineering through the power of machine learning. In Systems and Control Transactions , 2024; p 157792.Google ScholarThere is no corresponding record for this reference.
- 97Liao, H.-C.; Lin, Y.-H.; Peng, C.-H.; Li, Y.-P. Directed message passing neural networks for accurate prediction of polymer-solvent interaction parameters. ACS Engineering Au 2025, 5, 530, DOI: 10.1021/acsengineeringau.5c00027Google ScholarThere is no corresponding record for this reference.
- 98Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B. A.; Thiessen, P. A.; Yu, B. Pubchem 2023 update. Nucleic Acids Res. 2023, 51 (D1), D1373– D1380, DOI: 10.1093/nar/gkac956Google ScholarThere is no corresponding record for this reference.
- 99Gao, Z.-Y.; Liu, G.-Q. Recent progress of web-enable material database and a case study of nims and matweb. J. Mater. Eng. 2013, 3 (11), 89– 96, DOI: 10.3969/j.issn.1001-4381.2013.06.001Google ScholarThere is no corresponding record for this reference.
- 100Wypych, G. Handbook of Polymers; Elsevier, 2022.Google ScholarThere is no corresponding record for this reference.
- 101Patra, A.; Batra, R.; Chandrasekaran, A.; Kim, C.; Huan, T. D.; Ramprasad, R. A multi-fidelity information-fusion approach to machine learn and predict polymer bandgap. Comput. Mater. Sci. 2020, 172, 109286 DOI: 10.1016/j.commatsci.2019.109286Google ScholarThere is no corresponding record for this reference.
- 102Dhamankar, S.; Webb, M. A. Chemically specific coarse-graining of polymers: methods and prospects. J. Polym. Sci. 2021, 59 (22), 2613– 2643, DOI: 10.1002/pol.20210555Google ScholarThere is no corresponding record for this reference.
- 103Müller-Plathe, F. Coarse-graining in polymer simulation: from the atomistic to the mesoscopic scale and back. ChemPhysChem 2002, 3 (9), 754– 769, DOI: 10.1002/1439-7641(20020916)3:9<754::AID-CPHC754>3.0.CO;2-UGoogle ScholarThere is no corresponding record for this reference.
- 104Harmandaris, V. A.; Reith, D.; Van der Vegt, N. F.; Kremer, K. Comparison between coarse-graining models for polymer systems: Two mapping schemes for polystyrene. Macromol. Chem. Phys. 2007, 208 (19–20), 2109– 2120, DOI: 10.1002/macp.200700245Google ScholarThere is no corresponding record for this reference.
- 105Padding, J. T.; Briels, W. J. Systematic coarse-graining of the dynamics of entangled polymer melts: the road from chemistry to rheology. J. Phys.: Condens. Matter 2011, 23 (23), 233101 DOI: 10.1088/0953-8984/23/23/233101Google ScholarThere is no corresponding record for this reference.
- 106Zhang, X.; Sheng, Y.; Liu, X.; Yang, J.; Goddard, W. A., III; Ye, C.; Zhang, W. Polymer-unit graph: Advancing interpretability in graph neural network machine learning for organic polymer semiconductor materials. J. Chem. Theory Comput. 2024, 20 (7), 2908– 2920, DOI: 10.1021/acs.jctc.3c01385Google ScholarThere is no corresponding record for this reference.
- 107Mohapatra, S.; An, J.; Gómez-Bombarelli, R. Chemistry-informed macromolecule graph representation for similarity computation, unsupervised and supervised learning. Machine Learning: Sci. Technol. 2022, 3 (1), 015028 DOI: 10.1088/2632-2153/ac545eGoogle ScholarThere is no corresponding record for this reference.
- 108Wang, X.; Xu, Y.; Zheng, H.; Yu, K. A scalable graph neural network method for developing an accurate force field of large flexible organic molecules. J. Phys. Chem. Lett. 2021, 12 (33), 7982– 7987, DOI: 10.1021/acs.jpclett.1c02214Google ScholarThere is no corresponding record for this reference.
- 109Qiu, H.; Qiu, X.; Dai, X.; Sun, Z.-Y. Design of polyimides with targeted glass transition temperature using a graph neural network. J. Mater. Chem. C 2023, 11 (8), 2930– 2940, DOI: 10.1039/D2TC05174EGoogle ScholarThere is no corresponding record for this reference.
- 110Zeng, M.; Kumar, J. N.; Zeng, Z.; Savitha, R.; Chandrasekhar, V. R.; Hippalgaonkar, K. “ Graph convolutional neural networks for polymers property prediction, 2018, arXiv:1811.06231. arXiv.org e-Print archive https://arxiv.org/abs/1811.06231.Google ScholarThere is no corresponding record for this reference.
- 111Queen, O.; McCarver, G. A.; Thatigotla, S.; Abolins, B. P.; Brown, C. L.; Maroulas, V.; Vogiatzis, K. D. Polymer graph neural networks for multitask property learning. npj Computat. Mater. 2023, 9, 90, DOI: 10.1038/s41524-023-01034-3Google ScholarThere is no corresponding record for this reference.
- 112Bongini, P.; Bianchini, M.; Scarselli, F. Molecular generative graph neural networks for drug discovery. Neurocomputing 2021, 450, 242– 252, DOI: 10.1016/j.neucom.2021.04.039Google ScholarThere is no corresponding record for this reference.
- 113Scarselli, F.; Gori, M.; Tsoi, A. C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Transact. Neural Networks 2009, 20 (1), 61– 80, DOI: 10.1109/TNN.2008.2005605Google ScholarThere is no corresponding record for this reference.
- 114Dwivedi, V. P.; Joshi, C. K.; Luu, A. T.; Laurent, T.; Bengio, Y.; Bresson, X. Benchmarking graph neural networks. J. Machine Learning Res. 2023, 24 (43), 1– 48Google ScholarThere is no corresponding record for this reference.
- 115Veličković, P. Everything is connected: Graph neural networks. Curr. Opin. Struct. Biol. 2023, 79, 102538 DOI: 10.1016/j.sbi.2023.102538Google ScholarThere is no corresponding record for this reference.
- 116Jegelka, S. Theory of graph neural networks: Representation and learning. In International Congress of Mathematicians , 2022; pp 1– 23.Google ScholarThere is no corresponding record for this reference.
- 117Liu, Z.; Zhou, J. Introduction to Graph Neural Networks; Springer Nature, 2022.Google ScholarThere is no corresponding record for this reference.
- 118Tang, M.; Li, B.; Chen, H. Application of message passing neural networks for molecular property prediction. Curr. Opin. Struct. Biol. 2023, 81, 102616 DOI: 10.1016/j.sbi.2023.102616Google ScholarThere is no corresponding record for this reference.
- 119Hasebe, T. Knowledge-embedded message-passing neural networks: improving molecular property prediction with human knowledge. ACS omega 2021, 6 (42), 27955– 27967, DOI: 10.1021/acsomega.1c03839Google ScholarThere is no corresponding record for this reference.
- 120Zhang, H.; Lin, Y.; Li, S.; Dai, M.; Zhang, Y.; Huang, L.; Pang, J.; Wu, P.; Peng, J.; Tang, Z.; Ding, P.; Xiao, W.; Song, N.; Dongbo, D. Substructure-enhanced mpnn for polymer discovery and knowledge: A study in predicting glass transition temperature. Macromolecules 2025, 58, 9515, DOI: 10.1021/acs.macromol.4c02859Google ScholarThere is no corresponding record for this reference.
- 121Aldeghi, M.; Coley, C. W. A graph representation of molecular ensembles for polymer property prediction. Chem. Sci. 2022, 13 (35), 10486– 10498, DOI: 10.1039/D2SC02839EGoogle ScholarThere is no corresponding record for this reference.
- 122Heid, E.; Greenman, K. P.; Chung, Y.; Li, S.-C.; Graff, D. E.; Vermeire, F. H.; Wu, H.; Green, W. H.; McGill, C. J. Chemprop: a machine learning package for chemical property prediction. J. Chem. Inf. Model. 2024, 64 (1), 9– 17, DOI: 10.1021/acs.jcim.3c01250Google ScholarThere is no corresponding record for this reference.
- 123Antoniuk, E. R.; Li, P.; Kailkhura, B.; Hiszpanski, A. M. Representing polymers as periodic graphs with learned descriptors for accurate polymer property predictions. J. Chem. Inf. Model. 2022, 62, 5435– 5445, DOI: 10.1021/acs.jcim.2c00875Google ScholarThere is no corresponding record for this reference.
- 124Sanchez Medina, E. I.; Kunchapu, S.; Sundmacher, K. Gibbs-helmholtz graph neural network for the prediction of activity coefficients of polymer solutions at infinite dilution. J. Phys. Chem. A 2023, 127 (46), 9863– 9873, DOI: 10.1021/acs.jpca.3c05892Google ScholarThere is no corresponding record for this reference.
- 125Battaglia, P. W.; Hamrick, J. B.; Bapst, V.; Sanchez-Gonzalez, A.; Zambaldi, V.; Malinowski, M.; Tacchetti, A.; Raposo, D.; Santoro, A.; Faulkner, R.; C, G.; F, S.; Ballard, A.; Justin, G.; George, D.; Ashish, V.; Kelsey, A.; Charles, N.; Victoria, L.; Chris, D.; Nicolas, H.; Dann, W.; Pushmeet, K.; Matt, B.; Oriol, V.; Yujia, L.; Razvan, P. Relational inductive biases, deep learning, and graph networks, 2018, arXiv:1806.01261. arXiv.org e-Print archive https://arxiv.org/abs/1806.01261.Google ScholarThere is no corresponding record for this reference.
- 126Gurnani, R.; Kuenneth, C.; Toland, A.; Ramprasad, R. Polymer informatics at scale with multitask graph neural networks. Chem. Mater. 2023, 35 (6), 1560– 1567, DOI: 10.1021/acs.chemmater.2c02991Google ScholarThere is no corresponding record for this reference.
- 127St John, P. C.; Phillips, C.; Kemper, T. W.; Wilson, A. N.; Guan, Y.; Crowley, M. F.; Nimlos, M. R.; Larsen, R. E. Message-passing neural networks for high-throughput polymer screening. J. Chem. Phys. 2019, 150, 234111 DOI: 10.1063/1.5099132Google ScholarThere is no corresponding record for this reference.
- 128Li, Y.; Zemel, R.; Brockschmidt, M.; Tarlow, D. Gated graph sequence neural networks. In Proceedings of ICLR16 , 2016.Google ScholarThere is no corresponding record for this reference.
- 129Dey, R.; Salem, F. M. Gate-variants of gated recurrent unit (gru) neural networks. In 2017 IEEE 60th international midwest symposium on circuits and systems (MWSCAS); IEEE, 2017; pp 1597– 1600.Google ScholarThere is no corresponding record for this reference.
- 130Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Briefings Bioinf. 2020, 21 (3), 919– 935, DOI: 10.1093/bib/bbz042Google ScholarThere is no corresponding record for this reference.
- 131Xu, X.; Zhao, X.; Wei, M.; Li, Z. A comprehensive review of graph convolutional networks: approaches and applications. Electronic Res. Archive 2023, 31 (7), 4185– 4215, DOI: 10.3934/era.2023213Google ScholarThere is no corresponding record for this reference.
- 132Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph neural networks: A review of methods and applications. IEEE Transactions on Pattern Analysis and Machine Intelligence 2019, 42, 283– 300Google ScholarThere is no corresponding record for this reference.
- 133Hu, J.; Li, Z.; Lin, J.; Zhang, L. Prediction and interpretability of glass transition temperature of homopolymers by data-augmented graph convolutional neural networks. ACS Appl. Mater. Interfaces 2023, 15 (46), 54006– 54017, DOI: 10.1021/acsami.3c13698Google ScholarThere is no corresponding record for this reference.
- 134Hickey, K.; Feinstein, J.; Sivaraman, G.; MacDonell, M.; Yan, E.; Matherson, C.; Coia, S.; Xu, J.; Picel, K. Applying machine learning and quantum chemistry to predict the glass transition temperatures of polymers. Comput. Mater. Sci. 2024, 238, 112933 DOI: 10.1016/j.commatsci.2024.112933Google ScholarThere is no corresponding record for this reference.
- 135Kimmig, J.; Schuett, T.; Vollrath, A.; Zechel, S.; Schubert, U. S. Prediction of nanoparticle sizes for arbitrary methacrylates using artificial neuronal networks. Adv. Sci. 2021, 8 (23), 2102429 DOI: 10.1002/advs.202102429Google ScholarThere is no corresponding record for this reference.
- 136Tian, Y.; Zhang, Y.; Zhang, H. Recent advances in stochastic gradient descent in deep learning. Mathematics 2023, 11 (3), 682, DOI: 10.3390/math11030682Google ScholarThere is no corresponding record for this reference.
- 137Merity, S. “ Single headed attention rnn: Stop thinking with your head, 2019, arXiv:1911.11423. arXiv.org e-Print archive https://arxiv.org/abs/1911.11423.Google ScholarThere is no corresponding record for this reference.
- 138Inoue, H. Multi-sample dropout for accelerated training and better generalization, 2019, arXiv:1905.09788. arXiv.org e-Print archive https://arxiv.org/abs/1905.09788.Google ScholarThere is no corresponding record for this reference.
- 139Cho, K.; Van Merriënboer, B.; Bahdanau, D.; Bengio, Y. On the properties of neural machine translation: Encoder-decoder approaches, 2014, arXiv:1409.1259. arXiv.org e-Print archive https://arxiv.org/abs/1409.1259.Google ScholarThere is no corresponding record for this reference.
- 140Morris, C.; Ritzert, M.; Fey, M.; Hamilton, W. L.; Lenssen, J. E.; Rattan, G.; Grohe, M. Weisfeiler and leman go neural: Higher-order graph neural networks. Proceedings of the AAAI Conference on Artificial Intelligence 2019, 33, 4602– 4609, DOI: 10.1609/aaai.v33i01.33014602Google ScholarThere is no corresponding record for this reference.
- 141Ye, X.-b.; Guan, Q.; Luo, W.; Fang, L.; Lai, Z.-R.; Wang, J. Molecular substructure graph attention network for molecular property identification in drug discovery. Pattern Recognition 2022, 128, 108659 DOI: 10.1016/j.patcog.2022.108659Google ScholarThere is no corresponding record for this reference.
- 142Vrahatis, A. G.; Lazaros, K.; Kotsiantis, S. Graph attention networks: a comprehensive review of methods and applications. Future Internet 2024, 16 (9), 318, DOI: 10.3390/fi16090318Google ScholarThere is no corresponding record for this reference.
- 143Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H.; Z M Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 2020, 63 (16), 8749– 8760, DOI: 10.1021/acs.jmedchem.9b00959Google ScholarThere is no corresponding record for this reference.
- 144Li, D.; Ru, Y.; Liu, J. Gatboost: Mining graph attention networks-based important substructures of polymers for a better property prediction. Mater. Today Commun. 2024, 38, 107577 DOI: 10.1016/j.mtcomm.2023.107577Google ScholarThere is no corresponding record for this reference.
- 145Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining , 2016; pp 785– 794.Google ScholarThere is no corresponding record for this reference.
- 146Liu, T.; Jiang, A.; Zhou, J.; Li, M.; Kwan, H. K. Graphsage-based dynamic spatial-temporal graph convolutional network for traffic prediction. IEEE Transactions on Intelligent Transportation Systems 2023, 24 (10), 11210– 11224, DOI: 10.1109/TITS.2023.3279929Google ScholarThere is no corresponding record for this reference.
- 147Xie, T.; France-Lanord, A.; Wang, Y.; Lopez, J.; Stolberg, M. A.; Hill, M.; Leverick, G. M.; Gomez-Bombarelli, R.; Johnson, J. A.; Shao-Horn, Y.; Grossman, J. C. Accelerating amorphous polymer electrolyte screening by learning to reduce errors in molecular dynamics simulated properties. Nat. Commun. 2022, 13 (1), 3415 DOI: 10.1038/s41467-022-30994-1Google ScholarThere is no corresponding record for this reference.
- 148Qiu, H.; Wang, J.; Qiu, X.; Dai, X.; Sun, Z.-Y. Heat-resistant polymer discovery by utilizing interpretable graph neural network with small data. Macromolecules 2024, 57 (8), 3515– 3528, DOI: 10.1021/acs.macromol.4c00508Google ScholarThere is no corresponding record for this reference.
- 149Park, J.; Shim, Y.; Lee, F.; Rammohan, A.; Goyal, S.; Shim, M.; Jeong, C.; Kim, D. S. Prediction and interpretation of polymer properties using the graph convolutional network. ACS Polymers Au 2022, 2, 213, DOI: 10.1021/acspolymersau.1c00050Google ScholarThere is no corresponding record for this reference.
- 150Burkholz, R.; Quackenbush, J.; Bojar, D. Using graph convolutional neural networks to learn a representation for glycans. Cell Rep. 2021, 35 (11), 109251, DOI: 10.1016/j.celrep.2021.109251Google ScholarThere is no corresponding record for this reference.
- 151Volgin, I. V.; Batyr, P. A.; Matseevich, A. V.; Dobrovskiy, A. Y.; Andreeva, M. V.; Nazarychev, V. M.; Larin, S. V.; Goikhman, M. Y.; Vizilter, Y. V.; Askadskii, A. A.; Lyulin, S. V. Machine learning with enormous “synthetic” data sets: Predicting glass transition temperature of polyimides using graph convolutional neural networks. ACS Omega 2022, 7 (48), 43678– 43691, DOI: 10.1021/acsomega.2c04649Google ScholarThere is no corresponding record for this reference.
- 152Otsuka, S.; Kuwajima, I.; Hosoya, J.; Xu, Y.; Yamazaki, M. Polyinfo: Polymer database for polymeric materials design. In 2011 International Conference on Emerging Intelligent Data and Web Technologies; IEEE, 2011; pp 22– 29.Google ScholarThere is no corresponding record for this reference.
- 153Brandrup, J.; Immergut, E. H.; Grulke, E. A.; Abe, A.; Bloch, D. R. Polymer Handbook; Wiley: New York, 1999; Vol. 89.Google ScholarThere is no corresponding record for this reference.
- 154Mark, J. E. Physical Properties of Polymers Handbook; Springer, 2007; Vol. 1076.Google ScholarThere is no corresponding record for this reference.
- 155Ma, R.; Luo, T. Pi1m: a benchmark database for polymer informatics. J. Chem. Inf. Model. 2020, 60 (10), 4684– 4690, DOI: 10.1021/acs.jcim.0c00726Google ScholarThere is no corresponding record for this reference.
- 156Walsh, D. J.; Zou, W.; Schneider, L.; Mello, R.; Deagen, M. E.; Mysona, J.; Lin, T.-S.; de Pablo, J. J.; Jensen, K. F.; Audus, D. J. Community resource for innovation in polymer technology (cript): a scalable polymer material data structure, 2023.Google ScholarThere is no corresponding record for this reference.
- 157Yan, C.; Li, G. The rise of machine learning in polymer discovery. Advanced Intelligent Systems 2023, 5 (4), 2200243 DOI: 10.1002/aisy.202200243Google ScholarThere is no corresponding record for this reference.
- 158Levine, D. S.; Shuaibi, M.; Spotte-Smith, E. W. C.; Taylor, M. G.; Hasyim, M. R.; Michel, K.; Batatia, I.; Csányi, G.; Dzamba, M.; Eastman, P. The open molecules 2025 (omol25) dataset, evaluations, and models, 2025, arXiv:2505.08762. arXiv.org e-Print archive https://arxiv.org/abs/2505.08762.Google ScholarThere is no corresponding record for this reference.
- 159Mark, J. E. Polymer Data Handbook, Oxford University Press, New York, NY, 2009.Google ScholarThere is no corresponding record for this reference.
- 160Hayashi, Y.; Shiomi, J.; Morikawa, J.; Yoshida, R. Radonpy: automated physical property calculation using all-atom classical molecular dynamics simulations for polymer informatics. npj Computat. Mater. 2022, 8 (1), 222, DOI: 10.1038/s41524-022-00906-4Google ScholarThere is no corresponding record for this reference.
- 161Ellis, B.; Smith, R. Polymers: A Property Database. CRC Press, 2008.Google ScholarThere is no corresponding record for this reference.
- 162Huan, T. D.; Mannodi-Kanakkithodi, A.; Kim, C.; Sharma, V.; Pilania, G.; Ramprasad, R. A polymer dataset for accelerated property prediction and design. Scientific data 2016, 3 (1), 1– 10, DOI: 10.1038/sdata.2016.12Google ScholarThere is no corresponding record for this reference.
- 163Pence, H. E.; Williams, A. Chemspider: an online chemical information resource, 2010.Google ScholarThere is no corresponding record for this reference.
- 164Polymer property predictor and database. https://pppdb.uchicago.edu/, 2019–2025.Google ScholarThere is no corresponding record for this reference.
- 165Campus plastic s database: Computer aided material preselection by uniform standards. https://www.campusplastics.com/, 1988–2025.Google ScholarThere is no corresponding record for this reference.
- 166Miccio, L. A.; Schwartz, G. A. From chemical structure to quantitative polymer properties prediction through convolutional neural networks. Polymer 2020, 193, 122341 DOI: 10.1016/j.polymer.2020.122341Google ScholarThere is no corresponding record for this reference.
- 167Xu, P.; Lu, T.; Ju, L.; Tian, L.; Li, M.; Lu, W. Machine learning aided design of polymer with targeted band gap based on dft computation. J. Phys. Chem. B 2021, 125 (2), 601– 611, DOI: 10.1021/acs.jpcb.0c08674Google ScholarThere is no corresponding record for this reference.
- 168Xiong, J.; Xiong, Z.; Chen, K.; Jiang, H.; Zheng, M. Graph neural networks for automated de novo drug design. Drug Discovery Today 2021, 26 (6), 1382– 1393, DOI: 10.1016/j.drudis.2021.02.011Google ScholarThere is no corresponding record for this reference.
- 169Medina, H.; Drake, R.; Farmer, C. Accelerated prediction of molecular properties for per-and polyfluoroalkyl substances using graph neural networks with adjacency-free message passing. Environ. Pollut. 2025, 382, 126705 DOI: 10.1016/j.envpol.2025.126705Google ScholarThere is no corresponding record for this reference.
- 170Tozzini, V. Coarse-grained models for proteins. Curr. Opin. Struct. Biol. 2005, 15 (2), 144– 150, DOI: 10.1016/j.sbi.2005.02.005Google ScholarThere is no corresponding record for this reference.
- 171Joshi, S. Y.; Deshmukh, S. A. A review of advancements in coarse-grained molecular dynamics simulations. Mol. Simul. 2021, 47 (10–11), 786– 803, DOI: 10.1080/08927022.2020.1828583Google ScholarThere is no corresponding record for this reference.
- 172Noid, W. G.; Liu, P.; Wang, Y.; Chu, J.-W.; Ayton, G. S.; Izvekov, S.; Andersen, H. C.; Voth, G. A. The multiscale coarse-graining method. ii. numerical implementation for coarse-grained molecular models. J. Chem. Phys. 2008, 128, 244115, DOI: 10.1063/1.2938857Google ScholarThere is no corresponding record for this reference.
- 173Pak, A. J.; Voth, G. A. Advances in coarse-grained modeling of macromolecular complexes. Curr. Opin. Struct. Biol. 2018, 52, 119– 126, DOI: 10.1016/j.sbi.2018.11.005Google ScholarThere is no corresponding record for this reference.
- 174Noid, W. G. Perspective: Coarse-grained models for biomolecular systems. J. Chem. Phys. 2013, 139, 090901, DOI: 10.1063/1.4818908Google ScholarThere is no corresponding record for this reference.
- 175Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A. E.; Kolinski, A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116 (14), 7898– 7936, DOI: 10.1021/acs.chemrev.6b00163Google ScholarThere is no corresponding record for this reference.
- 176Ingólfsson, H. I.; Lopez, C. A.; Uusitalo, J. J.; de Jong, D. H.; Gopal, S. M.; Periole, X.; Marrink, S. J. The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4 (3), 225– 248, DOI: 10.1002/wcms.1169Google ScholarThere is no corresponding record for this reference.
- 177Noid, W.; Szukalo, R. J.; Kidder, K. M.; Lesniewski, M. C. Rigorous progress in coarse-graining. Annu. Rev. Phys. Chem. 2024, 75 (1), 21– 45, DOI: 10.1146/annurev-physchem-062123-010821Google ScholarThere is no corresponding record for this reference.
- 178Guenza, M. G. Everything you want to know about coarse-graining and never dared to ask: Macromolecules as a key example. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2025, 15 (2), e70022 DOI: 10.1002/wcms.70022Google ScholarThere is no corresponding record for this reference.
- 179Harmandaris, V. A.; Kremer, K. Predicting polymer dynamics at multiple length and time scales. Soft Matter 2009, 5 (20), 3920– 3926, DOI: 10.1039/b905361aGoogle ScholarThere is no corresponding record for this reference.
- 180Ye, H.; Xian, W.; Li, Y. Machine learning of coarse-grained models for organic molecules and polymers: Progress, opportunities, and challenges. ACS Omega 2021, 6 (3), 1758– 1772, DOI: 10.1021/acsomega.0c05321Google ScholarThere is no corresponding record for this reference.
- 181Noid, W. G. Perspective: Advances, challenges, and insight for predictive coarse-grained models. J. Phys. Chem. B 2023, 127 (19), 4174– 4207, DOI: 10.1021/acs.jpcb.2c08731Google ScholarThere is no corresponding record for this reference.
- 182Vettorel, T.; Besold, G.; Kremer, K. Fluctuating soft-sphere approach to coarse-graining of polymer models. Soft Matter 2010, 6 (10), 2282– 2292, DOI: 10.1039/b921159dGoogle ScholarThere is no corresponding record for this reference.
- 183Español, P.; Serrano, M.; Pagonabarraga, I.; Zúñiga, I. Energy-conserving coarse-graining of complex molecules. Soft Matter 2016, 12 (21), 4821– 4837, DOI: 10.1039/C5SM03038BGoogle ScholarThere is no corresponding record for this reference.
- 184Husic, B. E.; Charron, N. E.; Lemm, D.; Wang, J.; Pérez, A.; Majewski, M.; Krämer, A.; Chen, Y.; Olsson, S.; De Fabritiis, G.; Noe, F.; Clementi, C. Coarse graining molecular dynamics with graph neural networks J. Chem. Phys. 2020; Vol. 153, DOI: 10.1063/5.0026133 .Google ScholarThere is no corresponding record for this reference.
- 185Wang, X.; Wu, Y.; Zhang, A.; He, X.; Chua, T.-S. Towards multi-grained explainability for graph neural networks. Adv. Neural Inf. Process. Syst. 2021, 34, 18446– 18458Google ScholarThere is no corresponding record for this reference.
- 186Peter, C.; Kremer, K. Multiscale simulation of soft matter systems-from the atomistic to the coarse-grained level and back. Soft Matter 2009, 5 (22), 4357– 4366, DOI: 10.1039/b912027kGoogle ScholarThere is no corresponding record for this reference.
- 187Zhao, Y.; Chen, G.; Liu, J. Polymer data challenges in the ai era: Bridging gaps for next-generation energy materials, 2025, arXiv:2505.13494. arXiv.org e-Print archive https://arxiv.org/abs/2505.13494.Google ScholarThere is no corresponding record for this reference.
- 188Orio, M.; Pantazis, D. A.; Neese, F. Density functional theory. Photosynth. Res. 2009, 102, 443– 453, DOI: 10.1007/s11120-009-9404-8Google ScholarThere is no corresponding record for this reference.
- 189Joshi, S. P.; Bucsek, A.; Pagan, D. C.; Daly, S.; Ravindran, S.; Marian, J.; Bessa, M. A.; Kalidindi, S. R.; Admal, N. C.; Reina, C.; Ghosh, S.; Warren, J. A.; Vinals, J.; Tadmor, E. B. “ Integrated experiment and simulation co-design: A key infrastructure for predictive mesoscale materials modeling, 2025, arXiv:2503.09793. arXiv.org e-Print archive https://arxiv.org/abs/2503.09793.Google ScholarThere is no corresponding record for this reference.
- 190Zaporozhets, I.; Clementi, C. Multibody terms in protein coarse-grained models: A top-down perspective. J. Phys. Chem. B 2023, 127 (31), 6920– 6927, DOI: 10.1021/acs.jpcb.3c04493Google ScholarThere is no corresponding record for this reference.
- 191Jin, J.; Pak, A. J.; Durumeric, A. E.; Loose, T. D.; Voth, G. A. Bottom-up coarse-graining: Principles and perspectives. J. Chem. Theory Comput. 2022, 18 (10), 5759– 5791, DOI: 10.1021/acs.jctc.2c00643Google ScholarThere is no corresponding record for this reference.
- 192Praprotnik, M.; Delle Site, L.; Kremer, K. Multiscale simulation of soft matter: From scale bridging to adaptive resolution. Annu. Rev. Phys. Chem. 2008, 59, 545– 571, DOI: 10.1146/annurev.physchem.59.032607.093707Google ScholarThere is no corresponding record for this reference.
- 193Milano, G.; Kawakatsu, T. Hybrid particle-field molecular dynamics simulations for dense polymer systems J. Chem. Phys. 2009; Vol. 130, DOI: 10.1063/1.3142103 .Google ScholarThere is no corresponding record for this reference.
- 194Wang, J.; Han, Y.; Xu, Z.; Yang, X.; Ramakrishna, S.; Liu, Y. Dissipative particle dynamics simulation: A review on investigating mesoscale properties of polymer systems. Macromol. Mater. Eng. 2021, 306 (4), 2000724 DOI: 10.1002/mame.202000724Google ScholarThere is no corresponding record for this reference.
- 195Santo, K. P.; Neimark, A. V. Dissipative particle dynamics simulations in colloid and interface science: A review. Adv. Colloid Interface Sci. 2021, 298, 102545 DOI: 10.1016/j.cis.2021.102545Google ScholarThere is no corresponding record for this reference.
- 196Hoogerbrugge, P. J.; Koelman, J. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys. Lett. 1992, 19 (3), 155, DOI: 10.1209/0295-5075/19/3/001Google ScholarThere is no corresponding record for this reference.
- 197Español, P.; Warren, P. Statistical mechanics of dissipative particle dynamics. Europhys. Lett. 1995, 30 (4), 191, DOI: 10.1209/0295-5075/30/4/001Google ScholarThere is no corresponding record for this reference.
- 198Marrink, S. J.; Monticelli, L.; Melo, M. N.; Alessandri, R.; Tieleman, D. P.; Souza, P. C. Two decades of martini: Better beads, broader scope. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2023, 13 (1), e1620 DOI: 10.1002/wcms.1620Google ScholarThere is no corresponding record for this reference.
- 199Alessandri, R.; Grünewald, F.; Marrink, S. J. The martini model in materials science. Adv. Mater. 2021, 33 (24), 2008635 DOI: 10.1002/adma.202008635Google ScholarThere is no corresponding record for this reference.
- 200Souza, P. C. T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B. M.; Wassenaar, T. A.; Kroon, P.; Melcr, K.; Nieto, V.; Corradi, V.; Khan, H. M.; Domanski, J.; Javanainen, M.; Martinez-Seara, H.; Reuter, N.; Best, R. B.; Vattulainen, I.; Monticelli, L.; Periole, X.; Peter, T. D.; Vries, dH.; Marrink, S. J. Martini 3: a general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18 (4), 382– 388, DOI: 10.1038/s41592-021-01098-3Google ScholarThere is no corresponding record for this reference.
- 201Fredrickson, G. H. The Equilibrium Theory of Inhomogeneous Polymers. Oxford University Press, 2006.Google ScholarThere is no corresponding record for this reference.
- 202Schmid, F. Self-consistent-field theories for complex fluids. J. Phys.: Condens. Matter 1998, 10 (37), 8105, DOI: 10.1088/0953-8984/10/37/002Google ScholarThere is no corresponding record for this reference.
- 203Maerzke, K. A.; Siepmann, J. I. Transferable potentials for phase equilibria- coarse-grain description for linear alkanes. J. Phys. Chem. B 2011, 115 (13), 3452– 3465, DOI: 10.1021/jp1063935Google ScholarThere is no corresponding record for this reference.
- 204Liwo, A.; Ołdziej, S.; Pincus, M. R.; Wawak, R. J.; Rackovsky, S.; Scheraga, H. A. A united-residue force field for off-lattice protein-structure simulations. i. functional forms and parameters of long-range side-chain interaction potentials from protein crystal data. J. Comput. Chem. 1997, 18 (7), 849– 873, DOI: 10.1002/(SICI)1096-987X(199705)18:7<849::AID-JCC1>3.0.CO;2-RGoogle ScholarThere is no corresponding record for this reference.
- 205Maisuradze, G. G.; Senet, P.; Czaplewski, C.; Liwo, A.; Scheraga, H. A. Investigation of protein folding by coarse-grained molecular dynamics with the unres force field. J. Phys. Chem. A 2010, 114 (13), 4471– 4485, DOI: 10.1021/jp9117776Google ScholarThere is no corresponding record for this reference.
- 206Reith, D.; Pütz, M.; Müller-Plathe, F. Deriving effective mesoscale potentials from atomistic simulations. J. Comput. Chem. 2003, 24 (13), 1624– 1636, DOI: 10.1002/jcc.10307Google ScholarThere is no corresponding record for this reference.
- 207Agrawal, V.; Arya, G.; Oswald, J. Simultaneous iterative boltzmann inversion for coarse-graining of polyurea. Macromolecules 2014, 47 (10), 3378– 3389, DOI: 10.1021/ma500320nGoogle ScholarThere is no corresponding record for this reference.
- 208Tschöp, W.; Kremer, K.; Batoulis, J.; Bürger, T.; Hahn, O. Simulation of polymer melts. i. coarse-graining procedure for polycarbonates. Acta Polym. 1998, 49 (2–3), 61– 74, DOI: 10.1002/(SICI)1521-4044(199802)49:2/3<61::AID-APOL61>3.0.CO;2-VGoogle ScholarThere is no corresponding record for this reference.
- 209McCoy, J. D.; Curro, J. G. Mapping of explicit atom onto united atom potentials. Macromolecules 1998, 31 (26), 9362– 9368, DOI: 10.1021/ma981060gGoogle ScholarThere is no corresponding record for this reference.
- 210Baschnagel, J.; Binder, K.; Doruker, P.; Gusev, A. A.; Hahn, O.; Kremer, K.; Mattice, W. L.; Müller-Plathe, F.; Murat, M.; Paul, W.; Santos, S.; Suter, U. W.; Tries, V. Bridging the gap between atomistic and coarse-grained models of polymers: Status and perspectives. Viscoelasticity, Atomistic Models, Statistical Chemistry 2000, 152, 41– 156, DOI: 10.1007/3-540-46778-5_2Google ScholarThere is no corresponding record for this reference.
- 211Akkermans, R. L. C.; Briels, W. J. A structure-based coarse-grained model for polymer melts. J. Chem. Phys. 2001, 114 (2), 1020– 1031, DOI: 10.1063/1.1330744Google ScholarThere is no corresponding record for this reference.
- 212Uneyama, T.; Masubuchi, Y. Plateau moduli of several single-chain slip-link and slip-spring models. Macromolecules 2021, 54 (3), 1338– 1353, DOI: 10.1021/acs.macromol.0c01790Google ScholarThere is no corresponding record for this reference.
- 213Behbahani, A. F.; Schneider, L.; Rissanou, A.; Chazirakis, A.; Bacova, P.; Jana, P. K.; Li, W.; Doxastakis, M.; Polinska, P.; Burkhart, C.; Muller, M.; Harmandaris, V. A. Dynamics and rheology of polymer melts via hierarchical atomistic, coarse-grained, and slip-spring simulations. Macromolecules 2021, 54 (6), 2740– 2762, DOI: 10.1021/acs.macromol.0c02583Google ScholarThere is no corresponding record for this reference.
- 214Wu, Z.; Müller-Plathe, F. Slip-spring hybrid particle-field molecular dynamics for coarse-graining branched polymer melts: Polystyrene melts as an example. J. Chem. Theory Comput. 2022, 18 (6), 3814– 3828, DOI: 10.1021/acs.jctc.2c00107Google ScholarThere is no corresponding record for this reference.
- 215Underhill, P. T.; Doyle, P. S. On the coarse-graining of polymers into bead-spring chains. J. Non-Newtonian Fluid Mech. 2004, 122 (1–3), 3– 31, DOI: 10.1016/j.jnnfm.2003.10.006Google ScholarThere is no corresponding record for this reference.
- 216Kremer, K.; Grest, G. S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92 (8), 5057– 5086, DOI: 10.1063/1.458541Google ScholarThere is no corresponding record for this reference.
- 217Everaers, R.; Karimi-Varzaneh, H. A.; Fleck, F.; Hojdis, N.; Svaneborg, C. Kremer-grest models for commodity polymer melts: Linking theory, experiment, and simulation at the kuhn scale. Macromolecules 2020, 53 (6), 1901– 1916, DOI: 10.1021/acs.macromol.9b02428Google ScholarThere is no corresponding record for this reference.
- 218Nitta, H.; Ozawa, T.; Yasuoka, K. Construction of full-atomistic polymer amorphous structures using reverse-mapping from kremer–grest models J. Chem. Phys. , 2023 159 no. 19.Google ScholarThere is no corresponding record for this reference.
- 219Miwatani, R.; Takahashi, K. Z.; Arai, N. Performance of coarse graining in estimating polymer properties: Comparison with the atomistic model. Polymers 2020, 12 (2), 382, DOI: 10.3390/polym12020382Google ScholarThere is no corresponding record for this reference.
- 220Praprotnik, M.; Delle Site, L.; Kremer, K. Adaptive resolution scheme for efficient hybrid atomistic-mesoscale molecular dynamics simulations of dense liquids. Phys. Rev. E 2006, 73 (6), 066701 DOI: 10.1103/PhysRevE.73.066701Google ScholarThere is no corresponding record for this reference.
- 221Darré, L.; Machado, M. R.; Brandner, A. F.; González, H. C.; Ferreira, S.; Pantano, S. Sirah: a structurally unbiased coarse-grained force field for proteins with aqueous solvation and long-range electrostatics. J. Chem. Theory Comput. 2015, 11 (2), 723– 739, DOI: 10.1021/ct5007746Google ScholarThere is no corresponding record for this reference.
- 222Das, R.; Baker, D. Macromolecular modeling with rosetta. Annu. Rev. Biochem. 2008, 77, 363– 382, DOI: 10.1146/annurev.biochem.77.062906.171838Google ScholarThere is no corresponding record for this reference.
- 223Leman, J. K.; Weitzner, B. D.; Lewis, S. M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R. F.; Aprahamian, M.; Baker, D.; Barlow, K. A.; Barth, P.; Basanta, B.; Bender, B. J.; Blacklock, K.; Bonet, J.; Boyken, S. E.; Bradley, P.; Bystroff, C.; Conway, P.; Cooper, S.; Correia, B. E.; Coventry, B.; Das, R.; De Jong, R. M.; DiMaio, F.; Dsilva, L.; Dunbrack, R.; Ford, A. S.; Frenz, B.; Fu, D. Y.; Geniesse, C.; Goldschmidt, L.; Gowthaman, R.; Gray, J. J.; Gront, D.; Guffy, S.; Horowitz, S.; Huang, P.-S.; Huber, T.; Jacobs, T. M.; Jeliazkov, J. R.; Johnson, D. K.; Kappel, K.; Karanicolas, J.; Khakzad, H.; Khar, K. R.; Khare, S. D.; Khatib, F.; Khramushin, A.; King, I. C.; Kleffner, R.; Koepnick, B.; Kortemme, T.; Kuenze, G.; Kuhlman, B.; Kuroda, D.; Labonte, J. W.; Lai, J. K.; Lapidoth, G.; Leaver-Fay, A.; Lindert, S.; Linsky, T.; London, N.; Lubin, J. H.; Lyskov, S.; Maguire, J.; Malmström, L.; Marcos, E.; Marcu, O.; Marze, N. A.; Meiler, J.; Moretti, R.; Mulligan, V. K.; Nerli, S.; Norn, C.; Ó’Conchúir, S.; Ollikainen, N.; Ovchinnikov, S.; Pacella, M. S.; Pan, X.; Park, H.; Pavlovicz, R. E.; Pethe, M.; Pierce, B. G.; Pilla, K. B.; Raveh, B.; Renfrew, P. D.; Roy Burman, S. S.; Rubenstein, A.; Sauer, M. F.; Scheck, A.; Schief, W.; Schueler-Furman, O.; Sedan, Y.; Sevy, A. M.; Sgourakis, N. G.; Shi, L.; Siegel, J. B.; Silva, D.-A.; Smith, S.; Song, Y.; Stein, A.; Szegedy, M.; Teets, F. D.; Thyme, S. B.; Wang, R. Y.-R.; Watkins, A.; Zimmerman, L.; Bonneau, R. Macromolecular modeling and design in rosetta: recent methods and frameworks. Nat. Methods 2020, 17 (7), 665– 680, DOI: 10.1038/s41592-020-0848-2Google ScholarThere is no corresponding record for this reference.
- 224Baldassarre, F.; Azizpour, H. Explainability techniques for graph convolutional networks, 2019, arXiv:1905.13686 arXiv.org e-Print archive https://arxiv.org/abs/1905.13686.Google ScholarThere is no corresponding record for this reference.
- 225Bai, Y.; Wilbraham, L.; Slater, B. J.; Zwijnenburg, M. A.; Sprick, R. S.; Cooper, A. I. Accelerated discovery of organic polymer photocatalysts for hydrogen evolution from water through the integration of experiment and theory. J. Am. Chem. Soc. 2019, 141 (22), 9063– 9071, DOI: 10.1021/jacs.9b03591Google ScholarThere is no corresponding record for this reference.
- 226Vosko, S. H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58 (8), 1200– 1211, DOI: 10.1139/p80-159Google ScholarThere is no corresponding record for this reference.
- 227Becke, A. D. Density-functional thermochemistry. iii. the role of exact exchange. J. Chem. Phys. 1993, 98 (7), 5648– 5652, DOI: 10.1063/1.464913Google ScholarThere is no corresponding record for this reference.
- 228Lee, C.; Yang, W.; Parr, R. G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37 (2), 785, DOI: 10.1103/PhysRevB.37.785Google ScholarThere is no corresponding record for this reference.
- 229Grimme, S.; Bannwarth, C.; Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (z= 1–86). J. Chem. Theory Comput. 2017, 13 (5), 1989– 2009, DOI: 10.1021/acs.jctc.7b00118Google ScholarThere is no corresponding record for this reference.
- 230Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. A 1994, 98 (45), 11623– 11627, DOI: 10.1021/j100096a001Google ScholarThere is no corresponding record for this reference.
- 231Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M. Analyzing learned molecular representations for property prediction. J. Chem. Inf. Model. 2019, 59 (8), 3370– 3388, DOI: 10.1021/acs.jcim.9b00237Google ScholarThere is no corresponding record for this reference.
- 232Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50 (5), 742– 754, DOI: 10.1021/ci100050tGoogle ScholarThere is no corresponding record for this reference.
- 233Landrum, G. Rdkit: open-source cheminformatics from machine learning to chemical registration. In Abstracts of Papers of the American Chemical Society; Vol. 258, AMER CHEMICAL SOC 1155 16TH ST, NW, WASHINGTON, DC 20036 USA, 2019.Google ScholarThere is no corresponding record for this reference.
- 234Mannodi-Kanakkithodi, A.; Chandrasekaran, A.; Kim, C.; Huan, T. D.; Pilania, G.; Botu, V.; Ramprasad, R. Scoping the polymer genome: A roadmap for rational polymer dielectrics design and beyond. Mater. Today 2018, 21, 785– 796, DOI: 10.1016/j.mattod.2017.11.021Google ScholarThere is no corresponding record for this reference.
- 235Kim, C.; Chandrasekaran, A.; Huan, T. D.; Das, D.; Ramprasad, R. Polymer genome: A data-powered polymer informatics platform for property predictions. J. Phys. Chem. C 2018, 122, 17575– 17585, DOI: 10.1021/acs.jpcc.8b02913Google ScholarThere is no corresponding record for this reference.
- 236Ward, L.; Dunn, A.; Faghaninia, A.; Zimmermann, N. E.; Bajaj, S.; Wang, Q.; Montoya, J.; Chen, J.; Bystrom, K.; Dylla, M.; Chard, K.; Asta, M.; Persson, K. A.; Snyder, G. J.; Foster, I.; Jain, A. Matminer: An open source toolkit for materials data mining. Comput. Mater. Sci. 2018, 152, 60– 69, DOI: 10.1016/j.commatsci.2018.05.018Google ScholarThere is no corresponding record for this reference.
- 237Lightstone, J. P.; Chen, L.; Kim, C.; Batra, R.; Ramprasad, R. Refractive index prediction models for polymers using machine learning J. Appl. Phys. 2020; Vol. 127, DOI: 10.1063/5.0008026 .Google ScholarThere is no corresponding record for this reference.
- 238Chen, L.; Kim, C.; Batra, R.; Lightstone, J. P.; Wu, C.; Li, Z.; Deshmukh, A. A.; Wang, Y.; Tran, H. D.; Vashishta, P.; Sotzing, G. A.; Cao, Y.; Ramprasad, R. Frequency-dependent dielectric constant prediction of polymers using machine learning. npj Computat. Mater. 2020, 6 (1), 61, DOI: 10.1038/s41524-020-0333-6Google ScholarThere is no corresponding record for this reference.
- 239Doan Tran, H.; Kim, C.; Chen, L.; Chandrasekaran, A.; Batra, R.; Venkatram, S.; Kamal, D.; Lightstone, J. P.; Gurnani, R.; Shetty, P. Machine-learning predictions of polymer properties with polymer genome. J. Appl. Phys. 2020, 128 (17), 171104 DOI: 10.1063/5.0023759Google ScholarThere is no corresponding record for this reference.
- 240Godwin, J.; Schaarschmidt, M.; Gaunt, A.; Sanchez-Gonzalez, A.; Rubanova, Y.; Veličković, P.; Kirkpatrick, J.; Battaglia, P. Simple gnn regularisation for 3d molecular property prediction & beyond, 2021, arXiv:2106.07971. arXiv.org e-Print archive https://arxiv.org/abs/2106.07971.Google ScholarThere is no corresponding record for this reference.
- 241Chen, D.; Lin, Y.; Li, W.; Li, P.; Zhou, J.; Sun, X. Measuring and relieving the over-smoothing problem for graph neural networks from the topological view. Proc. AAAI Conf. Artificial Intelligence 2020, 34, 3438– 3445, DOI: 10.1609/aaai.v34i04.5747Google ScholarThere is no corresponding record for this reference.
- 242Frisch, M. Gaussian 09, Revision D.01, Gaussian , 2009.Google ScholarThere is no corresponding record for this reference.
- 243Kuenneth, C.; Schertzer, W.; Ramprasad, R. Copolymer informatics with multitask deep neural networks. Macromolecules 2021, 54 (13), 5957– 5961, DOI: 10.1021/acs.macromol.1c00728Google ScholarThere is no corresponding record for this reference.
- 244You, J.; Ying, Z.; Leskovec, J. Design space for graph neural networks. In Advances in Neural Information Processing Systems (NeurIPS) 2020.Google ScholarThere is no corresponding record for this reference.
- 245Rupp, M.; Tkatchenko, A.; Müller, K.-R.; Von Lilienfeld, O. A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012, 108 (5), 058301 DOI: 10.1103/PhysRevLett.108.058301Google ScholarThere is no corresponding record for this reference.
- 246Bartók, A. P.; Kondor, R.; Csányi, G. On representing chemical environments. Phys. Rev. B 2013, 87 (18), 184115 DOI: 10.1103/PhysRevB.87.184115Google ScholarThere is no corresponding record for this reference.
- 247Townsend, J.; Micucci, C. P.; Hymel, J. H.; Maroulas, V.; Vogiatzis, K. D. Representation of molecular structures with persistent homology for machine learning applications in chemistry. Nat. Commun. 2020, 11 (1), 3230 DOI: 10.1038/s41467-020-17035-5Google ScholarThere is no corresponding record for this reference.
- 248Huo, H.; Rupp, M. Unified representation of molecules and crystals for machine learning. Machine Learning: Sci. Technol. 2022, 3 (4), 045017 DOI: 10.1088/2632-2153/aca005Google ScholarThere is no corresponding record for this reference.
- 249Sanchez Medina, E. I.; Linke, S.; Stoll, M.; Sundmacher, K. Gibbs-helmholtz graph neural network: capturing the temperature dependency of activity coefficients at infinite dilution. Digital Discovery 2023, 2 (3), 781– 798, DOI: 10.1039/D2DD00142JGoogle ScholarThere is no corresponding record for this reference.
- 250Zhong, C.; Sato, Y.; Masuoka, H.; Chen, X. Improvement of predictive accuracy of the unifac model for vapor-liquid equilibria of polymer solutions. Fluid Phase Equilib. 1996, 123 (1–2), 97– 106, DOI: 10.1016/S0378-3812(96)90015-1Google ScholarThere is no corresponding record for this reference.
- 251Kontogeorgis, G. M.; Fredenslund, A.; Tassios, D. P. Simple activity coefficient model for the prediction of solvent activities in polymer solutions. Ind. Eng. Chem. Res. 1993, 32 (2), 362– 372, DOI: 10.1021/ie00014a013Google ScholarThere is no corresponding record for this reference.
- 252Pappa, G. D.; Voutsas, E. C.; Tassios, D. P. Prediction of activity coefficients in polymer and copolymer solutions using simple activity coefficient models. Ind. Eng. Chem. Res. 1999, 38 (12), 4975– 4984, DOI: 10.1021/ie990265qGoogle ScholarThere is no corresponding record for this reference.
- 253Cremer, D. Møller-plesset perturbation theory: from small molecule methods to methods for thousands of atoms. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1 (4), 509– 530, DOI: 10.1002/wcms.58Google ScholarThere is no corresponding record for this reference.
- 254Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: moving beyond fingerprints. J. Comput.-Aided Mol. Des. 2016, 30, 595– 608, DOI: 10.1007/s10822-016-9938-8Google ScholarThere is no corresponding record for this reference.
- 255Wang, M. Y. Deep graph library: Towards efficient and scalable deep learning on graphs. In ICLR Workshop on Representation Learning on Graphs and Manifolds , 2019.Google ScholarThere is no corresponding record for this reference.
- 256Bojar, D.; Powers, R. K.; Camacho, D. M.; Collins, J. J. Deep-learning resources for studying glycan-mediated host-microbe interactions. Cell Host Microbe 2021, 29, 132– 144.e3, DOI: 10.1016/j.chom.2020.10.004Google ScholarThere is no corresponding record for this reference.
- 257Montavon, G.; Binder, A.; Lapuschkin, S.; Samek, W.; Müller, K.-R. Layer-wise relevance propagation: an overview. In Lecture Notes in Computer Science 2019; pp 193– 209.Google ScholarThere is no corresponding record for this reference.
- 258Binder, A.; Bach, S.; Montavon, G.; Müller, K.-R.; Samek, W. Layer-wise relevance propagation for deep neural network architectures. In Information Science and Applications (ICISA) 2016; Springer, 2016; pp 913– 922.Google ScholarThere is no corresponding record for this reference.
- 259Adebayo, J.; Gilmer, J.; Muelly, M.; Goodfellow, I.; Hardt, M.; Kim, B. Sanity checks for saliency maps Advances in Neural Information Processing Systems , 2018 31.Google ScholarThere is no corresponding record for this reference.
- 260Qi, Z.; Khorram, S.; Li, F. Visualizing deep networks by optimizing with integrated gradients. AAAI 2020, 34, 11890– 11898, DOI: 10.1609/aaai.v34i07.6863Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
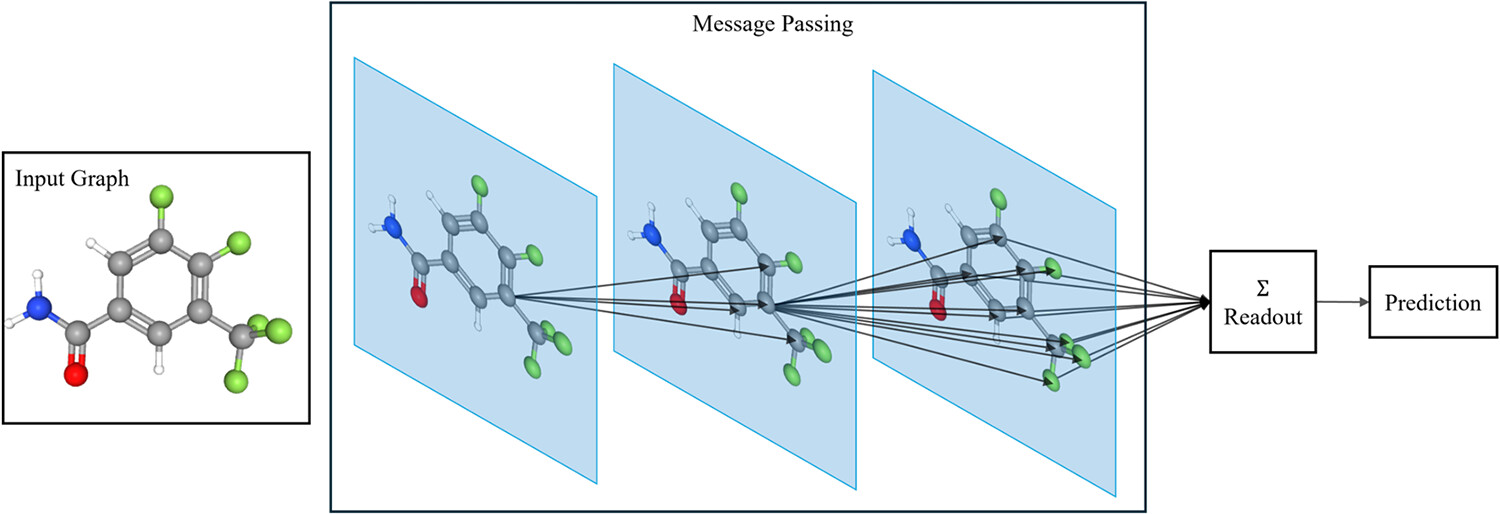{kind=link}
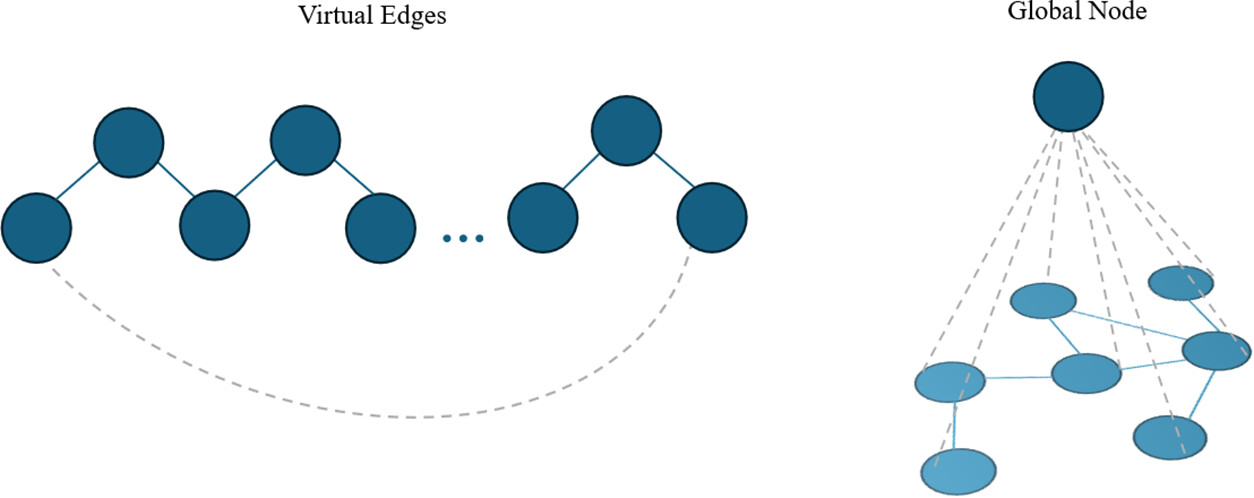{kind=link}
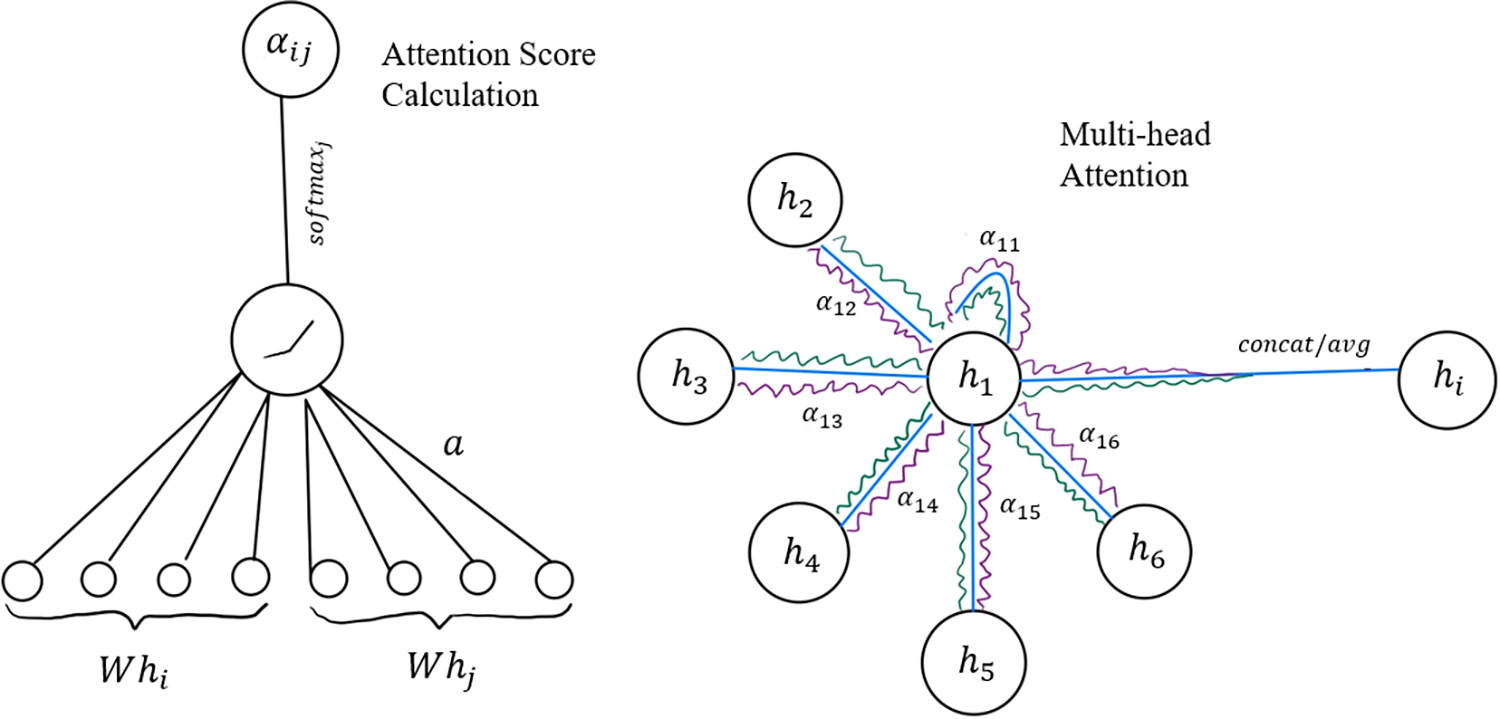{kind=link}
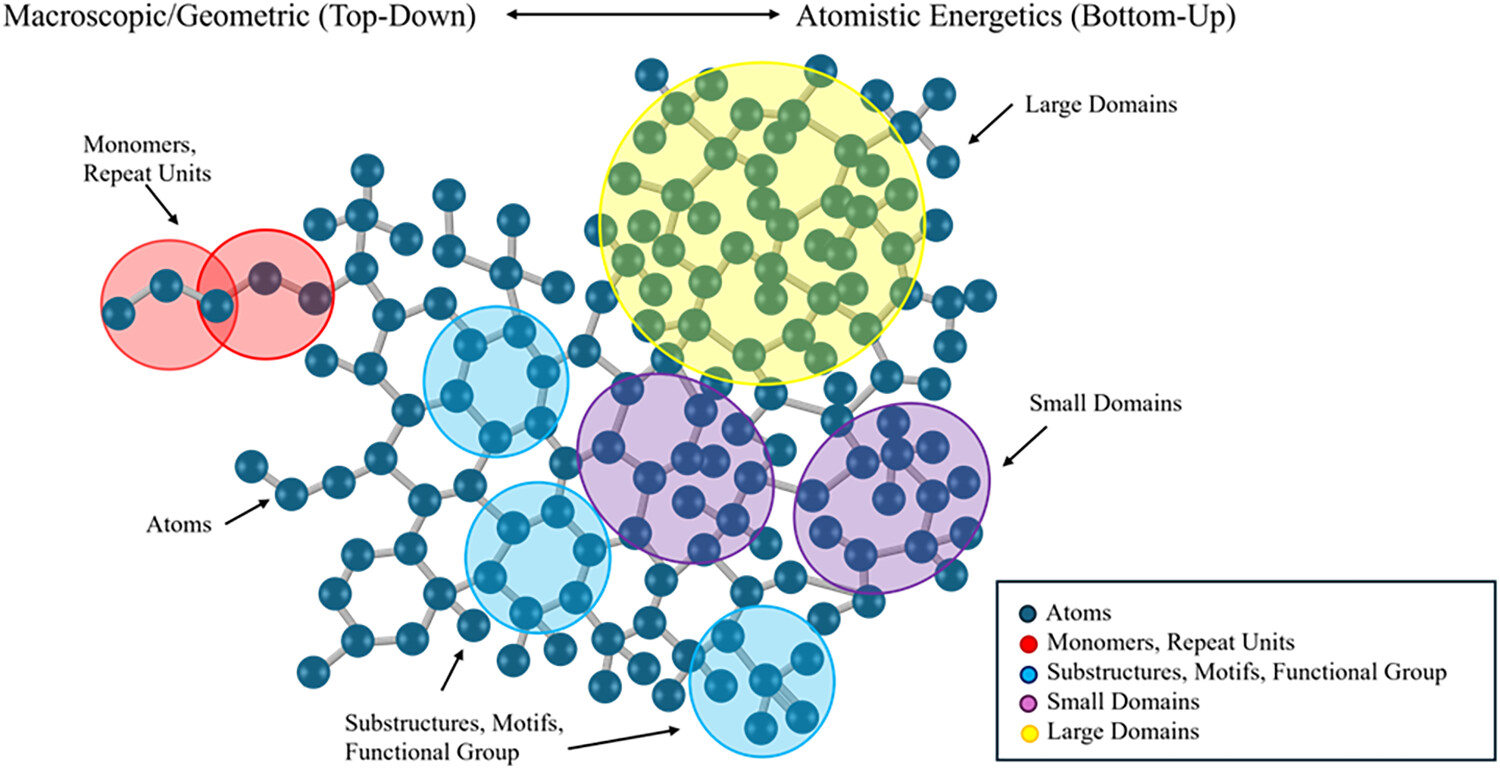{kind=link}
Journal of Chemical Information and Modeling
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
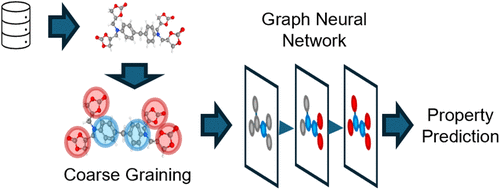
Figure 1

Figure 1. General architecture of a GNN for molecular property prediction. The input molecular graph is processed through multiple message passing layers, where the node embeddings are iteratively updated by propagating information from neighboring atoms. These updated representations are then combined by a readout function (e.g., average or sum) to produce a graph-level embedding, which is fed into a downstream prediction layer.
Figure 2

Figure 2. Two graph augmentations in polymer GNN architectures: a virtual edges connecting distant nodes to encode periodic or long-range interactions (left), and a global node connected to all nodes to capture system-level or graph-wide information (right).
Figure 3

Figure 3. An illustration of GAT operations. The left portion of the diagram shows how attention coefficients αij are computed: the transformed node features Whi and Whj are concatenated and passed through a shared attention mechanism parametrized by the vector a, followed by a LeakyReLU nonlinearity and a softmax normalization over all neighbors j ∈ Ni. The resulting coefficient αij encodes the learned importance of neighbor j when updating node i. The right portion illustrates multihead attention for a specific node (node 1 in the drawing): each neighbor contributes messages through multiple attention heads (depicted by the green, blue and purple lines), producing head-specific weights α1j(k) and transformed messages W(k)hj. The index k refers to the attention head number, where each head has its own learnable weight matrix W(k) and produces its own attention score. These per-head aggregations are combined via concatenation (for intermediate layers) or averaging (for final layers) to yield the updated node embedding hi. The diagram highlights both key components of GATs: attention-based neighborhood weighting and multihead message aggregation.
Figure 4

Figure 4. Conceptual depiction of CG in polymer informatics. The top bar represents the (abstract) spectrum of CG strategies from macroscopic, geometry-based top-down strategies to atomistic, energetics-based bottom-up approaches. The molecular structure is overlaid with color-coded regions representing multiple levels of abstraction: atoms (gray), monomer/repeat units (red), substructures, functional groups or motifs (blue), small domains (purple), and large domains (yellow). This hierarchy also illustrates the conceptual transition from atomistic to increasingly coarse-grained graph representations, by grouping chemically or structurally important substructures so that scalable and interpretable ML pipelines are achievable.
References
This article references 260 other publications.
- 1Liang, Y.; Yu, L. Development of semiconducting polymers for solar energy harvesting. Polym. Rev. 2010, 50 (4), 454– 473, DOI: 10.1080/15583724.2010.515765There is no corresponding record for this reference.
- 2Baytekin, B.; Baytekin, H. T.; Grzybowski, B. A. Retrieving and converting energy from polymers: deployable technologies and emerging concepts. Energy Environ. Sci. 2013, 6 (12), 3467– 3482, DOI: 10.1039/c3ee41360hThere is no corresponding record for this reference.
- 3Abdelhamid, M. E.; O’Mullane, A. P.; Snook, G. A. Storing energy in plastics: a review on conducting polymers & their role in electrochemical energy storage. RSC Adv. 2015, 5 (15), 11611– 11626, DOI: 10.1039/C4RA15947KThere is no corresponding record for this reference.
- 4Fan, J.; Njuguna, J. An introduction to lightweight composite materials and their use in transport structures. In Lightweight Composite Structures in Transport; Elsevier, 2016; pp 3– 34.There is no corresponding record for this reference.
- 5Singh, J.; Srivastawa, K.; Jana, S.; Dixit, C.; Ravichandran, S. Advancements in lightweight materials for aerospace structures: A comprehensive review. Acceleron Aerosp. J. 2024, 2 (3), 173– 183, DOI: 10.61359/11.2106-2409There is no corresponding record for this reference.
- 6Yeo, S. J.; Oh, M. J.; Yoo, P. J. Structurally controlled cellular architectures for high-performance ultra-lightweight materials. Adv. Mater. 2019, 31 (34), 1803670 DOI: 10.1002/adma.201803670There is no corresponding record for this reference.
- 7Cui, H.; Zhao, Q.; Zhang, L.; Du, X. Intelligent polymer-based bioinspired actuators: from monofunction to multifunction. Adv. Intell. Syst. 2020, 2 (11), 2000138 DOI: 10.1002/aisy.202000138There is no corresponding record for this reference.
- 8Weng, M.; Ding, M.; Zhou, P.; Ye, Y.; Luo, Z.; Ye, X.; Guo, Q.; Chen, L. Multi-functional and integrated actuators made with bio-inspired cobweb carbon nanotube-polymer composites. Chem. Eng. J. 2023, 452, 139146 DOI: 10.1016/j.cej.2022.139146There is no corresponding record for this reference.
- 9Lendlein, A.; Rehahn, M.; Buchmeiser, M. R.; Haag, R. Polymers in biomedicine and electronics. Macromol. Rapid Commun. 2010, 31 (17), 1487– 1491, DOI: 10.1002/marc.201000426There is no corresponding record for this reference.
- 10Medina, H.; Child, N. A review of developments in carbon-based nanocomposite electrodes for noninvasive electroencephalography. Sensors 2025, 25 (7), 2274 DOI: 10.3390/s25072274There is no corresponding record for this reference.
- 11Farmer, C.; Medina, H. Effects of electrostriction on the bifurcated electro-mechanical performance of conical dielectric elastomer actuators and sensors. Robotica 2023, 41 (1), 215– 235, DOI: 10.1017/S0263574722001254There is no corresponding record for this reference.
- 12Medina, H.; Farmer, C.; Liu, I. Dielectric elastomer-based actuators: a modeling and control review for non-experts. In Actuators; MDPI, 2024; Vol. 13, p 151.There is no corresponding record for this reference.
- 13Korn, D.; Farmer, C.; Medina, H. A detailed solution framework for the out-of-plane displacement of circular dielectric elastomer actuators. Eng. Rep. 2022, 4 (1), e12442 DOI: 10.1002/eng2.12442There is no corresponding record for this reference.
- 14Peterson, G. I.; Choi, T.-L. Cascade polymerizations: recent developments in the formation of polymer repeat units by cascade reactions. Chem. Sci. 2020, 11 (19), 4843– 4854, DOI: 10.1039/D0SC01475CThere is no corresponding record for this reference.
- 15Cao, C.; Lin, Y. Correlation between the glass transition temperatures and repeating unit structure for high molecular weight polymers. J. Chem. Inf. Comput. Sci. 2003, 43 (2), 643– 650, DOI: 10.1021/ci0202990There is no corresponding record for this reference.
- 16Tezuka, Y.; Oike, H. Topological polymer chemistry: systematic classification of nonlinear polymer topologies. J. Am. Chem. Soc. 2001, 123 (47), 11570– 11576, DOI: 10.1021/ja0114409There is no corresponding record for this reference.
- 17Gu, Y.; Zhao, J.; Johnson, J. A. A (macro) molecular-level understanding of polymer network topology. Trends Chem. 2019, 1 (3), 318– 334, DOI: 10.1016/j.trechm.2019.02.017There is no corresponding record for this reference.
- 18Li, Y.; Abberton, B. C.; Kröger, M.; Liu, W. K. Challenges in multiscale modeling of polymer dynamics. Polymers 2013, 5 (2), 751– 832, DOI: 10.3390/polym5020751There is no corresponding record for this reference.
- 19Schmid, F. Understanding and modeling polymers: The challenge of multiple scales. ACS Polymers Au 2023, 3 (1), 28– 58, DOI: 10.1021/acspolymersau.2c00049There is no corresponding record for this reference.
- 20Jones, R. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87 (3), 897, DOI: 10.1103/RevModPhys.87.897There is no corresponding record for this reference.
- 21Parr, R. G. Density-Functional Theory of Atoms and Molecules; Oxford University Press, 1989.There is no corresponding record for this reference.
- 22Karplus, M.; McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9 (9), 646– 652, DOI: 10.1038/nsb0902-646There is no corresponding record for this reference.
- 23Hollingsworth, S. A.; Dror, R. O. Molecular dynamics simulation for all. Neuron 2018, 99 (6), 1129– 1143, DOI: 10.1016/j.neuron.2018.08.011There is no corresponding record for this reference.
- 24Tuckerman, M. E.; Martyna, G. J. Understanding modern molecular dynamics: Techniques and applications. J. Phys. Chem. B 2000, 104, 159– 178, DOI: 10.1021/jp992433yThere is no corresponding record for this reference.
- 25Bhavikatti, S. Finite Element Analysis. New Age International, 2005.There is no corresponding record for this reference.
- 26Kim, N.-H.; Sankar, B. V.; Kumar, A. V. Introduction to Finite Element Analysis and Design; John Wiley & Sons, 2018.There is no corresponding record for this reference.
- 27Farmer, C.; Medina, H. Hyperelastics. jl: A julia package for hyperelastic material modelling with a large collection of models. J. Open Source Softw. 2024, 9 (96), 6314 DOI: 10.21105/joss.06314There is no corresponding record for this reference.
- 28Farmer, C.; Medina, H. Impact-4ccs: Integrated modeling and prediction using ab initio and trained potentials for collision cross sections. J. Comput. Chem. 2025, 46 (11), e70106 DOI: 10.1002/jcc.70106There is no corresponding record for this reference.
- 29Medina, H.; Farmer, C. Current challenges in monitoring low contaminant levels of per-and polyfluoroalkyl substances in water matrices in the field. Toxics 2024, 12 (8), 610 DOI: 10.3390/toxics12080610There is no corresponding record for this reference.
- 30Farmer, C.; Medina, H. Universal interatomic potentials with dft for understanding orbital localization in polydimethylsiloxane-amorphous silica nanocomposites. ACS Omega 2025, 10 (49), 60812– 60818, DOI: 10.1021/acsomega.5c09273There is no corresponding record for this reference.
- 31McCormick, T. M.; Bridges, C. R.; Carrera, E. I.; DiCarmine, P. M.; Gibson, G. L.; Hollinger, J.; Kozycz, L. M.; Seferos, D. S. Conjugated polymers: Evaluating dft methods for more accurate orbital energy modeling. Macromolecules 2013, 46 (10), 3879– 3886, DOI: 10.1021/ma4005023There is no corresponding record for this reference.
- 32Adekoya, O. C.; Adekoya, G. J.; Sadiku, E. R.; Hamam, Y.; Ray, S. S. Application of dft calculations in designing polymer-based drug delivery systems: An overview. Pharmaceutics 2022, 14 (9), 1972, DOI: 10.3390/pharmaceutics14091972There is no corresponding record for this reference.
- 33Cohen, A. J.; Mori-Sánchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112 (1), 289– 320, DOI: 10.1021/cr200107zThere is no corresponding record for this reference.
- 34Kohn, W.; Becke, A. D.; Parr, R. G. Density functional theory of electronic structure. J. Phys. Chem. A 1996, 100 (31), 12974– 12980, DOI: 10.1021/jp960669lThere is no corresponding record for this reference.
- 35Dawson, W.; Degomme, A.; Stella, M.; Nakajima, T.; Ratcliff, L. E.; Genovese, L. Density functional theory calculations of large systems: Interplay between fragments, observables, and computational complexity. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2022, 12 (3), e1574 DOI: 10.1002/wcms.1574There is no corresponding record for this reference.
- 36Bishop, M.; Kalos, M.; Frisch, H. Molecular dynamics of polymeric systems. J. Chem. Phys. 1979, 70 (3), 1299– 1304, DOI: 10.1063/1.437567There is no corresponding record for this reference.
- 37Han, J.; Gee, R. H.; Boyd, R. H. Glass transition temperatures of polymers from molecular dynamics simulations. Macromolecules 1994, 27 (26), 7781– 7784, DOI: 10.1021/ma00104a036There is no corresponding record for this reference.
- 38Lau, D.; Jian, W.; Yu, Z.; Hui, D. Nano-engineering of construction materials using molecular dynamics simulations: Prospects and challenges. Compos. Part B 2018, 143, 282– 291, DOI: 10.1016/j.compositesb.2018.01.014There is no corresponding record for this reference.
- 39Mackerle, J. Finite element analysis and simulation of polymersan addendum: a bibliography (1996–2002). Modell. Simul. Mater. Sci. Eng. 2003, 11 (2), 195 DOI: 10.1088/0965-0393/11/2/307There is no corresponding record for this reference.
- 40Zeng, X.; Brown, L. P.; Endruweit, A.; Matveev, M.; Long, A. C. Geometrical modelling of 3d woven reinforcements for polymer composites: Prediction of fabric permeability and composite mechanical properties. Compos. Part A: Appl. Sci. Manuf. 2014, 56, 150– 160, DOI: 10.1016/j.compositesa.2013.10.004There is no corresponding record for this reference.
- 41Alshahrani, H. Characterization and finite element modeling of coupled properties during polymer composites forming processes. Mech. Mater. 2020, 144, 103370 DOI: 10.1016/j.mechmat.2020.103370There is no corresponding record for this reference.
- 42Thompson, A. P.; Aktulga, H. M.; Berger, R.; Bolintineanu, D. S.; Brown, W. M.; Crozier, P. S.; In’t Veld, P. J.; Kohlmeyer, A.; Moore, S. G.; Nguyen, T. D.; Shan, R.; Stevens, M. J.; Tranchida, J.; Trott, C.; Plimpton, S. J. Lammps-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171 DOI: 10.1016/j.cpc.2021.108171There is no corresponding record for this reference.
- 43Ratcliff, L. E.; Mohr, S.; Huhs, G.; Deutsch, T.; Masella, M.; Genovese, L. Challenges in large scale quantum mechanical calculations. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2017, 7 (1), e1290 DOI: 10.1002/wcms.1290There is no corresponding record for this reference.
- 44Fredrickson, G. H. Computational field theory of polymers: opportunities and challenges. Soft Matter 2007, 3 (11), 1329– 1334, DOI: 10.1039/b710604aThere is no corresponding record for this reference.
- 45Sindu, B.; Hamaekers, J. Feature-based prediction of properties of cross-linked epoxy polymers by molecular dynamics and machine learning techniques. Modell. Simul. Mater. Sci. Eng. 2025, 33 (6), 065010 DOI: 10.1088/1361-651X/adf56cThere is no corresponding record for this reference.
- 46Chew, A. K.; Afzal, M. A. F.; Chandrasekaran, A.; Kamps, J. H.; Ramakrishnan, V. Designing the next generation of polymers with machine learning and physics-based models. Mach. Learn. Sci. Technol. 2024, 5 (4), 045031 DOI: 10.1088/2632-2153/ad88d7There is no corresponding record for this reference.
- 47He, J.; Rong, F. Prediction of molecularly imprinted polymer binding affinity based on graph neural networks. J. Phys.: Conference Series 2025, 3030, 012009There is no corresponding record for this reference.
- 48Nagasawa, S.; Al-Naamani, E.; Saeki, A. Computer-aided screening of conjugated polymers for organic solar cell: classification by random forest. J. Phys. Chem. Lett. 2018, 9 (10), 2639– 2646, DOI: 10.1021/acs.jpclett.8b00635There is no corresponding record for this reference.
- 49Kishino, M.; Matsumoto, K.; Kobayashi, Y.; Taguchi, R.; Akamatsu, N.; Shishido, A. Fatigue life prediction of bending polymer films using random forest. Int. J. Fatigue 2023, 166, 107230 DOI: 10.1016/j.ijfatigue.2022.107230There is no corresponding record for this reference.
- 50Arora, A.; Lin, T.-S.; Rebello, N. J.; Av-Ron, S. H.; Mochigase, H.; Olsen, B. D. Random forest predictor for diblock copolymer phase behavior. ACS Macro Lett. 2021, 10 (11), 1339– 1345, DOI: 10.1021/acsmacrolett.1c00521There is no corresponding record for this reference.
- 51Malashin, I.; Tynchenko, V.; Gantimurov, A.; Nelyub, V.; Borodulin, A. Support vector machines in polymer science: a review. Polymers 2025, 17 (4), 491, DOI: 10.3390/polym17040491There is no corresponding record for this reference.
- 52Ziaee, H.; Hosseini, S. M.; Sharafpoor, A.; Fazavi, M.; Ghiasi, M. M.; Bahadori, A. Prediction of solubility of carbon dioxide in different polymers using support vector machine algorithm. J. Taiwan Inst. Chem. Eng. 2015, 46, 205– 213, DOI: 10.1016/j.jtice.2014.09.015There is no corresponding record for this reference.
- 53Chen, F.-C. Virtual screening of conjugated polymers for organic photovoltaic devices using support vector machines and ensemble learning. Int. J. Polym. Sci. 2019, 2019 (1), 4538514 DOI: 10.1155/2019/4538514There is no corresponding record for this reference.
- 54Brereton, R. G.; Lloyd, G. R. Support vector machines for classification and regression. Analyst 2010, 135 (2), 230– 267, DOI: 10.1039/B918972FThere is no corresponding record for this reference.
- 55Youshia, J.; Ali, M. E.; Lamprecht, A. Artificial neural network based particle size prediction of polymeric nanoparticles. Eur. J. Pharm. Biopharm. 2017, 119, 333– 342, DOI: 10.1016/j.ejpb.2017.06.030There is no corresponding record for this reference.
- 56Zhang, Z.; Friedrich, K. Artificial neural networks applied to polymer composites: a review. Compos. Sci. Technol. 2003, 63 (14), 2029– 2044, DOI: 10.1016/S0266-3538(03)00106-4There is no corresponding record for this reference.
- 57El Kadi, H. Modeling the mechanical behavior of fiber-reinforced polymeric composite materials using artificial neural networksa review. Compos. Struct. 2006, 73 (1), 1– 23, DOI: 10.1016/j.compstruct.2005.01.020There is no corresponding record for this reference.
- 58Liu, W.; Cao, C. Artificial neural network prediction of glass transition temperature of polymers. Colloid Polym. Sci. 2009, 287 (7), 811– 818, DOI: 10.1007/s00396-009-2035-yThere is no corresponding record for this reference.
- 59Chen, L.; Pilania, G.; Batra, R.; Huan, T. D.; Kim, C.; Kuenneth, C.; Ramprasad, R. Polymer informatics: Current status and critical next steps. Mater. Sci. Eng. R 2021, 144, 100595 DOI: 10.1016/j.mser.2020.100595There is no corresponding record for this reference.
- 60Butler, K. T.; Davies, D. W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547– 555, DOI: 10.1038/s41586-018-0337-2There is no corresponding record for this reference.
- 61Schleder, G. R.; Padilha, A. C. M.; Acosta, C. M.; Costa, M.; Fazzio, A. From dft to machine learning: Recent approaches to materials science-a review. J. Chem. Inf. Model. 2019, 2 (4), 1347– 1356, DOI: 10.1088/2515-7639/ab084bThere is no corresponding record for this reference.
- 62Sha, W.; Li, Y.; Tang, S.; Tian, J.; Zhao, Y.; Guo, Y.; Zhang, W.; Zhang, X.; Lu, S.; Cao, Y.-C.; Cheng, S. Machine learning in polymer informatics. InfoMat 2021, 3 (4), 353– 361, DOI: 10.1002/inf2.12167There is no corresponding record for this reference.
- 63Martin, T. B.; Audus, D. J. Emerging trends in machine learning: a polymer perspective. ACS Polymers Au 2023, 3 (3), 239– 258, DOI: 10.1021/acspolymersau.2c00053There is no corresponding record for this reference.
- 64Ramprasad, R.; Batra, R.; Pilania, G.; Mannodi-Kanakkithodi, A.; Kim, C. Machine learning in materials informatics: recent applications and prospects. npj Comput. Mater. 2017, 3 (1), 54, DOI: 10.1038/s41524-017-0056-5There is no corresponding record for this reference.
- 65Chen, G.; Shen, Z.; Iyer, A.; Ghumman, U. F.; Tang, S.; Bi, J.; Chen, W.; Li, Y. Machine-learning-assisted de novo design of organic molecules and polymers: opportunities and challenges. Polymers 2020, 12 (1), 163, DOI: 10.3390/polym12010163There is no corresponding record for this reference.
- 66Wu, S.; Yamada, H.; Hayashi, Y.; Zamengo, M.; Yoshida, R. Potentials and challenges of polymer informatics: exploiting machine learning for polymer design, 2020, arXiv:2010.07683. arXiv.org e-Print archive https://arxiv.org/abs/2010.07683.There is no corresponding record for this reference.
- 67Kumar, J. N.; Li, Q.; Jun, Y. Challenges and opportunities of polymer design with machine learning and high throughput experimentation. MRS Commun. 2019, 9 (2), 537– 544, DOI: 10.1557/mrc.2019.54There is no corresponding record for this reference.
- 68Wieder, O.; Kohlbacher, S.; Kuenemann, M.; Garon, A.; Ducrot, P.; Seidel, T.; Langer, T. A compact review of molecular property prediction with graph neural networks. Drug Discovery Today: Technol. 2020, 37, 1– 12, DOI: 10.1016/j.ddtec.2020.11.009There is no corresponding record for this reference.
- 69Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Philip, S. Y. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2021, 32 (1), 4– 24, DOI: 10.1109/TNNLS.2020.2978386There is no corresponding record for this reference.
- 70Gao, Q.; Dukker, T.; Schweidtmann, A. M.; Weber, J. M. Self-supervised graph neural networks for polymer property prediction. Mol. Syst. Des. Eng. 2024, 9 (11), 1130– 1143, DOI: 10.1039/D4ME00088AThere is no corresponding record for this reference.
- 71Inae, E.; Liu, Y.; Zhu, Y.; Xu, J.; Liu, G.; Zhang, R.; Luo, T.; Jiang, M. Modeling Polymers with Neural Networks; American Chemical Society, 2025.There is no corresponding record for this reference.
- 72Gurney, K. An Introduction to Neural Networks; CRC Press, 2018.There is no corresponding record for this reference.
- 73Haykin, S.; Network, N. A comprehensive foundation. Neural Netw. 2004, 2, 41There is no corresponding record for this reference.
- 74LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86 (11), 2278– 2324, DOI: 10.1109/5.726791There is no corresponding record for this reference.
- 75Li, Z.; Liu, F.; Yang, W.; Peng, S.; Zhou, J. A survey of convolutional neural networks: analysis, applications, and prospects. IEEE Trans. Neural Netw. Learn. Syst. 2022, 33 (12), 6999– 7019, DOI: 10.1109/TNNLS.2021.3084827There is no corresponding record for this reference.
- 76Gu, J.; Wang, Z.; Kuen, J.; Ma, L.; Shahroudy, A.; Shuai, B.; Liu, T.; Wang, X.; Wang, G.; Cai, J.; Chen, T. Recent advances in convolutional neural networks. Pattern Recognit. 2018, 77, 354– 377, DOI: 10.1016/j.patcog.2017.10.013There is no corresponding record for this reference.
- 77Ge, W.; De Silva, R.; Fan, Y.; Sisson, S. A.; Stenzel, M. H. Machine learning in polymer research. Adv. Mater. 2025, 37 (11), 2413695 DOI: 10.1002/adma.202413695There is no corresponding record for this reference.
- 78Platonov, O.; Kuznedelev, D.; Diskin, M.; Babenko, A.; Prokhorenkova, L. A critical look at the evaluation of gnns under heterophily: Are we really making progress? 2023, arXiv:2302.11640. arXiv.org e-Print archive https://arxiv.org/abs/2302.11640.There is no corresponding record for this reference.
- 79Wang, Y.; Li, Z.; Barati Farimani, A. Graph neural networks for molecules. In Machine Learning in Molecular Sciences; Springer, 2023; pp 21– 66.There is no corresponding record for this reference.
- 80Hamilton, W.; Ying, Z.; Leskovec, J. Inductive representation learning on large graphs. In NIPS’17: Proceedings of the 31st International Conference on Neural Information Processing Systems; Department of Computer Science: Stanford, CA, 2017; pp 1025– 1035.There is no corresponding record for this reference.
- 81Xu, K.; Hu, W.; Leskovec, J.; Jegelka, S. How powerful are graph neural networks? 2018, arXiv:1810.00826. arXiv.org e-Print archive https://arxiv.org/abs/1810.00826.There is no corresponding record for this reference.
- 82Sharma, K.; Lee, Y.-C.; Nambi, S.; Salian, A.; Shah, S.; Kim, S.-W.; Kumar, S. A survey of graph neural networks for social recommender systems. ACM Computing Surveys 2024, 56 (10), 1– 34, DOI: 10.1145/3661821There is no corresponding record for this reference.
- 83Kumar, S.; Mallik, A.; Khetarpal, A.; Panda, B. S. Influence maximization in social networks using graph embedding and graph neural network. Informat. Sci. 2022, 607, 1617– 1636, DOI: 10.1016/j.ins.2022.06.075There is no corresponding record for this reference.
- 84Zhang, X.-M.; Liang, L.; Liu, L.; Tang, M.-J. Graph neural networks and their current applications in bioinformatics. Front. Genetics 2021, 12, 690049 DOI: 10.3389/fgene.2021.690049There is no corresponding record for this reference.
- 85Malla, A. M.; Banka, A. A. A systematic review of deep graph neural networks: Challenges, classification, architectures, applications & potential utility in bioinformatics, 2023, arXiv:2311.02127. arXiv.org e-Print archive https://arxiv.org/abs/2311.02127.There is no corresponding record for this reference.
- 86Zheng, P.; Wang, S.; Wang, X.; Zeng, X. Artificial intelligence in bioinformatics and drug repurposing: methods and applications. In Frontiers Research Topics , 2022.There is no corresponding record for this reference.
- 87Reiser, P.; Neubert, M.; Eberhard, A.; Torresi, L.; Zhou, C.; Shao, C.; Metni, H.; van Hoesel, C.; Schopmans, H.; Sommer, T.; Friederich, P. Graph neural networks for materials science and chemistry. Commun. Mater. 2022, 3 (1), 93, DOI: 10.1038/s43246-022-00315-6There is no corresponding record for this reference.
- 88Fung, V.; Zhang, J.; Juarez, E.; Sumpter, B. G. Benchmarking graph neural networks for materials chemistry. npj Computat. Mater. 2021, 7 (1), 84, DOI: 10.1038/s41524-021-00554-0There is no corresponding record for this reference.
- 89Maurizi, M.; Gao, C.; Berto, F. Predicting stress, strain and deformation fields in materials and structures with graph neural networks. Sci. Rep. 2022, 12 (1), 21834 DOI: 10.1038/s41598-022-26424-3There is no corresponding record for this reference.
- 90Kipf, T. N.; Welling, M. Semi-supervised classification with graph convolutional networks, 2016, arXiv:1609.02907. arXiv.org e-Print archive https://arxiv.org/abs/1609.02907.There is no corresponding record for this reference.
- 91Gilmer, J.; Schoenholz, S. S.; Riley, P. F.; Vinyals, O.; Dahl, G. E. Neural message passing for quantum chemistry. In International Conference on Machine Learning; PMLR, 2017; pp 1263– 1272.There is no corresponding record for this reference.
- 92Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. “ Graph attention networks, 2017, arXiv:1710.10903. arXiv.org e-Print archive https://arxiv.org/abs/1710.10903.There is no corresponding record for this reference.
- 93Liu, G.; Inae, E.; Jiang, M. Deep learning for polymer property prediction. In Deep Learning for Polymer Discovery: Foundation and Advances; Springer, 2025; pp 17– 35.There is no corresponding record for this reference.
- 94Dong, C.; Li, D.; Liu, J. Glass transition temperature prediction of polymers via graph reinforcement learning. Langmuir 2024, 40 (35), 18568– 18580, DOI: 10.1021/acs.langmuir.4c01906There is no corresponding record for this reference.
- 95Wang, X.; Liao, L.; Huang, C.; Wu, X. Prediction of mechanical properties of cross-linked polymer interface by graph convolution network. Acta Mechanica Sinica 2026, 42 (3), 424627 DOI: 10.1007/s10409-024-24627-xThere is no corresponding record for this reference.
- 96Safari, H.; Bavarian, M. Enhancing polymer reaction engineering through the power of machine learning. In Systems and Control Transactions , 2024; p 157792.There is no corresponding record for this reference.
- 97Liao, H.-C.; Lin, Y.-H.; Peng, C.-H.; Li, Y.-P. Directed message passing neural networks for accurate prediction of polymer-solvent interaction parameters. ACS Engineering Au 2025, 5, 530, DOI: 10.1021/acsengineeringau.5c00027There is no corresponding record for this reference.
- 98Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B. A.; Thiessen, P. A.; Yu, B. Pubchem 2023 update. Nucleic Acids Res. 2023, 51 (D1), D1373– D1380, DOI: 10.1093/nar/gkac956There is no corresponding record for this reference.
- 99Gao, Z.-Y.; Liu, G.-Q. Recent progress of web-enable material database and a case study of nims and matweb. J. Mater. Eng. 2013, 3 (11), 89– 96, DOI: 10.3969/j.issn.1001-4381.2013.06.001There is no corresponding record for this reference.
- 100Wypych, G. Handbook of Polymers; Elsevier, 2022.There is no corresponding record for this reference.
- 101Patra, A.; Batra, R.; Chandrasekaran, A.; Kim, C.; Huan, T. D.; Ramprasad, R. A multi-fidelity information-fusion approach to machine learn and predict polymer bandgap. Comput. Mater. Sci. 2020, 172, 109286 DOI: 10.1016/j.commatsci.2019.109286There is no corresponding record for this reference.
- 102Dhamankar, S.; Webb, M. A. Chemically specific coarse-graining of polymers: methods and prospects. J. Polym. Sci. 2021, 59 (22), 2613– 2643, DOI: 10.1002/pol.20210555There is no corresponding record for this reference.
- 103Müller-Plathe, F. Coarse-graining in polymer simulation: from the atomistic to the mesoscopic scale and back. ChemPhysChem 2002, 3 (9), 754– 769, DOI: 10.1002/1439-7641(20020916)3:9<754::AID-CPHC754>3.0.CO;2-UThere is no corresponding record for this reference.
- 104Harmandaris, V. A.; Reith, D.; Van der Vegt, N. F.; Kremer, K. Comparison between coarse-graining models for polymer systems: Two mapping schemes for polystyrene. Macromol. Chem. Phys. 2007, 208 (19–20), 2109– 2120, DOI: 10.1002/macp.200700245There is no corresponding record for this reference.
- 105Padding, J. T.; Briels, W. J. Systematic coarse-graining of the dynamics of entangled polymer melts: the road from chemistry to rheology. J. Phys.: Condens. Matter 2011, 23 (23), 233101 DOI: 10.1088/0953-8984/23/23/233101There is no corresponding record for this reference.
- 106Zhang, X.; Sheng, Y.; Liu, X.; Yang, J.; Goddard, W. A., III; Ye, C.; Zhang, W. Polymer-unit graph: Advancing interpretability in graph neural network machine learning for organic polymer semiconductor materials. J. Chem. Theory Comput. 2024, 20 (7), 2908– 2920, DOI: 10.1021/acs.jctc.3c01385There is no corresponding record for this reference.
- 107Mohapatra, S.; An, J.; Gómez-Bombarelli, R. Chemistry-informed macromolecule graph representation for similarity computation, unsupervised and supervised learning. Machine Learning: Sci. Technol. 2022, 3 (1), 015028 DOI: 10.1088/2632-2153/ac545eThere is no corresponding record for this reference.
- 108Wang, X.; Xu, Y.; Zheng, H.; Yu, K. A scalable graph neural network method for developing an accurate force field of large flexible organic molecules. J. Phys. Chem. Lett. 2021, 12 (33), 7982– 7987, DOI: 10.1021/acs.jpclett.1c02214There is no corresponding record for this reference.
- 109Qiu, H.; Qiu, X.; Dai, X.; Sun, Z.-Y. Design of polyimides with targeted glass transition temperature using a graph neural network. J. Mater. Chem. C 2023, 11 (8), 2930– 2940, DOI: 10.1039/D2TC05174EThere is no corresponding record for this reference.
- 110Zeng, M.; Kumar, J. N.; Zeng, Z.; Savitha, R.; Chandrasekhar, V. R.; Hippalgaonkar, K. “ Graph convolutional neural networks for polymers property prediction, 2018, arXiv:1811.06231. arXiv.org e-Print archive https://arxiv.org/abs/1811.06231.There is no corresponding record for this reference.
- 111Queen, O.; McCarver, G. A.; Thatigotla, S.; Abolins, B. P.; Brown, C. L.; Maroulas, V.; Vogiatzis, K. D. Polymer graph neural networks for multitask property learning. npj Computat. Mater. 2023, 9, 90, DOI: 10.1038/s41524-023-01034-3There is no corresponding record for this reference.
- 112Bongini, P.; Bianchini, M.; Scarselli, F. Molecular generative graph neural networks for drug discovery. Neurocomputing 2021, 450, 242– 252, DOI: 10.1016/j.neucom.2021.04.039There is no corresponding record for this reference.
- 113Scarselli, F.; Gori, M.; Tsoi, A. C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Transact. Neural Networks 2009, 20 (1), 61– 80, DOI: 10.1109/TNN.2008.2005605There is no corresponding record for this reference.
- 114Dwivedi, V. P.; Joshi, C. K.; Luu, A. T.; Laurent, T.; Bengio, Y.; Bresson, X. Benchmarking graph neural networks. J. Machine Learning Res. 2023, 24 (43), 1– 48There is no corresponding record for this reference.
- 115Veličković, P. Everything is connected: Graph neural networks. Curr. Opin. Struct. Biol. 2023, 79, 102538 DOI: 10.1016/j.sbi.2023.102538There is no corresponding record for this reference.
- 116Jegelka, S. Theory of graph neural networks: Representation and learning. In International Congress of Mathematicians , 2022; pp 1– 23.There is no corresponding record for this reference.
- 117Liu, Z.; Zhou, J. Introduction to Graph Neural Networks; Springer Nature, 2022.There is no corresponding record for this reference.
- 118Tang, M.; Li, B.; Chen, H. Application of message passing neural networks for molecular property prediction. Curr. Opin. Struct. Biol. 2023, 81, 102616 DOI: 10.1016/j.sbi.2023.102616There is no corresponding record for this reference.
- 119Hasebe, T. Knowledge-embedded message-passing neural networks: improving molecular property prediction with human knowledge. ACS omega 2021, 6 (42), 27955– 27967, DOI: 10.1021/acsomega.1c03839There is no corresponding record for this reference.
- 120Zhang, H.; Lin, Y.; Li, S.; Dai, M.; Zhang, Y.; Huang, L.; Pang, J.; Wu, P.; Peng, J.; Tang, Z.; Ding, P.; Xiao, W.; Song, N.; Dongbo, D. Substructure-enhanced mpnn for polymer discovery and knowledge: A study in predicting glass transition temperature. Macromolecules 2025, 58, 9515, DOI: 10.1021/acs.macromol.4c02859There is no corresponding record for this reference.
- 121Aldeghi, M.; Coley, C. W. A graph representation of molecular ensembles for polymer property prediction. Chem. Sci. 2022, 13 (35), 10486– 10498, DOI: 10.1039/D2SC02839EThere is no corresponding record for this reference.
- 122Heid, E.; Greenman, K. P.; Chung, Y.; Li, S.-C.; Graff, D. E.; Vermeire, F. H.; Wu, H.; Green, W. H.; McGill, C. J. Chemprop: a machine learning package for chemical property prediction. J. Chem. Inf. Model. 2024, 64 (1), 9– 17, DOI: 10.1021/acs.jcim.3c01250There is no corresponding record for this reference.
- 123Antoniuk, E. R.; Li, P.; Kailkhura, B.; Hiszpanski, A. M. Representing polymers as periodic graphs with learned descriptors for accurate polymer property predictions. J. Chem. Inf. Model. 2022, 62, 5435– 5445, DOI: 10.1021/acs.jcim.2c00875There is no corresponding record for this reference.
- 124Sanchez Medina, E. I.; Kunchapu, S.; Sundmacher, K. Gibbs-helmholtz graph neural network for the prediction of activity coefficients of polymer solutions at infinite dilution. J. Phys. Chem. A 2023, 127 (46), 9863– 9873, DOI: 10.1021/acs.jpca.3c05892There is no corresponding record for this reference.
- 125Battaglia, P. W.; Hamrick, J. B.; Bapst, V.; Sanchez-Gonzalez, A.; Zambaldi, V.; Malinowski, M.; Tacchetti, A.; Raposo, D.; Santoro, A.; Faulkner, R.; C, G.; F, S.; Ballard, A.; Justin, G.; George, D.; Ashish, V.; Kelsey, A.; Charles, N.; Victoria, L.; Chris, D.; Nicolas, H.; Dann, W.; Pushmeet, K.; Matt, B.; Oriol, V.; Yujia, L.; Razvan, P. Relational inductive biases, deep learning, and graph networks, 2018, arXiv:1806.01261. arXiv.org e-Print archive https://arxiv.org/abs/1806.01261.There is no corresponding record for this reference.
- 126Gurnani, R.; Kuenneth, C.; Toland, A.; Ramprasad, R. Polymer informatics at scale with multitask graph neural networks. Chem. Mater. 2023, 35 (6), 1560– 1567, DOI: 10.1021/acs.chemmater.2c02991There is no corresponding record for this reference.
- 127St John, P. C.; Phillips, C.; Kemper, T. W.; Wilson, A. N.; Guan, Y.; Crowley, M. F.; Nimlos, M. R.; Larsen, R. E. Message-passing neural networks for high-throughput polymer screening. J. Chem. Phys. 2019, 150, 234111 DOI: 10.1063/1.5099132There is no corresponding record for this reference.
- 128Li, Y.; Zemel, R.; Brockschmidt, M.; Tarlow, D. Gated graph sequence neural networks. In Proceedings of ICLR16 , 2016.There is no corresponding record for this reference.
- 129Dey, R.; Salem, F. M. Gate-variants of gated recurrent unit (gru) neural networks. In 2017 IEEE 60th international midwest symposium on circuits and systems (MWSCAS); IEEE, 2017; pp 1597– 1600.There is no corresponding record for this reference.
- 130Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Briefings Bioinf. 2020, 21 (3), 919– 935, DOI: 10.1093/bib/bbz042There is no corresponding record for this reference.
- 131Xu, X.; Zhao, X.; Wei, M.; Li, Z. A comprehensive review of graph convolutional networks: approaches and applications. Electronic Res. Archive 2023, 31 (7), 4185– 4215, DOI: 10.3934/era.2023213There is no corresponding record for this reference.
- 132Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph neural networks: A review of methods and applications. IEEE Transactions on Pattern Analysis and Machine Intelligence 2019, 42, 283– 300There is no corresponding record for this reference.
- 133Hu, J.; Li, Z.; Lin, J.; Zhang, L. Prediction and interpretability of glass transition temperature of homopolymers by data-augmented graph convolutional neural networks. ACS Appl. Mater. Interfaces 2023, 15 (46), 54006– 54017, DOI: 10.1021/acsami.3c13698There is no corresponding record for this reference.
- 134Hickey, K.; Feinstein, J.; Sivaraman, G.; MacDonell, M.; Yan, E.; Matherson, C.; Coia, S.; Xu, J.; Picel, K. Applying machine learning and quantum chemistry to predict the glass transition temperatures of polymers. Comput. Mater. Sci. 2024, 238, 112933 DOI: 10.1016/j.commatsci.2024.112933There is no corresponding record for this reference.
- 135Kimmig, J.; Schuett, T.; Vollrath, A.; Zechel, S.; Schubert, U. S. Prediction of nanoparticle sizes for arbitrary methacrylates using artificial neuronal networks. Adv. Sci. 2021, 8 (23), 2102429 DOI: 10.1002/advs.202102429There is no corresponding record for this reference.
- 136Tian, Y.; Zhang, Y.; Zhang, H. Recent advances in stochastic gradient descent in deep learning. Mathematics 2023, 11 (3), 682, DOI: 10.3390/math11030682There is no corresponding record for this reference.
- 137Merity, S. “ Single headed attention rnn: Stop thinking with your head, 2019, arXiv:1911.11423. arXiv.org e-Print archive https://arxiv.org/abs/1911.11423.There is no corresponding record for this reference.
- 138Inoue, H. Multi-sample dropout for accelerated training and better generalization, 2019, arXiv:1905.09788. arXiv.org e-Print archive https://arxiv.org/abs/1905.09788.There is no corresponding record for this reference.
- 139Cho, K.; Van Merriënboer, B.; Bahdanau, D.; Bengio, Y. On the properties of neural machine translation: Encoder-decoder approaches, 2014, arXiv:1409.1259. arXiv.org e-Print archive https://arxiv.org/abs/1409.1259.There is no corresponding record for this reference.
- 140Morris, C.; Ritzert, M.; Fey, M.; Hamilton, W. L.; Lenssen, J. E.; Rattan, G.; Grohe, M. Weisfeiler and leman go neural: Higher-order graph neural networks. Proceedings of the AAAI Conference on Artificial Intelligence 2019, 33, 4602– 4609, DOI: 10.1609/aaai.v33i01.33014602There is no corresponding record for this reference.
- 141Ye, X.-b.; Guan, Q.; Luo, W.; Fang, L.; Lai, Z.-R.; Wang, J. Molecular substructure graph attention network for molecular property identification in drug discovery. Pattern Recognition 2022, 128, 108659 DOI: 10.1016/j.patcog.2022.108659There is no corresponding record for this reference.
- 142Vrahatis, A. G.; Lazaros, K.; Kotsiantis, S. Graph attention networks: a comprehensive review of methods and applications. Future Internet 2024, 16 (9), 318, DOI: 10.3390/fi16090318There is no corresponding record for this reference.
- 143Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H.; Z M Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 2020, 63 (16), 8749– 8760, DOI: 10.1021/acs.jmedchem.9b00959There is no corresponding record for this reference.
- 144Li, D.; Ru, Y.; Liu, J. Gatboost: Mining graph attention networks-based important substructures of polymers for a better property prediction. Mater. Today Commun. 2024, 38, 107577 DOI: 10.1016/j.mtcomm.2023.107577There is no corresponding record for this reference.
- 145Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining , 2016; pp 785– 794.There is no corresponding record for this reference.
- 146Liu, T.; Jiang, A.; Zhou, J.; Li, M.; Kwan, H. K. Graphsage-based dynamic spatial-temporal graph convolutional network for traffic prediction. IEEE Transactions on Intelligent Transportation Systems 2023, 24 (10), 11210– 11224, DOI: 10.1109/TITS.2023.3279929There is no corresponding record for this reference.
- 147Xie, T.; France-Lanord, A.; Wang, Y.; Lopez, J.; Stolberg, M. A.; Hill, M.; Leverick, G. M.; Gomez-Bombarelli, R.; Johnson, J. A.; Shao-Horn, Y.; Grossman, J. C. Accelerating amorphous polymer electrolyte screening by learning to reduce errors in molecular dynamics simulated properties. Nat. Commun. 2022, 13 (1), 3415 DOI: 10.1038/s41467-022-30994-1There is no corresponding record for this reference.
- 148Qiu, H.; Wang, J.; Qiu, X.; Dai, X.; Sun, Z.-Y. Heat-resistant polymer discovery by utilizing interpretable graph neural network with small data. Macromolecules 2024, 57 (8), 3515– 3528, DOI: 10.1021/acs.macromol.4c00508There is no corresponding record for this reference.
- 149Park, J.; Shim, Y.; Lee, F.; Rammohan, A.; Goyal, S.; Shim, M.; Jeong, C.; Kim, D. S. Prediction and interpretation of polymer properties using the graph convolutional network. ACS Polymers Au 2022, 2, 213, DOI: 10.1021/acspolymersau.1c00050There is no corresponding record for this reference.
- 150Burkholz, R.; Quackenbush, J.; Bojar, D. Using graph convolutional neural networks to learn a representation for glycans. Cell Rep. 2021, 35 (11), 109251, DOI: 10.1016/j.celrep.2021.109251There is no corresponding record for this reference.
- 151Volgin, I. V.; Batyr, P. A.; Matseevich, A. V.; Dobrovskiy, A. Y.; Andreeva, M. V.; Nazarychev, V. M.; Larin, S. V.; Goikhman, M. Y.; Vizilter, Y. V.; Askadskii, A. A.; Lyulin, S. V. Machine learning with enormous “synthetic” data sets: Predicting glass transition temperature of polyimides using graph convolutional neural networks. ACS Omega 2022, 7 (48), 43678– 43691, DOI: 10.1021/acsomega.2c04649There is no corresponding record for this reference.
- 152Otsuka, S.; Kuwajima, I.; Hosoya, J.; Xu, Y.; Yamazaki, M. Polyinfo: Polymer database for polymeric materials design. In 2011 International Conference on Emerging Intelligent Data and Web Technologies; IEEE, 2011; pp 22– 29.There is no corresponding record for this reference.
- 153Brandrup, J.; Immergut, E. H.; Grulke, E. A.; Abe, A.; Bloch, D. R. Polymer Handbook; Wiley: New York, 1999; Vol. 89.There is no corresponding record for this reference.
- 154Mark, J. E. Physical Properties of Polymers Handbook; Springer, 2007; Vol. 1076.There is no corresponding record for this reference.
- 155Ma, R.; Luo, T. Pi1m: a benchmark database for polymer informatics. J. Chem. Inf. Model. 2020, 60 (10), 4684– 4690, DOI: 10.1021/acs.jcim.0c00726There is no corresponding record for this reference.
- 156Walsh, D. J.; Zou, W.; Schneider, L.; Mello, R.; Deagen, M. E.; Mysona, J.; Lin, T.-S.; de Pablo, J. J.; Jensen, K. F.; Audus, D. J. Community resource for innovation in polymer technology (cript): a scalable polymer material data structure, 2023.There is no corresponding record for this reference.
- 157Yan, C.; Li, G. The rise of machine learning in polymer discovery. Advanced Intelligent Systems 2023, 5 (4), 2200243 DOI: 10.1002/aisy.202200243There is no corresponding record for this reference.
- 158Levine, D. S.; Shuaibi, M.; Spotte-Smith, E. W. C.; Taylor, M. G.; Hasyim, M. R.; Michel, K.; Batatia, I.; Csányi, G.; Dzamba, M.; Eastman, P. The open molecules 2025 (omol25) dataset, evaluations, and models, 2025, arXiv:2505.08762. arXiv.org e-Print archive https://arxiv.org/abs/2505.08762.There is no corresponding record for this reference.
- 159Mark, J. E. Polymer Data Handbook, Oxford University Press, New York, NY, 2009.There is no corresponding record for this reference.
- 160Hayashi, Y.; Shiomi, J.; Morikawa, J.; Yoshida, R. Radonpy: automated physical property calculation using all-atom classical molecular dynamics simulations for polymer informatics. npj Computat. Mater. 2022, 8 (1), 222, DOI: 10.1038/s41524-022-00906-4There is no corresponding record for this reference.
- 161Ellis, B.; Smith, R. Polymers: A Property Database. CRC Press, 2008.There is no corresponding record for this reference.
- 162Huan, T. D.; Mannodi-Kanakkithodi, A.; Kim, C.; Sharma, V.; Pilania, G.; Ramprasad, R. A polymer dataset for accelerated property prediction and design. Scientific data 2016, 3 (1), 1– 10, DOI: 10.1038/sdata.2016.12There is no corresponding record for this reference.
- 163Pence, H. E.; Williams, A. Chemspider: an online chemical information resource, 2010.There is no corresponding record for this reference.
- 164Polymer property predictor and database. https://pppdb.uchicago.edu/, 2019–2025.There is no corresponding record for this reference.
- 165Campus plastic s database: Computer aided material preselection by uniform standards. https://www.campusplastics.com/, 1988–2025.There is no corresponding record for this reference.
- 166Miccio, L. A.; Schwartz, G. A. From chemical structure to quantitative polymer properties prediction through convolutional neural networks. Polymer 2020, 193, 122341 DOI: 10.1016/j.polymer.2020.122341There is no corresponding record for this reference.
- 167Xu, P.; Lu, T.; Ju, L.; Tian, L.; Li, M.; Lu, W. Machine learning aided design of polymer with targeted band gap based on dft computation. J. Phys. Chem. B 2021, 125 (2), 601– 611, DOI: 10.1021/acs.jpcb.0c08674There is no corresponding record for this reference.
- 168Xiong, J.; Xiong, Z.; Chen, K.; Jiang, H.; Zheng, M. Graph neural networks for automated de novo drug design. Drug Discovery Today 2021, 26 (6), 1382– 1393, DOI: 10.1016/j.drudis.2021.02.011There is no corresponding record for this reference.
- 169Medina, H.; Drake, R.; Farmer, C. Accelerated prediction of molecular properties for per-and polyfluoroalkyl substances using graph neural networks with adjacency-free message passing. Environ. Pollut. 2025, 382, 126705 DOI: 10.1016/j.envpol.2025.126705There is no corresponding record for this reference.
- 170Tozzini, V. Coarse-grained models for proteins. Curr. Opin. Struct. Biol. 2005, 15 (2), 144– 150, DOI: 10.1016/j.sbi.2005.02.005There is no corresponding record for this reference.
- 171Joshi, S. Y.; Deshmukh, S. A. A review of advancements in coarse-grained molecular dynamics simulations. Mol. Simul. 2021, 47 (10–11), 786– 803, DOI: 10.1080/08927022.2020.1828583There is no corresponding record for this reference.
- 172Noid, W. G.; Liu, P.; Wang, Y.; Chu, J.-W.; Ayton, G. S.; Izvekov, S.; Andersen, H. C.; Voth, G. A. The multiscale coarse-graining method. ii. numerical implementation for coarse-grained molecular models. J. Chem. Phys. 2008, 128, 244115, DOI: 10.1063/1.2938857There is no corresponding record for this reference.
- 173Pak, A. J.; Voth, G. A. Advances in coarse-grained modeling of macromolecular complexes. Curr. Opin. Struct. Biol. 2018, 52, 119– 126, DOI: 10.1016/j.sbi.2018.11.005There is no corresponding record for this reference.
- 174Noid, W. G. Perspective: Coarse-grained models for biomolecular systems. J. Chem. Phys. 2013, 139, 090901, DOI: 10.1063/1.4818908There is no corresponding record for this reference.
- 175Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A. E.; Kolinski, A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116 (14), 7898– 7936, DOI: 10.1021/acs.chemrev.6b00163There is no corresponding record for this reference.
- 176Ingólfsson, H. I.; Lopez, C. A.; Uusitalo, J. J.; de Jong, D. H.; Gopal, S. M.; Periole, X.; Marrink, S. J. The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4 (3), 225– 248, DOI: 10.1002/wcms.1169There is no corresponding record for this reference.
- 177Noid, W.; Szukalo, R. J.; Kidder, K. M.; Lesniewski, M. C. Rigorous progress in coarse-graining. Annu. Rev. Phys. Chem. 2024, 75 (1), 21– 45, DOI: 10.1146/annurev-physchem-062123-010821There is no corresponding record for this reference.
- 178Guenza, M. G. Everything you want to know about coarse-graining and never dared to ask: Macromolecules as a key example. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2025, 15 (2), e70022 DOI: 10.1002/wcms.70022There is no corresponding record for this reference.
- 179Harmandaris, V. A.; Kremer, K. Predicting polymer dynamics at multiple length and time scales. Soft Matter 2009, 5 (20), 3920– 3926, DOI: 10.1039/b905361aThere is no corresponding record for this reference.
- 180Ye, H.; Xian, W.; Li, Y. Machine learning of coarse-grained models for organic molecules and polymers: Progress, opportunities, and challenges. ACS Omega 2021, 6 (3), 1758– 1772, DOI: 10.1021/acsomega.0c05321There is no corresponding record for this reference.
- 181Noid, W. G. Perspective: Advances, challenges, and insight for predictive coarse-grained models. J. Phys. Chem. B 2023, 127 (19), 4174– 4207, DOI: 10.1021/acs.jpcb.2c08731There is no corresponding record for this reference.
- 182Vettorel, T.; Besold, G.; Kremer, K. Fluctuating soft-sphere approach to coarse-graining of polymer models. Soft Matter 2010, 6 (10), 2282– 2292, DOI: 10.1039/b921159dThere is no corresponding record for this reference.
- 183Español, P.; Serrano, M.; Pagonabarraga, I.; Zúñiga, I. Energy-conserving coarse-graining of complex molecules. Soft Matter 2016, 12 (21), 4821– 4837, DOI: 10.1039/C5SM03038BThere is no corresponding record for this reference.
- 184Husic, B. E.; Charron, N. E.; Lemm, D.; Wang, J.; Pérez, A.; Majewski, M.; Krämer, A.; Chen, Y.; Olsson, S.; De Fabritiis, G.; Noe, F.; Clementi, C. Coarse graining molecular dynamics with graph neural networks J. Chem. Phys. 2020; Vol. 153, DOI: 10.1063/5.0026133 .There is no corresponding record for this reference.
- 185Wang, X.; Wu, Y.; Zhang, A.; He, X.; Chua, T.-S. Towards multi-grained explainability for graph neural networks. Adv. Neural Inf. Process. Syst. 2021, 34, 18446– 18458There is no corresponding record for this reference.
- 186Peter, C.; Kremer, K. Multiscale simulation of soft matter systems-from the atomistic to the coarse-grained level and back. Soft Matter 2009, 5 (22), 4357– 4366, DOI: 10.1039/b912027kThere is no corresponding record for this reference.
- 187Zhao, Y.; Chen, G.; Liu, J. Polymer data challenges in the ai era: Bridging gaps for next-generation energy materials, 2025, arXiv:2505.13494. arXiv.org e-Print archive https://arxiv.org/abs/2505.13494.There is no corresponding record for this reference.
- 188Orio, M.; Pantazis, D. A.; Neese, F. Density functional theory. Photosynth. Res. 2009, 102, 443– 453, DOI: 10.1007/s11120-009-9404-8There is no corresponding record for this reference.
- 189Joshi, S. P.; Bucsek, A.; Pagan, D. C.; Daly, S.; Ravindran, S.; Marian, J.; Bessa, M. A.; Kalidindi, S. R.; Admal, N. C.; Reina, C.; Ghosh, S.; Warren, J. A.; Vinals, J.; Tadmor, E. B. “ Integrated experiment and simulation co-design: A key infrastructure for predictive mesoscale materials modeling, 2025, arXiv:2503.09793. arXiv.org e-Print archive https://arxiv.org/abs/2503.09793.There is no corresponding record for this reference.
- 190Zaporozhets, I.; Clementi, C. Multibody terms in protein coarse-grained models: A top-down perspective. J. Phys. Chem. B 2023, 127 (31), 6920– 6927, DOI: 10.1021/acs.jpcb.3c04493There is no corresponding record for this reference.
- 191Jin, J.; Pak, A. J.; Durumeric, A. E.; Loose, T. D.; Voth, G. A. Bottom-up coarse-graining: Principles and perspectives. J. Chem. Theory Comput. 2022, 18 (10), 5759– 5791, DOI: 10.1021/acs.jctc.2c00643There is no corresponding record for this reference.
- 192Praprotnik, M.; Delle Site, L.; Kremer, K. Multiscale simulation of soft matter: From scale bridging to adaptive resolution. Annu. Rev. Phys. Chem. 2008, 59, 545– 571, DOI: 10.1146/annurev.physchem.59.032607.093707There is no corresponding record for this reference.
- 193Milano, G.; Kawakatsu, T. Hybrid particle-field molecular dynamics simulations for dense polymer systems J. Chem. Phys. 2009; Vol. 130, DOI: 10.1063/1.3142103 .There is no corresponding record for this reference.
- 194Wang, J.; Han, Y.; Xu, Z.; Yang, X.; Ramakrishna, S.; Liu, Y. Dissipative particle dynamics simulation: A review on investigating mesoscale properties of polymer systems. Macromol. Mater. Eng. 2021, 306 (4), 2000724 DOI: 10.1002/mame.202000724There is no corresponding record for this reference.
- 195Santo, K. P.; Neimark, A. V. Dissipative particle dynamics simulations in colloid and interface science: A review. Adv. Colloid Interface Sci. 2021, 298, 102545 DOI: 10.1016/j.cis.2021.102545There is no corresponding record for this reference.
- 196Hoogerbrugge, P. J.; Koelman, J. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys. Lett. 1992, 19 (3), 155, DOI: 10.1209/0295-5075/19/3/001There is no corresponding record for this reference.
- 197Español, P.; Warren, P. Statistical mechanics of dissipative particle dynamics. Europhys. Lett. 1995, 30 (4), 191, DOI: 10.1209/0295-5075/30/4/001There is no corresponding record for this reference.
- 198Marrink, S. J.; Monticelli, L.; Melo, M. N.; Alessandri, R.; Tieleman, D. P.; Souza, P. C. Two decades of martini: Better beads, broader scope. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2023, 13 (1), e1620 DOI: 10.1002/wcms.1620There is no corresponding record for this reference.
- 199Alessandri, R.; Grünewald, F.; Marrink, S. J. The martini model in materials science. Adv. Mater. 2021, 33 (24), 2008635 DOI: 10.1002/adma.202008635There is no corresponding record for this reference.
- 200Souza, P. C. T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B. M.; Wassenaar, T. A.; Kroon, P.; Melcr, K.; Nieto, V.; Corradi, V.; Khan, H. M.; Domanski, J.; Javanainen, M.; Martinez-Seara, H.; Reuter, N.; Best, R. B.; Vattulainen, I.; Monticelli, L.; Periole, X.; Peter, T. D.; Vries, dH.; Marrink, S. J. Martini 3: a general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18 (4), 382– 388, DOI: 10.1038/s41592-021-01098-3There is no corresponding record for this reference.
- 201Fredrickson, G. H. The Equilibrium Theory of Inhomogeneous Polymers. Oxford University Press, 2006.There is no corresponding record for this reference.
- 202Schmid, F. Self-consistent-field theories for complex fluids. J. Phys.: Condens. Matter 1998, 10 (37), 8105, DOI: 10.1088/0953-8984/10/37/002There is no corresponding record for this reference.
- 203Maerzke, K. A.; Siepmann, J. I. Transferable potentials for phase equilibria- coarse-grain description for linear alkanes. J. Phys. Chem. B 2011, 115 (13), 3452– 3465, DOI: 10.1021/jp1063935There is no corresponding record for this reference.
- 204Liwo, A.; Ołdziej, S.; Pincus, M. R.; Wawak, R. J.; Rackovsky, S.; Scheraga, H. A. A united-residue force field for off-lattice protein-structure simulations. i. functional forms and parameters of long-range side-chain interaction potentials from protein crystal data. J. Comput. Chem. 1997, 18 (7), 849– 873, DOI: 10.1002/(SICI)1096-987X(199705)18:7<849::AID-JCC1>3.0.CO;2-RThere is no corresponding record for this reference.
- 205Maisuradze, G. G.; Senet, P.; Czaplewski, C.; Liwo, A.; Scheraga, H. A. Investigation of protein folding by coarse-grained molecular dynamics with the unres force field. J. Phys. Chem. A 2010, 114 (13), 4471– 4485, DOI: 10.1021/jp9117776There is no corresponding record for this reference.
- 206Reith, D.; Pütz, M.; Müller-Plathe, F. Deriving effective mesoscale potentials from atomistic simulations. J. Comput. Chem. 2003, 24 (13), 1624– 1636, DOI: 10.1002/jcc.10307There is no corresponding record for this reference.
- 207Agrawal, V.; Arya, G.; Oswald, J. Simultaneous iterative boltzmann inversion for coarse-graining of polyurea. Macromolecules 2014, 47 (10), 3378– 3389, DOI: 10.1021/ma500320nThere is no corresponding record for this reference.
- 208Tschöp, W.; Kremer, K.; Batoulis, J.; Bürger, T.; Hahn, O. Simulation of polymer melts. i. coarse-graining procedure for polycarbonates. Acta Polym. 1998, 49 (2–3), 61– 74, DOI: 10.1002/(SICI)1521-4044(199802)49:2/3<61::AID-APOL61>3.0.CO;2-VThere is no corresponding record for this reference.
- 209McCoy, J. D.; Curro, J. G. Mapping of explicit atom onto united atom potentials. Macromolecules 1998, 31 (26), 9362– 9368, DOI: 10.1021/ma981060gThere is no corresponding record for this reference.
- 210Baschnagel, J.; Binder, K.; Doruker, P.; Gusev, A. A.; Hahn, O.; Kremer, K.; Mattice, W. L.; Müller-Plathe, F.; Murat, M.; Paul, W.; Santos, S.; Suter, U. W.; Tries, V. Bridging the gap between atomistic and coarse-grained models of polymers: Status and perspectives. Viscoelasticity, Atomistic Models, Statistical Chemistry 2000, 152, 41– 156, DOI: 10.1007/3-540-46778-5_2There is no corresponding record for this reference.
- 211Akkermans, R. L. C.; Briels, W. J. A structure-based coarse-grained model for polymer melts. J. Chem. Phys. 2001, 114 (2), 1020– 1031, DOI: 10.1063/1.1330744There is no corresponding record for this reference.
- 212Uneyama, T.; Masubuchi, Y. Plateau moduli of several single-chain slip-link and slip-spring models. Macromolecules 2021, 54 (3), 1338– 1353, DOI: 10.1021/acs.macromol.0c01790There is no corresponding record for this reference.
- 213Behbahani, A. F.; Schneider, L.; Rissanou, A.; Chazirakis, A.; Bacova, P.; Jana, P. K.; Li, W.; Doxastakis, M.; Polinska, P.; Burkhart, C.; Muller, M.; Harmandaris, V. A. Dynamics and rheology of polymer melts via hierarchical atomistic, coarse-grained, and slip-spring simulations. Macromolecules 2021, 54 (6), 2740– 2762, DOI: 10.1021/acs.macromol.0c02583There is no corresponding record for this reference.
- 214Wu, Z.; Müller-Plathe, F. Slip-spring hybrid particle-field molecular dynamics for coarse-graining branched polymer melts: Polystyrene melts as an example. J. Chem. Theory Comput. 2022, 18 (6), 3814– 3828, DOI: 10.1021/acs.jctc.2c00107There is no corresponding record for this reference.
- 215Underhill, P. T.; Doyle, P. S. On the coarse-graining of polymers into bead-spring chains. J. Non-Newtonian Fluid Mech. 2004, 122 (1–3), 3– 31, DOI: 10.1016/j.jnnfm.2003.10.006There is no corresponding record for this reference.
- 216Kremer, K.; Grest, G. S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92 (8), 5057– 5086, DOI: 10.1063/1.458541There is no corresponding record for this reference.
- 217Everaers, R.; Karimi-Varzaneh, H. A.; Fleck, F.; Hojdis, N.; Svaneborg, C. Kremer-grest models for commodity polymer melts: Linking theory, experiment, and simulation at the kuhn scale. Macromolecules 2020, 53 (6), 1901– 1916, DOI: 10.1021/acs.macromol.9b02428There is no corresponding record for this reference.
- 218Nitta, H.; Ozawa, T.; Yasuoka, K. Construction of full-atomistic polymer amorphous structures using reverse-mapping from kremer–grest models J. Chem. Phys. , 2023 159 no. 19.There is no corresponding record for this reference.
- 219Miwatani, R.; Takahashi, K. Z.; Arai, N. Performance of coarse graining in estimating polymer properties: Comparison with the atomistic model. Polymers 2020, 12 (2), 382, DOI: 10.3390/polym12020382There is no corresponding record for this reference.
- 220Praprotnik, M.; Delle Site, L.; Kremer, K. Adaptive resolution scheme for efficient hybrid atomistic-mesoscale molecular dynamics simulations of dense liquids. Phys. Rev. E 2006, 73 (6), 066701 DOI: 10.1103/PhysRevE.73.066701There is no corresponding record for this reference.
- 221Darré, L.; Machado, M. R.; Brandner, A. F.; González, H. C.; Ferreira, S.; Pantano, S. Sirah: a structurally unbiased coarse-grained force field for proteins with aqueous solvation and long-range electrostatics. J. Chem. Theory Comput. 2015, 11 (2), 723– 739, DOI: 10.1021/ct5007746There is no corresponding record for this reference.
- 222Das, R.; Baker, D. Macromolecular modeling with rosetta. Annu. Rev. Biochem. 2008, 77, 363– 382, DOI: 10.1146/annurev.biochem.77.062906.171838There is no corresponding record for this reference.
- 223Leman, J. K.; Weitzner, B. D.; Lewis, S. M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R. F.; Aprahamian, M.; Baker, D.; Barlow, K. A.; Barth, P.; Basanta, B.; Bender, B. J.; Blacklock, K.; Bonet, J.; Boyken, S. E.; Bradley, P.; Bystroff, C.; Conway, P.; Cooper, S.; Correia, B. E.; Coventry, B.; Das, R.; De Jong, R. M.; DiMaio, F.; Dsilva, L.; Dunbrack, R.; Ford, A. S.; Frenz, B.; Fu, D. Y.; Geniesse, C.; Goldschmidt, L.; Gowthaman, R.; Gray, J. J.; Gront, D.; Guffy, S.; Horowitz, S.; Huang, P.-S.; Huber, T.; Jacobs, T. M.; Jeliazkov, J. R.; Johnson, D. K.; Kappel, K.; Karanicolas, J.; Khakzad, H.; Khar, K. R.; Khare, S. D.; Khatib, F.; Khramushin, A.; King, I. C.; Kleffner, R.; Koepnick, B.; Kortemme, T.; Kuenze, G.; Kuhlman, B.; Kuroda, D.; Labonte, J. W.; Lai, J. K.; Lapidoth, G.; Leaver-Fay, A.; Lindert, S.; Linsky, T.; London, N.; Lubin, J. H.; Lyskov, S.; Maguire, J.; Malmström, L.; Marcos, E.; Marcu, O.; Marze, N. A.; Meiler, J.; Moretti, R.; Mulligan, V. K.; Nerli, S.; Norn, C.; Ó’Conchúir, S.; Ollikainen, N.; Ovchinnikov, S.; Pacella, M. S.; Pan, X.; Park, H.; Pavlovicz, R. E.; Pethe, M.; Pierce, B. G.; Pilla, K. B.; Raveh, B.; Renfrew, P. D.; Roy Burman, S. S.; Rubenstein, A.; Sauer, M. F.; Scheck, A.; Schief, W.; Schueler-Furman, O.; Sedan, Y.; Sevy, A. M.; Sgourakis, N. G.; Shi, L.; Siegel, J. B.; Silva, D.-A.; Smith, S.; Song, Y.; Stein, A.; Szegedy, M.; Teets, F. D.; Thyme, S. B.; Wang, R. Y.-R.; Watkins, A.; Zimmerman, L.; Bonneau, R. Macromolecular modeling and design in rosetta: recent methods and frameworks. Nat. Methods 2020, 17 (7), 665– 680, DOI: 10.1038/s41592-020-0848-2There is no corresponding record for this reference.
- 224Baldassarre, F.; Azizpour, H. Explainability techniques for graph convolutional networks, 2019, arXiv:1905.13686 arXiv.org e-Print archive https://arxiv.org/abs/1905.13686.There is no corresponding record for this reference.
- 225Bai, Y.; Wilbraham, L.; Slater, B. J.; Zwijnenburg, M. A.; Sprick, R. S.; Cooper, A. I. Accelerated discovery of organic polymer photocatalysts for hydrogen evolution from water through the integration of experiment and theory. J. Am. Chem. Soc. 2019, 141 (22), 9063– 9071, DOI: 10.1021/jacs.9b03591There is no corresponding record for this reference.
- 226Vosko, S. H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58 (8), 1200– 1211, DOI: 10.1139/p80-159There is no corresponding record for this reference.
- 227Becke, A. D. Density-functional thermochemistry. iii. the role of exact exchange. J. Chem. Phys. 1993, 98 (7), 5648– 5652, DOI: 10.1063/1.464913There is no corresponding record for this reference.
- 228Lee, C.; Yang, W.; Parr, R. G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37 (2), 785, DOI: 10.1103/PhysRevB.37.785There is no corresponding record for this reference.
- 229Grimme, S.; Bannwarth, C.; Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (z= 1–86). J. Chem. Theory Comput. 2017, 13 (5), 1989– 2009, DOI: 10.1021/acs.jctc.7b00118There is no corresponding record for this reference.
- 230Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. A 1994, 98 (45), 11623– 11627, DOI: 10.1021/j100096a001There is no corresponding record for this reference.
- 231Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M. Analyzing learned molecular representations for property prediction. J. Chem. Inf. Model. 2019, 59 (8), 3370– 3388, DOI: 10.1021/acs.jcim.9b00237There is no corresponding record for this reference.
- 232Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50 (5), 742– 754, DOI: 10.1021/ci100050tThere is no corresponding record for this reference.
- 233Landrum, G. Rdkit: open-source cheminformatics from machine learning to chemical registration. In Abstracts of Papers of the American Chemical Society; Vol. 258, AMER CHEMICAL SOC 1155 16TH ST, NW, WASHINGTON, DC 20036 USA, 2019.There is no corresponding record for this reference.
- 234Mannodi-Kanakkithodi, A.; Chandrasekaran, A.; Kim, C.; Huan, T. D.; Pilania, G.; Botu, V.; Ramprasad, R. Scoping the polymer genome: A roadmap for rational polymer dielectrics design and beyond. Mater. Today 2018, 21, 785– 796, DOI: 10.1016/j.mattod.2017.11.021There is no corresponding record for this reference.
- 235Kim, C.; Chandrasekaran, A.; Huan, T. D.; Das, D.; Ramprasad, R. Polymer genome: A data-powered polymer informatics platform for property predictions. J. Phys. Chem. C 2018, 122, 17575– 17585, DOI: 10.1021/acs.jpcc.8b02913There is no corresponding record for this reference.
- 236Ward, L.; Dunn, A.; Faghaninia, A.; Zimmermann, N. E.; Bajaj, S.; Wang, Q.; Montoya, J.; Chen, J.; Bystrom, K.; Dylla, M.; Chard, K.; Asta, M.; Persson, K. A.; Snyder, G. J.; Foster, I.; Jain, A. Matminer: An open source toolkit for materials data mining. Comput. Mater. Sci. 2018, 152, 60– 69, DOI: 10.1016/j.commatsci.2018.05.018There is no corresponding record for this reference.
- 237Lightstone, J. P.; Chen, L.; Kim, C.; Batra, R.; Ramprasad, R. Refractive index prediction models for polymers using machine learning J. Appl. Phys. 2020; Vol. 127, DOI: 10.1063/5.0008026 .There is no corresponding record for this reference.
- 238Chen, L.; Kim, C.; Batra, R.; Lightstone, J. P.; Wu, C.; Li, Z.; Deshmukh, A. A.; Wang, Y.; Tran, H. D.; Vashishta, P.; Sotzing, G. A.; Cao, Y.; Ramprasad, R. Frequency-dependent dielectric constant prediction of polymers using machine learning. npj Computat. Mater. 2020, 6 (1), 61, DOI: 10.1038/s41524-020-0333-6There is no corresponding record for this reference.
- 239Doan Tran, H.; Kim, C.; Chen, L.; Chandrasekaran, A.; Batra, R.; Venkatram, S.; Kamal, D.; Lightstone, J. P.; Gurnani, R.; Shetty, P. Machine-learning predictions of polymer properties with polymer genome. J. Appl. Phys. 2020, 128 (17), 171104 DOI: 10.1063/5.0023759There is no corresponding record for this reference.
- 240Godwin, J.; Schaarschmidt, M.; Gaunt, A.; Sanchez-Gonzalez, A.; Rubanova, Y.; Veličković, P.; Kirkpatrick, J.; Battaglia, P. Simple gnn regularisation for 3d molecular property prediction & beyond, 2021, arXiv:2106.07971. arXiv.org e-Print archive https://arxiv.org/abs/2106.07971.There is no corresponding record for this reference.
- 241Chen, D.; Lin, Y.; Li, W.; Li, P.; Zhou, J.; Sun, X. Measuring and relieving the over-smoothing problem for graph neural networks from the topological view. Proc. AAAI Conf. Artificial Intelligence 2020, 34, 3438– 3445, DOI: 10.1609/aaai.v34i04.5747There is no corresponding record for this reference.
- 242Frisch, M. Gaussian 09, Revision D.01, Gaussian , 2009.There is no corresponding record for this reference.
- 243Kuenneth, C.; Schertzer, W.; Ramprasad, R. Copolymer informatics with multitask deep neural networks. Macromolecules 2021, 54 (13), 5957– 5961, DOI: 10.1021/acs.macromol.1c00728There is no corresponding record for this reference.
- 244You, J.; Ying, Z.; Leskovec, J. Design space for graph neural networks. In Advances in Neural Information Processing Systems (NeurIPS) 2020.There is no corresponding record for this reference.
- 245Rupp, M.; Tkatchenko, A.; Müller, K.-R.; Von Lilienfeld, O. A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012, 108 (5), 058301 DOI: 10.1103/PhysRevLett.108.058301There is no corresponding record for this reference.
- 246Bartók, A. P.; Kondor, R.; Csányi, G. On representing chemical environments. Phys. Rev. B 2013, 87 (18), 184115 DOI: 10.1103/PhysRevB.87.184115There is no corresponding record for this reference.
- 247Townsend, J.; Micucci, C. P.; Hymel, J. H.; Maroulas, V.; Vogiatzis, K. D. Representation of molecular structures with persistent homology for machine learning applications in chemistry. Nat. Commun. 2020, 11 (1), 3230 DOI: 10.1038/s41467-020-17035-5There is no corresponding record for this reference.
- 248Huo, H.; Rupp, M. Unified representation of molecules and crystals for machine learning. Machine Learning: Sci. Technol. 2022, 3 (4), 045017 DOI: 10.1088/2632-2153/aca005There is no corresponding record for this reference.
- 249Sanchez Medina, E. I.; Linke, S.; Stoll, M.; Sundmacher, K. Gibbs-helmholtz graph neural network: capturing the temperature dependency of activity coefficients at infinite dilution. Digital Discovery 2023, 2 (3), 781– 798, DOI: 10.1039/D2DD00142JThere is no corresponding record for this reference.
- 250Zhong, C.; Sato, Y.; Masuoka, H.; Chen, X. Improvement of predictive accuracy of the unifac model for vapor-liquid equilibria of polymer solutions. Fluid Phase Equilib. 1996, 123 (1–2), 97– 106, DOI: 10.1016/S0378-3812(96)90015-1There is no corresponding record for this reference.
- 251Kontogeorgis, G. M.; Fredenslund, A.; Tassios, D. P. Simple activity coefficient model for the prediction of solvent activities in polymer solutions. Ind. Eng. Chem. Res. 1993, 32 (2), 362– 372, DOI: 10.1021/ie00014a013There is no corresponding record for this reference.
- 252Pappa, G. D.; Voutsas, E. C.; Tassios, D. P. Prediction of activity coefficients in polymer and copolymer solutions using simple activity coefficient models. Ind. Eng. Chem. Res. 1999, 38 (12), 4975– 4984, DOI: 10.1021/ie990265qThere is no corresponding record for this reference.
- 253Cremer, D. Møller-plesset perturbation theory: from small molecule methods to methods for thousands of atoms. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1 (4), 509– 530, DOI: 10.1002/wcms.58There is no corresponding record for this reference.
- 254Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: moving beyond fingerprints. J. Comput.-Aided Mol. Des. 2016, 30, 595– 608, DOI: 10.1007/s10822-016-9938-8There is no corresponding record for this reference.
- 255Wang, M. Y. Deep graph library: Towards efficient and scalable deep learning on graphs. In ICLR Workshop on Representation Learning on Graphs and Manifolds , 2019.There is no corresponding record for this reference.
- 256Bojar, D.; Powers, R. K.; Camacho, D. M.; Collins, J. J. Deep-learning resources for studying glycan-mediated host-microbe interactions. Cell Host Microbe 2021, 29, 132– 144.e3, DOI: 10.1016/j.chom.2020.10.004There is no corresponding record for this reference.
- 257Montavon, G.; Binder, A.; Lapuschkin, S.; Samek, W.; Müller, K.-R. Layer-wise relevance propagation: an overview. In Lecture Notes in Computer Science 2019; pp 193– 209.There is no corresponding record for this reference.
- 258Binder, A.; Bach, S.; Montavon, G.; Müller, K.-R.; Samek, W. Layer-wise relevance propagation for deep neural network architectures. In Information Science and Applications (ICISA) 2016; Springer, 2016; pp 913– 922.There is no corresponding record for this reference.
- 259Adebayo, J.; Gilmer, J.; Muelly, M.; Goodfellow, I.; Hardt, M.; Kim, B. Sanity checks for saliency maps Advances in Neural Information Processing Systems , 2018 31.There is no corresponding record for this reference.
- 260Qi, Z.; Khorram, S.; Li, F. Visualizing deep networks by optimizing with integrated gradients. AAAI 2020, 34, 11890– 11898, DOI: 10.1609/aaai.v34i07.6863There is no corresponding record for this reference.