

About the Cover:
Application Notes

Selector: A General Python Library for Diverse Subset Selection
Fanwang Meng *- ,
Marco Martínez González - ,
Valerii Chuiko - ,
Alireza Tehrani - ,
Abdul Rahman Al Nabulsi - ,
Abigail Broscius - ,
Hasan Khaleel - ,
Kenneth López-Pérez - ,
Ramón Alain Miranda-Quintana *- ,
Paul W. Ayers *- , and
Farnaz Heidar-Zadeh *
Selector is a free, open-source Python library for selecting diverse subsets from any dataset, making it a versatile tool across a wide range of application domains. Selector implements different subset sampling algorithms based on sample distance, similarity, and spatial partitioning along with metrics to quantify subset diversity. It is flexible and integrates seamlessly with popular Python libraries such as Scikit-Learn, demonstrating the interoperability of the implemented algorithms with data analysis workflows. Selector is an operating-system-agnostic, accessible, and easily extensible package designed with modern software development practices, including version control, unit testing, and continuous integration. Interactive quick-start notebooks, which are also web-accessible, provide user-friendly tutorials for all skill levels, showcasing applications in computational chemistry, drug discovery, and chemical library design. Additionally, a web interface has been developed that allows users to easily upload datasets, configure sampling settings, and run subset selection algorithms with no programming required. This work serves as the official release note for the Selector package, offering a technical overview of its features, use cases, and development practices that ensure its quality and maintainability.

EasyHybrid: An Interactive Graphical Environment for Quantum, Classical and Hybrid Simulations with pDynamo3
Jose Fernando R. Bachega *- ,
Gustavo Hagen - ,
Carlos Sequeiros-Borja - ,
Kai Nikklas - ,
Jorge Chahine - ,
Luis Fernando M. S. Timmers - , and
Martin J. Field *

This publication is Open Access under the license indicated. Learn More
We present EasyHybrid, a free and open-source graphical interface for hybrid quantum chemical/molecular mechanical (QC/MM) simulations built on the pDynamo3 library. The software provides an intuitive environment for preparing, inspecting, and editing molecular systems, while supporting a broad range of simulations, including reaction coordinate scans, molecular dynamics, normal-mode analysis, Nudged Elastic Band, and umbrella sampling. Key features include advanced 3D visualization of large biomolecular systems, interactive editing, flexible atom selection, system pruning for efficient QC/MM setup, orbital and electrostatic potential surfaces, automated log parsing, and trajectory analysis. EasyHybrid integrates these tools into a single platform, offering a familiar yet specialized environment for quantum chemistry and hybrid QC/MM simulations.

ToxFCDB: Toxicity Database for Forever Chemicals
Meetali Sinha - ,
Deepak Kumar Sachan - ,
Joy Chakraborty - ,
Anamta Ali - ,
Anshika Gupta - ,
Tanya Jamal - , and
Ramakrishnan Parthasarathi *
Per- and polyfluoroalkyl substances (PFAS)/forever chemicals are persistent synthetic chemicals with widespread use in a variety of consumer and industrial products. Some of these chemicals have undergone exhaustive research regarding experimental toxicity testing and human epidemiological inference; however, most compounds contain little or no information about their hazards or safety. ToxFCDB prioritizes these data-poor compounds for detailed toxicity investigations by constructing an effective web-based database for in silico preliminary evaluations employing more than 50 QSAR models/databases. The database compiles 8204 PFAS with their molecular structures, chemical classification, physicochemical and toxicokinetic properties, molecular descriptors, toxicological data, chemical genes, and human targets. This database aims to assist industrialists, policymakers, and researchers in assessing state-of-the-art data-centric information to make informed decisions to safeguard public health and the environment. In addition, the ToxFCDB could be a valuable tool for encouraging additional toxicological research in the domain of redesigning chemicals and polymers. The ToxFCDB is accessible online at http://ctf.iitr.res.in/toxfcdb/.

PharmGEO: A Curated Atlas of Drug-Response Transcriptomes Enabling Cross-Study Comparisons
Haoran Chen - ,
Mujin Li - ,
Jie Geng - ,
Chen Cao *- , and
Jianxin Chen *
Drug-induced transcriptomic profiles capture how compounds reprogram genes and pathways across dose and time, supporting drug–gene associations and interaction analysis. The field faces a practical bottleneck: GEO drug studies are scattered, metadata for dose, exposure time, and cell type are inconsistent, and processing pipelines differ, which limits reuse and fair comparison. We built PharmGEO, an interactive Shiny resource that curates and standardizes 7,931 pharmaco-transcriptomic experiments across 1,334 drugs. Key variables are harmonized, and all data sets are reprocessed through a single pipeline, yielding transcriptome-wide results with a mean of 17,776 genes per experiment. PharmGEO supports interactive differential expression and pathway enrichment, prioritizes high-confidence drug–gene associations using cross-data set consistency, and provides a directionally annotated drug–drug interaction network with 115,264 interactions derived from transcriptomic overlap. Case studies in target validation and combination assessment show how PharmGEO enables drug repurposing and interaction evaluation by turning heterogeneous studies into a coherent, searchable atlas of drug responses. PharmGEO is available at http://www.pharmgeo.net.
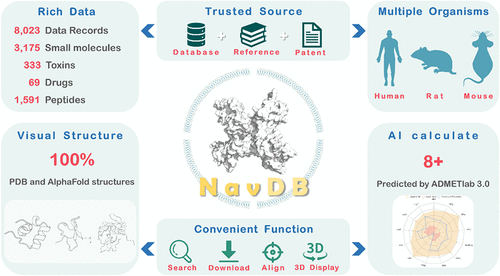
NavDB: A Comprehensive Database for Voltage-Gated Sodium Channels Modulators and Targets
Gaoang Wang - ,
Jiahui Yu - ,
Haiyi Chen - ,
Hao Luo - ,
Peichen Pan *- , and
Tingjun Hou *
Voltage-gated sodium channels (VGSCs/Navs) are essential targets for the treatment of numerous neurological, muscular, and cardiac disorders. Despite the increasing clinical interest in subtype-selective modulators, current public databases provide fragmented and inconsistent information on VGSC-related compounds and targets, particularly lacking coverage on peptides. To address this limitation, we developed NavDB, a specialized and open-access database focusing on VGSC modulators and targets. NavDB integrates 8023 curated data records covering 5168 compounds, including small molecules, toxins, drugs, and peptides, along with comprehensive annotations on biological activity, druggability, and structural feature. NavDB also features advanced functions such as text-based and structure-based search, peptide similarity matching, and AI-powered property prediction. Moreover, the database offers high-quality 3D visualizations of targets and peptides, with disulfide bond and signal peptide annotations. All data are freely downloadable to support both experimental and computational drug discovery. NavDB is publicly available at: http://cadd.zju.edu.cn/navdb/.
Reviews
Graph Neural Networks for Polymer Characterization and Property Prediction: Opportunities and Challenges
Hector Medina *- and
Rachel Drake
This publication is Open Access under the license indicated. Learn More
Using machine learning to accelerate the characterization and prediction of properties of many-molecule systems, such as polymers, is appealing, yet challenging. Polymers are large, complex molecules that have unique properties and potential applications in a wide range of industries. Their potential in advancing fields such as ion-transport polymer for energy storage, lightweighting of structural materials, bioinspired multifunctional materials, etc., provide enough impetus for accelerating the discovery of novel polymeric materials. However, mathematical mapping and the consequent manipulation of polymer structures are still challenging tasks due to their complex configuration and the smorgasbord of motifs encountered naturally and in engineering materials. Traditional methods of polymer structure mapping and property prediction at multiscale domains can include approaches such as Density Functional Theory, Molecular Dynamics, and Finite Element Analysis, which can be time-consuming and computationally expensive. The promise of machine learning to accelerate these tasks is appealing, and currently, researchers are pursuing the development of architectures and composition approaches to accomplish this. Here we discuss the current state of the knowledge on the use of Graph Neural Networks, and related architectures, being developed and/or used for the characterization and prediction of properties of polymers. Many challenges still exist such as the lack of sufficient and comprehensive data sets. To address these issues, efforts are being pursued─such as the so-called CRIPT (Community Resource for Innovation in Polymer Technology) led by a lab consortium that includes representations from private industry, academia, government, and others. We conclude that even though this field is young it has both momentum and promise. The current challenges that must be overcome are also addressed.
Letters

Evidence for Epibatidine Binding to the Desensitization Gate in α7 nAChR from Molecular Dynamics Simulations and Cryo-EM
Jesko Kaiser - ,
Christoph G. W. Gertzen - ,
Daniel Mann - ,
Carsten Sachse - , and
Holger Gohlke *
The homopentameric α7-nicotinic acetylcholine receptor (nAChR) is a ligand-gated ion channel widely expressed in the human nervous system and susceptible to allosteric modulation. A recent cryo-EM structure (EMD 22983; PDB ID 7KOX) revealed unassigned Coulomb density. Unbiased molecular dynamics simulations of buffer components around α7-nAChR show that (±)-epibatidine can occupy not only the orthosteric site but also the pore near the desensitization gate, consistent with the unmodeled Coulomb density and expanding the receptor’s pocketome.
Machine Learning and Deep Learning
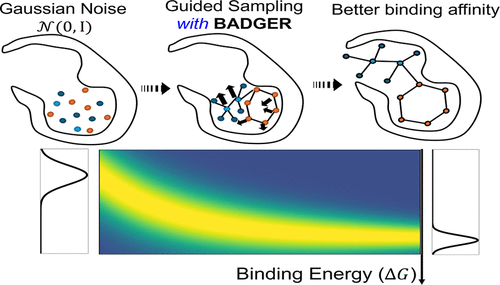
General Binding Affinity Guidance for Diffusion Models in Structure-Based Drug Design
Yue Jian - ,
Curtis Wu - ,
Danny Reidenbach - , and
Aditi S. Krishnapriyan *
Structure-based drug design (SBDD) aims to generate ligands that bind strongly and specifically to target protein pockets. Recent diffusion models have advanced SBDD by capturing the distributions of atomic positions and types, yet they often underemphasize binding affinity control during generation. To address this limitation, we introduce BADGER, a general binding-affinity guidance framework for diffusion models in SBDD. BADGER incorporates binding affinity awareness through two complementary strategies: (1) classifier guidance, which applies gradient-based affinity signals during sampling in a plug-and-play fashion, and (2) classifier-free guidance, which integrates affinity conditioning directly into diffusion model training. Together, these approaches enable controllable ligand generation guided by binding affinity. BADGER achieves up to a 60% improvement in ligand–protein binding affinity of sampled molecules over prior methods. Furthermore, we extend the framework to multiconstraint diffusion guidance, jointly optimizing for binding affinity, drug-likeness (QED), and synthetic accessibility (SA) to design realistic and synthesizable drug candidates.
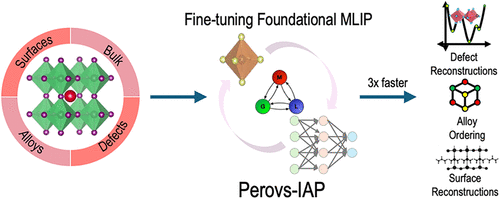
Unified Graph-Based Interatomic Potential for Perovskite Structure Optimization
Maitreyo Biswas - ,
Rushik Desai - ,
Gavin Bidna - , and
Arun Mannodi-Kanakkithodi *
Halide perovskites (HaPs) hold immense potential for applications such as optoelectronics and catalysis. Their vast compositional space, spanning bulk alloys, defects, impurities, surfaces, and surface defects, poses significant challenges for efficient exploration and optimization. To address this, we trained a unified graph-based deep learned interatomic potential capable of optimizing and predicting energetics across these diverse structural motifs and navigating the complex potential energy surface (PES). Using a comprehensive density functional theory data set of HaP structures, which includes bulk alloys, native and impurity defects, and surface slabs, we rigorously trained and benchmarked the M3GNet-based machine learning interatomic potential (IAP). The M3GNet-IAP framework, trained on DFT-calculated energies, forces, and stresses, enables gradient-based optimization and efficient exploration of the PES. Our models, trained on a data set of ∼12,000 HaP structures across diverse structural domains, demonstrated robust generalizability across the complex PES, achieving low errors (energies (E): 3.7 meV/atom; forces (f): 16.5 meV/Å; stresses (σ): 5.5 MPa), and accurately predicting formation energies, decomposition energies, defect energies, and surface energies. Our unified surrogate model provides a holistic approach to geometry optimization across different structural variations in HaPs and will be transformative for the discovery of promising new compositions, important defects and dopants, and surface properties.

Density Estimation Based on Mixtures of Gaussians for Perovskite Solar Cells Modeling
F. Alexander Sepúlveda *- ,
Daniel Cerro-Ramos - , and
T. Jesper Jacobsson *
This publication is Open Access under the license indicated. Learn More
Accurately modeling the complex relationships among synthesis parameters, material compositions, and performance metrics is essential for accelerating the development of perovskite solar cells (PSCs). In this context, machine learning (ML) has proven to be a valuable tool. While most ML applications in PSC research rely on discriminative “black-box” models, this study adopts a generative approach by modeling the joint probability density function. We employ Gaussian Mixture Models (GMMs), a pragmatic and interpretable choice well-suited for the scarce, low-dimensional tabular data typical of PSC research. This single GMM framework is evaluated on five distinct tasks: discovering clusters, regression, generating novel configurations, training on data sets with missing data and, inverse design of the experimental (synthesis) conditions. That is, assuming we have the perovskite material composition and a target PCE, we infer the experimental conditions. For this latter task we use a novel “GMM-Assisted Optimization” method, which demonstrates to be more effective than standard random-start optimization, achieving an RMSE of 1.52 against target PCEs, more than halving the 3.32 RMSE of the baseline. These findings highlight the power of probabilistic modeling for data-driven discovery in PSC research.
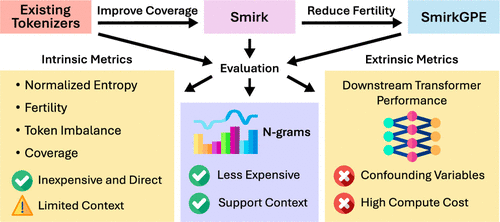
Tokenization for Molecular Foundation Models
Alexius Wadell - ,
Anoushka Bhutani - , and
Venkatasubramanian Viswanathan *
Text-based foundation models have become an important part of scientific discovery, with molecular foundation models accelerating advancements in material science and molecular design. However, existing models are constrained by closed-vocabulary tokenizers that capture only a fraction of molecular space. In this work, we systematically evaluate 35 tokenizers, including 20 chemistry-specific tokenizers, and reveal significant gaps in their coverage of the SMILES molecular representation. To assess the impact of tokenizer choice, we introduce n-gram language models as a low-cost proxy and validate their effectiveness by pretraining and finetuning 18 RoBERTa-style encoders for molecular property prediction. To overcome the limitations of existing tokenizers, we propose two new tokenizers─Smirk and Smirk-GPE─with full coverage of the OpenSMILES specification. The proposed tokenizers systematically integrate nuclear, electronic, and geometric degrees of freedom, facilitating applications in pharmacology, agriculture, biology, and energy storage. Our results highlight the need for open-vocabulary modeling and chemically diverse benchmarks in cheminformatics.
Optimizing Prediction of Chemical Bonds in Interfacial Dynamics through Local Uncertainty Estimates with Neural Network Ensembles
Suman Bhasker-Ranganath - ,
Filippo Balzaretti - , and
Johannes Voss *
We present a framework for data-efficient training of machine-learning interatomic potentials for interfacial chemistry, especially heterogeneous catalytic systems. We establish strategies for density functional theory training data generation consisting of procedurally generated bulk, surface, and gas-phase atomic geometries, as well as moderately randomized structures. We show how ensembles of neural network machine-learning interatomic potentials trained on different splits of these training structures yield reliable uncertainty estimates at the atomic node energy level. Our models can thus identify which atomic sites and chemical bonds in a system lead to uncertainties in the predicted potential energy surface. Using hydrogen interacting with platinum as a test case, we find that the atomic uncertainty estimates identify both unphysical bonding scenarios and physically relevant interactions that are underrepresented in the original training data, such as surface diffusion, bond breaking, and bond formation. Building on these insights, we propose local uncertainty-informed strategies that flag outliers via statistical correlations, thereby improving active learning efficiency and enhancing the reliability of neural network-based potentials for extended-scale reactive dynamics.
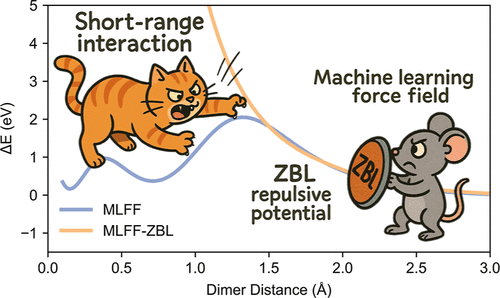
Improving Robustness and Training Efficiency of Machine-Learned Potentials by Incorporating Short-Range Empirical Potentials
Zihan Yan - ,
Zheyong Fan - , and
Yizhou Zhu *
Machine learning force fields (MLFFs) are powerful tools for materials modeling, but their performance is often limited by training data set quality, particularly the lack of rare event configurations. This limitation undermines their accuracy and robustness in long- and large-scale molecular dynamics simulations. In this work, we demonstrate a simple yet general approach to improving robustness and training efficiency by utilizing a hybrid MLFF framework that integrates an empirical short-range repulsive potential. Using solid electrolyte Li7La3Zr2O12 (LLZO) as a model system, we show that purely data-driven MLFFs fail to prevent unphysical atomistic clustering in extended simulations due to inadequate short-range repulsion. In contrast, the hybrid force field eliminates these artifacts, enabling stable long-time simulations, which are critical for studying various properties of LLZO. Our approach also reduces the need for extensive active learning and performs well with just 25 training configurations. By combining physics-driven constraints with data-driven flexibility, this approach is compatible with most existing MLFF architectures and establishes a universal paradigm for developing robust, training-efficient force fields for complex material systems.
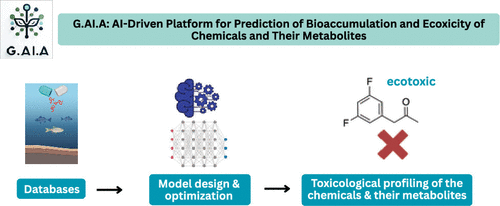
G.AI.A: An Integrated Machine-Learning Platform for Predicting Bioaccumulation and Ecotoxicity of Pharmaceuticals
Evangelos Tsoukas - ,
Michail Papadourakis - ,
Eleni Chontzopoulou - ,
Spyridon Vythoulkas - ,
Christos Didachos - ,
Dionisis Cavouras - ,
Panagiotis Zoumpoulakis *- , and
Minos-Timotheos Matsoukas *
This publication is Open Access under the license indicated. Learn More
Pharmaceutical pollution in aquatic environments poses a significant ecological threat due to the accumulation of bioactive compounds from human and veterinary sources. In support of the EU Green Deal’s Chemicals Strategy for Sustainability, this study presents a computational framework for predicting two key environmental risk indicators in fish: bioconcentration and ecotoxicity. Bioconcentration, quantified by the bioconcentration factor (BCF), reflects a chemical’s tendency to accumulate in organisms, while ecotoxicity is assessed via the median lethal concentration (LC50) over defined exposure periods. We developed two high-performing machine learning (ML) models, achieving ROC AUC scores of 94.60% for bioconcentration and 96.06% for ecotoxicity, validated across both internal and external data sets. To expand the scope of risk evaluation, we incorporated metabolite prediction using the SyGMa tool, selected after benchmarking multiple alternatives. This enables the assessment of both parent compounds and their potentially toxic metabolites. Model interpretability was enhanced through molecular fingerprint analysis, which identified structural features associated with toxicity and accumulation, informing the early stages of drug design. To support practical implementation, we introduced G.AI.A (https://gaiatox.eu/), an intuitive web platform that allows users to input Simplified Molecular Input Line Entry System (SMILES) strings for rapid prediction of environmental risk end points. The application domain of G.AI.A lies in predictive toxicology, enabling researchers and regulatory bodies to assess the toxicological profiles of small organic compounds, excluding those containing heavy metals, by analyzing their chemical structures. The platform supports batch processing and offers interactive visualizations, facilitating compound screening and early stage environmental risk assessment. By integrating predictive modeling with interpretability and usability, our framework advances green-by-design pharmaceutical development and contributes to sustainable chemical management.
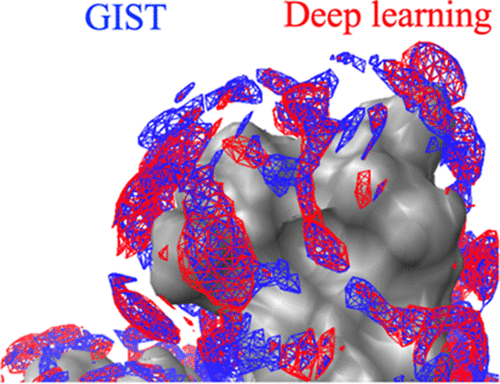
Deep GIST: Deep Learning Models for Predicting the Distribution of Hydration Thermodynamics around Proteins
Yusaku Fukushima - and
Takashi Yoshidome *
This publication is Open Access under the license indicated. Learn More
Hydration thermodynamic quantities are essential for understanding protein function from a free-energy perspective. The grid inhomogeneous solvation theory (GIST) enables the computation of spatial distributions of hydration energy, ΔEW(r), and hydration entropy, ΔSW(r), using molecular dynamics (MD) simulations, from which the distribution of the hydration free energy, ΔGW(r), is obtained as ΔGW(r) = ΔEW(r) – TΔSW(r), where T is the absolute temperature. However, GIST is computationally demanding, requiring tens of hours to compute these distributions. To overcome this bottleneck, we developed a set of deep learning models capable of predicting ΔEW(r), TΔSW(r), and ΔGW(r). Our deep learning models completed these predictions within tens of seconds using a single graphics processing unit. The resulting distributions achieved coefficient of determination values of 0.76–0.84 for ΔGW(r) when compared to GIST results, and lower values were obtained for ΔEW(r) and TΔSW(r). As a practical application, we examined the free energy change required for a water molecule to move from the bulk region to the ligand-binding site, ΔGW,replace, using both our deep learning model and GIST. A high correlation coefficient of 0.78 was observed between the predictions of our model and GIST, confirming its reliability. Furthermore, the results for a representative protein were consistent with experimental data of the corresponding protein–ligand complex: Water molecules with low ΔGW,replace values located near crystallographic waters, suggesting retention upon ligand binding, whereas those with unfavorable values overlapped with the ligand, indicating displacement upon the ligand binding. These findings demonstrate that our deep learning models provide an efficient and accurate alternative to GIST for predicting hydration thermodynamics and enable the consideration of protein conformational fluctuations, which is difficult to achieve with conventional GIST. The program called “Deep GIST” is available under the GNU General Public License from https://github.com/YoshidomeGroup-Hydration/Deep-GIST.
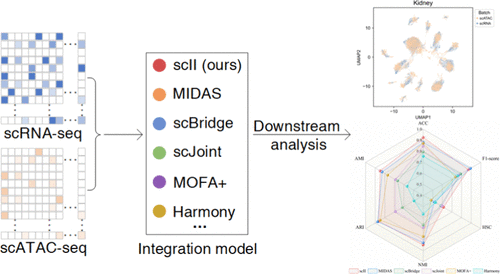
scII: Dual-Threshold Adaptive Integration of Single-Cell Multiomics Data Driven by Imputation
Yi Zhang - ,
Yuru Li *- ,
Zhicheng Jin - ,
Ye Tian - , and
Chen Su
This publication is Open Access under the license indicated. Learn More
Single-cell multiomics technologies provide unprecedented opportunities to dissect cellular heterogeneity by capturing multidimensional information on complex cellular states and regulatory networks. However, challenges such as high dimensionality, extreme data sparsity, and modality-specific discrepancies hinder the accuracy, interpretability, and scalability of the existing integration methods. Existing integration paradigms, including horizontal, vertical, and diagonal strategies, are further limited by their inability to fully capture nonlinear biological relationships, their reliance on high-quality data, and their substantial computational demands. Here, we present scII (Dual-Threshold Adaptive Integration of Single-Cell Multiomics Data Driven by Imputation), an adaptive framework designed to integrate gene expression (scRNA-seq) and chromatin accessibility (scATAC-seq) data. Our approach is built on several key conceptual innovations: (i) scRNA-seq–guided signal imputation to enhance information integrity in scATAC-seq; (ii) a multilayer perceptron with the Maxout activation function to improve the modeling of complex nonlinear relationships and mitigate the vanishing gradient problem; (iii) a dynamic dual-threshold adaptive selection mechanism that jointly evaluates cross-modality feature similarity and classification reliability to select high-quality cells; and (iv) Bayesian Information Criterion (BIC)-based optimization to dynamically determine the number of Gaussian Mixture Model components according to data distribution, thereby eliminating reliance on manually preset parameters. Extensive experiments on multiple real-world and simulated data sets demonstrate that scII not only enables efficient integration of unpaired scRNA-seq and scATAC-seq data but also achieves accurate transfer of cell-type annotations, allowing high-precision cell-type prediction for scATAC-seq.
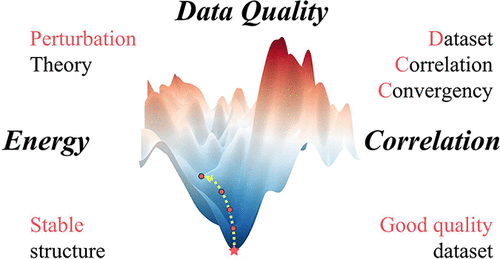
DCC: A Model-Free Frame to Evaluate Data Set Quality
Chunhui Xie - and
Yunqi Li *
Inspired by perturbation theory to locate thermodynamic equilibrium, we propose the data correlation convergence (DCC) framework to evaluate the quality of a data set accounting for completeness and representativeness, an alternative to conventionally computation-intensive and model-dependent approaches. The core hypothesis is that a high-quality data set should maintain stable correlation patterns under perturbations, and DCC can quantify such stability through integrating multiple correlation functions to quantify numeric correlations and distributional similarities. Based on hypothetical data sets generated by linear/determinative and random correlations, the lowest DCC was found at 10–20% linear correlations, which monotonously increased with more determinative correlations. It also revealed that a data set with around 1000 samples approaches the central limit theorem. Based on seven benchmark data sets released in material science, DCC values for the whole data set or each individual feature are informative, either to predict performance metrics including accuracies and determination coefficients for classification and regression models or to predict feature importance quantified by SHAP values. It can also efficiently compress raw data sets through recording their inherent correlation patterns and ignoring their exactly composed data. The DCC framework offers a theoretically grounded, widely applicable, and extensible approach to evaluate data set quality and annotate multiple data sets.
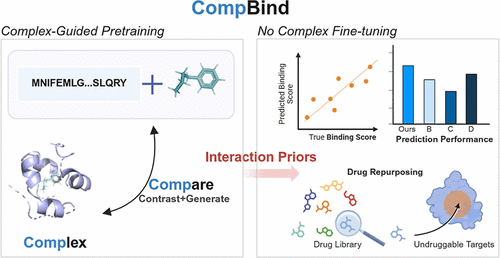
CompBind: Complex Guided Pretraining-Based Structure-Free Protein–Ligand Affinity Prediction
Duoyun Yi - ,
Yanpeng Zhao - ,
Huiyan Xu - ,
Yixin Zhang - ,
Mengxuan Wan - ,
Peng Zan *- ,
Song He *- , and
Xiaochen Bo *
Accurate prediction of protein–ligand binding affinity is essential in drug discovery. However, the limited availability and high cost of experimentally resolved protein–ligand complex structures significantly hinder the generalizability and broad applicability of current structure-based deep learning approaches. To address this challenge, we present CompBind, a novel framework for binding affinity prediction that leverages latent interaction patterns learned from existing complex structures while eliminating the need for 3D structural inputs during inference. Specifically, CompBind integrates bidirectional cross-attention with a dual-objective pretraining strategy, where contrastive learning enforces feature-space consistency between monomer pairs and their corresponding complex structures, while generative learning reconstructs interaction features to model the bidirectional mapping between monomeric and complex representations. This enables the model to infer binding representations directly from protein and ligand sequences alone. Across challenging affinity prediction scenarios, including cold-start and sparse-label conditions, CompBind not only outperforms noncomplex-based methods but also competitively rivals complex-based prediction approaches. In a drug repurposing case study targeting glutathione peroxidase 4 (GPX4), a clinically relevant but traditionally undruggable protein, CompBind successfully ranked known inhibitors among the top candidates. Furthermore, the built-in attention mechanism enhances model interpretability by identifying key binding residues. By decoupling predictive accuracy from the availability of experimental complex structures, CompBind offers a scalable, generalizable, and practical solution for accelerating drug discovery pipelines.
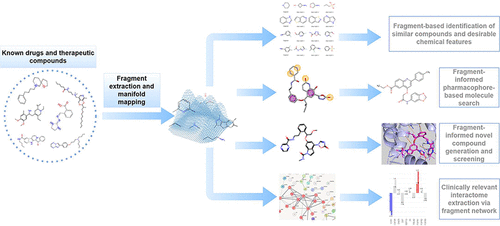
Fragment-Guided New Therapeutic Molecule Discovery and Mapping of Clinically Relevant Interactomes
Austė Kanapeckaitė *- ,
Sarper Okuyan - ,
David James Wagg - ,
Jan Koster - ,
Ligita Jančorienė - ,
Indrė Sakalauskaitė - ,
Birutė Brasiu̅nienė - , and
Andrea Townsend-Nicholson *
Therapeutic interventions for complex diseases depend on the targeted modulation of key pathological pathways. While growing clinical needs continue to drive advancements in the drug discovery space, current strategies primarily rely on searching large volumes of chemical data without addressing the specific contributions of molecular features. Moreover, both clinicians and researchers recognize the need for improved drug discovery methods and characterization that could aid in clinical strategy selection. To address these challenges, we propose a new perspective on targeted therapy development as well as interactome mapping, utilizing molecular fragments. The present study focuses on therapeutic areas that represent emerging targets, namely JAK2 and GLP-1R, both of which have broad clinical potential. We developed a new self-adjusting neural network that enabled us to discover novel therapeutic candidates with improved in silico binding profiles, gain additional insights into drug-target binding that were not previously reported, and identify new metabolic trajectories. Importantly, our work revealed that even a small compound library can effectively generate lead candidates, expediting the search and exploration process. In addition, the fragment-guided bridging of chemical and biological spaces has revealed new opportunities for drug repurposing efforts and a means of improving the prediction of side effects. We concluded our study with insights into the recent high-profile clinical trial failure of danuglipron and how this could have been prevented with our methodology. Thus, building a robust in silico pipeline with integrated screening data can significantly reduce costs and guide therapy adoption. Furthermore, our proposed strategy highlights promising avenues for the discovery of new therapeutics and the development of clinical interventions.
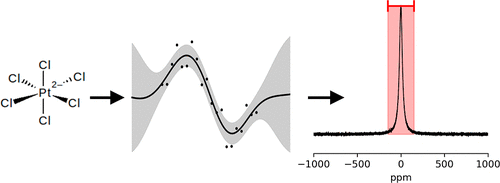
Uncertainty-Aware Prediction of 195Pt Chemical Shifts from Limited Data
Alexander Meßler - and
Hilke Bahmann *
Platinum (Pt) complexes are highly relevant for medicinal chemistry and homogeneous catalysis. In the development of novel Pt-based chemotherapeutic agents and catalysts, characterization of the compounds using nuclear magnetic resonance (NMR) spectroscopy of the 195Pt nucleus is standard. However, measuring 195Pt-NMR signals can be tedious due to the large chemical shift range and limited resolution. To facilitate experimental measurements by narrowing down the shift range, reliable predictions of the chemical shift are needed. Especially for lighter nuclei such as 1H and 13C, machine learning (ML) methods predict chemical shifts accurately, while analogous models for heavier nuclei are scarce. In this work, we propose Gaussian Process Regression (GPR) models for the prediction of 195Pt chemical shifts. The underlying data set comprises 292 structures and three different descriptors were used to encode structural and chemical features of the molecules. Based on the prediction uncertainties derived from the posterior variance of the models, a reasonably narrow shift range can be estimated for a given Pt complex. The most robust model yields a mean absolute error (MAE) of 114 ppm on the holdout test set, which is significantly more accurate than relativistic DFT calculations.
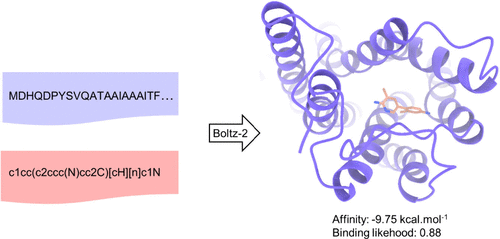
Assessing Boltz-2 Performance for the Binding Classification of Docking Hits
Guillaume Bret - ,
François Sindt - , and
Didier Rognan *
The recently released Boltz-2 cofolding model is generating high expectations by enabling both protein–ligand structure and binding affinity predictions. When applied to a recently described and challenging data set of ultralarge-virtual-screening hits, Boltz-2 excels at discriminating true from false positives, overcoming by a large margin all scoring functions tested so far on raw docking poses. Strikingly, affinity predictions seem to be relatively independent of pose quality but are not biased by obvious chemical similarity to known compounds sharing comparable binding potencies. To ascertain that Boltz-2 truly relies on the physics of intermolecular interactions, we challenged affinity predictions with biologically meaningful challenges (target mutation and target shuffling). Binary classification of active vs inactive compounds remains insensitive to key binding site mutations and even in some cases to target exchange, raising concerns on the hidden features governing Boltz-2 affinity predictions.
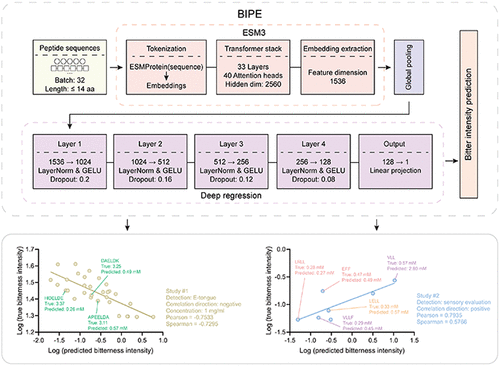
BIPE: Artificial Intelligence-Driven Peptide Bitterness Intensity Prediction Engine
Jianda Yue - ,
Hua Tan - ,
Jiawei Xu - ,
Tingting Li - ,
Zihui Chen - ,
Xie Li - ,
Zhaoyang Tang - ,
Songping Liang - ,
Zhonghua Liu *- , and
Ying Wang *
Bitterness, alongside sour, sweet, umami, and salty tastes, constitutes one of the five basic tastes and serves as a key dimension in shaping food flavor profiles. Food protein processing readily generates bitter peptides, whose intense bitterness often leads to consumer rejection, yet these peptides frequently carry beneficial bioactivities, necessitating a trade-off between flavor and functionality. This necessitates the quantitative assessment of bitterness intensity in the early stages of product development. However, experimental assays relying on sensory evaluation and electronic tongue instruments are complex, costly, and limited in throughput, constraining the systematic identification of bitter peptides and process optimization. Here, we present BIPE (Bitterness Intensity Prediction Engine), an end-to-end regression model that integrates ESM3 protein language model representations with a multilayer perceptron readout, performing regression of bitterness thresholds in log space to directly assess bitterness intensity from sequence alone. BIPE achieves R2 = 0.9050 under 10-fold cross-validation and R2 = 0.9449 on an independent test set. BIPE accurately reproduces trends in both electronic tongue readouts and human sensory scores, demonstrating a consistent external validity across assays. Besides, BIPE accurately differentiates the bitterness intensities of soybean protein hydrolysates produced by multiple commercial proteases. Finally, systematic scanning of the complete pentapeptide sequence space by BIPE further reveals amino acid compositional patterns associated with bitterness, providing mechanistic insights. By advancing from classification to quantitative regression, BIPE enables rational design of low-bitterness peptides, supports flavor engineering and process optimization, and establishes a reusable baseline for taste modeling.
NeuroTDPi: Interpretable Deep Learning Models with Multimodal Fusion for Identifying Neurotoxic Compounds
Baodi Liu - ,
Zhaoyang Chen - ,
Nianlu Li - ,
Na Li - ,
Wenhui Zhang - ,
Yan Li - ,
Xin Huang - ,
Meng Li *- , and
Xiao Li *
Chemical neurotoxicity remains a critical safety concern in the domains of drug development and environmental risk assessment. In these contexts, reliable early stage prediction can significantly reduce experimental costs. In this study, we developed NeuroTDPi, a multilayer fully connected deep neural network model designed to identify neurotoxic compounds. The model employs a multimodal fusion strategy, integrating molecular characterization with feature representations tailored to three specific neurotoxicity end points: blood-brain barrier permeability, neuronal toxicity, and mammalian neurotoxicity. In order to enhance the interpretability of the model, the SHapley Additive Explanations (SHAP) method was employed to elucidate the contributions of various physical and chemical properties. NeuroTDPi exhibited a commendable performance, attaining area under the receiver operating characteristic curve values of 0.97, 0.84, and 0.82 for the three end points, respectively. Furthermore, a comprehensive mining and visualization workflow identified structural alerts associated with neurotoxicity, offering mechanistic insights into the observed toxic effects. These resources, which provide a robust platform for neurotoxicity evaluation and actionable structural insights for risk assessment, are freely available at https://www.sapredictor.cn/.
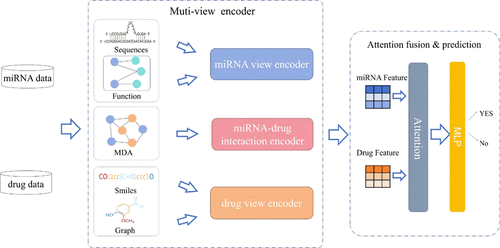
Attention-Guided Multiview Deep Learning Framework Uncovers miRNA-Drug Associations for Therapeutic Discovery
Yan Wang - ,
Yunzhi Liu - ,
Chenxu Si - ,
Jie Hong - ,
Lei Wang - ,
Lan Huang *- , and
Nan Sheng *
MicroRNAs (miRNAs) play critical roles in regulating various biological processes and offer significant potential for treating human diseases. Aberrant expression of miRNAs is known to contribute to drug resistance/sensitivity, posing a significant challenge to miRNA-based therapeutic approaches. Currently, traditional biological experiments to detect miRNA-drug associations (MDAs) are costly and time-consuming, while sequence- or topology-based deep learning methods have gained recognition for their efficiency and accuracy. Nevertheless, existing computational methods tend to ignore multiple sources of information and are overly reliant on known MDAs. We introduce an attention-guided multiview deep learning framework (DLMVF) for predicting MDAs. Our innovative approach fully integrates multisource information about miRNAs and drugs rather than relying exclusively on interaction graph data. DLMVF contains miRNA attribute view encoder, drug attribute view encoder, and miRNA-drug interactions encoder modules, enabling the extraction of miRNA and drug features from multiple perspectives. Moreover, the DLMVF can enhance the learned latent representations for association prediction through view-level attention, which adaptively learns the importance of different features. To evaluate the effectiveness of DLMVF, we manually constructed an experimental benchmark data set based on the latest database. DLMVF achieves an AUROC of 0.9611 and an AUPRC of 0.9543 on the benchmark data set. Extensive benchmarking demonstrates that the DLMVF outperforms existing methods with good robustness and generalization. In addition, a case study of three common anticancer drugs demonstrates its effectiveness in discovering novel MDAs. Data and source code will be published at https://github.com/Lgubig/DLMVF_model.
Chemical Information
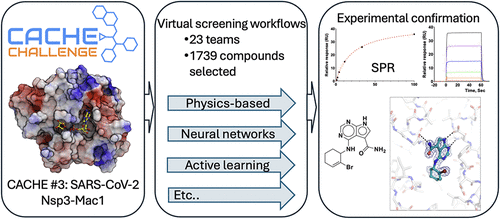
CACHE Challenge #3: Targeting the Nsp3 Macrodomain of SARS-CoV-2
Oleksandra Herasymenko - ,
Madhushika Silva - ,
Galen J. Correy - ,
Abd Al-Aziz A. Abu-Saleh - ,
Suzanne Ackloo - ,
Cheryl Arrowsmith - ,
Alan Ashworth - ,
Fuqiang Ban - ,
Hartmut Beck - ,
Kevin P. Bishop - ,
Hugo J. Bohórquez - ,
Albina Bolotokova - ,
Marko Breznik - ,
Irene Chau - ,
Yu Chen - ,
Artem Cherkasov - ,
Wim Dehaen - ,
Dennis Della Corte - ,
Katrin Denzinger - ,
Niklas P. Doering - ,
Kristina Edfeldt - ,
Aled Edwards - ,
Darren Fayne - ,
Francesco Gentile - ,
Elisa Gibson - ,
Ozan Gokdemir - ,
Anders Gunnarsson - ,
Judith Günther - ,
John J. Irwin - ,
Jan Halborg Jensen - ,
Rachel J. Harding - ,
Alexander Hillisch - ,
Laurent Hoffer - ,
Anders Hogner - ,
Ashley Hutchinson - ,
Shubhangi Kandwal - ,
Andrea Karlova - ,
Kushal Koirala - ,
Sergei Kotelnikov - ,
Dima Kozakov - ,
Juyong Lee - ,
Soowon Lee - ,
Uta Lessel - ,
Sijie Liu - ,
Xuefeng Liu - ,
Peter Loppnau - ,
Jens Meiler - ,
Rocco Moretti - ,
Yurii S. Moroz - ,
Charuvaka Muvva - ,
Tudor I. Oprea - ,
Brooks Paige - ,
Amit Pandit - ,
Keunwan Park - ,
Gennady Poda - ,
Mykola V. Protopopov - ,
Vera Pütter - ,
Rahul Ravichandran - ,
Didier Rognan - ,
Edina Rosta - ,
Yogesh Sabnis - ,
Thomas Scott - ,
Almagul Seitova - ,
Purshotam Sharma - ,
François Sindt - ,
Minghu Song - ,
Casper Steinmann - ,
Rick Stevens - ,
Valerij Talagayev - ,
Valentyna V. Tararina - ,
Olga Tarkhanova - ,
Damon Tingey - ,
John F. Trant - ,
Dakota Treleaven - ,
Alexander Tropsha - ,
Patrick Walters - ,
Jude Wells - ,
Yvonne Westermaier - ,
Gerhard Wolber - ,
Lars Wortmann - ,
Shuangjia Zheng - ,
James S. Fraser *- , and
Matthieu Schapira *
This publication is Open Access under the license indicated. Learn More
The third Critical Assessment of Computational Hit-finding Experiments (CACHE) challenged computational teams to identify chemically novel ligands targeting the macrodomain 1 of SARS-CoV-2 Nsp3, a promising coronavirus drug target. Twenty-three groups deployed diverse design strategies to collectively select 1739 ligand candidates. While over 85% of the designed molecules were chemically novel, the best experimentally confirmed hits were structurally similar to previously published compounds. Confirming a trend observed in CACHE #1 and #2, two of the best-performing workflows used compounds selected by physics-based computational screening methods to train machine learning models able to rapidly screen large chemical libraries, while four others used exclusively physics-based approaches. Three pharmacophore searches and one fragment growing strategy were also part of the seven winning workflows. While active molecules discovered by CACHE #3 participants largely mimicked the adenine ring of the endogenous substrate, ADP-ribose, preserving the canonical chemotype commonly observed in previously reported Nsp3-Mac1 ligands, they still provide novel structure–activity relationship insights that may inform the development of future antivirals. Collectively, these results show that multiple molecular design strategies can efficiently converge on similar potent molecules.
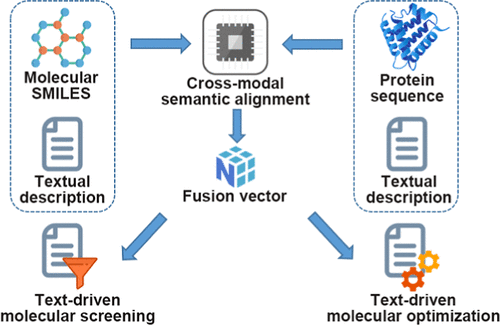
CoDrug: A Text-Driven Molecular Virtual Screening and Multiproperty Optimization Framework via Multimodal Language Model
Rui Gu - ,
Yingxu Liu - ,
Bingxing Zhu - ,
Li Liang - ,
Haichun Liu *- ,
Yanmin Zhang *- , and
Yadong Chen *
Traditional molecular screening methods are often limited by high computational cost, long design cycles, and a strong reliance on high-quality 3D protein structures, which are not always available or reliable. To address these limitations, we propose CoDrug, an innovative multimodal fusion framework that integrates textual information with structural representations of proteins and compounds. CoDrug employs two complementary fusion strategies─text–protein sequence fusion, in which SciBERT encodes functional descriptions and ESM extracts sequence-level features, and text–compound structure fusion, in which ChemFormer encodes SMILES and SciBERT processes compound-related textual descriptions. Using contrastive learning, CoDrug aligns textual and structural embeddings in a shared latent space, enabling effective cross-modal representation learning. This architecture supports novel functionalities, including text-driven virtual screening and text-driven molecular optimization, enhancing representation expressiveness and generalization while delivering strong performance under zero-shot settings. Evaluations on diverse benchmarks demonstrate that CoDrug achieves competitive or superior results compared with state-of-the-art baselines, particularly when 3D structural data are incomplete or unavailable. The framework’s natural language interface lowers the technical barrier for AI-assisted drug discovery, allowing chemists to efficiently navigate and optimize chemical space without specialized computational expertise. By bridging language-driven hypotheses and structure-guided molecular design, CoDrug offers a scalable and flexible paradigm for accelerating the early stages of drug discovery.

Agentic Knowledge Graphs of the LiFePO4 Cathode for Lithium Ion Battery: Balancing Discovery and Stability with LLMs
Lee Bin Choi - ,
Ohyeon Lee - , and
Sanghun Lee *
Lithium iron phosphate (LiFePO4, LFP) has regained prominence as a cathode for lithium ion batteries thanks to its intrinsic safety, thermal stability, long cycle life, and cost advantages. We present an agentic knowledge-graph pipeline that converts titles/abstracts into directed, signed agent → property relations. Using a Scopus corpus of the 9500 most-cited LFP journal articles (2000–present), we benchmark three matched modes: A, rules with a closed vocabulary; B, LLM-only with an open vocabulary; and mixed LLM with a hybrid vocabulary. A yields a compact, high-precision core; B expands recall but increases label dispersion; C preserves much of B’s breadth while maintaining schema alignment via canonicalization and role gating. Robustness tests with eight bootstrap passes show rapid convergence: requiring recurrence across ∼6 passes plus a modest publication-support threshold yields a compact, high-confidence backbone. The resulting network is predominantly positive and centers on transport and interfacial outcomes, with a small number of mixed and negative ties indicating condition dependence. Beyond LFP, the workflow can be adapted to other battery chemistries with modest retuning of vocabularies and projection rules alongside routine validation on held-out annotations, enabling a stability-aware, literature-scale synthesis of direction-of-effect relations.
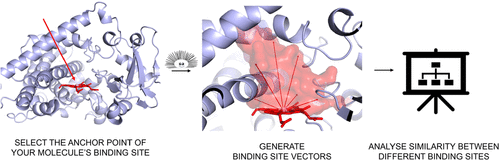
Binding Site Vectors Enable Mapping of Cytochrome P450 Functional Landscapes
Tea Kuvek - ,
Zuzana Jandová - ,
Klaus-Juergen Schleifer - , and
Chris Oostenbrink *
This publication is Open Access under the license indicated. Learn More
Understanding similarities between protein binding sites has long been of great interest, as such comparisons can reveal functional relationships that transcend sequence or fold. However, systematic comparison remains challenging due to the difficulty of defining active sites consistently and developing descriptors that are both general and discriminative. We present binding site vectors, a computational framework for a high-resolution comparison of macromolecular binding sites that integrates both structural and electrostatic properties. The vectors extend spherically from the center of the pocket, terminating at its surface to capture shape and electrostatic features in a multidimensional manner. Geometrically anchored, they enable a systematic comparison of binding sites across diverse systems. We applied this approach to cytochrome P450 (CYP) enzymes, analyzing over 600 human and plant CYP structures and a subset of 23 extensive structural ensembles obtained through molecular dynamics (MD) simulation. Comparisons based on binding site vectors reveal structural–functional relationships missed by sequence- or backbone-based groupings, particularly when full conformational ensembles are included. This demonstrates that binding site vectors provide a robust framework for both functional classification and deep mechanistic insights into macromolecular systems.
Computational Chemistry
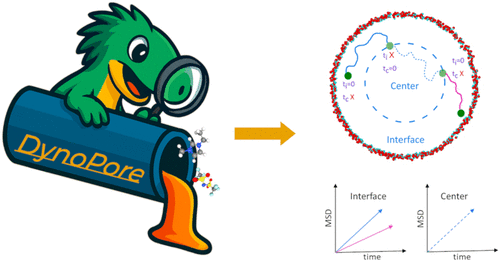
DynoPore─A Package to Analyze Molecular Dynamics Trajectories of Confined Liquids
Samanvitha Kunigal Vijaya Shankar - ,
Christopher P. Ewels - , and
Yann Claveau *
We present a Python package, DynoPore, to study the liquids confined in cylindrical and slit-like geometries. Structural analysis functions such as density profiles and radial distribution functions are included to facilitate the understanding of the environment and local structure of liquid molecules within the confined systems. For dynamics, DynoPore includes region-resolved mean-squared displacement and lifetime functions to investigate molecular motion in different regions of the pore. For ionic systems, Dynopore also offer Nernst–Einstein and Einstein–Helfand conductivity analysis functions. By combining these structural and dynamical analysis tools in a single, user-friendly framework, DynoPore delivers a convenient and comprehensive package to analyze confined liquids.
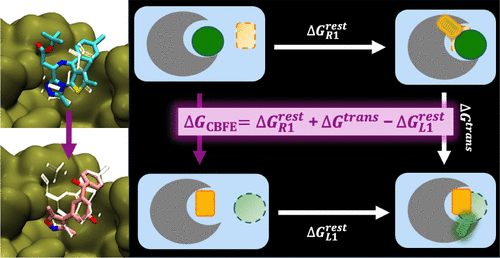
A Relative Binding Free Energy Framework for Structurally Dissimilar Molecules
Hsu-Chun Tsai - ,
Shi Zhang - ,
Tai-Sung Lee - ,
Timothy J. Giese - ,
Charles Lin - ,
James Xu - ,
Yinhui Yi - ,
Darrin M. York *- ,
Abir Ganguly *- , and
Albert C. Pan *
Relative binding free energy (RBFE) calculations, widely used to predict the potencies of congeneric small molecules binding to a protein receptor, can greatly increase the efficiency of the hit-to-lead and lead optimization stages of the drug discovery process. Traditional RBFE methods, however, cannot be easily applied to small molecules lacking a common core or binding mode, precluding their use in a challenging but crucial component of many drug discovery campaigns. In principle, an absolute binding free energy (ABFE) method can be applied to such molecules, but ABFE often suffers from high computational cost and poor statistical convergence due to the large amount of additional sampling required when compared to RBFE. Here, we introduce core-hopping binding free energy (CBFE) calculations, a computationally efficient framework for the accurate determination of relative binding free energies between small molecules with different cores, leveraging several recently developed techniques such as Alchemical Enhanced Sampling (ACES) with optimized transformation pathways and flexible λ-spacing, as well as λ-dependent Boresch restraints. We benchmark the performance of CBFE across 4 protein systems consisting of 56 small molecules, and find that the results are consistent with RBFE for a congeneric series of ligands and offer considerable improvement in computational cost and precision relative to ABFE results for a series of small molecules with diverse cores and binding modes. All CBFE-related developments are fully implemented in the GPU-accelerated AMBER free energy module (pmemd.cuda) and are available as part of the latest official AMBER release.

A Sensitivity Analysis Methodology for Rule-Based Stochastic Chemical Systems
Erika M. Herrera Machado *- ,
Jakob L. Andersen *- ,
Rolf Fagerberg *- , and
Daniel Merkle *
In this study, we introduce a sensitivity analysis methodology for stochastic systems in chemistry, where dynamics are often governed by random processes. Our approach is based on gradient estimation via finite differences, averaging simulation outcomes, and analyzing variability under intrinsic noise. We characterize gradient uncertainty as an angular range within which all plausible gradient directions are expected to lie. A key feature of our approach is that this uncertainty measure adaptively guides the number of simulations performed for each nominal-perturbation pair of points in order to minimize unnecessary computations while maintaining robustness. Systematically exploring a range of parameter values across the parameter space, rather than focusing on a single value, allows us to identify not only sensitive parameters but also regions of parameter space associated with different levels of sensitivity. These results are visualized through vector field plots to offer an intuitive representation of local sensitivity across parameter space. Additionally, global sensitivity coefficients over sampled points in the parameter space are computed to capture overall trends. Flexibility regarding the choice of output observable measures is another key feature of our method: while traditional sensitivity analyses often focus on species concentrations, our framework allows for the definition of a large range of problem-specific observables. This makes it broadly applicable in diverse chemical and biochemical scenarios. We demonstrate our approach on two systems: classical Michaelis–Menten kinetics and a rule-based model of the formose reaction, using the cheminformatics software MØD for Gillespie-based stochastic simulations.
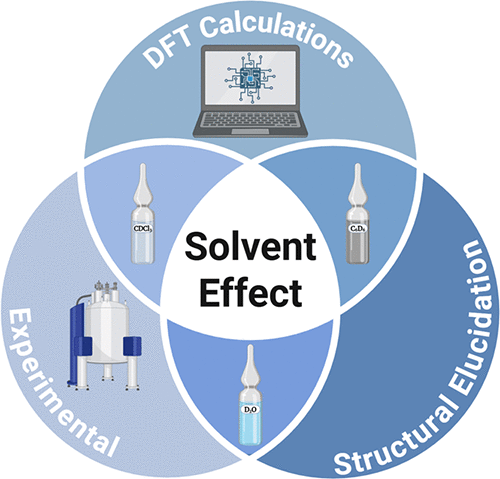
Solvent Matters: Bridging Theory and Experiment in Quantum-Mechanical NMR Structural Elucidation
Iván Cortés - ,
Cristina Cuadrado - ,
José A. Gavín - ,
María Marta Zanardi - ,
Antonio Hernández Daranas *- , and
Ariel M. Sarotti *
Quantum-mechanical NMR (QM-NMR) is widely used in structure elucidation. A long-sought holey grail in this field is solving structures from a simple 1H NMR spectrum with AI-driven workflows. Yet, solvent effects on chemical shifts, though long recognized, remain overlooked. We show in a theory–experiment study that implicit solvation models miss solvent-induced variations and introduce a Python tool to quantify solvent sensitivity, aiding more reliable QM-NMR structural assignments.
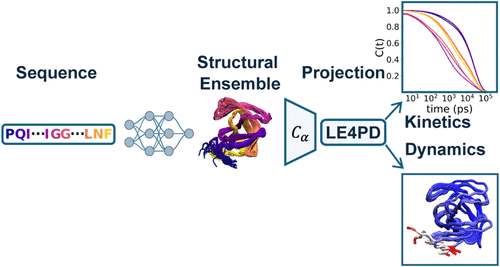
Applied Causality to Infer Protein Dynamics and Kinetics
Akashnathan Aranganathan - and
Eric R. Beyerle *
The use of generative machine learning models, trained on the experimentally resolved structures deposited in the protein data bank, is an attractive approach to sampling conformational ensembles of proteins. However, the ensembles generated by these models lack time scale or causal information. We use the structural ensembles generated from AlphaFold2 at a range of MSA depths to parametrize the potential of mean force of an overdamped, memory-free, coarse-grained Langevin equation. This approach couples the AlphaFold2 ensembles to a causal model, allowing us to estimate the time scales spanned by the ensembles generated at each MSA depth. Performing this analysis on six variants of HIV-1 protease, we confirm an inverse relationship between MSA depth and the time scale of an ensemble’s conformational fluctuations. The MSA depth essentially serves as a conformational restraint, and AlphaFold2 is generally able to probe time scales at or below those seen in microsecond-long, unbiased molecular dynamics simulations. We conclude by generalizing this approach to other generative structural ensemble-prediction methods as well as cofolding models, in this case, the biologically functional HIV-1 protease dimer.
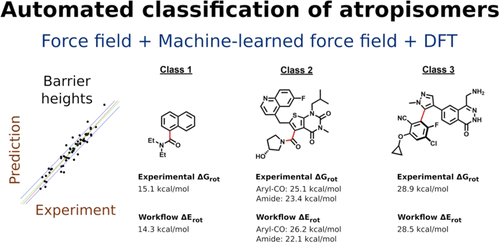
Prediction of Atropisomerism for Drug-like Molecules
Ty Balduf - ,
Philip A. Gerken - ,
Mee Y. Shelley - ,
Mark A. Watson - ,
M. Chandler Bennett - ,
Mats Svensson - ,
Abba E. Leffler - , and
Art Bochevarov *
A multistep computational workflow that accurately assigns organic drug-like molecules to one of three atropisomer classes on the basis of computed barrier heights has been developed. The workflow identifies rotatable bonds and applies progressively more accurate types of calculations to the eligible rotational degrees of freedom. An initial energy scan with a force field (OPLS4) is followed by a similar scan that uses an energy function driven by a neural network model (QRNN-TB) trained on density functional theory (DFT) energies. The maxima corresponding to the potentially stereogenic rotatable bonds identified at this point are further processed by applying a transition state search at the QRNN-TB level of theory. Finally, ωB97X-D3/def2-TZVP(-f) DFT energies are computed for all located extrema. The accuracy of the predicted rotational barriers was benchmarked against ωB97M-V/cc-pVTZ and DLPNO-CCSD(T)/def2-TZVPP energies with excellent correlations. The automated protocol classifies organic molecules into atropisomeric classes with a greater than 90% success rate when applied to a test set of 65 molecules containing rotationally restricted torsions (68 torsions in total). We anticipate that the balance of speed and accuracy in this method will make it conducive to production use in drug discovery programs.
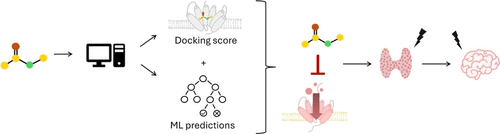
Combined Modeling Approaches for Assessing Sodium-Iodide Symporter Inhibition
Julia Kandler - ,
Ayse Sıla Kantarçeken - ,
Aljoša Smajić - , and
Gerhard F. Ecker *
This publication is Open Access under the license indicated. Learn More
The sodium-iodide symporter (NIS, SLC5A5) plays a crucial role in thyroid hormone synthesis. Especially during brain development, correct thyroid signaling is of critical importance. Hence, inhibition of this transporter can lead to neurodevelopmental disorders, such as lowered IQ or autism. In order to uncover environmental chemicals with the potential of causing developmental neurotoxicity (DNT), NIS was selected for modeling. To support next-generation risk assessment, in silico-based methods were utilized. Docking-based virtual screening workflows of a library of compounds with experimentally determined inhibitory activity on NIS were applied. In addition, machine learning (ML) models based on random forest (RF), extreme gradient boosting (XGB), and support vector machines (SVM) were trained using extended-connectivity fingerprints 4 (ECFP4) and continuous and data-driven descriptors (CDDDs) with 9-fold cross validation to discriminate between NIS inhibiting and noninhibiting compounds. Ultimately, combining ML and docking predictions improved discrimination, achieving an area under the receiver operating characteristic curve (ROC AUC) of 0.77. Thresholds for optimal discrimination between actives and inactives were determined using kernel density estimate plots, at which a Matthews correlation coefficient (MCC) of 0.32, and a balanced accuracy (BA) of 0.78 were achieved on the internal test set. By combining ML predictions with docking scores and training on a larger, more diverse data set of 1412 compounds, this study provides a novel and robust framework for NIS inhibition prediction, which constitutes a new approach method in toxicological risk assessment.
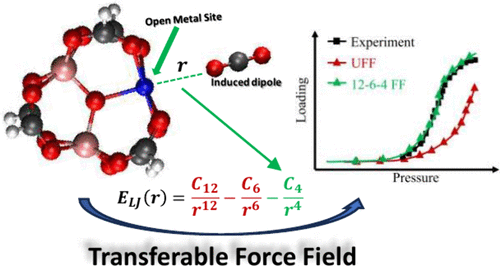
A Transferable Force Field for Simulating Adsorption in Metal–Organic Frameworks with Open Metal Sites Based on the 12–6–4 Lennard-Jones Potential
Meng Du - ,
Alan Rodriguez - ,
Matthew Z. Lin - , and
Haoyuan Chen *
This publication is Open Access under the license indicated. Learn More
Metal–organic frameworks (MOFs) that contain coordinatively unsaturated open metal sites (OMSs) provide strong host–guest interactions, making them promising sorbents for low-concentration gas adsorption applications such as direct air capture and atmospheric water harvesting. However, accurately modeling host–guest interactions involving OMSs remains challenging for classical force fields (FFs) based on the 12–6 Lennard–Jones (LJ) potential, as the polarization effect of the guest molecule induced by the positively charged OMS is not considered. Here, we introduce an FF based on the 12–6–4 LJ potential, which incorporates charge–induced dipole interactions and is parametrized against a diverse set of host–guest potential energy surfaces (PESs) obtained from density functional theory (DFT). The resulting FF, trained on a generic trimetallic cluster, performs well in both host–guest binding energetics and gas adsorption isotherms across different OMS-containing MOFs, including MOF-74 series and Cu-BTC. These results highlight the excellent transferability of our approach and its potential to enhance the accuracy and robustness of high-throughput MOF discovery workflows, particularly for gas adsorption and separation in large and diverse MOF databases.
Noncovalent Interactions in Solvated Proteins and Protein Crystals Studied with the Fragment Molecular Orbital Method
Dmitri G. Fedorov *- ,
Katarzyna J. Zator - ,
Julia Contreras-García - , and
Seiji Mori
A new formulation of the many-body expansion of the electron density expressed in terms of the wave function data is developed in the framework of the fragment molecular orbital (FMO) method for the purpose of visualizing noncovalent interactions (NCI) in large systems. This expansion can also be used for a selected site of interest, such as a ligand binding site in a protein. The site formulation is shown to be both accurate and efficient, as demonstrated for a small protein–ligand complex (Trp-cage protein, PDB: 1L2Y) and a large complex of prostaglandin H2 synthase-1 (1EQG) with ibuprofen. In addition, the FMO/NCI methodology is extended to treat periodic boundary conditions, with an application to study packing effects in the crystal of crambin (1CBN).
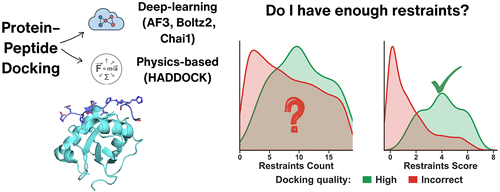
Restraint Quality, Not Quantity, Predicts Peptide–Protein Docking Outcomes
Miriam Gulman - ,
Jordan Chill - , and
Dan Thomas Major *
Understanding protein-peptide interactions is essential for uncovering cellular signaling mechanisms and advancing therapeutic development, as these interactions play central roles in numerous biological processes. Gaining structural insight into such complexes is crucial, yet traditional methods like nuclear magnetic resonance (NMR) and X-ray crystallography are often time-consuming and experimentally demanding. Computational approaches─including physics-based docking and deep-learning (DL) structure predictors such as AlphaFold3, Boltz-2, and Chai-1─offer powerful alternatives. Accurately modeling flexible peptides that bind to shallow, surface-exposed regions remains difficult for physics-based methods, and although multiple sequence alignment-driven DL models can achieve excellent performance in well-behaved systems, they too can struggle when the peptide adopts noncanonical conformations or when sequence identity is low. In such cases, distance restraints are often required to guide the docking toward accurate and biologically meaningful solutions, yet acquiring multiple high-quality restraints is often difficult. To address the limitation of physics and DL approaches, we developed a restraint scoring function that integrates evolutionary conservation, spatial proximity, and geometric distribution to assess the informativeness of restraint sets. This enables a more accurate evaluation of docking inputs and overcomes the shortcomings of relying solely on restraint count. Building on this framework, we introduce a minimal-restraint docking strategy, capable of identifying optimized subsets of restraints that lead to high-quality structural models. We evaluate a comprehensive set of protein–peptide systems, including 43 SH3 domain complexes, 8 WW domain complexes, and 19 medium-difficulty cases from the PepPCBench benchmark. Our approach shows that model quality improves as the restraint score increases, supporting restraint score as a simple, interpretable indicator of docking success. We further identify clear, domain-specific restraint-score thresholds for the SH3 and WW systems that enable accurate model selection. Together, these results offer a scalable and efficient strategy for structure prediction in data-limited contexts and lay the groundwork for restraint-informed modeling with quantifiable confidence, as well as a powerful foundation for data-efficient machine learning-based peptide–protein docking.
Computational Biochemistry

Graph-Based Deep Learning Models for Predicting pKa Values of Protein-Ionizable Residues via Physically Inspired Feature Engineering
Ziyu Song - ,
Ruixuan Wang - ,
Xun Jiao - , and
Zuyi Huang *
This publication is Open Access under the license indicated. Learn More
The pKa value of a protein-ionizable residue reflects its potency to donate a proton at a given pH value, which is essential for understanding a wide range of biological activity. Therefore, the accurate prediction of pKa values of protein residues is crucial for understanding enzymatic activity and protein–ligand binding, which are fundamental to drug discovery. Despite significant time and resources being invested to develop computational methods for protein residue pKa prediction, the accuracy of existing tools, such as the widely used PROPKA, remains limited. In this study, an integrated framework that fuses molecular dynamics simulations and deep learning models is proposed to improve the predictive accuracy of pKa values for ionizable residues. Specifically, we employ high-throughput molecular modeling using the AMOEBA polarized force field to construct a protein structure data set enriched with atomic electrostatics and other physics-inspired features. Using the experimentally determined pKa values from the PKAD-2 data set, we trained three graph-based neural network models. All three models demonstrated substantial improvements in prediction accuracy across four ionizable residue types, aspartic acid, glutamic acid, lysine, and histidine, when compared to PROPKA3.5.1, with the graph attention networks-based model exhibiting both high accuracy and strong generalizability when benchmarking against several recently published machine learning models. Beyond these improvements in predictive performance, feature importance analysis of the best-performing models revealed physically meaningful patterns of the descriptive features that aligned with the underlying biophysical principles governing protein residue pKa values, most notably, the complexity of the local microenvironment and the atomic geometric arrangement within the protein structure. Together, the trained pKa models and the curated dipole moment-enhanced data set based on a polarizable FF offer a valuable resource for the research community, with potential applications in early-stage drug target identification and protein engineering.
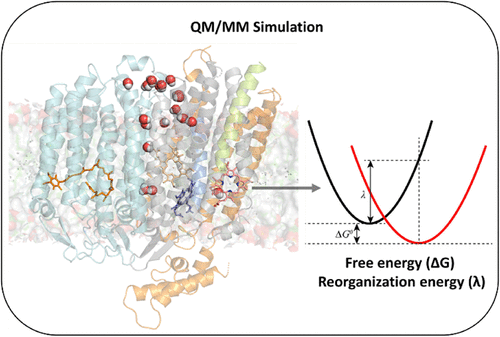
Computational Study of Heme b595 to Heme d Electron Transfer in E. coli Cytochrome bd-I Oxidase
Raaif Siddeeque - ,
Baptiste Etcheverry - ,
Côme Cattin - ,
Jean Deviers - ,
Frédéric Melin - ,
Petra Hellwig - ,
Fabien Cailliez *- , and
Aurélien de la Lande *
Cytochrome bd is a distinctive family of terminal oxidases present in the respiratory chains of many prokaryotes. Despite their biological importance, the redox chemistry of these proteins remains poorly understood, largely due to the presence of two b-type hemes and one d-type heme. Here, we report the first computational study of interheme electron transfer in the cytochrome bd family. We performed 10 μs of molecular dynamics simulations of E. coli cytochrome bd-I embedded in realistic membranes, combined with quantum chemical calculations to estimate the thermodynamic parameters of electron transfer from heme b595 to heme d within the framework of Marcus theory. We further identify the respective contributions of the hemes, protein scaffold, lipid bilayer, water, and counterions to the driving force and reorganization energy. The interheme electronic coupling was calculated using the Projected Orbital Diabatization (POD) method in a hybrid Quantum Mechanics/Molecular Mechanics scheme and rationalized through electron transfer pathway analysis. This study provides fundamental insights into how electron transfer steps are orchestrated in the catalytic cycle of E. coli cytochrome bd-I.

HelixSide: A Comprehensive Method for Local and Global Orientational Analysis of Proteins
Sakari Pirnes - ,
Veera Hägg - ,
Mykhailo Girych - ,
Ilpo Vattulainen - , and
Giray Enkavi *
This publication is Open Access under the license indicated. Learn More
Understanding the relative orientation of protein secondary structure elements is crucial for elucidating their tertiary organization, function, and interactions. Here, we introduce HelixSide, a comprehensive method for systematically quantifying geometrical metrics of helical secondary structures, including widely used measures, such as tilt and kink angles. Additionally, to characterize the orientation of secondary structure motifs relative to each other or to the helical axis, we introduce a new quantity, the side angle. HelixSide computes these metrics at both single-residue and whole-protein levels, revealing local and global conformational features of the system. We demonstrate the method’s utility through case studies of two well-characterized single-pass transmembrane proteins: insulin receptor and glycophorin A. These analyses showcase HelixSide’s ability to capture tertiary structural characteristics and compare conformational states. HelixSide is open source and available on GitHub at https://github.com/SakariPirnes/helixside. It is applicable to experimental structures, theoretical models, and molecular dynamics trajectories of membrane and soluble proteins, and can be used as a featurization tool for machine learning.
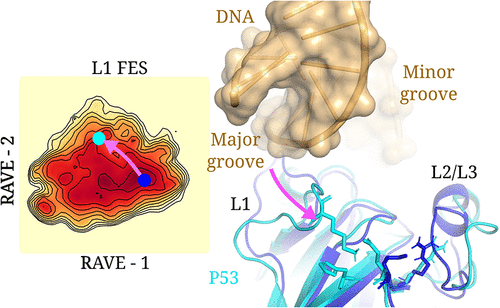
AI-Guided Conformational Dynamics of p53 L1 Loop Reveal an Allosteric Switch Regulating DNA Binding and Cancer Hotspots
Pablo Navarro Acero - ,
Ming-Hong Hao - , and
Karan Kapoor *
The tumor suppressor p53 regulates transcription in response to cellular stress, with mutations in its DNA-binding domain (DBD) found in most human cancers. The L1 loop within the DBD is believed to play a critical role in DNA recognition, yet its conformational dynamics remain poorly understood. Using enhanced molecular dynamics simulations combined with machine learning-derived collective variables, we reveal a novel conformational switch mechanism governing p53’s DNA-binding activity. Our analysis identifies two distinct transition pathways between extended (DNA-binding competent) and recessed conformations, each characterized by specific hydrogen bond networks and high energy barriers. We discovered a potential allosteric mechanism regulating the DNA-p53 binding interface that could provide an atomistic basis for gene-specific transcription regulation. This mechanism would explain the prevalence of certain cancer mutations, particularly at residue R282. Finally, we provide a mechanistic rationale for how compounds targeting a reactivation pocket near the L1 loop may restore p53 function by modulating DNA binding kinetics rather than affinity, thereby reconciling previously observed rescue effects.
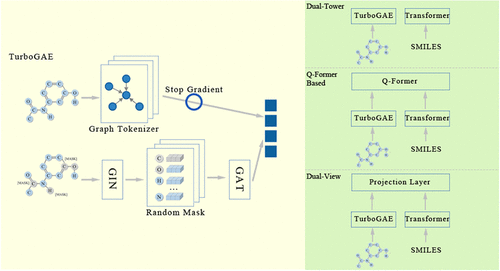
Multi-Modal Fusion Frameworks of Subgraph-Optimized Graph Autoencoder for Molecular Property Prediction
Kaiyuan Zhang - ,
Congyu Han - ,
Fenghua Zhang - ,
Cheng Lin - ,
Quanlong Li - ,
Tianyi Zang *- , and
Yanli Zhao *
This publication is Open Access under the license indicated. Learn More
Molecular property prediction refers to predicting the properties of a given molecular representation. This task is of great significance in fields such as drug design and has garnered widespread attention from researchers. For molecular property prediction, the quality of feature learning plays a decisive role in model performance. Although existing molecular graph models can extract effective feature representations from graph structures, how to better utilize these features across different learning tasks remains an important challenge. This paper proposes a subgraph-optimized Graph Autoencoder (TurboGAE) and several multimodal fusion strategies. By introducing a subgraph-level graph tokenizer, TurboGAE more effectively captures the impact of substructure features (within molecular structures) on molecular properties. For cross-modal molecular features, a rational and effective multimodal feature fusion strategy can align intermodal features during the pretraining phase, leveraging the unique strengths of each modality. The proposed methods demonstrate excellent performance in experiments on downstream tasks.

Unravelling the Role Played by Non-covalent Interactions in the Action Mechanism of PCDDs within Cells
Lorena Ruano - ,
Álvaro Pérez-Barcia - ,
Vito F. Palmisano - ,
Juan J. Nogueira *- ,
Marcos Mandado *- , and
Nicolás Ramos-Berdullas *
This publication is Open Access under the license indicated. Learn More
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that mediates biological signals and regulates diverse cellular functions. Of particular concern are the effects triggered by dioxins and dioxin-like compounds (DLCs), whose toxicological outcomes arise through both canonical and noncanonical pathways, leading to the designation of AhR as the “dioxin receptor”. However, conventional risk assessment approaches based on toxic equivalency factors (TEFs), which primarily reflect the capacity of these compounds to bind and activate AhR, do not fully account for critical aspects such as environmental concentration and bioavailability, potentially underestimating their true impact. In this work, we present a comparative analysis of polychlorinated dibenzo-p-dioxins (PCDDs) with varying degrees of chlorination, focusing on their interactions with the AhR at the ligand-binding domain and on their permeation abilities across a model lipid membrane. To this end, we combine classical molecular dynamics (CMD) simulations with a hybrid quantum mechanics/molecular mechanics energy decomposition analysis (QM/MM-EDA) framework. This integrated approach enables a molecular-level characterization of receptor binding affinities and membrane permeation efficiencies. Our findings provide novel insights into the mechanisms underlying the relative toxicity of DLCs and highlight the need for integrative assessment strategies that encompass both receptor–ligand interactions and physicochemical behavior in biological environments. It is noteworthy that the toxicity of these compounds, as quantified by the pEC50 index, correlates with the membrane permeation barrier rather than with AhR binding affinity, identifying permeation as the key mechanistic step in the toxicological process of these compounds.

Metadynamics Simulation Reveals Allosteric Communication Effects of the Flipping Process of the Atypical DLG Motif in RIPK1
Bo Liu - ,
Likun Zhao - ,
Lingling Wang - ,
Xiaoqing Gong - ,
Xiaojun Yao - ,
Huanxiang Liu - , and
Qianqian Zhang *
Kinases are key molecules in cell signal transduction. Their abnormal activation is closely related to cancer, inflammation, and metabolic diseases, making them important drug targets. However, the high sequence conservation of the kinase family limits the selectivity of inhibitors, and the dynamic conformational changes of kinases profoundly affect drug binding. Receptor-interacting protein kinase 1 (RIPK1) is a core factor in regulating cell necrosis. Its unique DLG (Asp-Leu-Gly) motif replaces the DFG (Asp-Phe-Gly) motif in traditional kinases. It is worth noting that, in the known RIPK1 crystal structure, the DLG motif is always in the “out” conformation, while its flipping mechanism and its regulatory mechanism on drug activity have not yet been elucidated. This study combined conventional molecular dynamics simulation and metadynamics simulation to deeply explore the conformational flipping process of the DLG motif in RIPK1 and its effect on protein conformation and drug binding. The results show that the flipping of the DLG motif occurs in coordination with the rotation of the αC helix, significantly changing the hydrophobicity and spatial volume of the ATP binding pocket, thereby regulating the affinity of drug molecules. In addition, the DLG flipping also reshapes the allosteric communication network of RIPK1, especially affecting the allosteric connection of the hinge region. The study further revealed the differential effects of different types of inhibitors on the conformational flipping of the DLG motif. This work not only provides a new structural perspective and theoretical basis for the design of highly selective RIPK1 inhibitors, but also provides important insights for the development of inhibitors targeting other kinases containing atypical DLG motifs.
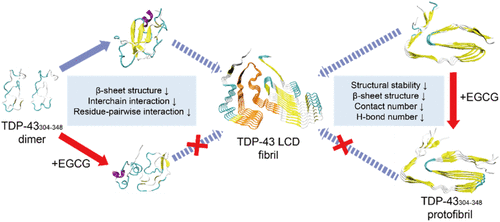
Computational Exploration of the Molecular Mechanism of Epigallocatechin Gallate against TDP-43 Aggregation
Wenjuan Yi - ,
Zhengdong Xu - ,
Dushuo Feng - ,
Lulu Guan - ,
Jiaxing Tang - , and
Yu Zou *
Cytoplasmic accumulation of the transactive response deoxyribonucleic acid (DNA)-binding protein of 43 kDa (TDP-43) aggregates represents the primary pathological hallmark of TDP-43 proteinopathies including amyotrophic lateral sclerosis (ALS) and chronic traumatic encephalopathy (CTE). Inhibiting TDP-43 aggregation or disrupting its preformed fibrils might be promising strategies to prevent or delay the development of TDP-43 proteinopathies. Recently, the green tea polyphenol, epigallocatechin gallate (EGCG), was observed to prevent the formation of TDP-43 oligomeric species and fibrillar aggregates. Nevertheless, the atomic-level mechanism of this inhibition has been incompletely characterized. In this study, we performed a multitude of replica exchange with solute tempering 2 (REST2) and all-atom molecular dynamics (MD) simulations of 46.8 μs in total on TDP-43 models with and without EGCG. The REST2 simulation results revealed that EGCG impedes the β-sheet structure formation and interferes the interchain interaction of TDP-43304–348 dimer. Subsequent analyses show that EGCG could alter the distribution of free energy landscape and hinder the residue–residue interaction of the dimer. The binding analyses confirmed that EGCG preferentially bound to M307, F313, F316, W334, M339, Q344, and Q346 residues, and hydrophobic, polar, and π–π stacking interactions dominate the binding of EGCG on the dimer. Additional conventional molecular dynamics (MD) simulations demonstrated that the protofibrillar tetramer is the minimal stable TDP-43304–348 protofibril. Taking the tetramer as a protofibril model, we found that EGCG could reduce the structural stability and disrupt the β-sheet structure of TDP-43304–348 protofibril, thus possessing a destabilization effect on its higher-order structure. This investigation unveils the atomic-level mechanism by which EGCG against TDP-43 aggregation, which may provide potential fundamental knowledge of therapeutic strategies for TDP-43 proteinopathies.

Recognition of Coexisting Phases in Model Membranes via an Unsupervised Method
Yuzhuo Dai - ,
Jianwei Zhao - ,
Beibei Wang *- ,
Qing Liang *- , and
Ruo-Xu Gu *
Phase separation in bilayers composed of a few lipid species is widely used as a model for exploring the lateral heterogeneity of complex cell membranes. Molecular dynamics (MD) simulations offer atomistic insights into coexisting lipid phases. But identifying these phases from trajectories remains challenging. Here, we present an unsupervised method for lipid phase recognition in phase-separated bilayers. In this method, the membrane plane is first discretized into pixels. For each pixel, the local lipid packing degree, which is defined as the atomic density within that pixel, is calculated and assigned to the corresponding pixel. A threshold is then determined by fitting a two-component Gaussian mixture model (GMM) to the distribution of lipid packing degree, enabling phase state assignment to pixels and subsequent mapping back to lipids. Our method is applicable to different systems, regardless of their compositions or temperatures, thus minimizing potential artifacts. Tests on bilayers with diverse lipid compositions and temperatures show that our method outperforms the commonly used hidden Markov model (HMM) in both accuracy and robustness. Notably, in this method, phase recognition relies solely on bilayer-intrinsic properties (lipid packing degree), without requiring temporal information, labeled data, or assumptions about the local lipid environment. This makes our method broadly applicable to various tasks, including characterizing the phase transformation process before the system reaches equilibration and identifying coexisting phases in protein-containing bilayers. In summary, we provide a robust and accurate framework for identifying coexisting phases in bilayers and tracking their dynamic transitions in simulations.
Computational Insights into the N-Migration and Oxidative Rearrangement Involved in the N-Nitrosourea Formation Catalyzed by the Cupin Domain of Multidomain Metalloenzyme SznF
Hong Li *- ,
Yongjun Liu - , and
Yongqing Zhang
The multidomain metalloenzyme SznF can specifically catalyze the conversion of Nω-methyl-l-arginine (l-NMA) to Nδ-hydroxy-Nω-methyl-Nω-nitroso-l-citrulline (l-NHMA), which is the key step for the biosynthesis of the N-nitrosourea pharmacophore, a precursor to the pancreatic cancer drug streptozotocin (SZN). The central domain of SznF is responsible for mediating the two sequential hydroxylations of l-NMA at Nδ and Nω positions to first generate Nδ,Nω-dihydroxy-Nω-methyl-l-arginine (l-DHMA), and the cupin domain of SznF promotes the N-migration and oxidative rearrangement of l-DHMA. This structural rearrangement contains both the C═N bond cleavage and N–N bond formation, and it is very challenging for chemical synthesis. To illuminate the catalytic mechanism of the cupin domain of SznF, we constructed the reactant models and performed a series of QM/MM calculations. We first determined the protonated states of two hydroxyls and imino of l-DHMA by calculating their pKa values, which are considered to be a crucial factor for theoretically exploring the reaction rhythm. The estimated pKa values revealed that the two hydroxyls and imino of l-DHMA should be in protonated states, and the previously proposed reaction mechanism in which superoxo addition to the unsaturated carbon as the first step is unlikely. Instead, the FeII–O2•– unit should first abstract a hydrogen from the Nω-hydroxyl group to trigger the reaction, and then the generated FeIII–OOH attacks the unsaturated carbon to form the peroxide-bridged intermediate, followed by the concerted O–O and N–C bond cleavage leading to the formation of the Fe-coordinated NO radical, which is the precondition for N-migration. During the reaction, the iron ion plays important roles, not only as a central ion to coordinate with the substrate to mediate the H-abstraction, Fe-OOH attack as well as the bond cleavage and formation but also in stabilizing the NO radical and promoting the final N–N bond formation. These results may deepen the understanding of the catalysis of nonheme iron enzymes.
Pharmaceutical Modeling

Understanding the Kinetic Mechanism of Ligands Stabilizing the RAS–CYPA Interaction
Kexin Xu - ,
Mingyun Shen - ,
Zhe Wang - ,
Sutong Xiang - ,
Qirui Deng - ,
Kaimo Yang - ,
Zhiliang Jiang - ,
Zihao Wang - ,
Chen Yin - ,
Tingjun Hou *- , and
Huiyong Sun *
Molecular glues, including protein degraders and protein–protein interaction (PPI) stabilizers, have emerged as a new paradigm of drug design for regulating interactions between biomacromolecules; yet it is still a challenge for rational design of molecular glues. KRAS, as a prevalent oncogenic driver, is notoriously difficult to target by traditional small molecular drugs due to its challenging binding surface and frequent mutations. Although the small molecular drug RMC7977 has been designed as a PPI stabilizer for stabilizing the inherently weak RAS–CYPA interaction, the precise molecular mechanism underlying its stabilization effect and selectivity difference requires a deeper understanding. To this end, we leverage an integrated computational strategy combining molecular dynamics (MD) simulation, end-point binding free-energy calculation, and enhanced sampling technologies to elucidate the dynamic characteristics of RAS–ligand–CYPA interactions. Our result exhibits a high correlation between the predicted binding affinities and the experimental observations, demonstrating that RMC7977, acting as a strong PPI stabilizer, significantly enhances the stability of the KRAS–CYPA interaction, where, by delicately remodeling the protein–protein interface, the drug optimizes various interactions. Moreover, the results also uncover the dynamic process of stabilizer-mediated KRAS–CYPA stabilization and the mechanistic origin of the binding selectivity. This study provides essential molecular-level insights into RMC7977’s function and offers a valuable computational framework for evaluating the stabilization effect of ligands targeting the KRAS–CYPA and other challenging PPI systems.
Bioinformatics
Allosteric Insights into TCR–pMHC Dynamics: Understanding the Effects of Melanoma-Associated Epitopes
Elif Naz Bingol - and
Pemra Ozbek *
Allostery, a crucial phenomenon for comprehending protein function, interactions, and regulation, involves the transmission of perturbations induced by ligand binding to distant sites within a molecule. Understanding the mechanisms of allostery holds the key to elucidating signal transmission failures and diseases resulting from such disruptions. This study focuses on contributing to this understanding by delving into the intricate dynamics of T-cell receptor and peptide-major histocompatibility complex interactions, which are essential components in the communication network of biological systems. Aiming to reveal the effect of melanoma-associated peptides on allosteric signaling, valuable insights are provided. Molecular dynamics simulations were performed on melanoma-associated-epitope-bound TCR–pMHC complexes, followed by machine learning clustering and network analysis, where this innovative combination facilitated the identification of critical TCR–pMHC contacts that modulate global dynamics and stability, presenting novel insights into the complex dynamics of TCR–pMHC interactions. The results not only contributed molecular understanding to TCR–pMHC interactions but also offered valuable information for the fields of immunotherapy and protein engineering. The findings serve as a guide for future experimental investigations and advance our understanding of the immune response in the context of melanoma.
Ab-SELDON: Leveraging Diversity Data for an Efficient Automated Computational Pipeline for Antibody Design
Jean V. Sampaio - ,
Andrielly H. S. Costa - ,
Aline O. Albuquerque - ,
Júlia S. Souza - ,
Diego S. Almeida - ,
Eduardo M. Gaieta - ,
Matheus V. Almeida - ,
Geraldo R. Sartori *- , and
João H. M. Silva *
This publication is Open Access under the license indicated. Learn More
The utilization of predictive tools has become increasingly prevalent in the development of biopharmaceuticals, reducing the time and cost of research. However, most methods for computational antibody design are hampered by their reliance on scarcely available antibody structures, potential for immunogenic modifications, and a restricted exploration of the paratope’s potential chemical and conformational space. We propose Ab-SELDON, a modular and easily customizable antibody design pipeline capable of iteratively optimizing an antibody–antigen (Ab–Ag) interaction in five different modification steps, including CDR and framework grafting, and mutagenesis. The optimization process is guided by diversity data collected from millions of publicly available human antibody sequences. This approach enhanced the exploration of the chemical and conformational space of the paratope during computational tests involving the optimization of an anti-HER2 antibody. Optimization of another antibody against Gal-3BP stabilized the Ab-Ag interaction in molecular dynamics simulations at lower runtime than alternative pipelines. Tests with SKEMPI’s Ab-Ag mutations also demonstrated the pipeline’s ability to correctly identify the effect of the majority of mutations, especially multipoint and those that increased binding affinity. This freely available pipeline presents a new approach for computationally efficient and automated in silico antibody design, thereby facilitating the development of new biopharmaceuticals.
Geometry-Enhanced Multiscale Joint Representation Learning for Drug-Target Interaction Prediction
Qiao Ning *- ,
Shaohang Qiao - ,
Yawen Cai - ,
Yanpeng Liu - ,
Hui Li *- ,
Qian Ma - , and
Shikai Guo
Drug-target interactions (DTIs) are the basis of the therapeutic effect of drugs, whose accurate prediction helps reduce the cost and time of experimental screening in drug development process. Present methods for DTIs prediction often focus on the study of molecular topological structure, which weakens spatial information such as the relative position of atoms and bond angle, and fail to effectively integrate molecular information with association network information. To address this issue, we propose a novel Geometry-enhanced Multiscale Joint Representation Learning method for drug-target interaction prediction (GMJRL). GMJRL not only considers the global information in the drug-target network from the macro-scale, but also extracts the geometric structure information on the drug and the target from the microscale, including the bond angle information on the drug and the atomic coordinate information on the target. To effectively fuse different scale representations, we develop a joint representation learning method with self-attention, which can capture correlations within the same scale and consider the interscale relationships, thus achieving effective fusion of the macro-scale and microscale representations. Finally, this study introduces a negative sampling algorithm to select reliable negative samples from unlabeled drug-target pairs. Extensive experiments validate that GMJRL yields promising outcomes in predicting drug-target interactions.
DeepDBPI: DNA-Binding Protein Identifier Using a Deep Learning Model with Transformed Denoised Features
Kamran Arshad - ,
Muhammad Arif - , and
Dong-Jun Yu *
Motivation: DNA-binding proteins (DBPs) play a significant role in the entire biological system. Many DNA-related studies actively investigate to understand whether a protein binds to DNA. Conventionally, wet-lab experiments are conducted to characterize DBP functions. However, these methods are often expensive and time-intensive. With the rapid advancement of bioinformatics, there is a growing demand for efficient computational protocols to predict DBPs. Several sequence-based computational tools have been designed to predict DBPs; however, research gaps persist for further improvement. Method: We developed a novel deep learning (DL)-based predictor, called DeepDBPI, for enhancing DBP prediction. The proposed DeepDBPI model leverages the evolutionary and graphical-based properties of protein sequences using novel descriptors, namely covariance correlation-based position-specific scoring matrix (CC-PSSM), binary-profile-based (BP-PSSM), Trigram (TRG-PSSM), and feature encoding based on graphical and statistical (FEGS) methods. Then, we applied the wavelet denoising (WD) algorithm to remove the noise from sequence-derived features. We fed the filtered features to ResNet, LSTM, BiLSTM, RNN, BiRNN, and BiGRU. Results: The DeepDBPI model achieved the best prediction performance with Bi-GRU using the denoised-based FEGS encoding method under 5-fold cross-validation, evaluated by ACC, SN, SP, and MCC. Our proposed model achieved 92.13% ACC, 93.07% SN, 91.19% SP, and 0.8427 MCC on the independent test. We believe the effectiveness of the developed bioinformatics protocol provides insights for drug discovery and other proteomic problems. All data, including the dataset, feature extraction techniques, and models, are available at: https://doi.org/10.5281/zenodo.17496063
scACAN: An Adaptive Learning Framework Aggregating Local Graph Structure Context for Rare Cell Type Identification
Shijia Yan - ,
Junliang Shang *- ,
Shoujia Jiang - ,
Xiaohan Zhang - ,
Fanyu Zhang - ,
Yan Sun - , and
Jin-Xing Liu
Single-cell RNA sequencing (scRNA-seq) technology has become an essential tool for dissecting cellular heterogeneity and elucidating complex biological systems. Nevertheless, the uneven distribution of cell types and the limited representation of rare cell populations present substantial challenges for effective modeling and accurate identification. Most existing methods primarily focus on the annotation of abundant cell types, often overlooking rare, yet biologically significant subpopulations. In addition, the variability of cellular distributions across different biological contexts highlights the need for models with greater adaptability and a stronger capacity for contextual information integration. To overcome these challenges, we introduced scACAN, an adaptive graph construction framework that leverages aggregated local graph context information to design a positive sample selection strategy. By incorporating adaptive sampling and iterative optimization based on clustering results, scACAN effectively enhances the identification of both the major and rare cell types. Comprehensive experiments on multiple real-world scRNA-seq data sets demonstrate that scACAN achieves superior performance and reveals additional biologically meaningful rare cell subpopulations, providing a robust and generalizable solution for single-cell data analysis.
Mastheads
Issue Editorial Masthead

This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More