



This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
The CP2K Program Package Made SimpleClick to copy article linkArticle link copied!
- Marcella IannuzziMarcella IannuzziDepartment of Chemistry, University of Zurich, CH-8057 Zürich, SwitzerlandMore by Marcella Iannuzzi
- Jan WilhelmJan WilhelmRegensburg Center for Ultrafast Nanoscopy (RUN) and Institute of Theoretical Physics, University of Regensburg, D-93053 Regensburg, GermanyMore by Jan Wilhelm
- Frederick SteinFrederick SteinCenter for Advanced Systems Understanding (CASUS), Helmholtz Zentrum Dresden-Rossendorf, D-02826 Görlitz, GermanyMore by Frederick Stein
- Augustin BussyAugustin BussySwiss National Supercomputing Centre (CSCS), ETH Zurich, CH-6900 Lugano, SwitzerlandMore by Augustin Bussy
- Hossam ElgabartyHossam ElgabartyDepartment of Chemistry, Paderborn University, D-33098 Paderborn, GermanyMore by Hossam Elgabarty
- Dorothea GolzeDorothea GolzeDepartment of Chemistry and Food Chemistry, Technische Universität Dresden, D-01069 Dresden, GermanyMore by Dorothea Golze
- Anna-Sophia HehnAnna-Sophia HehnDepartment of Chemistry, Christian-Albrechts-University Kiel, D-24118 Kiel, GermanyMore by Anna-Sophia Hehn
- Maximilian GramlMaximilian GramlRegensburg Center for Ultrafast Nanoscopy (RUN) and Institute of Theoretical Physics, University of Regensburg, D-93053 Regensburg, GermanyMore by Maximilian Graml
- Stepan MarekStepan MarekRegensburg Center for Ultrafast Nanoscopy (RUN) and Institute of Theoretical Physics, University of Regensburg, D-93053 Regensburg, GermanyMore by Stepan Marek
- Beliz Sertcan GökmenBeliz Sertcan GökmenDepartment of Chemistry, University of Zurich, CH-8057 Zürich, SwitzerlandMore by Beliz Sertcan Gökmen
- Christoph SchranChristoph SchranCavendish Laboratory, Department of Physics, University of Cambridge, CB3 0HE Cambridge, U.K.More by Christoph Schran
- Harald ForbertHarald ForbertCenter for Solvation Science ZEMOS, Ruhr-Universität Bochum, D-44801 Bochum, GermanyMore by Harald Forbert
- Rustam Z. KhaliullinRustam Z. KhaliullinDepartment of Chemistry, McGill University, Montreal QC H3A 0B8, CanadaMore by Rustam Z. Khaliullin
- Anton KozhevnikovAnton KozhevnikovSwiss National Supercomputing Centre (CSCS), ETH Zurich, CH-6900 Lugano, SwitzerlandMore by Anton Kozhevnikov
- Mathieu TaillefumierMathieu TaillefumierSwiss National Supercomputing Centre (CSCS), ETH Zurich, CH-6900 Lugano, SwitzerlandMore by Mathieu Taillefumier
- Rocco MeliRocco MeliSwiss National Supercomputing Centre (CSCS), ETH Zurich, CH-6900 Lugano, SwitzerlandMore by Rocco Meli
- Vladimir V. RybkinVladimir V. RybkinHQS Quantum Simulations GmbH, D-76131 Karlsruhe, GermanyMore by Vladimir V. Rybkin
- Martin BrehmMartin BrehmDepartment of Chemistry, Paderborn University, D-33098 Paderborn, GermanyMore by Martin Brehm
- Robert SchadeRobert SchadePaderborn Center for Parallel Computing (PC2), Paderborn University, D-33098 Paderborn, GermanyMore by Robert Schade
- Ole Schütt
- Johann V. PototschnigJohann V. PototschnigCenter for Advanced Systems Understanding (CASUS), Helmholtz Zentrum Dresden-Rossendorf, D-02826 Görlitz, GermanyMore by Johann V. Pototschnig
- Hossein MirhosseiniHossein MirhosseiniCenter for Advanced Systems Understanding (CASUS), Helmholtz Zentrum Dresden-Rossendorf, D-02826 Görlitz, GermanyMore by Hossein Mirhosseini
- Andreas KnüpferAndreas KnüpferCenter for Advanced Systems Understanding (CASUS), Helmholtz Zentrum Dresden-Rossendorf, D-02826 Görlitz, GermanyMore by Andreas Knüpfer
- Dominik MarxDominik MarxLehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, D-44780 Bochum, GermanyMore by Dominik Marx
- Matthias KrackMatthias KrackPSI Center for Scientific Computing, Theory and Data, Paul Scherrer Institute, CH-5232 Villigen PSI, SwitzerlandMore by Matthias Krack
- Jürg HutterJürg HutterDepartment of Chemistry, University of Zurich, CH-8057 Zürich, SwitzerlandMore by Jürg Hutter
- Thomas D. Kühne*Thomas D. Kühne*Email: [email protected]Center for Advanced Systems Understanding (CASUS), Helmholtz Zentrum Dresden-Rossendorf, D-02826 Görlitz, GermanyInstitute of Artificial Intelligence, Technische Universität Dresden, D-01187 Dresden, GermanyCluster of Excellence “Physics of Life”, Technische Universität Dresden, D-01307 Dresden, GermanyMore by Thomas D. Kühne
The Journal of Physical Chemistry B
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
CP2K is a versatile open-source software package for simulations across a wide range of atomistic systems, from isolated molecules in the gas phase to low-dimensional functional materials and interfaces, as well as highly symmetric crystalline solids, disordered amorphous glasses, and weakly interacting soft-matter systems in the liquid state and in solution. This review highlights CP2K’s capabilities for computing both static and dynamical properties using quantum-mechanical and classical simulation methods. In contrast to the accompanying theory and code paper [J. Chem. Phys. 152, 194103 (2020)], the focus here is on the practical usage and applications of CP2K, with underlying theoretical concepts introduced only as needed.
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
1. Introduction
2. Total Energy and Force Methods
2.1. Density Functional Theory
2.1.1. Basis Set Convergence Using the Quickstep Method

2.1.2. Pseudopotentials and Basis Sets

Figure 1
Figure 1. Values of the comparison metric ε for CP2K/Quickstep using the “UZH protocol” with respect to all-electron FP-LAPW calculations using the CP2K/SIRIUS code. For each element up to Rn four monoelemental cubic crystals were considered. (55)
2.1.3. All-Electron Calculations Using the GAPW Method
large-core PP ([86 core] 6 valence electrons)
[Rn] 7s2 5f3 6d1
medium-core PP ([78 core] 14 valence electrons)
[Xe 4f14 5d10] 6s2 6p6 7s2 5f3 6d1
small-core PP ([60 core] 32 valence electrons)
[Kr 4d10 4f14] 5s2 5p6 5d10 6s2 6p6 7s2 5f3 6d1


2.1.4. Wave Function Optimization


2.1.5. Brillouin-Zone Sampling Using k-Points


2.2. Hartree–Fock and Hybrid Density Functional Theory




2.2.1. Auxiliary Density Matrix Method


2.2.2. Resolution-of-the-Identity Hartree–Fock Exchange


2.2.3. Resolution-of-the-Identity Hartree–Fock Exchange with k-Point Brillouin-Zone Sampling
&HF%INTERACTION_POTENTIAL: As for any periodic HF calculation, a short-range potential must be selected. The L/2 requirement now refers to the Born–von Kármán supercell, i.e. a supercell obtained by multiplying the unit cell by the number of k-points in each direction. As for Γ-point HFX, the potential range should be long enough for convergence, while kept as short as possible for performance.
&RI%RI_METRIC: In the special case of RI-HFXk, the range of the RI metric barely impacts the performance. It is hence recommended to always take it to be the same as the interaction potential for maximal accuracy (default option).
&QS%PW_GRID_BLOCKED: It is recommended to set this keyword to FALSE. This ensures that calculations of small unit cells with a lot of CPUs can go ahead without crashing.
&SCF%EPS_DIIS: The default threshold value to start DIIS is 0.1. Experience has shown that better convergence is achieved when this value is lower, typically around 0.05.
&RI%KP_NGROUPS: There is a lot of computational work to be done, and most of it can be efficiently parallelized over MPI subgroups. This is controlled with the KP_NGROUPS keyword, which should be a divisor of the total number of ranks. Using N subgroups should accelerate the calculations by a factor ∼N and increase its memory usage by the same factor.
&RI%KP_STACK_SIZE: Most of the computational effort is spent on contracting tensors. In some cases, more efficient batched contractions can take place, at the cost of greater memory usage. The default value of 32 is meant for small systems of a few atoms. Larger systems will require more memory overall and might not have enough room for the storage of temporary tensors (in which case, use a smaller stack size).
2.3. Post-Hartree–Fock and Double-Hybrid Density Functional Theory
2.3.1. Second-Order Møller–Plesset Perturbation Theory
&WF_CORRELATION%MEMORY: the allowed memory for the MP2 calculation.
&WF_CORRELATION%GROUP_SIZE: it needs to be a divisor of the total number of processes. Lower values reduce communication costs, but increase memory requirements.
&WF_CORRELATION%INTEGRALS: optional section to configure the ERI method via the ERI_METHOD keyword and by the INTERACTION_POTENTIAL subsection the corresponding interaction potential (default: Coulomb potential).


&XC_FUNCTIONAL: set up the respective DFT part. In addition to the natively implemented XC functionals, an even larger selection of functionals is available through the LibXC library. (94)
&HF: set up the respective HF interaction operator. For simple functionals, the adjustment of the FRACTION parameter is sufficient. For more elaborate functionals consider the parameters in the &INTERACTION_POTENTIAL subsection.
&WF_CORRELATION: the keywords SCALE_S and SCALE_T determine the weight of singlet and triplet contributions. The &INTEGRALS%INTERACTION_POTENTIAL subsection allows the adjustment of the operator.
2.3.2. Random Phase Approximation and Laplace-Transformed Scaled Opposite-Spin Second-Order Møller–Plesset Perturbation Theory
 or the RI_SOS_MP2 section
or the RI_SOS_MP2 section replaces the &MP2 or &RI_MP2-sections, respectively. Note that the Minimax quadrature has to be turned on manually for RPA calculations. This difference is related to RPA calculations being the precursor of GW bandstructure calculations, which are described in Section 3.2, requiring quadratures suitable for different kinds of integration kernels.
replaces the &MP2 or &RI_MP2-sections, respectively. Note that the Minimax quadrature has to be turned on manually for RPA calculations. This difference is related to RPA calculations being the precursor of GW bandstructure calculations, which are described in Section 3.2, requiring quadratures suitable for different kinds of integration kernels.
2.3.3. Low-Scaling Post-Hartree–Fock

2.4. Density Functional Theory plus Hubbard U
 and the DFT + U specific printout is controlled via the corresponding &PRINT section within &PLUS_U. The Ueff parameter U_MINUS_J and the angular momentum number L must be defined in an atomic &KIND section. Here is an example that applies an effective Hubbard Ueff of 2 eV to the 5f orbitals (l = 3) of the uranium atoms assigned to the atomic kind Ua:
and the DFT + U specific printout is controlled via the corresponding &PRINT section within &PLUS_U. The Ueff parameter U_MINUS_J and the angular momentum number L must be defined in an atomic &KIND section. Here is an example that applies an effective Hubbard Ueff of 2 eV to the 5f orbitals (l = 3) of the uranium atoms assigned to the atomic kind Ua:
 to create a spin-up on-site 5f2 triplet for a U4+ cation. For the corresponding spin-down 5f2 cation, the definitions in the sections &ALPHA and &BETA for spin-up and spin-down electrons have to be exchanged. The MULTIPLICITY in the &DFT section must be adapted if the number of spin-up and spin-down U atoms is not equal, which is needed for ferri- and ferromagnetic materials. The corresponding initial orbital occupation for the O2– anion in UO2 can be specified with
to create a spin-up on-site 5f2 triplet for a U4+ cation. For the corresponding spin-down 5f2 cation, the definitions in the sections &ALPHA and &BETA for spin-up and spin-down electrons have to be exchanged. The MULTIPLICITY in the &DFT section must be adapted if the number of spin-up and spin-down U atoms is not equal, which is needed for ferri- and ferromagnetic materials. The corresponding initial orbital occupation for the O2– anion in UO2 can be specified with
3. Electronic Band Structure
3.1. SIRIUS
APW (117) and LAPW (20) basis sets with an arbitrary number of local orbitals composed of up to third-order energy derivatives of radial functions
evaluation of stress tensor and nuclear forces
collinear and noncollinear magnetism
symmetrization of lattice-periodic functions and on-site matrices
generation of irreducible k-point meshes
local-density approximation (LDA) (118) and GGA flavours of XC potentials via the LibXC library (94)


ELECTRONIC_STRUCTURE_METHOD: type of calculation, i.e. FULL_POTENTIAL_LAPWLO, or PSEUDOPOTENTIAL.
CORE_RELATIVITY: specifies if the core states in FP-LAPW are treated relativistically via the DIRAC equation or not.
VALENCE_RELATIVITY: type of relativistic treatment of valence FP-LAPW radial basis functions and local orbitals. Possible values are NONE, KOELLING_HARMON, ZORA, or IORA.
NUM_BANDS: number of ”bands” (KS states) to compute during Hamiltonian diagonalization.
SMEARING: type of smearing used to compute the Fermi level, such as GAUSSIAN, COLD, FERMI_DIRAC, GAUSSIAN_SPLINE and METHFESEL_PAXTON.
SMEARING_WIDTH: Width (in Ha) of the smearing function.
PW_CUTOFF: PW cutoff (in a.u.–1) for expansion of electron density and potential.
GK_CUTOFF: PW cutoff (in a.u.–1) for |G + k|.
NUM_MAG_DIMS: number of magnetic dimensions in the system, i.e. 0─nonmagnetic calculations, 1─spin-collinear case, 3─noncollinear magnetic calculation.
LMAX_APW: maximum orbital quantum number for LAPW basis functions.
LMAX_RHO: maximum orbital quantum number for charge density expansion in muffin-tins (LAPW only).
LMAX_POT: maximum orbital quantum number for KS potential expansion in muffin-tins (LAPW only).
NGRIDK: dimensions of the k-point mesh. In other words, this is a division of the first Brillouin zone into microcells for k-point integration.
ENERGY_TOL: SCF convergence tolerance of total energy.
DENSITY_TOL: SCF convergence tolerance of charge density.
GAMMA_POINT: Specifies if this is a Γ-point calculation (PP case only). NGRIDK must be set to 1 1 1 in that case.
TYPE: denotes the type of the density mixer, i.e. LINEAR, ANDERSON, ANDERSON_STABLE, or BROYDEN2.
BETA: determines mixing parameter, which is a real value in the interval [0, 1].
USE_HARTREE: logical variable that controls what is used as estimation of charge density difference. TRUE: Hartree energy of density residual is used as a measure of density convergence (works only for PP calculation). FALSE: normalized inner product of density residuals is used as a measure of density convergence.
MPI_GRID_DIMS: A 2-dimensional array that defines how the band parallelization is performed. The first dimension of the grid specifies the size of the FFT communicator used in the transformation of individual WFs. The second dimension defines the number of independent FFT communicator groups used to parallelize FFTs across different WFs. The product of the two dimensions defines the total number of MPI ranks used for the band parallelization. An orthogonal communicator will be built from the total available number of MPI ranks to perform k-point parallelization. Additionally, if the MPI grid is square, for example {3, 3}, a parallel eigensolver will be used to diagonalize the subspace Hamiltonian matrix.
3.2. The GW Method
3.2.1. The Band Gap Problem of Density Functional Theory
3.2.2. Theory of GW Band Structure Calculations
3.2.3. GW for Molecules

QUADRATURE_POINTS: number of imaginary-frequency points for computing the self-energy, see eq 21 in ref (104). Usually, 100 points converge the quasiparticle energies within 10 meV.
SELF_CONSISTENCY: determines which GW self-consistency variant (G0W0, evGW0 or evGW) is used to calculate the GW quasiparticle energies.
XC_FUNCTIONAL: starting XC functional for the G0W0, evGW0 or evGW calculation, respectively. We recommend to use evGW0@PBE, as discussed in ref (125). For further guidance on selecting an appropriate DFT starting functional and self-consistency scheme for your system, you may consult ref (119).
BASIS_SET: the basis set is of Gaussian type and can have a strong influence on the quasiparticle energies. For computing quasiparticle energies, a basis set extrapolation is necessary, (104) and we recommend all-electron GAPW calculations with correlation-consistent basis sets cc-pVDZ, cc-pVTZ, cc-pVQZ from the EMSL Basis Set Exchange. (44) For computing the HOMO – LUMO gap from GW, we recommend the usage of augmented basis sets, for example aug-cc-pVDZ and aug-cc-pVTZ. As RI_AUX basis set, we recommend the RIFIT basis sets from the EMSL database, for example aug-cc-pVDZ-RIFIT.
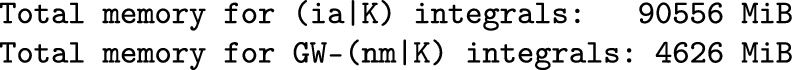
3.2.4. GW for Small Unit Cells with k-Point Sampling


NUM_TIME_FREQ_POINTS: number of imaginary-time and imaginary-frequency points used for computing the self-energy. Between 20 and 30 points are usually enough for converging quasiparticle energies within 10 meV. Grids up to 34 points are available.
MEMORY_PER_PROC: specifies the available memory per MPI process. A larger MEMORY_PER_PROC can increase performance.
EPS_FILTER: filter for three-center integrals, 10–11 should be well-converged.
REGULARIZATION_RI: regularization parameter for RI basis set. For a big RI basis set (>50 RI function per atom), we recommend 10–2 to prevent linear dependencies. For a small RI basis set, one can turn RI regularization off by setting 0.0.
CUTOFF_RADIUS_RI: cutoff radius of truncated Coulomb metric in Å. A larger cutoff leads to faster RI basis set convergence, but also the computational cost increases. A cutoff of 7 Å is an accurate choice.
&SOC: activates spin–orbit coupling (SOC) from GTH PPs. (32) But, the usage of SOC also needs POTENTIAL_FILE_NAME GTH_SOC_POTENTIALS.
&BANDSTRUCTURE_PATH: specify the k-path in the Brillouin zone for computing the band structure. Relative k-coordinates are needed, which you can retrieve for your crystal structure from ref (129).
3.2.5. GW for Large Cells in Γ-Only Approach
4. Embedding Methods
4.1. Implicit Solvation Methods

 while keeping the periodicity defined in the &CELL section identical, i.e.
while keeping the periodicity defined in the &CELL section identical, i.e.
 allowing for printing the polarization potential and the dielectric function in cube file format.
allowing for printing the polarization potential and the dielectric function in cube file format.4.2. Quantum Mechanics/Molecular Mechanics Methods

ECOUPL: defines the type of electrostatic treatment. On the one hand GAUSS enables the electrostatic embedding for KS-DFT/MM simulations based on GEEP (see next section). On the other hand COULOMB enables electrostatic embedding for SQC methods, or SCC-DFTB/xTB. Mechanical embedding schemes can be used by setting the keyword to NONE.
CELL: defines the cell for the QM calculation, which must be orthorhombic.
QM_KIND: defines the QM system and the corresponding indices are given for each atomic type separately.
PERIODIC: applies the periodic potential. The MULTIPOLE section turns on the coupling/recoupling of the QM periodic images, as described later. In the previous example, it was turned off because the QM box and the MM box are identical in size.
4.2.1. Electrostatic Embedding by the Gaussian Expansion of the Electrostatic Potential Method


4.2.2. Image-Charge Augmented Quantum Mechanics/Molecular Mechanics Method

MM_ATOM_LIST: defines the list of MM atoms that carry the Gaussian charge ga. These should be the atoms of the metallic slab.
EXT_POTENTIAL: sets the external potential V0.
4.2.3. Partial Atomic Charges from Restrained Electrostatic Potential Fitting

SPHERE_SAMPLING: the real-space points rk are sampled in spherical shells around each atom. The shells are defined by a minimal and maximal radius, which can be set by the RMIN and RMAX keywords in this section. This option should be used for molecules, molecular liquids, or porous periodic systems like metal–organic frameworks.
SLAB_SAMPLING: the option should be used for slab-like systems, where it is important to reproduce the potential well above the surface, e.g. to study adsorption processes. The rk grid is then sampled as a thin slice above the surface. In that case, keywords defining the surface atoms, the direction, and the thickness of the slice need to be set.

 to the &RESP section. This is to say that the atoms with indices 1, 2, and 3 carry the same charge. More details on how to set constraints can be found in the online tutorial. (162)
to the &RESP section. This is to say that the atoms with indices 1, 2, and 3 carry the same charge. More details on how to set constraints can be found in the online tutorial. (162)4.3. Density Functional Embedding Theory

4.3.1. General Procedure
4.3.2. Implementation



 in the &QS section. In this force evaluation, the options for optimizing the embedding potential are to be specified. This is done by inserting a &OPT_EMBED section within the &QS section:
in the &QS section. In this force evaluation, the options for optimizing the embedding potential are to be specified. This is done by inserting a &OPT_EMBED section within the &QS section:


5. Nuclear Magnetic and Electron Paramagnetic Resonance Spectroscopy
5.1. Density Functional Perturbation Theory

&CURRENT: the induced current density in response to an external homogeneous magnetic field. (168)
&LOCALIZE: computes localized orbitals, which are required if the perturbation includes the position operator.
&NMR: the nuclear magnetic shielding tensors, or more generally, the nucleus-independent chemical shifts (NICS). (171)
&EPR: the electronic g-tensor. (172)
&POLAR: computes the polarizability. (173)
&VCD: enables the calculation of vibrational circular dichroism (VCD). (174)
&DCDR: analytical gradients of the dipole moments, e.g. for Born effective charges and atomic polar tensors.
5.2. The Magnetic Shielding Tensor

 one can further choose to perform the response calculation using only the Wannier functions that are within a certain distance from some chosen list of atoms. If only the magnetic shieldings of some particular atoms are of interest, this can lead to enormous reductions in computational time. Naturally, one needs to ascertain that the selected radius provides acceptable results for the atoms of interest, which depends on the respective chemical environments.
one can further choose to perform the response calculation using only the Wannier functions that are within a certain distance from some chosen list of atoms. If only the magnetic shieldings of some particular atoms are of interest, this can lead to enormous reductions in computational time. Naturally, one needs to ascertain that the selected radius provides acceptable results for the atoms of interest, which depends on the respective chemical environments.

5.3. The Electron Paramagnetic Resonance g-Tensor

5.4. Hyperfine Couplings

6. Optical Spectroscopy
6.1. Linear-Response Time-Dependent Density Functional Theory



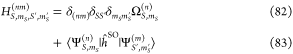

6.2. Bethe–Salpeter Equation
6.2.1. Electronic Excitation Energies
6.2.2. Optical Absorption Spectrum
6.2.3. Measures for the Size of an Excited State

SELF_CONSISTENCY: determines which GW self-consistency (G0W0, evGW0 or evGW) is used to calculate the single-particle GW energies εpGW necessary in the BSE calculation of eq 84. We recommend using evGW0 for BSE runs. (125)
TDA: specifies if the TDA and/or diagonalization of the full ABBA-matrix is employed. OFF: Generalized diagonalization of ABBA of eq 78. ON: use the TDA of eq 85 and diagonalize only A. TDA+ABBA: compute excitation energies Ω(n) and ΩTDA(n) from eqs 78 and 85, respectively.
SPIN_CONFIG: choose between SINGLET for computing singlet excitation energies (αS = 2) and TRIPLET for computing triplet excitation energies (αT = 0). The standard is SINGLET as an electronic excitation directly after photoexcitation is a singlet due to angular momentum conservation; triplet excited states can form by intersystem crossings.
NUM_PRINT_EXC: number of excitation energies Ω(n) to be printed.
ENERGY_CUTOFF_OCC (in eV): only use indices i of occupied MOs in the interval εiDFT ∈ to set up the matrices A and B in eq 84. A small ENERGY_CUTOFF_OCC reduces computation time and memory consumption, but can affect the computed excitation energies Ω(n). Usage of ENERGY_CUTOFF_OCC is recommended for molecules with more than 30 atoms. We recommend a convergence test by increasing ENERGY_CUTOFF_OCC and observing the effect on Ω(n). (202)
ENERGY_CUTOFF_EMPTY (in eV): analogously to ENERGY_CUTOFF_OCC, but for the empty states, i.e. only empty states in the interval εiDFT ∈ are used in the BSE calculation.
NUM_PRINT_EXC_DESCR: number of excitations n, for which the exciton descriptors are printed, e.g. dexc(n) of eq 93.
BSE_SPECTRUM: Activates the computation and printing of the photoabsorption cross section tensor, i.e. its spatial average σ̅(ω) of eq 89 and its elements σμ,μ′(ω) (μ, μ′ ∈ {x, y, z}) of eq 87 are printed.
XC_FUNCTIONAL: choose between one of the available XC functionals. The starting point can have a profound influence on the excitation energies. (208) Motivated by the discussion in ref (125), we strongly recommend to use BSE@evGW0@PBE, i.e. the PBE functional (35) as the DFT starting point.
BASIS_SET: the all-electron aug-cc-pVDZ basis set (44,209) should be sufficient for most organic molecules, but needs to be checked with respect to convergence.
7. Excited State Dynamics
7.1. Real-Time Propagation and Ehrenfest Dynamics
7.2. Real-Time Bethe−Salpeter Propagation

set RUN_TYPE to RT_PROPAGATION
include a &MD section to set the size of the TIMESTEP and the total number of STEPS
specify all options necessary to run an accurate G0W0 calculation for molecules (in particular the subsection &PROPERTIES%BANDSTRUCTURE%GW should be specified)
include the &REAL_TIME_PROPAGATION section with all relevant entries and add subsection &RTBSE.

EPS_ITER: the equation of motion of eq 109 is solved by exponential evolution operators applied to the single-particle density matrix ρ̂. These are applied self-consistently by an enforced time reversal symmetry scheme described previously, (212,214,224) so that

MAX_ITER: limits the maximum number of the self-consistent iterations in the ETRS scheme.
MAT_EXP: the matrix exponentiation in RT-BSE is implemented by two approaches─the (Baker–Campbell–Hausdorff) BCH (212) and the EXACT diagonalization. The keyword MAT_EXP controls the choice between these two.
EXP_ACCURACY: for the BCH method, it is also necessary to specify the matrix exponentiation accuracy, which should be stricter than the ETRS threshold EPS_ITER.
APPLY_DELTA_PULSE: in order to observe any oscillations, the external field Û(t) needs to be specified. Either the explicit time-dependent field can be supplied in the &DFT%EFIELD subsection, or a delta pulse can be triggered by inclusion of this keyword. (225)
DELTA_PULSE_DIRECTION: sets the Cartesian direction of the delta pulse. The resulting vector is always normalized by the code.
DELTA_PULSE_SCALE: sets the magnitude of the delta pulse directly (in atomic units). The change in the metric of the single-particle density matrix after the application of the delta pulse is reported by the code - if the change is larger than 1 in atomic units, then the ETRS loop might struggle to converge─we recommend decreasing the DELTA_PULSE_SCALE in such cases.
&MOMENTS: activates printing of the time-dependent dipole moment
where μj is the jth component of the dipole moment μ(t) and rCC is the reference point─in RT-BSE, this is the center of atomic charges.(112)FT_DAMPING: Damping γ applied during Fourier transform to stabilize it. (226) Example is given in the &MOMENTS_FT section. If not given explicitly, γ is determined so that factor of e–4 is applied at the end of the time trace of the observables, starting from FT_START_TIME.
FT_START_TIME: offset along the time axis for the purposes of Fourier transform. Defaults to zero. Useful for real-time pulses defined in &EFIELD section. For example, for GAUSSIAN_ENV with nonzero T0 parameter, setting FT_START_TIME to the same value will result in the envelope being a real function in frequency space.
&MOMENTS_FT: activates printing of the Fourier transform of the dipole moment μ(t), which is stabilized by the application of FT_DAMPING γ, so that
where t0 is the defined by the START_TIME.(113)&FIELD: activates printing of the time-dependent trace of the external field. When no &EFIELD subsection within the &DFT section is present, outputs just zeros (for example when only the delta pulse is applied.)
&POLARIZABILITY: activates printing of the polarizability tensor, defined in terms of the Fourier transform of the dipole moment and the Fourier transform of the electric field as (227,228)
(114)
&PADE_FT: section controlling the Padé interpolation (229−231) of the Fourier transforms. A new frequency grid spanning from E_MIN to E_MAX with E_STEP steps is created, on which the Fourier transforms are reevaluated using the Padé fitting of the original transforms. The original transforms are restricted to energies within FIT_E_MIN and FIT_E_MAX. By default, no restriction is applied─entire frequency range of the original transform is used to construct the Padé parameters.
7.3. Surface Hopping via NEWTON-X


8. X-ray Spectroscopy
8.1. Transition Potential Method

8.2. Linear-Response Time-Dependent Density Functional Theory


 would define grids with 100 angular points (Lebedev scheme) and 250 radial grid points for excited oxygen atoms. Note that the GAPW default is a 50 × 50 grid.
would define grids with 100 angular points (Lebedev scheme) and 250 radial grid points for excited oxygen atoms. Note that the GAPW default is a 50 × 50 grid. would turn on SOC for LR-TDDFT-based XAS calculations on top of a restricted closed-shell ground state.
would turn on SOC for LR-TDDFT-based XAS calculations on top of a restricted closed-shell ground state.8.3. Real-Time Time-Dependent Density Functional Theory


9. Energy Decomposition Analysis
9.1. Implementation
9.1.1. Step 1. Define Fragments

9.1.2. Step 2. Assign Electrons to Fragments

9.1.3. Step 3. Turn ALMO SCF On
9.1.4. Step 4. Compute ΔEFRZ
| state | CP2K print out | definition | EDA terms |
|---|---|---|---|
| (0) | “single-molecule energy” | ∑xE(Rx) in eq (132) | ΔEFRZ = (1) – (0) |
| (1) | “energy of the initial guess” | E(RFRZ) in eq (132) | ΔEPOL = (2) – (1) |
| (2) | “ENERGY OF BLOCK-DIAGONAL ALMOs” | E(RALMO) in eq (133) | ΔECT(pair) = (3) – (2) |
| (3) | “CORRECTED ENERGY” | eq (135) | ΔEHO = (4) – (3) |
| (4)a | “ENERGY|” | E(RSCF) in eq (134) | ΔECT = (4) – (2) |
If FULL_X or XALMO_X methods are used then the “ENERGY|” line contains the energy of state (3).
9.1.5. Step 5. Compute ΔEPOL
9.1.6. Step 6. Compute ΔECT
9.1.7. Input File Example

9.1.8. Illustrative Applications
10. Finite Temperature Effects
10.1. Neural Network and Machine Learning Interaction Potentials




Figure 2
Figure 2. Comparison of NNP and AIMD results for liquid water at 300 K as obtained from CP2K. (a) Time per MD step for 64, 512, and 4096 water molecules with the C-NNP model (solid lines with circles) and 64 water molecules with the revPBE0-D3 hybrid functional (dashed line with crosses) reported on a logarithmic scale. (b) Radial distribution functions and (c) vibrational density of states of the hydrogen atoms. Data in panel (b,c) are partially reused from ref (322). Copyright 2020, American Institute of Physics.
10.2. Nuclear Quantum Effects


10.3. Quantum Convergence

Figure 3
Figure 3. Impact of NQEs on the structure and dynamics of hexagonal ice at 250 K. (a) Bead convergence of the virial kinetic energy estimator using the PILE and PIQTB thermostat. (b) Radial distribution functions with classical and quantum nuclei and (b) vibrational density of states obtained with classical MD, RPMD and TRPMD, respectively. All simulations rely on using the C-NNP trained with revPBE0-D3 reference data, (322) as available in the CP2K data repository (see also Section 10.1).
10.4. Approximate Quantum Dynamics

10.5. Bosonic Quantum Solvation at Ultralow Temperatures



Figure 4
Figure 4. Temperature dependence of (a) the heat capacity and (b) the superfluid fraction of bulk 4He obtained using the canonical worm algorithm to sample bosonic exchange and the winding number estimator to analyze superfluidity (see text). The numerical pair density matrix is the one provided with the CP2K package and has been computed at 80 K using the He···He potential from ref (389), thus P × T = 80 K is kept constant for the temperature scan. The present bosonic PIMC data were obtained from only 32 atoms (in a truncated octahedral supercell according to the sample input provided in the text) and are compared to published results of ref (386) generated from 64, 128, and 512 atoms in the corresponding periodic truncated octahedron using CP2K and the same methodology. For reference, the respective experimental data (391,392) are shown by gray lines; note that deviations of the computed properties from experiment are mainly due to finite-size effects close to the superfluid phase transition given such small system sizes. (390)
Figure 5
Figure 5. For an illustration of the HPIMD/MC method, as implemented in CP2K. (386) The fully flexible and reactive solute species are propagated using PIMD techniques (such as PIQTB in conjunction with the one-body density matrix), while the bosonic solvent (for instance many 4He atoms either forming a finite cluster as illustrated here, or hosted within a periodic supercell) is sampled using bosonic PIMC (for instance based on the canonical worm algorithm in conjunction with the numerical pair density matrix) at a common temperature T (see text). The interactions can be described using high-dimensional NNPs with CCSD(T) accuracy, as described in Section 10.1.
Figure 6
Figure 6. Potential energy Umol along one Trotter replica of quantum PIMD trajectories of (from top to bottom) CH4, CH5+, H3O+, H5O2+, respectively, all at 1.67 K using NNPs trained with CCSD(T*)-F12a/aug-cc-pVTZ reference energies obtained from Molpro. The corresponding CC single-point data were obtained by recomputing the energies at each and every step of the depicted NNP trajectories and are shown as red dotted lines. All energies are reported relative to the equilibrium structures of the respective global minima. Reproduced from Figure 6 of ref (386). Copyright 2020, American Institute of Physics.





Figure 7
Figure 7. Spatial distributions functions stemming from all N helium atoms around a fixed molecular impurity (i.e., X·HeN), as sampled from PIMC simulations (without bosonic exchange) at 1.67 K. The solute···helium interaction energies Uint used in the PIMC sampling of the helium atoms are interpolated from values that have been precomputed on a grid using either the NNP (left column) or single-point CC calculations (right column) on the identical grid both based on counterpoise-corrected CCSD(T*)-F12a/aug-cc-pVTZ energy differences obtained from Molpro. Reproduced from Figure 7 of ref (386). Copyright 2020, American Institute of Physics.
Figure 8
Figure 8. Illustration of PI striding for bosonic HPIMD/MC simulations, as implemented in CP2K: coupling of the PI representation of one helium atom (left) discretized with three Trotter replica (in practice sampled using PIMC based on the pair density matrix representation) to the PI representation of the solute molecule (right) discretized with nine beads (sampled using PIMD based on the one-body density matrix in conjunction with PIQTB thermostatting, as detailed in Section 10.3), thus corresponding to a striding length of three, Pstride = 3. Reproduced from Figure 3 of ref (386). Copyright 2020, American Institute of Physics.
Figure 9
Figure 9. Distance distributions functions of H3O+·4He8 at 1 K with BE sampling of the helium microsolvation environment. The intramolecular O–H, as well as the intermolecular O–He and H–He distance distributions are presented. The inset depicts the beads of the hydronium ion in terms of balls (red/gray for O/H), while all 4He atoms involved in a bosonic exchange cycle are visualized by green strings and all others by green balls.
10.6. Vibrational Spectroscopy
10.6.1. Static-Harmonic Approach

Due to the harmonic approximation of the potential energy surface, all anharmonic effects are neglected. (419) If the system possesses features such as strong hydrogen bonds or hindered rotations, the harmonic approximation of certain modes will be poor, and so will the quality of the predicted spectrum.
The spectrum can only be computed for one minimum energy structure at a time. If there exist several conformers of the same molecule, they need to be considered separately. If the system can hardly be described by minimum-energy structures, such as bulk phase liquids, it will be hard to obtain reasonable spectra from the outset.
The method works best for molecules or small clusters in vacuum. Solvent effects on the spectrum can be crudely approximated either via continuum solvation models such as COSMO, (420) PCM, (421) or the SCCS implicit solvation method described in Section 4.1, (135−137) as well as by microsolvation, but the solvent effect cannot be captured comprehensively.
The approach only yields a discrete line spectrum; no line widths or band shapes can be obtained. Hence, to predict realistic spectra, empirical line broadening needs to be applied.
10.6.2. Time-Correlation Function Approach
Condensed phase systems can be handled; it is possible to explicitly capture the effects of solvent and entropy on the spectrum.
Some anharmonic effects, such as line broadening, approximate overtones, and combination bands, are reproduced.
Realistic band shapes are obtained instead of a discrete line spectrum.
Intrinsic conformer sampling takes place during the MD simulation.
No minimum energy structure is required to compute the spectrum.
10.6.3. Computing Electromagnetic Moments

10.6.4. Total vs Molecular Moments
10.6.5. Orbital Localization

METHOD: how to minimize the spread functional. Default is JACOBI (slow, but very robust), but should be set to CRAZY (a lot faster, but less robust) in most cases. See next keyword.
JACOBI_FALLBACK: do a JACOBI minimization (“fallback”), if CRAZY did not converge.
MAX_ITER: maximum number of iterations for minimizing the spread functional. Default is 10 000, which is almost always enough. Maybe reduce it to not waste time if it is not going to converge anyway.
&WANNIER_CENTERS: outputs the “Wannier centers”, i.e. the centroids of the MLWFs to a XYZ file with the specified name. Also works in AIMD runs, where it just writes a consecutive Wannier center trajectory. If the IONS+CENTERS keyword is set, the atom coordinates are added to that file, which can make analysis easier.
&WANNIER_SPREADS: outputs the remaining spread of the Wannier orbitals. This can be useful to, e.g. approximate polarizabilities, (450−452) as shown below.
&WANNIER_CUBES: outputs the actual Wannier orbitals on a Cartesian grid as cube files. Useful for visualization.
&WANNIER_STATES: outputs the Wannier orbitals in the basis of the AOs, i.e. the corresponding coefficient matrix.
10.6.6. Voronoi Integration
the CPU time required for localization can be substantial and scales unfavorably with system size. For a 1000 atom system, the localization typically takes almost as long as the energy and force calculation, i.e. slowing down the simulation by approximately a factor of 2.
All known methods for computing the Wannier localization are iterative and are not guaranteed to converge. It happens in practice that for certain AIMD timesteps, neither the crazy angle algorithm, nor the Jacobi fallback converge, so that the electric moments are missing for these timesteps.
Only molecular electric dipoles can be derived from the Wannier centers; higher-order momenta such as quadrupoles, as for instance required for Raman optical activity (ROA) spectra, are not directly accessible.
Wannier localization enforces integer molecular charges. Any charge transfer effects between molecules cannot be captured, which leads to artificially increased dipole moments in some cases.
Systems with delocalized electrons, such as aromatic molecules or metals, may entail convergence issues. Also, for example, the simulated IR spectrum of liquid benzene contains artificial bands that are a consequence from the Wannier localization. (453)

VORONOI_RADII: chooses the radii for the radical Voronoi tessellation. Can be set to UNITY (all radii are 1, i.e. classical Voronoi), VdW (vdW radii are used), COVALENT (covalent radii are employed), (466) or USER. Default is VdW.
USER_RADII: in case of VORONOI_RADII USER, specifies the radii (one number per atom in the system).
OUTPUT_TEXT: outputs a .voronoi text file with all properties in a human-readable format. The file name can be set via FILENAME. On by default.
OUTPUT_EMP: outputs a binary .emp file with all electromagnetic properties. Can be read by TRAVIS (418,467) to compute vibrational spectra. Off by default.
JITTER: randomly displaces all Voronoi sites a tiny bit to avoid problems with highly symmetric structures. The amount can be controlled by the keyword JITTER_AMPLITUDE. On by default.
10.6.7. Polarizabilities


10.6.8. Frequency-Dependent Polarizabilities

10.6.9. Supported Types of Spectra
10.6.9.1. Storing Compressed Density Cubes

10.6.9.2. Normal Mode Analysis from Ab Initio Molecular Dynamics
11. Technical Aspects
11.1. Installation
11.1.1. Prerequisites and Dependencies
| library | req ? | GPU | purposes |
|---|---|---|---|
| BLAS/LAPACK | req | general | |
| DBCSR (90) | req | C,H,O | sparse matrix |
| DBM | int. | C, H | sparse matrix |
| grid | int. | C, H | integration |
| MPI | par. | general | |
| ScaLAPACK | par. | general | |
| FFTW3 (499) | opt. | FFT | |
| FPGA (500−503) | opt. | O | PW-FFT |
| COSMA (103) | opt. | C, H | matrix multipl. |
| SPLA (91) | opt. | C, H | matrix multipl. |
| LIBXSMM (504) | opt. | DBCSR, DBM | |
| LIBINT (105) | opt. | HF exchange | |
| LIBXC (94) | opt. | XC functionals | |
| ELPA (505) | opt. | C | diagonalization |
| cuSOLVERMp (506) | opt. | C | diagonalization |
| DLA-Future (507) | opt. | C, H | diagonalization |
| SIRIUS (508) | opt. | C, H | separate PW code |
| libvori (464) | opt. | Voronoi integration | |
| DFT-D4 (509) | opt. | dispersion corr. | |
| tblite (510) | opt. | GFN2-xTB | |
| PW | int. | C, H | solvation models |
| libgrpp (511) | int. | ECPs | |
| PLUMED (512) | opt. | sampling methods | |
| spglib (513) | opt. | symmetry detection | |
| LibTorch | opt. | ML library | |
| DeePMD-kit (319) | opt. | ML potentials | |
| ACE (318) | opt. | ML potentials | |
| NequIP (320) | opt. | ML potentials | |
| Allegro (321) | opt. | ML potentials | |
| Smeagol (514) | opt. | NEGF transport | |
| TREXIO (515) | opt. | IO formats | |
| GreenX (516) | opt. | Green’s functions |
In order to use GPUs, either CUDA (C), HIP (H), or OpenCL (O) can be employed.
11.1.2. Building CP2K
11.1.3. Sanity Checks after Installation
11.2. Performance Aspects


Data Availability
The data underlying this study are openly available at https://github.com/cp2k/cp2k-examples
Author Information
- Thomas D. Kühne - Center for Advanced Systems Understanding (CASUS), Helmholtz Zentrum Dresden-Rossendorf, D-02826 Görlitz, Germany; Institute of Artificial Intelligence, Technische Universität Dresden, D-01187 Dresden, Germany; Cluster of Excellence “Physics of Life”, Technische Universität Dresden, D-01307 Dresden, Germany;
 https://orcid.org/0000-0001-5471-2407;
https://orcid.org/0000-0001-5471-2407;
- Marcella Iannuzzi - Department of Chemistry, University of Zurich, CH-8057 Zürich, Switzerland;https://orcid.org/0000-0001-9717-2527
- Jan Wilhelm - Regensburg Center for Ultrafast Nanoscopy (RUN) and Institute of Theoretical Physics, University of Regensburg, D-93053 Regensburg, Germany;https://orcid.org/0000-0001-8678-8246
- Frederick Stein - Center for Advanced Systems Understanding (CASUS), Helmholtz Zentrum Dresden-Rossendorf, D-02826 Görlitz, Germany;https://orcid.org/0000-0003-3689-4926
- Dorothea Golze - Department of Chemistry and Food Chemistry, Technische Universität Dresden, D-01069 Dresden, Germany;https://orcid.org/0000-0002-2196-9350
- Anna-Sophia Hehn - Department of Chemistry, Christian-Albrechts-University Kiel, D-24118 Kiel, Germany;https://orcid.org/0000-0002-0498-740X
- Maximilian Graml - Regensburg Center for Ultrafast Nanoscopy (RUN) and Institute of Theoretical Physics, University of Regensburg, D-93053 Regensburg, Germany;https://orcid.org/0000-0002-4279-8511
- Stepan Marek - Regensburg Center for Ultrafast Nanoscopy (RUN) and Institute of Theoretical Physics, University of Regensburg, D-93053 Regensburg, Germany;https://orcid.org/0000-0002-8806-1670
- Christoph Schran - Cavendish Laboratory, Department of Physics, University of Cambridge, CB3 0HE Cambridge, U.K.;https://orcid.org/0000-0003-4595-5073
- Rustam Z. Khaliullin - Department of Chemistry, McGill University, Montreal QC H3A 0B8, Canada;https://orcid.org/0000-0002-9073-6753
- Rocco Meli - Swiss National Supercomputing Centre (CSCS), ETH Zurich, CH-6900 Lugano, Switzerland;https://orcid.org/0000-0002-2845-3410
- Vladimir V. Rybkin - HQS Quantum Simulations GmbH, D-76131 Karlsruhe, Germany;https://orcid.org/0000-0001-5136-6035
- Johann V. Pototschnig - Center for Advanced Systems Understanding (CASUS), Helmholtz Zentrum Dresden-Rossendorf, D-02826 Görlitz, Germany;https://orcid.org/0000-0002-9982-0556
- Dominik Marx - Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, D-44780 Bochum, Germany;https://orcid.org/0009-0005-0865-1419
- Matthias Krack - PSI Center for Scientific Computing, Theory and Data, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland;https://orcid.org/0000-0002-2082-7027
Acknowledgments
J.W. acknowledges the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for funding via the Emmy Noether Programme (project number 503985532), CRC1277 (project number 314695032) and RTG 2905 (project number 502572516). D.G. acknowledges funding by the Emmy Noether Programme of the DFG (project number 453275048). R.Z.K. gratefully acknowledges funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) through the Discovery Grant program (RGPIN-2025-06117), and computing resources provided by the Digital Research Alliance of Canada. H.M. and T.D.K. would like to thank the European Union’s Just Transition Fund (JTF), administered by the Sächsische Aufbaubank (SAB), under the InfraProNet Research 2021–2027 programme. A.B., A.K., R.M., and M.T. were supported by the Swiss Platform for Advanced Scientific Computing (PASC), and the Swiss National Center of Competence in Research (NCCR) MARVEL. Part of the research was funded by the DFG (project number 519869949) and via the CRC1415 (project number 417590517). D.M. acknowledges funding by DFG under Germany’s Excellence Strategy -- EXC2033 -- 390677874 -- RESOLV. The authors gratefully acknowledge the computing time provided to them on the high-performance computers at the NHR Center PC2. These are funded by the Federal Ministry of Education and Research and the state governments participating on the basis of the resolutions of the GWK for national high-performance computing at universities (www.nhr-verein.de/unsere-partner).
Additional Notes
a https://www.cp2k.org/dev:compiler_support.
b https://manual.cp2k.org/trunk/getting-started/distributions.html.
c https://github.com/cp2k/cp2k-containers.
d Docker hub https://hub.docker.com/r/cp2k/cp2k.
e NVIDIA NGC catalog https://catalog.ngc.nvidia.com/orgs/hpc/containers/cp2k.
f https://manual.cp2k.org/trunk/getting-started/build-from-source.html.
g https://manual.cp2k.org/trunk/getting-started/build-with-spack.html.
h CP2K issue tracker: https://github.com/cp2k/cp2k/issues.
i CP2K Google group: https://groups.google.com/g/cp2k.
j CP2K GitHub discussions: https://github.com/cp2k/cp2k/discussions.
References
This article references 549 other publications.
- 1Dirac, P. A. M. Quantum mechanics of many-electron systems. Math. Proc. Cambridge Philos. Soc. 1929, 123, 714– 733, DOI: 10.1098/rspa.1929.0094Google ScholarThere is no corresponding record for this reference.
- 2Billeter, S. R.; Curioni, A.; Andreoni, W. Efficient linear scaling geometry optimization and transition-state search for direct wavefunction optimization schemes in density functional theory using a plane-wave basis. Comput. Mater. Sci. 2003, 27, 437– 445, DOI: 10.1016/S0927-0256(03)00043-0Google ScholarThere is no corresponding record for this reference.
- 3Schlegel, H. B. Geometry Optimization. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 790– 809, DOI: 10.1002/wcms.34Google ScholarThere is no corresponding record for this reference.
- 4Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087– 1092, DOI: 10.1063/1.1699114Google ScholarThere is no corresponding record for this reference.
- 5Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12562– 12566, DOI: 10.1073/pnas.202427399Google ScholarThere is no corresponding record for this reference.
- 6Rahman, A. Correlations in the Motion of Atoms in Liquid Argon. Phys. Rev. 1964, 136, A405– A411, DOI: 10.1103/PhysRev.136.A405Google ScholarThere is no corresponding record for this reference.
- 7Tuckerman, M. E. Statistical Mechanics: Theory and Molecular Simulation; Oxford University Press, 2010.Google ScholarThere is no corresponding record for this reference.
- 8Frenkel, D.; Smit, B. Understanding Molecular Simulation From Algorithms to Applications; Academic Press, 2023.Google ScholarThere is no corresponding record for this reference.
- 9Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press, 2009.Google ScholarThere is no corresponding record for this reference.
- 10Hutter, J. Car-Parrinello molecular dynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 604– 612, DOI: 10.1002/wcms.90Google ScholarThere is no corresponding record for this reference.
- 11Kühne, T. D.; Krack, M.; Mohamed, F. R.; Parrinello, M. Efficient and accurate Car-Parrinello-like approach to Born-Oppenheimer molecular dynamics. Phys. Rev. Lett. 2007, 98, 066401, DOI: 10.1103/PhysRevLett.98.066401Google ScholarThere is no corresponding record for this reference.
- 12Kühne, T. D.; Prodan, E. Disordered crystals from first principles I: Quantifying the configuration space. Ann. Phys. 2018, 391, 120– 149, DOI: 10.1016/j.aop.2018.01.016Google ScholarThere is no corresponding record for this reference.
- 13Kühne, T. D. Second generation Car-Parrinello molecular dynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 391– 406, DOI: 10.1002/wcms.1176Google ScholarThere is no corresponding record for this reference.
- 14Kühne, T. D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V. V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R. Z.; Schütt, O.; Schiffmann, F. CP2K: An electronic structure and molecular dynamics software package-Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103, DOI: 10.1063/5.0007045Google ScholarThere is no corresponding record for this reference.
- 15Hutter, J.; Iannuzzi, M.; Kühne, T. D.. In Comprehensive Computational Chemistry; Yáñez, M., Boyed, R. J., Eds.; Elsevier, 2024; pp 493– 517.Google ScholarThere is no corresponding record for this reference.
- 16Kohn, W. Nobel Lecture: Electronic structure of matter─wave functions and density functionals. Rev. Mod. Phys. 1999, 71, 1253– 1266, DOI: 10.1103/RevModPhys.71.1253Google ScholarThere is no corresponding record for this reference.
- 17Pople, J. A. Nobel Lecture: Quantum chemical models. Rev. Mod. Phys. 1999, 71, 1267– 1274, DOI: 10.1103/RevModPhys.71.1267Google ScholarThere is no corresponding record for this reference.
- 18Ihm, J.; Zunger, A.; Cohen, M. L. Momentum-space formalism for the total energy of solids. J. Phys. C: Solid State Phys. 1979, 12, 4409– 4422, DOI: 10.1088/0022-3719/12/21/009Google ScholarThere is no corresponding record for this reference.
- 19Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953– 17979, DOI: 10.1103/PhysRevB.50.17953Google ScholarThere is no corresponding record for this reference.
- 20Andersen, O. K. Linear methods in band theory. Phys. Rev. B 1975, 12, 3060– 3083, DOI: 10.1103/PhysRevB.12.3060Google ScholarThere is no corresponding record for this reference.
- 21Aryasetiawan, F.; Gunnarsson, O. The GW method. Rep. Prog. Phys. 1998, 61, 237, DOI: 10.1088/0034-4885/61/3/002Google ScholarThere is no corresponding record for this reference.
- 22Krack, M.; Parrinello, M. High Performance Computing in Chemistry; NIC Series; NIC-Directors, 2004; Vol. 25, pp 29– 51.Google ScholarThere is no corresponding record for this reference.
- 23VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103– 128, DOI: 10.1016/j.cpc.2004.12.014Google ScholarThere is no corresponding record for this reference.
- 24Lippert, G.; Hutter, J.; Parrinello, M. A hybrid Gaussian and plane wave density functional scheme. Mol. Phys. 1997, 92, 477– 488, DOI: 10.1080/00268979709482119Google ScholarThere is no corresponding record for this reference.
- 25Lippert, G.; Hutter, J.; Parrinello, M. The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations. Theor. Chem. Acc. 1999, 103, 124– 140, DOI: 10.1007/s002140050523Google ScholarThere is no corresponding record for this reference.
- 26Slater, J. C. A simplification of the Hartree-Fock method. Phys. Rev. 1951, 81, 385, DOI: 10.1103/PhysRev.81.385Google ScholarThere is no corresponding record for this reference.
- 27Møller, C.; Plesset, M. S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618, DOI: 10.1103/PhysRev.46.618Google ScholarThere is no corresponding record for this reference.
- 28Bohm, D.; Pines, D. A. collective description of electron interactions: III. Coulomb interactions in a degenerate electron gas. Phys. Rev. 1953, 92, 609, DOI: 10.1103/PhysRev.92.609Google ScholarThere is no corresponding record for this reference.
- 29Gell-Mann, M.; Brueckner, K. A. Correlation energy of an electron gas at high density. Phys. Rev. 1957, 106, 364, DOI: 10.1103/PhysRev.106.364Google ScholarThere is no corresponding record for this reference.
- 30VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105, DOI: 10.1063/1.2770708Google ScholarThere is no corresponding record for this reference.
- 31Goedecker, S.; Teter, M.; Hutter, J. Separable dual–space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703– 1710, DOI: 10.1103/PhysRevB.54.1703Google ScholarThere is no corresponding record for this reference.
- 32Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641– 3662, DOI: 10.1103/PhysRevB.58.3641Google ScholarThere is no corresponding record for this reference.
- 33Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 2005, 114, 145– 152, DOI: 10.1007/s00214-005-0655-yGoogle ScholarThere is no corresponding record for this reference.
- 34Reiher, M. Relativistic douglas–kroll–hess theory. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 139– 149, DOI: 10.1002/wcms.67Google ScholarThere is no corresponding record for this reference.
- 35Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865– 3868, DOI: 10.1103/PhysRevLett.77.3865Google ScholarThere is no corresponding record for this reference.
- 36Sun, J.; Ruzsinszky, A.; Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 2015, 115, 036402, DOI: 10.1103/PhysRevLett.115.036402Google ScholarThere is no corresponding record for this reference.
- 37Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158– 6170, DOI: 10.1063/1.478522Google ScholarThere is no corresponding record for this reference.
- 38Lu, J.-B.; Cantu, D. C.; Nguyen, M.-T.; Li, J.; Glezakou, V.-A.; Rousseau, R. Norm-conserving pseudopotentials and basis sets to explore lanthanide chemistry in complex environments. J. Chem. Theory Comput. 2019, 15, 5987– 5997, DOI: 10.1021/acs.jctc.9b00553Google ScholarThere is no corresponding record for this reference.
- 39Lu, J.-B.; Cantu, D. C.; Xu, C.-Q.; Nguyen, M.-T.; Hu, H.-S.; Glezakou, V.-A.; Rousseau, R.; Li, J. Norm-conserving pseudopotentials and basis sets to explore actinide chemistry in complex environments. J. Chem. Theory Comput. 2021, 17, 3360– 3371, DOI: 10.1021/acs.jctc.1c00026Google ScholarThere is no corresponding record for this reference.
- 40Lu, J.-B.; Jiang, X.-L.; Hu, H.-S.; Li, J. Norm-conserving 4f-in-core pseudopotentials and basis sets optimized for trivalent lanthanides (Ln= Ce–Lu). J. Chem. Theory Comput. 2023, 19, 82– 96, DOI: 10.1021/acs.jctc.2c00922Google ScholarThere is no corresponding record for this reference.
- 41Lu, J.-B.; Zhang, Y.-Y.; Liu, J.-B.; Li, J. Norm-Conserving 5f-in-Core Pseudopotentials and Gaussian Basis Sets Optimized for Tri-and Tetra-Valent Actinides (An= Pa–Lr). J. Chem. Theory Comput. 2025, 21, 170– 182, DOI: 10.1021/acs.jctc.4c01189Google ScholarThere is no corresponding record for this reference.
- 42Willand, A.; Kvashnin, Y. O.; Genovese, L.; Vázquez-Mayagoitia, Á.; Deb, A. K.; Sadeghi, A.; Deutsch, T.; Goedecker, S. Norm-conserving pseudopotentials with chemical accuracy compared to all-electron calculations. J. Chem. Phys. 2013, 138, 104109, DOI: 10.1063/1.4793260Google ScholarThere is no corresponding record for this reference.
- 43Oleynichenko, A. V.; Zaitsevskii, A.; Mosyagin, N. S.; Petrov, A. N.; Eliav, E.; Titov, A. V. LIBGRPP: A library for the evaluation of molecular integrals of the generalized relativistic pseudopotential operator over Gaussian functions. Symmetry 2023, 15, 197, DOI: 10.3390/sym15010197Google ScholarThere is no corresponding record for this reference.
- 44Pritchard, B. P.; Altarawy, D.; Didier, B.; Gibson, T. D.; Windus, T. L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814– 4820, DOI: 10.1021/acs.jcim.9b00725Google ScholarThere is no corresponding record for this reference.
- 45Zijlstra, E. S.; Huntemann, N.; Kalitsov, A.; Garcia, M. E.; Von Barth, U. Optimized Gaussian basis sets for Goedecker–Teter–Hutter pseudopotentials. Model. Simul. Mater. Sci. Eng. 2009, 17, 015009, DOI: 10.1088/0965-0393/17/1/015009Google ScholarThere is no corresponding record for this reference.
- 46Peintinger, M. F.; Oliveira, D. V.; Bredow, T. Consistent Gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2013, 34, 451– 459, DOI: 10.1002/jcc.23153Google ScholarThere is no corresponding record for this reference.
- 47Vilela Oliveira, D.; Laun, J.; Peintinger, M. F.; Bredow, T. BSSE-correction scheme for consistent gaussian basis sets of double-and triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2019, 40, 2364– 2376, DOI: 10.1002/jcc.26013Google ScholarThere is no corresponding record for this reference.
- 48Laun, J.; Bredow, T. BSSE-corrected consistent Gaussian basis sets of triple-zeta valence with polarization quality of the sixth period for solid-state calculations. J. Comput. Chem. 2021, 42, 1064– 1072, DOI: 10.1002/jcc.26521Google ScholarThere is no corresponding record for this reference.
- 49Laun, J.; Bredow, T. BSSE-corrected consistent Gaussian basis sets of triple-zeta valence with polarization quality of the fifth period for solid-state calculations. J. Comput. Chem. 2022, 43, 839– 846, DOI: 10.1002/jcc.26839Google ScholarThere is no corresponding record for this reference.
- 50Guidon, M.; Hutter, J.; VandeVondele, J. Auxiliary Density Matrix Methods for Hartree-Fock Exchange Calculations. J. Chem. Theory Comput. 2010, 6, 2348– 2364, DOI: 10.1021/ct1002225Google ScholarThere is no corresponding record for this reference.
- 51Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 1965, 139, A796– A823, DOI: 10.1103/PhysRev.139.A796Google ScholarThere is no corresponding record for this reference.
- 52Aryasetiawan, F.; Gunnarsson, O. The GW method. Rep. Prog. Phys. 1998, 61, 237– 312, DOI: 10.1088/0034-4885/61/3/002Google ScholarThere is no corresponding record for this reference.
- 53Whitten, J. L. Coulombic potential energy integrals and approximations. J. Chem. Phys. 1973, 58, 4496– 4501, DOI: 10.1063/1.1679012Google ScholarThere is no corresponding record for this reference.
- 54Dunlap, B. I.; Connolly, J.; Sabin, J. On some approximations in applications of Xα theory. J. Chem. Phys. 1979, 71, 3396– 3402, DOI: 10.1063/1.438728Google ScholarThere is no corresponding record for this reference.
- 55Bosoni, E.; Beal, L.; Bercx, M.; Blaha, P.; Blügel, S.; Bröder, J.; Callsen, M.; Cottenier, S.; Degomme, A.; Dikan, V. How to verify the precision of density-functional-theory implementations via reproducible and universal workflows. Nat. Rev. Phys. 2024, 6, 45– 58, DOI: 10.1038/s42254-023-00655-3Google ScholarThere is no corresponding record for this reference.
- 56Krack, M.; Parrinello, M. All-electron ab-initio molecular dynamics. Phys. Chem. Chem. Phys. 2000, 2, 2105– 2112, DOI: 10.1039/b001167nGoogle ScholarThere is no corresponding record for this reference.
- 57Dorado, B.; Amadon, B.; Freyss, M.; Bertolus, M. DFT + U calculations of the ground state and metastable states of uranium dioxide. Phys. Rev. B 2009, 79, 235125, DOI: 10.1103/PhysRevB.79.235125Google ScholarThere is no corresponding record for this reference.
- 58Krack, M. On the ground state electronic structure of uranium dioxide. Phys. Scr. 2015, 90, 094014, DOI: 10.1088/0031-8949/90/9/094014Google ScholarThere is no corresponding record for this reference.
- 59Krack, M.; Gambirasio, A.; Parrinello, M. Ab initio X-ray scattering of liquid water. J. Chem. Phys. 2002, 117, 9409– 9412, DOI: 10.1063/1.1517040Google ScholarThere is no corresponding record for this reference.
- 60Müller, P.; Karhan, K.; Krack, M.; Gerstmann, U.; Schmidt, W. G.; Bauer, M.; Kühne, T. D. Impact of finite-temperature and condensed-phase effects on theoretical X-ray absorption spectra of transition metal complexes. J. Comput. Chem. 2019, 40, 712– 716, DOI: 10.1002/jcc.25641Google ScholarThere is no corresponding record for this reference.
- 61Krack, M.; Köster, A. M. An adaptive numerical integrator for molecular integrals. J. Chem. Phys. 1998, 108, 3226– 3234, DOI: 10.1063/1.475719Google ScholarThere is no corresponding record for this reference.
- 62VandeVondele, J.; Hutter, J. An efficient orbital transformation method for electronic structure calculations. J. Chem. Phys. 2003, 118, 4365– 4369, DOI: 10.1063/1.1543154Google ScholarThere is no corresponding record for this reference.
- 63Kerker, G. Efficient iteration scheme for self-consistent pseudopotential calculations. Phys. Rev. B 1981, 23, 3082, DOI: 10.1103/PhysRevB.23.3082Google ScholarThere is no corresponding record for this reference.
- 64Pulay, P. Convergence acceleration of iterative sequences. the case of scf iteration. Chem. Phys. Lett. 1980, 73, 393– 398, DOI: 10.1016/0009-2614(80)80396-4Google ScholarThere is no corresponding record for this reference.
- 65Marks, L. D.; Luke, D. Robust mixing for ab initio quantum mechanical calculations. Phys. Rev. B 2008, 78, 075114, DOI: 10.1103/PhysRevB.78.075114Google ScholarThere is no corresponding record for this reference.
- 66Broyden, C. G. A Class of Methods for Solving Nonlinear Simultaneous Equations. Math. Comput. 1965, 19, 577, DOI: 10.1090/s0025-5718-1965-0198670-6Google ScholarThere is no corresponding record for this reference.
- 67Mermin, N. D. Thermal Properties of the Inhomogeneous Electron Gas. Phys. Rev. 1965, 137, A1441– A1443, DOI: 10.1103/PhysRev.137.A1441Google ScholarThere is no corresponding record for this reference.
- 68Hutter, J.; Parrinello, M.; Vogel, S. Exponential transformation of molecular orbitals. J. Chem. Phys. 1994, 101, 3862– 3865, DOI: 10.1063/1.467504Google ScholarThere is no corresponding record for this reference.
- 69Gan, C.; Haynes, P.; Payne, M. Preconditioned conjugate gradient method for the sparse generalized eigenvalue problem in electronic structure calculations. Comput. Phys. Commun. 2001, 134, 33– 40, DOI: 10.1016/S0010-4655(00)00188-0Google ScholarThere is no corresponding record for this reference.
- 70Monkhorst, H. J.; Pack, J. D. Special points for Brillouin–zone integrations. Phys. Rev. B 1976, 13, 5188– 5192, DOI: 10.1103/PhysRevB.13.5188Google ScholarThere is no corresponding record for this reference.
- 71Guidon, M.; Hutter, J.; VandeVondele, J. Robust Periodic Hartree-Fock Exchange for Large-Scale Simulations Using Gaussian Basis Sets. J. Chem. Theory Comput. 2009, 5, 3010– 3021, DOI: 10.1021/ct900494gGoogle ScholarThere is no corresponding record for this reference.
- 72Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098– 3100, DOI: 10.1103/PhysRevA.38.3098Google ScholarThere is no corresponding record for this reference.
- 73Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785– 789, DOI: 10.1103/PhysRevB.37.785Google ScholarThere is no corresponding record for this reference.
- 74Merlot, P.; Izsák, R.; Borgoo, A.; Kjærgaard, T.; Helgaker, T.; Reine, S. Charge-constrained auxiliary-density-matrix methods for the Hartree–Fock exchange contribution. J. Chem. Phys. 2014, 141, 094104, DOI: 10.1063/1.4894267Google ScholarThere is no corresponding record for this reference.
- 75Jensen, F. Unifying general and segmented contracted basis sets. Segmented polarization consistent basis sets. J. Chem. Theory Comput. 2014, 10, 1074– 1085, DOI: 10.1021/ct401026aGoogle ScholarThere is no corresponding record for this reference.
- 76Kumar, C.; Fliegl, H.; Jensen, F.; Teale, A. M.; Reine, S.; Kjærgaard, T. Accelerating Kohn-Sham response theory using density fitting and the auxiliary-density-matrix method. Int. J. Quantum Chem. 2018, 118, e25639 DOI: 10.1002/qua.25639Google ScholarThere is no corresponding record for this reference.
- 77Gavini, V.; Baroni, S.; Blum, V.; Bowler, D. R.; Buccheri, A.; Chelikowsky, J. R.; Das, S.; Dawson, W.; Delugas, P.; Dogan, M. Roadmap on electronic structure codes in the exascale era. Model. Simul. Mater. Sci. Eng. 2023, 31, 063301, DOI: 10.1088/1361-651X/acdf06Google ScholarThere is no corresponding record for this reference.
- 78Bussy, A.; Schütt, O.; Hutter, J. Sparse tensor based nuclear gradients for periodic Hartree–Fock and low-scaling correlated wave function methods in the CP2K software package: A massively parallel and GPU accelerated implementation. J. Chem. Phys. 2023, 158, 164109, DOI: 10.1063/5.0144493Google ScholarThere is no corresponding record for this reference.
- 79Vahtras, O.; Almlöf, J.; Feyereisen, M. Integral approximations for LCAO-SCF calculations. Chem. Phys. Lett. 1993, 213, 514– 518, DOI: 10.1016/0009-2614(93)89151-7Google ScholarThere is no corresponding record for this reference.
- 80Weigend, F. A fully direct RI-HF algorithm: Implementation, optimized auxiliary basis sets, demonstration of accuracy and efficiency. Phys. Chem. Chem. Phys. 2002, 4, 4285– 4291, DOI: 10.1039/b204199pGoogle ScholarThere is no corresponding record for this reference.
- 81Stoychev, G. L.; Auer, A. A.; Neese, F. Automatic generation of auxiliary basis sets. J. Chem. Theory Comput. 2017, 13, 554– 562, DOI: 10.1021/acs.jctc.6b01041Google ScholarThere is no corresponding record for this reference.
- 82Bussy, A.; Hutter, J. Efficient periodic resolution-of-the-identity Hartree–Fock exchange method with k-point sampling and Gaussian basis sets. J. Chem. Phys. 2024, 160, 064116, DOI: 10.1063/5.0189659Google ScholarThere is no corresponding record for this reference.
- 83Del Ben, M.; Hutter, J.; VandeVondele, J. Second-Order Møller–Plesset Perturbation Theory in the Condensed Phase: An Efficient and Massively Parallel Gaussian and Plane Waves Approach. J. Chem. Theory Comput. 2012, 8, 4177– 4188, DOI: 10.1021/ct300531wGoogle ScholarThere is no corresponding record for this reference.
- 84Del Ben, M.; Hutter, J.; VandeVondele, J. Electron Correlation in the Condensed Phase from a Resolution of Identity Approach Based on the Gaussian and Plane Waves Scheme. J. Chem. Theory Comput. 2013, 9, 2654– 2671, DOI: 10.1021/ct4002202Google ScholarThere is no corresponding record for this reference.
- 85Weigend, F.; Häser, M. RI-MP2: first derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331– 340, DOI: 10.1007/s002140050269Google ScholarThere is no corresponding record for this reference.
- 86Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143– 152, DOI: 10.1016/S0009-2614(98)00862-8Google ScholarThere is no corresponding record for this reference.
- 87Del Ben, M.; Hutter, J.; VandeVondele, J. Forces and stress in second order Møller-Plesset perturbation theory for condensed phase systems within the resolution-of-identity Gaussian and plane waves approach. J. Chem. Phys. 2015, 143, 102803, DOI: 10.1063/1.4919238Google ScholarThere is no corresponding record for this reference.
- 88Rybkin, V. V.; VandeVondele, J. Spin-Unrestricted Second-Order Møller–Plesset (MP2) Forces for the Condensed Phase: From Molecular Radicals to F-Centers in Solids. J. Chem. Theory Comput. 2016, 12, 2214– 2223, DOI: 10.1021/acs.jctc.6b00015Google ScholarThere is no corresponding record for this reference.
- 89Schwartz, C. Importance of Angular Correlations between Atomic Electrons. Phys. Rev. 1962, 126, 1015– 1019, DOI: 10.1103/PhysRev.126.1015Google ScholarThere is no corresponding record for this reference.
- 90Borštnik, U.; VandeVondele, J.; Weber, V.; Hutter, J. Sparse matrix multiplication: The distributed block-compressed sparse row library. Parallel Comput. 2014, 40, 47– 58, DOI: 10.1016/j.parco.2014.03.012Google ScholarThere is no corresponding record for this reference.
- 91SPLA: Specialized Parallel Linear Algebra. https://github.com/eth-cscs/spla, (accessed, 2025 05 29).Google ScholarThere is no corresponding record for this reference.
- 92Stein, F.; Hutter, J. Double-hybrid density functionals for the condensed phase: Gradients, stress tensor, and auxiliary-density matrix method acceleration. J. Chem. Phys. 2022, 156, 074107, DOI: 10.1063/5.0082327Google ScholarThere is no corresponding record for this reference.
- 93Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108, DOI: 10.1063/1.2148954Google ScholarThere is no corresponding record for this reference.
- 94(a) Lehtola, S.; Steigemann, C.; Oliveira, M. J. T.; Marques, M. A. L. Recent developments in libxc ─ A comprehensive library of functionals for density functional theory. SoftwareX 2018, 7, 1– 5, DOI: 10.1016/j.softx.2017.11.002Google ScholarThere is no corresponding record for this reference.See. https://gitlab.com/libxc/libxc.Google ScholarThere is no corresponding record for this reference.
- 95Stein, F.; Hutter, J.; Rybkin, V. V. Double-hybrid DFT functionals for the condensed phase: Gaussian and plane waves implementation and evaluation. Molecules 2020, 25, 5174, DOI: 10.3390/molecules25215174Google ScholarThere is no corresponding record for this reference.
- 96Furche, F. Developing the random phase approximation into a practical post-Kohn–Sham correlation model. J. Chem. Phys. 2008, 129, 114105, DOI: 10.1063/1.2977789Google ScholarThere is no corresponding record for this reference.
- 97Boyd, J. P. Exponentially convergent Fourier-Chebshev quadrature schemes on bounded and infinite intervals. J. Sci. Comput. 1987, 2, 99– 109, DOI: 10.1007/BF01061480Google ScholarThere is no corresponding record for this reference.
- 98Braess, D.; Hackbusch, W. Approximation of 1/x by exponential sums in [1, ∞). IMA J. Numer. Anal. 2005, 25, 685– 697, DOI: 10.1093/imanum/dri015Google ScholarThere is no corresponding record for this reference.
- 99Ren, X.; Rinke, P.; Scuseria, G. E.; Scheffler, M. Renormalized second-order perturbation theory for the electron correlation energy: Concept, implementation, and benchmarks. Phys. Rev. B 2013, 88, 035120, DOI: 10.1103/PhysRevB.88.035120Google ScholarThere is no corresponding record for this reference.
- 100Bates, J. E.; Furche, F. Communication: Random phase approximation renormalized many-body perturbation theory. J. Chem. Phys. 2013, 139, 171103, DOI: 10.1063/1.4827254Google ScholarThere is no corresponding record for this reference.
- 101Freeman, D. L. Coupled-cluster expansion applied to the electron gas: Inclusion of ring and exchange effects. Phys. Rev. B 1977, 15, 5512– 5521, DOI: 10.1103/PhysRevB.15.5512Google ScholarThere is no corresponding record for this reference.
- 102Stein, F.; Hutter, J. Massively parallel implementation of gradients within the random phase approximation: Application to the polymorphs of benzene. J. Chem. Phys. 2024, 160, 024120, DOI: 10.1063/5.0180704Google ScholarThere is no corresponding record for this reference.
- 103Kwasniewski, G.; Kabić, M.; Besta, M.; VandeVondele, J.; Solcà, R.; Hoefler, T. Red-blue pebbling revisited: Near optimal parallel matrix-matrix multiplication. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; Association for Computing Machinery, 2019; pp 1– 22.Google ScholarThere is no corresponding record for this reference.
- 104Wilhelm, J.; Seewald, P.; Del Ben, M.; Hutter, J. Large-scale cubic-scaling random phase approximation correlation energy calculations using a Gaussian basis. J. Chem. Theory Comput. 2016, 12, 5851– 5859, DOI: 10.1021/acs.jctc.6b00840Google ScholarThere is no corresponding record for this reference.
- 105Valeev, E. F. Libint: A library for the evaluation of molecular integrals of many-body operators over Gaussian functions. http://libint.valeyev.net/, 2024; (accessed, 2025 05 29).Google ScholarThere is no corresponding record for this reference.
- 106Anisimov, V. I.; Zaanen, J.; Andersen, O. K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 1991, 44, 943– 954, DOI: 10.1103/PhysRevB.44.943Google ScholarThere is no corresponding record for this reference.
- 107Dudarev, S. L.; Manh, D. N.; Sutton, A. P. Effect of Mott-Hubbard correlations on the electronic structure and structural stability of uranium dioxide. Philos. Mag. B 1997, 75, 613– 628, DOI: 10.1080/13642819708202343Google ScholarThere is no corresponding record for this reference.
- 108Dudarev, S. L.; Botton, G. A.; Savrasov, S. Y.; Humphreys, C. J.; Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505– 1509, DOI: 10.1103/PhysRevB.57.1505Google ScholarThere is no corresponding record for this reference.
- 109Mulliken, R. S. Electronic Population Analysis on LCAO-MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833– 1840, DOI: 10.1063/1.1740588Google ScholarThere is no corresponding record for this reference.
- 110Löwdin, P.-O. On the Non-Orthogonality Problem Connected with the Use of Atomic Wave Functions in the Theory of Molecules and Crystals. J. Chem. Phys. 1950, 18, 365– 375, DOI: 10.1063/1.1747632Google ScholarThere is no corresponding record for this reference.
- 111Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G. K. H.; Marks, L. D. WIEN2k: An APW+lo program for calculating the properties of solids. J. Chem. Phys. 2020, 152, 074101, DOI: 10.1063/1.5143061Google ScholarThere is no corresponding record for this reference.
- 112The FLEUR project. https://www.flapw.de/, (accessed: 2025 10 17).Google ScholarThere is no corresponding record for this reference.
- 113The Elk Code. https://elk.sourceforge.io/, (accessed, 2025 10 17).Google ScholarThere is no corresponding record for this reference.
- 114Gulans, A.; Kontur, S.; Meisenbichler, C.; Nabok, D.; Pavone, P.; Rigamonti, S.; Sagmeister, S.; Werner, U.; Draxl, C. exciting: a full-potential all-electron package implementing density-functional theory and many-body perturbation theory. J. Phys.: Condens. Matter 2014, 26, 363202, DOI: 10.1088/0953-8984/26/36/363202Google ScholarThere is no corresponding record for this reference.
- 115Hamann, D. R.; Schlüter, M.; Chiang, C. Norm-Conserving Pseudopotentials. Phys. Rev. Lett. 1979, 43, 1494– 1497, DOI: 10.1103/PhysRevLett.43.1494Google ScholarThere is no corresponding record for this reference.
- 116Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892– 7895, DOI: 10.1103/PhysRevB.41.7892Google ScholarThere is no corresponding record for this reference.
- 117Slater, J. C. Wave functions in a periodic potential. Phys. Rev. 1937, 51, 846, DOI: 10.1103/PhysRev.51.846Google ScholarThere is no corresponding record for this reference.
- 118Ceperley, D. M.; Alder, B. J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566, DOI: 10.1103/PhysRevLett.45.566Google ScholarThere is no corresponding record for this reference.
- 119Golze, D.; Dvorak, M.; Rinke, P. The GW compendium: A practical guide to theoretical photoemission spectroscopy. Front. Chem. 2019, 7, 377, DOI: 10.3389/fchem.2019.00377Google ScholarThere is no corresponding record for this reference.
- 120Capelle, K. A. bird’s-eye view of density-functional theory. Braz. J. Phys. 2006, 36, 1318– 1343, DOI: 10.1590/S0103-97332006000700035Google ScholarThere is no corresponding record for this reference.
- 121Caravati, S.; Bernasconi, M.; Kühne, T.; Krack, M.; Parrinello, M. First-principles study of crystalline and amorphousGe2Sb2Te5 and the effects of stoichiometric defects. J. Phys.: Condens. Matter 2009, 21, 255501, DOI: 10.1088/0953-8984/21/25/255501Google ScholarThere is no corresponding record for this reference.
- 122Caravati, S.; Colleoni, D.; Mazzarello, R.; Kühne, T.; Krack, M.; Bernasconi, M.; Parrinello, M. First-principles study of nitrogen doping in cubic and amorphousGe2Sb2Te5. J. Phys.: Condens. Matter 2011, 23, 265801, DOI: 10.1088/0953-8984/23/26/265801Google ScholarThere is no corresponding record for this reference.
- 123Los, J. H.; Kühne, T. D.; Gabardi, S.; Bernasconi, M. First-principles study of the amorphous In 3 SbTe 2 phase change compound. Phys. Rev. B 2013, 88, 174203, DOI: 10.1103/PhysRevB.88.174203Google ScholarThere is no corresponding record for this reference.
- 124Gabardi, S.; Caravati, S.; Los, J. H.; Kühne, T. D.; Bernasconi, M. Influence of the exchange and correlation functional on the structure of amorphous InSb and In3SbTe2 compounds. J. Chem. Phys. 2016, 144, 204508, DOI: 10.1063/1.4950817Google ScholarThere is no corresponding record for this reference.
- 125Schambeck, M.; Golze, D.; Wilhelm, J. Solving multipole challenges in the GW100 benchmark enables precise low-scaling GW calculations. Phys. Rev. B 2024, 110, 125146, DOI: 10.1103/PhysRevB.110.125146Google ScholarThere is no corresponding record for this reference.
- 126Pasquier, R.; Camarasa-Gómez, M.; Hehn, A.-S.; Hernangómez-Pérez, D.; Wilhelm, J. Efficient GW band structure calculations using Gaussian basis functions and application to atomically thin transition-metal dichalcogenides. Phys. Rev. B 2025, 112, 205103, DOI: 10.1103/v4zv-1pf9Google ScholarThere is no corresponding record for this reference.
- 127Graml, M.; Zollner, K.; Hernangómez-Pérez, D.; Faria Junior, P. E.; Wilhelm, J. Low-Scaling GW Algorithm Applied to Twisted Transition-Metal Dichalcogenide Heterobilayers. J. Chem. Theory Comput. 2024, 20, 2202– 2208, DOI: 10.1021/acs.jctc.3c01230Google ScholarThere is no corresponding record for this reference.
- 128van Setten, M. J.; Caruso, F.; Sharifzadeh, S.; Ren, X.; Scheffler, M.; Liu, F.; Lischner, J.; Lin, L.; Deslippe, J. R.; Louie, S. G. GW100: Benchmarking G0W0 for Molecular Systems. J. Chem. Theory Comput. 2015, 11, 5665– 5687, DOI: 10.1021/acs.jctc.5b00453Google ScholarThere is no corresponding record for this reference.
- 129Setyawan, W.; Curtarolo, S. High-throughput electronic band structure calculations: Challenges and tools. Comput. Mater. Sci. 2010, 49, 299– 312, DOI: 10.1016/j.commatsci.2010.05.010Google ScholarThere is no corresponding record for this reference.
- 130Schusteritsch, G.; Kühne, T. D.; Guo, Z. X.; Kaxiras, E. The effect of Ag, Pb and Bi impurities on grain boundary sliding and intergranular decohesion in Copper. Philos. Mag. 2016, 96, 2868– 2886, DOI: 10.1080/14786435.2016.1217360Google ScholarThere is no corresponding record for this reference.
- 131Praprotnik, M.; Site, L. D.; Kremer, K. Multiscale simulation of soft matter: From scale bridging to adaptive resolution. Annu. Rev. Phys. Chem. 2008, 59, 545– 571, DOI: 10.1146/annurev.physchem.59.032607.093707Google ScholarThere is no corresponding record for this reference.
- 132Cortes-Huerto, R.; Praprotnik, M.; Kremer, K.; Delle Site, L. From adaptive resolution to molecular dynamics of open systems. Eur. Phys. J. B 2021, 94, 189, DOI: 10.1140/epjb/s10051-021-00193-wGoogle ScholarThere is no corresponding record for this reference.
- 133Panahian Jand, S.; Kühne, T. D.; Delle Site, L. On the physical consistency of an open quantum region with a classical reservoir in molecular simulation. Adv. Theory Simul. 2024, 7, 2400833, DOI: 10.1002/adts.202400833Google ScholarThere is no corresponding record for this reference.
- 134Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999– 3094, DOI: 10.1021/cr9904009Google ScholarThere is no corresponding record for this reference.
- 135Fattebert, J.-L.; Gygi, F. Density functional theory for efficient ab initio molecular dynamics simulations in solution. J. Comput. Chem. 2002, 23, 662– 666, DOI: 10.1002/jcc.10069Google ScholarThere is no corresponding record for this reference.
- 136Fattebert, J.-L.; Gygi, F. First-principles molecular dynamics simulations in a continuum solvent. Int. J. Quantum Chem. 2003, 93, 139– 147, DOI: 10.1002/qua.10548Google ScholarThere is no corresponding record for this reference.
- 137Andreussi, O.; Dabo, I.; Marzari, N. Revised self-consistent continuum solvation in electronic-structure calculations. J. Chem. Phys. 2012, 136, 064102, DOI: 10.1063/1.3676407Google ScholarThere is no corresponding record for this reference.
- 138Yin, W.-J.; Krack, M.; Li, X.; Chen, L.-Z.; Liu, L.-M. Periodic continuum solvation model integrated with first-principles calculations for solid surfaces. Prog. Nat. Sci. 2017, 27, 283– 288, DOI: 10.1016/j.pnsc.2017.03.003Google ScholarThere is no corresponding record for this reference.
- 139Yin, W.-J.; Krack, M.; Wen, B.; Ma, S.-Y.; Liu, L.-M. CO2 Capture and Conversion on Rutile TiO2(110) in the Water Environment: Insight by First-Principles Calculations. J. Phys. Chem. Lett. 2015, 6, 2538– 2545, DOI: 10.1021/acs.jpclett.5b00798Google ScholarThere is no corresponding record for this reference.
- 140Dupont, C.; Andreussi, O.; Marzari, N. Self-consistent continuum solvation (SCCS): The case of charged systems. J. Chem. Phys. 2013, 139, 214110, DOI: 10.1063/1.4832475Google ScholarThere is no corresponding record for this reference.
- 141Martyna, G. J.; Tuckerman, M. E. A reciprocal space based method for treating long range interactions inab initioand force-field-based calculations in clusters. J. Chem. Phys. 1999, 110, 2810– 2821, DOI: 10.1063/1.477923Google ScholarThere is no corresponding record for this reference.
- 142Cococcioni, M.; Mauri, F.; Ceder, G.; Marzari, N. Electronic-Enthalpy Functional for Finite Systems Under Pressure. Phys. Rev. Lett. 2005, 94, 145501, DOI: 10.1103/physrevlett.94.145501Google ScholarThere is no corresponding record for this reference.
- 143Warshel, A.; Levitt, M. Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme. J. Mol. Biol. 1976, 103, 227– 249, DOI: 10.1016/0022-2836(76)90311-9Google ScholarThere is no corresponding record for this reference.
- 144Stewart, J. J. P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173– 1213, DOI: 10.1007/s00894-007-0233-4Google ScholarThere is no corresponding record for this reference.
- 145Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260– 7268, DOI: 10.1103/PhysRevB.58.7260Google ScholarThere is no corresponding record for this reference.
- 146Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989– 2009, DOI: 10.1021/acs.jctc.7b00118Google ScholarThere is no corresponding record for this reference.
- 147Abraham, M. J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J. C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19– 25, DOI: 10.1016/j.softx.2015.06.001Google ScholarThere is no corresponding record for this reference.
- 148QM/MM Documentation - GROMACS 2022-beta1. https://manual.gromacs.org/documentation/2022-beta1/reference-manual/special/qmmm.html, 2022; (accessed, 2024 10 29).Google ScholarThere is no corresponding record for this reference.
- 149Senn, H. M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed. 2009, 48, 1198– 1229, DOI: 10.1002/anie.200802019Google ScholarThere is no corresponding record for this reference.
- 150Zhang, Y.; Lee, T.-S.; Yang, W. A pseudobond approach to combining quantum mechanical and molecular mechanical methods. J. Chem. Phys. 1999, 110, 46– 54, DOI: 10.1063/1.478083Google ScholarThere is no corresponding record for this reference.
- 151Ihrig, A. C.; Schiffmann, C.; Sebastiani, D. Specific quantum mechanical/molecular mechanical capping-potentials for biomolecular functional groups. J. Chem. Phys. 2011, 135, 214107, DOI: 10.1063/1.3664300Google ScholarThere is no corresponding record for this reference.
- 152Schiffmann, C.; Sebastiani, D. Artificial bee colony optimization of capping potentials for hybrid quantum mechanical/molecular mechanical calculations. J. Chem. Theory Comput. 2011, 7, 1307– 1315, DOI: 10.1021/ct1007108Google ScholarThere is no corresponding record for this reference.
- 153Laino, T.; Mohamed, F.; Laio, A.; Parrinello, M. An efficient real space multigrid QM/MM electrostatic coupling. J. Chem. Theory Comput. 2005, 1, 1176– 1184, DOI: 10.1021/ct050123fGoogle ScholarThere is no corresponding record for this reference.
- 154Laino, T.; Mohamed, F.; Laio, A.; Parrinello, M. An Efficient Linear-Scaling Electrostatic Coupling for treating periodic boundary conditions in QM/MM Simulations. J. Chem. Theory Comput. 2006, 2, 1370– 1378, DOI: 10.1021/ct6001169Google ScholarThere is no corresponding record for this reference.
- 155Golze, D.; Iannuzzi, M.; Nguyen, M.-T.; Passerone, D.; Hutter, J. Simulation of Adsorption Processes at Metallic Interfaces: An Image Charge Augmented QM/MM Approach. J. Chem. Theory Comput. 2013, 9, 5086– 5097, DOI: 10.1021/ct400698yGoogle ScholarThere is no corresponding record for this reference.
- 156Devynck, F.; Iannuzzi, M.; Krack, M. Frenkel pair recombinations in UO2: Importance of explicit description of polarizability in core-shell molecular dynamics simulations. Phys. Rev. B 2012, 85, 184103, DOI: 10.1103/PhysRevB.85.184103Google ScholarThere is no corresponding record for this reference.
- 157Blöchl, P. E. Electrostatic decoupling of periodic images of plane-wave-expanded densities and derived atomic point charges. J. Chem. Phys. 1995, 103, 7422– 7428, DOI: 10.1063/1.470314Google ScholarThere is no corresponding record for this reference.
- 158Laino, T.; Mohamed, F.; Laio, A.; Parrinello, M. An efficient linear-scaling electrostatic coupling for treating periodic boundary conditions in QM/MM simulations. J. Chem. Theory Comput. 2006, 2, 1370– 1378, DOI: 10.1021/ct6001169Google ScholarThere is no corresponding record for this reference.
- 159Image Charges. https://manual.cp2k.org/trunk/methods/qm_mm/image_charges.html, 2015; (accessed, 2025 02 05).Google ScholarThere is no corresponding record for this reference.
- 160Bayly, C. I.; Cieplak, P.; Cornell, W. D.; Kollman, P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269– 10280, DOI: 10.1021/j100142a004Google ScholarThere is no corresponding record for this reference.
- 161Golze, D.; Hutter, J.; Iannuzzi, M. Wetting of water on hexagonal boron nitrideRh(111): a QM/MM model based on atomic charges derived for nano-structured substrates. Phys. Chem. Chem. Phys. 2015, 17, 14307– 14316, DOI: 10.1039/C4CP04638BGoogle ScholarThere is no corresponding record for this reference.
- 162https://www.cp2k.org/howto:resp, 2015; (accessed, 2025 06 13).Google ScholarThere is no corresponding record for this reference.
- 163Campañá, C.; Mussard, B.; Woo, T. K. Electrostatic Potential Derived Atomic Charges for Periodic Systems Using a Modified Error Functional. J. Chem. Theory Comput. 2009, 5, 2866– 2878, DOI: 10.1021/ct9003405Google ScholarThere is no corresponding record for this reference.
- 164Huang, C.; Pavone, M.; Carter, E. A. Quantum mechanical embedding theory based on a unique embedding potential. J. Chem. Phys. 2011, 134, 154110, DOI: 10.1063/1.3577516Google ScholarThere is no corresponding record for this reference.
- 165Wu, Q.; Yang, W. A direct optimization method for calculating density functionals and exchange-correlation potentials from electron densities. J. Chem. Phys. 2003, 118, 2498– 2509, DOI: 10.1063/1.1535422Google ScholarThere is no corresponding record for this reference.
- 166Van Leeuwen, R.; Baerends, E. Exchange-correlation potential with correct asymptotic behavior. Phys. Rev. A 1994, 49, 2421, DOI: 10.1103/PhysRevA.49.2421Google ScholarThere is no corresponding record for this reference.
- 167Putrino, A.; Sebastiani, D.; Parrinello, M. Generalized variational density functional perturbation theory. J. Chem. Phys. 2000, 113, 7102– 7109, DOI: 10.1063/1.1312830Google ScholarThere is no corresponding record for this reference.
- 168Sebastiani, D.; Parrinello, M. A New ab-Initio Approach for NMR Chemical Shifts in Periodic Systems. J. Phys. Chem. A 2001, 105, 1951– 1958, DOI: 10.1021/jp002807jGoogle ScholarThere is no corresponding record for this reference.
- 169Sebastiani, D. Ab-initio calculations of NMR parameters in condensed phases. Mod. Phys. Lett. B 2003, 17, 1301– 1319, DOI: 10.1142/S0217984903006372Google ScholarThere is no corresponding record for this reference.
- 170Gonze, X. Adiabatic density-functional perturbation theory. Phys. Rev. A 1995, 52, 1096– 1114, DOI: 10.1103/PhysRevA.52.1096Google ScholarThere is no corresponding record for this reference.
- 171Sebastiani, D. Current Densities and Nucleus-Independent Chemical Shift Maps from Reciprocal-Space Density Functional Perturbation Theory Calculations. ChemPhysChem 2006, 7, 164– 175, DOI: 10.1002/cphc.200500438Google ScholarThere is no corresponding record for this reference.
- 172Weber, V.; Iannuzzi, M.; Giani, S.; Hutter, J.; Declerck, R.; Waroquier, M. Magnetic linear response properties calculations with the Gaussian and augmented-plane-wave method. J. Chem. Phys. 2009, 131, 014106, DOI: 10.1063/1.3156803Google ScholarThere is no corresponding record for this reference.
- 173Putrino, A.; Parrinello, M. Anharmonic Raman spectra in high-pressure ice from ab initio simulations. Phys. Rev. Lett. 2002, 88, 176401, DOI: 10.1103/physrevlett.88.176401Google ScholarThere is no corresponding record for this reference.
- 174Scherrer, A.; Vuilleumier, R.; Sebastiani, D. Vibrational circular dichroism from ab initio molecular dynamics and nuclear velocity perturbation theory in the liquid phase. J. Chem. Phys. 2016, 145, 084101, DOI: 10.1063/1.4960653Google ScholarThere is no corresponding record for this reference.
- 175Resta, R. Why are insulators insulating and metals conducting?. J. Phys.: Condens. Matter 2002, 14, R625– R656, DOI: 10.1088/0953-8984/14/20/201Google ScholarThere is no corresponding record for this reference.
- 176Mauri, F.; Louie, S. Magnetic Susceptibility of Insulators from First Principles. Phys. Rev. Lett. 1996, 76, 4246– 4249, DOI: 10.1103/PhysRevLett.76.4246Google ScholarThere is no corresponding record for this reference.
- 177Keith, T. A.; Bader, R. F. W. Calculation of magnetic response properties using atoms in molecules. Chem. Phys. Lett. 1992, 194, 1– 8, DOI: 10.1016/0009-2614(92)85733-QGoogle ScholarThere is no corresponding record for this reference.
- 178Keith, T. A.; Bader, R. F. W. Calculation of magnetic response properties using a continuous set of gauge transformations. Chem. Phys. Lett. 1993, 210, 223– 231, DOI: 10.1016/0009-2614(93)89127-4Google ScholarThere is no corresponding record for this reference.
- 179van Wüllen, C. Calculation of NMR and EPR Parameters: Theory and Applications; Wiley, 2004; pp 83– 100.Google ScholarThere is no corresponding record for this reference.
- 180Cowan, B. P. Nuclear Magnetic Resonance and Relaxation, online-ausg ed.; Cambridge University Press: Cambridge, 1997.Google ScholarThere is no corresponding record for this reference.
- 181Mondal, A.; Gaultois, M. W.; Pell, A. J.; Iannuzzi, M.; Grey, C. P.; Hutter, J.; Kaupp, M. Large-scale computation of nuclear magnetic resonance shifts for paramagnetic solids using CP2K. J. Chem. Theory Comput. 2018, 14, 377– 394, DOI: 10.1021/acs.jctc.7b00991Google ScholarThere is no corresponding record for this reference.
- 182Schreckenbach, G.; Ziegler, T. Calculation of NMR shielding tensors based on density functional theory and a scalar relativistic Pauli-type Hamiltonian. The application to transition metal complexes. Int. J. Quantum Chem. 1997, 61, 899– 918, DOI: 10.1002/(SICI)1097-461X(1997)61:6<899::AID-QUA3>3.0.CO;2-RGoogle ScholarThere is no corresponding record for this reference.
- 183Pickard, C.; Mauri, F. First-Principles Theory of the EPR G Tensor in Solids: Defects in Quartz. Phys. Rev. Lett. 2002, 88, 086403, DOI: 10.1103/physrevlett.88.086403Google ScholarThere is no corresponding record for this reference.
- 184Van Yperen-De Deyne, A.; Pauwels, E.; Van Speybroeck, V.; Waroquier, M. Accurate Spin–orbit and Spin–other-orbit Contributions to the G-tensor for Transition Metal Containing Systems. Phys. Chem. Chem. Phys. 2012, 14, 10690– 10704, DOI: 10.1039/c2cp41086aGoogle ScholarThere is no corresponding record for this reference.
- 185Neese, F. Efficient and Accurate Approximations to the Molecular Spin-orbit Coupling Operator and Their Use in Molecular G-tensor Calculations. J. Chem. Phys. 2005, 122, 034107, DOI: 10.1063/1.1829047Google ScholarThere is no corresponding record for this reference.
- 186Malkina, O. L.; Vaara, J.; Schimmelpfennig, B.; Munzarova, M.; Malkin, V. G.; Kaupp, M. Density Functional Calculations of Electronic G-tensors Using Spin-orbit Pseudopotentials and Mean-field All-electron Spin-orbit Operators. J. Am. Chem. Soc. 2000, 122, 9206– 9218, DOI: 10.1021/ja000984sGoogle ScholarThere is no corresponding record for this reference.
- 187Pigliapochi, R.; Pell, A. J.; Seymour, I. D.; Grey, C. P.; Ceresoli, D.; Kaupp, M. DFT investigation of the effect of spin-orbit coupling on the NMR shifts in paramagnetic solids. Phys. Rev. B 2017, 95, 054412, DOI: 10.1103/PhysRevB.95.054412Google ScholarThere is no corresponding record for this reference.
- 188Declerck, R.; Pauwels, E.; Van Speybroeck, V.; Waroquier, M. First-principles Calculations of Hyperfine Parameters with the Gaussian and Augmented-plane-wave Method: Application to Radicals Embedded in a Crystalline Environment. Phys. Rev. B 2006, 74, 245103, DOI: 10.1103/physrevb.74.245103Google ScholarThere is no corresponding record for this reference.
- 189Blase, X.; Duchemin, I.; Jacquemin, D. The Bethe–Salpeter equation in chemistry: relations with TD-DFT, applications and challenges. Chem. Soc. Rev. 2018, 47, 1022– 1043, DOI: 10.1039/C7CS00049AGoogle ScholarThere is no corresponding record for this reference.
- 190Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291, DOI: 10.1016/S0009-2614(99)01149-5Google ScholarThere is no corresponding record for this reference.
- 191Hutter, J. Excited state nuclear forces from the Tamm–Dancoff approximation to time–dependent density functional theory within the plane wave basis set framework. J. Chem. Phys. 2003, 118, 3928– 3934, DOI: 10.1063/1.1540109Google ScholarThere is no corresponding record for this reference.
- 192Grimme, S. A simplified Tamm-Dancoff density functional approach for the electronic excitation spectra of very large molecules. J. Chem. Phys. 2013, 138, 244104, DOI: 10.1063/1.4811331Google ScholarThere is no corresponding record for this reference.
- 193Hehn, A.-S.; Sertcan, B.; Belleflamme, F.; Chulkov, S. K.; Watkins, M. B.; Hutter, J. Excited-State Properties for Extended Systems: Efficient Hybrid Density Functional Methods. J. Chem. Theory Comput. 2022, 18, 4186– 4202, DOI: 10.1021/acs.jctc.2c00144Google ScholarThere is no corresponding record for this reference.
- 194Sertcan Gokmen, B.; Hutter, J.; Hehn, A.-S. Excited-State Forces with the Gaussian and Augmented Plane Wave Method for the Tamm–Dancoff Approximation of Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2024, 20, 8494– 8504, DOI: 10.1021/acs.jctc.4c00614Google ScholarThere is no corresponding record for this reference.
- 195Vogt, J.-R.; Wilhelm, J.; Hehn, A.-S. Perturbative spin–orbit couplings for the simulation of extended framework materials. J. Chem. Phys. 2025, 162, 082502, DOI: 10.1063/5.0242940Google ScholarThere is no corresponding record for this reference.
- 196van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392– 399, DOI: 10.1063/1.476576Google ScholarThere is no corresponding record for this reference.
- 197Bussy, A.; Hutter, J. Efficient and low-scaling linear-response time-dependent density functional theory implementation for core-level spectroscopy of large and periodic systems. Phys. Chem. Chem. Phys. 2021, 23, 4736– 4746, DOI: 10.1039/D0CP06164FGoogle ScholarThere is no corresponding record for this reference.
- 198Ljungberg, M. P.; Koval, P.; Ferrari, F.; Foerster, D.; Sánchez-Portal, D. Cubic-Scaling Iterative Solution of the Bethe-Salpeter Equation for Finite Systems. Phys. Rev. B 2015, 92, 075422, DOI: 10.1103/PhysRevB.92.075422Google ScholarThere is no corresponding record for this reference.
- 199Jacquemin, D.; Duchemin, I.; Blondel, A.; Blase, X. Assessment of the Accuracy of the Bethe–Salpeter (BSE/GW) Oscillator Strengths. J. Chem. Theory Comput. 2016, 12, 3969– 3981, DOI: 10.1021/acs.jctc.6b00419Google ScholarThere is no corresponding record for this reference.
- 200Bruneval, F.; Hamed, S. M.; Neaton, J. B. A systematic benchmark of the ab initio Bethe-Salpeter equation approach for low-lying optical excitations of small organic molecules. J. Chem. Phys. 2015, 142, 244101, DOI: 10.1063/1.4922489Google ScholarThere is no corresponding record for this reference.
- 201Sander, T.; Maggio, E.; Kresse, G. Beyond the Tamm-Dancoff approximation for extended systems using exact diagonalization. Phys. Rev. B 2015, 92, 045209, DOI: 10.1103/PhysRevB.92.045209Google ScholarThere is no corresponding record for this reference.
- 202Liu, C.; Kloppenburg, J.; Yao, Y.; Ren, X.; Appel, H.; Kanai, Y.; Blum, V. All-electron ab initio Bethe-Salpeter equation approach to neutral excitations in molecules with numeric atom-centered orbitals. J. Chem. Phys. 2020, 152, 044105, DOI: 10.1063/1.5123290Google ScholarThere is no corresponding record for this reference.
- 203Blase, X.; Duchemin, I.; Jacquemin, D.; Loos, P.-F. The Bethe–Salpeter Equation Formalism: From Physics to Chemistry. J. Phys. Chem. Lett. 2020, 11, 7371– 7382, DOI: 10.1021/acs.jpclett.0c01875Google ScholarThere is no corresponding record for this reference.
- 204Bechstedt, F. Many-Body Approach to Electronic Excitations: Concepts and Applications; Springer Series in Solid-State Sciences; Springer-Verlag: Berlin, Heidelberg, 2015; Vol. 181.Google ScholarThere is no corresponding record for this reference.
- 205Mewes, S. A.; Plasser, F.; Krylov, A.; Dreuw, A. Benchmarking Excited-State Calculations Using Exciton Properties. J. Chem. Theory Comput. 2018, 14, 710– 725, DOI: 10.1021/acs.jctc.7b01145Google ScholarThere is no corresponding record for this reference.
- 206Ullrich, C. A. Time-Dependent Density-Functional Theory: Concepts and Applications; Oxford University Press: New York, 2012.Google ScholarThere is no corresponding record for this reference.
- 207Bruneval, F.; Rangel, T.; Hamed, S. M.; Shao, M.; Yang, C.; Neaton, J. B. Molgw 1: Many-body Perturbation Theory Software for Atoms, Molecules, and Clusters. Comput. Phys. Commun. 2016, 208, 149– 161, DOI: 10.1016/j.cpc.2016.06.019Google ScholarThere is no corresponding record for this reference.
- 208Knysh, I.; Lipparini, F.; Blondel, A.; Duchemin, I.; Blase, X.; Loos, P.-F.; Jacquemin, D. Reference CC3 Excitation Energies for Organic Chromophores: Benchmarking TD-DFT, BSE/GW, and Wave Function Methods. J. Chem. Theory Comput. 2024, 20, 8152– 8174, DOI: 10.1021/acs.jctc.4c00906Google ScholarThere is no corresponding record for this reference.
- 209Kendall, R. A.; Dunning, T. H.; Harrison, R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796– 6806, DOI: 10.1063/1.462569Google ScholarThere is no corresponding record for this reference.
- 210Schreiber, M.; Silva-Junior, M. R.; Sauer, S. P. A.; Thiel, W. Benchmarks for Electronically Excited States: CASPT2, CC2, CCSD, and CC3. J. Chem. Phys. 2008, 128, 134110, DOI: 10.1063/1.2889385Google ScholarThere is no corresponding record for this reference.
- 211Kunert, T.; Schmidt, R. Non-adiabatic quantum molecular dynamics: General formalism and case study H2+ in strong laser fields. Eur. Phys. J. D 2003, 25, 15– 24, DOI: 10.1140/epjd/e2003-00086-8Google ScholarThere is no corresponding record for this reference.
- 212Castro, A.; Marques, M. A. L.; Rubio, A. Propagators for the time-dependent Kohn-Sham equations. J. Chem. Phys. 2004, 121, 3425– 3433, DOI: 10.1063/1.1774980Google ScholarThere is no corresponding record for this reference.
- 213Moler, C.; Loan, C. V. Nineteen Dubious Ways to Compute the Exponential of a Matrix, Twenty-Five Years Later. SIAM Rev. 2003, 45, 3– 49, DOI: 10.1137/s00361445024180Google ScholarThere is no corresponding record for this reference.
- 214Andermatt, S.; Bani-Hashemian, M. H.; Brück, S.; VandeVondele, J.; Luisier, M. Transport simulations with density-matrix-based real-time time-dependant density functional theory. In 2017 International Conference on Simulation of Semiconductor Processes and Devices (SISPAD); Institute of Electrical and Electronics Engineers, 2017, pp 177– 180.Google ScholarThere is no corresponding record for this reference.
- 215Bertsch, G. F.; Iwata, J.-I.; Rubio, A.; Yabana, K. Real-space, real-time method for the dielectric function. Phys. Rev. B 2000, 62, 7998– 8002, DOI: 10.1103/PhysRevB.62.7998Google ScholarThere is no corresponding record for this reference.
- 216Yabana, K.; Sugiyama, T.; Shinohara, Y.; Otobe, T.; Bertsch, G. F. Time-dependent density functional theory for strong electromagnetic fields in crystalline solids. Phys. Rev. B 2012, 85, 045134, DOI: 10.1103/physrevb.85.045134Google ScholarThere is no corresponding record for this reference.
- 217Pemmaraju, C.; Vila, F.; Kas, J.; Sato, S.; Rehr, J.; Yabana, K.; Prendergast, D. Velocity-gauge real-time TDDFT within a numerical atomic orbital basis set. Comput. Phys. Commun. 2018, 226, 30– 38, DOI: 10.1016/j.cpc.2018.01.013Google ScholarThere is no corresponding record for this reference.
- 218Onida, G.; Reining, L.; Rubio, A. Electronic excitations: density-functional versus many-body Green’s-function approaches. Rev. Mod. Phys. 2002, 74, 601– 659, DOI: 10.1103/RevModPhys.74.601Google ScholarThere is no corresponding record for this reference.
- 219Marek, S.; Wilhelm, J. Linear and Nonlinear Optical Properties of Molecules from Real-Time Propagation Based on the Bethe-Salpeter Equation. J. Chem. Theory Comput. 2025, 21, 9814– 9822, DOI: 10.1021/acs.jctc.5c01246Google ScholarThere is no corresponding record for this reference.
- 220Attaccalite, C.; Grüning, M.; Marini, A. Real-time approach to the optical properties of solids and nanostructures: Time-dependent Bethe-Salpeter equation. Phys. Rev. B 2011, 84, 245110, DOI: 10.1103/PhysRevB.84.245110Google ScholarThere is no corresponding record for this reference.
- 221Casida, M. E.; Chong, D. P. Physical interpretation and assessment of the Coulomb-hole and screened-exchange approximation for molecules. Phys. Rev. A 1989, 40, 4837– 4848, DOI: 10.1103/PhysRevA.40.4837Google ScholarThere is no corresponding record for this reference.
- 222Farid, B.; Daling, R.; Lenstra, D.; van Haeringen, W. GW approach to the calculation of electron self-energies in semiconductors. Phys. Rev. B 1988, 38, 7530– 7534, DOI: 10.1103/PhysRevB.38.7530Google ScholarThere is no corresponding record for this reference.
- 223Betzinger, M.; Friedrich, C.; Görling, A.; Blügel, S. Precise all-electron dynamical response functions: Application to COHSEX and the RPA correlation energy. Phys. Rev. B 2015, 92, 245101, DOI: 10.1103/PhysRevB.92.245101Google ScholarThere is no corresponding record for this reference.
- 224O’Rourke, C.; Bowler, D. R. Linear scaling density matrix real time TDDFT: Propagator unitarity and matrix truncation. J. Chem. Phys. 2015, 143, 102801, DOI: 10.1063/1.4919128Google ScholarThere is no corresponding record for this reference.
- 225Mattiat, J.; Luber, S. Comparison of Length, Velocity, and Symmetric Gauges for the Calculation of Absorption and Electric Circular Dichroism Spectra with Real-Time Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2022, 18, 5513– 5526, DOI: 10.1021/acs.jctc.2c00644Google ScholarThere is no corresponding record for this reference.
- 226Müller, C.; Sharma, M.; Sierka, M. Real-time time-dependent density functional theory using density fitting and the continuous fast multipole method. J. Comput. Chem. 2020, 41, 2573– 2582, DOI: 10.1002/jcc.26412Google ScholarThere is no corresponding record for this reference.
- 227Yabana, K.; Nakatsukasa, T.; Iwata, J.-I.; Bertsch, G. F. Real-time, real-space implementation of the linear response time-dependent density-functional theory. Phys. Status Solidi B 2006, 243, 1121– 1138, DOI: 10.1002/pssb.200642005Google ScholarThere is no corresponding record for this reference.
- 228Li, X.; Govind, N.; Isborn, C.; DePrince, A. E., III; Lopata, K. Real-time time-dependent electronic structure theory. Chem. Rev. 2020, 120, 9951– 9993, DOI: 10.1021/acs.chemrev.0c00223Google ScholarThere is no corresponding record for this reference.
- 229Mattiat, J.; Luber, S. Efficient calculation of (resonance) Raman spectra and excitation profiles with real-time propagation. J. Chem. Phys. 2018, 149, 174108, DOI: 10.1063/1.5051250Google ScholarThere is no corresponding record for this reference.
- 230Bruner, A.; LaMaster, D.; Lopata, K. Accelerated Broadband Spectra Using Transition Dipole Decomposition and Padé Approximants. J. Chem. Theory Comput. 2016, 12, 3741– 3750, DOI: 10.1021/acs.jctc.6b00511Google ScholarThere is no corresponding record for this reference.
- 231Leucke, M.; Panadés-Barrueta, R. L.; Bas, E. E.; Golze, D. Analytic continuation component of the GreenX library: robust Padé approximants with symmetry constraints. J. Open Source Softw. 2025, 10, 7859, DOI: 10.21105/joss.07859Google ScholarThere is no corresponding record for this reference.
- 232Barbatti, M.; Bondanza, M.; Crespo-Otero, R.; Demoulin, B.; Dral, P. O.; Granucci, G.; Kossoski, F.; Lischka, H.; Mennucci, B.; Mukherjee, S. Newton-X Platform: New Software Developments for Surface Hopping and Nuclear Ensembles. J. Chem. Theory Comput. 2022, 18, 6851– 6865, DOI: 10.1021/acs.jctc.2c00804Google ScholarThere is no corresponding record for this reference.
- 233Barbatti, M.; Ruckenbauer, M.; Plasser, F.; Pittner, J.; Granucci, G.; Persico, M.; Lischka, H. Newton-X: a surface-hopping program for nonadiabatic molecular dynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 26– 33, DOI: 10.1002/wcms.1158Google ScholarThere is no corresponding record for this reference.
- 234Barbatti, M.; Granucci, G.; Persico, M.; Ruckenbauer, M.; Vazdar, M.; Eckert-Maksić, M.; Lischka, H. The on-the-fly surface-hopping program system Newton-X: Application to ab initio simulation of the nonadiabatic photodynamics of benchmark systems. J. Photochem. Photobiol., A 2007, 190, 228– 240, DOI: 10.1016/j.jphotochem.2006.12.008Google ScholarThere is no corresponding record for this reference.
- 235Cederbaum, L. S.; Domcke, W.; Schirmer, J. Many-body theory of core holes. Phys. Rev. A 1980, 22, 206, DOI: 10.1103/PhysRevA.22.206Google ScholarThere is no corresponding record for this reference.
- 236DeBeer George, S.; Petrenko, T.; Neese, F. Time-dependent density functional calculations of ligand K-edge X-ray absorption spectra. Inorg. Chim. Acta 2008, 361, 965– 972, DOI: 10.1016/j.ica.2007.05.046Google ScholarThere is no corresponding record for this reference.
- 237Müller, P.; Karhan, K.; Krack, M.; Gerstmann, U.; Schmidt, W. G.; Bauer, M.; Kühne, T. D. Impact of finite-temperature and condensed-phase effects on theoretical X-ray absorption spectra of transition metal complexes. J. Comput. Chem. 2019, 40, 712– 716, DOI: 10.1002/jcc.25641Google ScholarThere is no corresponding record for this reference.
- 238Franco de Carvalho, F.; Curchod, B. F.; Penfold, T. J.; Tavernelli, I. Derivation of spin-orbit couplings in collinear linear-response TDDFT: A rigorous formulation. J. Chem. Phys. 2014, 140, 144103, DOI: 10.1063/1.4870010Google ScholarThere is no corresponding record for this reference.
- 239Bussy, A.; Hutter, J. Data for: Efficient and low-scaling linear-response time-dependent density functional theory implementation for core-level spectroscopy of large and periodic systems, 2021. accessed: 2025–08–23. DOI: 10.24435/materialscloud:js-me .Google ScholarThere is no corresponding record for this reference.
- 240Lopata, K.; Van Kuiken, B.; Khalil, M.; Govind, N. Linear-response and real-time time-dependent density functional theory studies of core-level near-edge X-ray absorption. J. Chem. Theory Comput. 2012, 8, 3284– 3292, DOI: 10.1021/ct3005613Google ScholarThere is no corresponding record for this reference.
- 241Khaliullin, R. Z.; Cobar, E. A.; Lochan, R. C.; Bell, A. T.; Head-Gordon, M. Unravelling the Origin of Intermolecular Interactions Using Absolutely Localized Molecular Orbitals. J. Phys. Chem. A 2007, 111, 8753– 8765, DOI: 10.1021/jp073685zGoogle ScholarThere is no corresponding record for this reference.
- 242Khaliullin, R. Z.; Kühne, T. D. Microscopic properties of liquid water from combined ab initio molecular dynamics and energy decomposition studies. Phys. Chem. Chem. Phys. 2013, 15, 15746– 15766, DOI: 10.1039/c3cp51039eGoogle ScholarThere is no corresponding record for this reference.
- 243Mo, Y.; Gao, J.; Peyerimhoff, S. D. Energy decomposition analysis of intermolecular interactions using a block-localized wave function approach. J. Chem. Phys. 2000, 112, 5530– 5538, DOI: 10.1063/1.481185Google ScholarThere is no corresponding record for this reference.
- 244Azar, R. J.; Horn, P. R.; Sundstrom, E. J.; Head-Gordon, M. Useful lower limits to polarization contributions to intermolecular interactions using a minimal basis of localized orthogonal orbitals: Theory and analysis of the water dimer. J. Chem. Phys. 2013, 138, 084102, DOI: 10.1063/1.4792434Google ScholarThere is no corresponding record for this reference.
- 245Stoll, H.; Wagenblast, G.; Preuß, H. On the use of local basis sets for localized molecular orbitals. Theor. Chim. Acta 1980, 57, 169– 178, DOI: 10.1007/BF00574903Google ScholarThere is no corresponding record for this reference.
- 246Khaliullin, R. Z.; Head-Gordon, M.; Bell, A. T. An efficient self-consistent field method for large systems of weakly interacting components. J. Chem. Phys. 2006, 124, 204105, DOI: 10.1063/1.2191500Google ScholarThere is no corresponding record for this reference.
- 247Khaliullin, R. Z.; Bell, A. T.; Head-Gordon, M. Analysis of charge transfer effects in molecular complexes based on absolutely localized molecular orbitals. J. Chem. Phys. 2008, 128, 184112, DOI: 10.1063/1.2912041Google ScholarThere is no corresponding record for this reference.
- 248Khaliullin, R. Z.; VandeVondele, J.; Hutter, J. Efficient Linear-Scaling Density Functional Theory for Molecular Systems. J. Chem. Theory Comput. 2013, 9, 4421– 4427, DOI: 10.1021/ct400595kGoogle ScholarThere is no corresponding record for this reference.
- 249Staub, R.; Iannuzzi, M.; Khaliullin, R. Z.; Steinmann, S. N. Energy Decomposition Analysis for Metal Surface,ÄìAdsorbate Interactions by Block Localized Wave Functions. J. Chem. Theory Comput. 2019, 15, 265– 275, DOI: 10.1021/acs.jctc.8b00957Google ScholarThere is no corresponding record for this reference.
- 250Shi, Y.; Scheiber, H.; Khaliullin, R. Z. Contribution of the Covalent Component of the Hydrogen-Bond Network to the Properties of Liquid Water. J. Phys. Chem. A 2018, 122, 7482– 7490, DOI: 10.1021/acs.jpca.8b06857Google ScholarThere is no corresponding record for this reference.
- 251Scheiber, H.; Shi, Y.; Khaliullin, R. Z. Communication: Compact orbitals enable low-cost linear-scaling ab initio molecular dynamics for weakly-interacting systems. J. Chem. Phys. 2018, 148, 231103, DOI: 10.1063/1.5029939Google ScholarThere is no corresponding record for this reference.
- 252Luo, Z.; Friscic, T.; Khaliullin, R. Z. Why pregnenolone and progesterone, two structurally similar steroids, exhibit remarkably different cocrystallization with aromatic molecules. Phys. Chem. Chem. Phys. 2018, 20, 898– 904, DOI: 10.1039/C7CP06828JGoogle ScholarThere is no corresponding record for this reference.
- 253Ojha, D.; Kaliannan, N. K.; Kühne, T. D. Time-dependent vibrational sum-frequency generation spectroscopy of the air-water interface. Commun. Chem. 2019, 2, 116, DOI: 10.1038/s42004-019-0220-6Google ScholarThere is no corresponding record for this reference.
- 254Ojha, D.; Kühne, T. D. Hydrogen bond dynamics of interfacial water molecules revealed from two-dimensional vibrational sum-frequency generation spectroscopy. Sci. Rep. 2021, 11, 2456, DOI: 10.1038/s41598-021-81635-4Google ScholarThere is no corresponding record for this reference.
- 255Kühne, T. D.; Khaliullin, R. Z. Electronic signature of the instantaneous asymmetry in the first coordination shell of liquid water. Nat. Commun. 2013, 4, 1450, DOI: 10.1038/ncomms2459Google ScholarThere is no corresponding record for this reference.
- 256Elgabarty, H.; Khaliullin, R. Z.; Kühne, T. D. Covalency of hydrogen bonds in liquid water can be probed by proton nuclear magnetic resonance experiments. Nat. Commun. 2015, 6, 8318, DOI: 10.1038/ncomms9318Google ScholarThere is no corresponding record for this reference.
- 257Elgabarty, H.; Kühne, T. D. Tumbling with a limp: local asymmetry in water’s hydrogen bond network and its consequences. Phys. Chem. Chem. Phys. 2020, 22, 10397– 10411, DOI: 10.1039/C9CP06960GGoogle ScholarThere is no corresponding record for this reference.
- 258Kühne, T. D.; Khaliullin, R. Z. Nature of the Asymmetry in the Hydrogen-Bond Networks of Hexagonal Ice and Liquid Water. J. Am. Chem. Soc. 2014, 136, 3395– 3399, DOI: 10.1021/ja411161aGoogle ScholarThere is no corresponding record for this reference.
- 259Yun, Y.; Khaliullin, R. Z.; Jung, Y. Low-Dimensional Confined Ice Has the Electronic Signature of Liquid Water. J. Phys. Chem. Lett. 2019, 10, 2008– 2016, DOI: 10.1021/acs.jpclett.9b00921Google ScholarThere is no corresponding record for this reference.
- 260Salem, M. A.; Kühne, T. D. Insight from energy decomposition analysis on a hydrogen-bond-mediated mechanism for on-water catalysis. Mol. Phys. 2020, 118, e1797920 DOI: 10.1080/00268976.2020.1797920Google ScholarThere is no corresponding record for this reference.
- 261Heske, J.; Kühne, T. D.; Antonietti, M. Water in PHI nanopores: modeling adsorption, solvent structure, and thermodynamics. ACS Omega 2023, 8, 26526– 26532, DOI: 10.1021/acsomega.3c03308Google ScholarThere is no corresponding record for this reference.
- 262Ramos-Cordoba, E.; Lambrecht, D. S.; Head-Gordon, M. Charge-transfer and the hydrogen bond: Spectroscopic and structural implications from electronic structure calculations. Faraday Discuss. 2011, 150, 345– 362, DOI: 10.1039/c1fd00004gGoogle ScholarThere is no corresponding record for this reference.
- 263Fransson, T.; Harada, Y.; Kosugi, N.; Besley, N. A.; Winter, B.; Rehr, J. J.; Pettersson, L. G. M.; Nilsson, A. X-ray and Electron Spectroscopy of Water. Chem. Rev. 2016, 116, 7551– 7569, DOI: 10.1021/acs.chemrev.5b00672Google ScholarThere is no corresponding record for this reference.
- 264Lenz, A.; Ojamäe, L. Theoretical IR Spectra for Water Clusters (H2O)n(n= 6–22, 28, 30) and Identification of Spectral Contributions from Different H-Bond Conformations in Gaseous and Liquid Water. J. Phys. Chem. A 2006, 110, 13388– 13393, DOI: 10.1021/jp066372xGoogle ScholarThere is no corresponding record for this reference.
- 265Ojha, D.; Karhan, K.; Kühne, T. D. On the hydrogen bond strength and vibrational spectroscopy of liquid water. Sci. Rep. 2018, 8, 16888, DOI: 10.1038/s41598-018-35357-9Google ScholarThere is no corresponding record for this reference.
- 266Elgabarty, H.; Kaliannan, N. K.; Kühne, T. D. Enhancement of the local asymmetry in the hydrogen bond network of liquid water by an ultrafast electric field pulse. Sci. Rep. 2019, 9, 10002, DOI: 10.1038/s41598-019-46449-5Google ScholarThere is no corresponding record for this reference.
- 267Yun, Y.; Khaliullin, R. Z.; Jung, Y. Correlated Local Fluctuations in the Hydrogen Bond Network of Liquid Water. J. Am. Chem. Soc. 2022, 144, 13127– 13136, DOI: 10.1021/jacs.2c02362Google ScholarThere is no corresponding record for this reference.
- 268Balos, V.; Kaliannan, N. K.; Elgabarty, H.; Wolf, M.; Kühne, T. D.; Sajadi, M. Time-resolved terahertz–Raman spectroscopy reveals that cations and anions distinctly modify intermolecular interactions of water. Nat. Chem. 2022, 14, 1031– 1037, DOI: 10.1038/s41557-022-00977-2Google ScholarThere is no corresponding record for this reference.
- 269McGrath, M. J.; Siepmann, J. I.; Kuo, I.-F. W.; Mundy, C. J.; VandeVondele, J.; Sprik, M.; Hutter, J.; Mohamed, F.; Krack, M.; Parrinello, M. Toward a Monte Carlo program for simulating vapor–liquid phase equilibria from first principles. Comput. Phys. Commun. 2005, 169, 289– 294, DOI: 10.1016/j.cpc.2005.03.065Google ScholarThere is no corresponding record for this reference.
- 270McGrath, M. J.; Siepmann, J. I.; Kuo, I.-F. W.; Mundy, C. J.; VandeVondele, J.; Hutter, J.; Mohamed, F.; Krack, M. Isobaric–isothermal Monte Carlo simulations from first principles: application to liquid water at ambient conditions. ChemPhysChem 2005, 6, 1894– 1901, DOI: 10.1002/cphc.200400580Google ScholarThere is no corresponding record for this reference.
- 271McGrath, M. J.; Siepmann, J. I.; Kuo, I.-F. W.; Mundy, C. J.; VandeVondele, J.; Hutter, J.; Mohamed, F.; Krack, M. Simulating fluid-phase equilibria of water from first principles. J. Phys. Chem. A 2006, 110, 640– 646, DOI: 10.1021/jp0535947Google ScholarThere is no corresponding record for this reference.
- 272Kuo, I. W.; Mundy, C. J.; Eggimann, B. L.; McGrath, M. J.; Siepmann, J. I.; Chen, B.; Vieceli, J.; Tobias, D. J. Structure and Dynamics of the Aqueous Liquid- Vapor Interface: A Comprehensive Particle-Based Simulation Study. J. Phys. Chem. B 2006, 110, 3738– 3746, DOI: 10.1021/jp056330tGoogle ScholarThere is no corresponding record for this reference.
- 273Iannuzzi, M.; Laio, A.; Parrinello, M. Efficient Exploration of Reactive Potential Energy Surfaces Using Car–Parrinello Molecular Dynamics. Phys. Rev. Lett. 2003, 90, 238302, DOI: 10.1103/PhysRevLett.90.238302Google ScholarThere is no corresponding record for this reference.
- 274Schade, R.; Kenter, T.; Elgabarty, H.; Lass, M.; Schütt, O.; Lazzaro, A.; Pabst, H.; Mohr, S.; Hutter, J.; Kühne, T. D. Towards electronic structure-based ab-initio molecular dynamics simulations with hundreds of millions of atoms. Parallel Comput. 2022, 111, 102920, DOI: 10.1016/j.parco.2022.102920Google ScholarThere is no corresponding record for this reference.
- 275Schade, R.; Kenter, T.; Elgabarty, H.; Lass, M.; Kühne, T. D.; Plessl, C. Breaking the exascale barrier for the electronic structure problem in ab-initio molecular dynamics. Int. J. High Perform. Comput. Appl. 2023, 37, 530– 538, DOI: 10.1177/10943420231177631Google ScholarThere is no corresponding record for this reference.
- 276Arias, T. A.; Payne, M. C.; Joannopoulos, J. D. Ab initio molecular dynamics: Analytically continued energy functionals and insights into iterative solutions. Phys. Rev. Lett. 1992, 69, 1077– 1080, DOI: 10.1103/PhysRevLett.69.1077Google ScholarThere is no corresponding record for this reference.
- 277Arias, T. A.; Payne, M. C.; Joannopoulos, J. D. Ab initio molecular-dynamics techniques extended to large-length-scale systems. Phys. Rev. B 1992, 45, 1538– 1549, DOI: 10.1103/PhysRevB.45.1538Google ScholarThere is no corresponding record for this reference.
- 278Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558, DOI: 10.1103/PhysRevB.47.558Google ScholarThere is no corresponding record for this reference.
- 279Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251, DOI: 10.1103/PhysRevB.49.14251Google ScholarThere is no corresponding record for this reference.
- 280Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471– 2474, DOI: 10.1103/PhysRevLett.55.2471Google ScholarThere is no corresponding record for this reference.
- 281Caravati, S.; Bernasconi, M.; Kühne, T.; Krack, M.; Parrinello, M. Coexistence of tetrahedral-and octahedral-like sites in amorphous phase change materials. Appl. Phys. Lett. 2007, 91, 171906, DOI: 10.1063/1.2801626Google ScholarThere is no corresponding record for this reference.
- 282Kühne, T. D.; Krack, M.; Parrinello, M. Static and Dynamical Properties of Liquid Water from First Principles by a Novel Car-Parrinello-like Approach. J. Chem. Theory Comput. 2009, 5, 235– 241, DOI: 10.1021/ct800417qGoogle ScholarThere is no corresponding record for this reference.
- 283Cucinotta, C. S.; Miceli, G.; Raiteri, P.; Krack, M.; Kühne, T. D.; Bernasconi, M.; Parrinello, M. Superionic Conduction in Substoichiometric LiAl Alloy: An Ab Initio Study. Phys. Rev. Lett. 2009, 103, 125901, DOI: 10.1103/PhysRevLett.103.125901Google ScholarThere is no corresponding record for this reference.
- 284Caravati, S.; Bernasconi, M.; Kühne, T. D.; Krack, M.; Parrinello, M. Unravelling the Mechanism of Pressure Induced Amorphization of Phase Change Materials. Phys. Rev. Lett. 2009, 102, 205502, DOI: 10.1103/PhysRevLett.102.205502Google ScholarThere is no corresponding record for this reference.
- 285Luduena, G. A.; Kühne, T. D.; Sebastiani, D. Mixed Grotthuss and Vehicle Transport Mechanism in Proton Conducting Polymers from Ab initio Molecular Dynamics Simulations. Chem. Mater. 2011, 23, 1424– 1429, DOI: 10.1021/cm102674uGoogle ScholarThere is no corresponding record for this reference.
- 286Kühne, T. D.; Pascal, T. A.; Kaxiras, E.; Jung, Y. New Insights into the Structure of the Vapor/Water Interface from Large-Scale First-Principles Simulations. J. Phys. Chem. Lett. 2011, 2, 105– 113, DOI: 10.1021/jz101391rGoogle ScholarThere is no corresponding record for this reference.
- 287Behler, J. Perspective: Machine learning potentials for atomistic simulations. J. Chem. Phys. 2016, 145, 170901, DOI: 10.1063/1.4966192Google ScholarThere is no corresponding record for this reference.
- 288Behler, J. First Principles Neural Network Potentials for Reactive Simulations of Large Molecular and Condensed Systems. Angew. Chem., Int. Ed. 2017, 56, 12828– 12840, DOI: 10.1002/anie.201703114Google ScholarThere is no corresponding record for this reference.
- 289Bartók, A. P.; De, S.; Poelking, C.; Bernstein, N.; Kermode, J. R.; Csányi, G.; Ceriotti, M. Machine learning unifies the modeling of materials and molecules. Sci. Adv. 2017, 3, e1701816 DOI: 10.1126/sciadv.1701816Google ScholarThere is no corresponding record for this reference.
- 290Butler, K. T.; Davies, D. W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547– 555, DOI: 10.1038/s41586-018-0337-2Google ScholarThere is no corresponding record for this reference.
- 291Deringer, V. L.; Caro, M. A.; Csányi, G. Machine Learning Interatomic Potentials as Emerging Tools for Materials Science. Adv. Mater. 2019, 31, 1902765, DOI: 10.1002/adma.201902765Google ScholarThere is no corresponding record for this reference.
- 292Kang, P. L.; Shang, C.; Liu, Z. P. Large-Scale Atomic Simulation via Machine Learning Potentials Constructed by Global Potential Energy Surface Exploration. Acc. Chem. Res. 2020, 53, 2119– 2129, DOI: 10.1021/acs.accounts.0c00472Google ScholarThere is no corresponding record for this reference.
- 293Behler, J. Four Generations of High-Dimensional Neural Network Potentials. Chem. Rev. 2021, 121, 10037– 10072, DOI: 10.1021/acs.chemrev.0c00868Google ScholarThere is no corresponding record for this reference.
- 294Fiedler, L.; Shah, K.; Bussmann, M.; Cangi, A. Deep dive into machine learning density functional theory for materials science and chemistry. Phys. Rev. Mater. 2022, 6, 040301, DOI: 10.1103/PhysRevMaterials.6.040301Google ScholarThere is no corresponding record for this reference.
- 295Behler, J.; Parrinello, M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 2007, 98, 146401, DOI: 10.1103/PhysRevLett.98.146401Google ScholarThere is no corresponding record for this reference.
- 296Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 2011, 134, 074106, DOI: 10.1063/1.3553717Google ScholarThere is no corresponding record for this reference.
- 297Natarajan, S. K.; Morawietz, T.; Behler, J. Representing the potential-energy surface of protonated water clusters by high-dimensional neural network potentials. Phys. Chem. Chem. Phys. 2015, 17, 8356– 8371, DOI: 10.1039/c4cp04751fGoogle ScholarThere is no corresponding record for this reference.
- 298Schran, C.; Behler, J.; Marx, D. Automated Fitting of Neural Network Potentials at Coupled Cluster Accuracy: Protonated Water Clusters as Testing Ground. J. Chem. Theory Comput. 2020, 16, 88– 99, DOI: 10.1021/acs.jctc.9b00805Google ScholarThere is no corresponding record for this reference.
- 299Morawietz, T.; Singraber, A.; Dellago, C.; Behler, J. How van der Waals interactions determine the unique properties of water. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 8368– 8373, DOI: 10.1073/pnas.1602375113Google ScholarThere is no corresponding record for this reference.
- 300Daru, J.; Forbert, H.; Behler, J.; Marx, D. Coupled Cluster Molecular Dynamics of Condensed Phase Systems Enabled by Machine Learning Potentials: Liquid Water Benchmark. Phys. Rev. Lett. 2022, 129, 226001, DOI: 10.1103/PhysRevLett.129.226001Google ScholarThere is no corresponding record for this reference.
- 301Quaranta, V.; Hellström, M.; Behler, J. Proton-Transfer Mechanisms at the Water-ZnO Interface: The Role of Presolvation. J. Phys. Chem. Lett. 2017, 8, 1476– 1483, DOI: 10.1021/acs.jpclett.7b00358Google ScholarThere is no corresponding record for this reference.
- 302Behler, J.; Martoňák, R.; Donadio, D.; Parrinello, M. Metadynamics simulations of the high-pressure phases of silicon employing a high-dimensional neural network potential. Phys. Rev. Lett. 2008, 100, 185501, DOI: 10.1103/PhysRevLett.100.185501Google ScholarThere is no corresponding record for this reference.
- 303Eshet, H.; Khaliullin, R. Z.; Kühne, T. D.; Behler, J.; Parrinello, M. Ab initio quality neural-network potential for sodium. Phys. Rev. B 2010, 81, 184107, DOI: 10.1103/PhysRevB.81.184107Google ScholarThere is no corresponding record for this reference.
- 304Eshet, H.; Khaliullin, R. Z.; Kühne, T. D.; Behler, J.; Parrinello, M. Microscopic origins of the anomalous melting behavior of sodium under high pressure. Phys. Rev. Lett. 2012, 108, 115701, DOI: 10.1103/PhysRevLett.108.115701Google ScholarThere is no corresponding record for this reference.
- 305Eckhoff, M.; Behler, J. From Molecular Fragments to the Bulk: Development of a Neural Network Potential for MOF-5. J. Chem. Theory Comput. 2019, 15, 3793– 3809, DOI: 10.1021/acs.jctc.8b01288Google ScholarThere is no corresponding record for this reference.
- 306Eckhoff, M.; Behler, J. Insights into lithium manganese oxide-water interfaces using machine learning potentials. J. Chem. Phys. 2021, 155, 244703, DOI: 10.1063/5.0073449Google ScholarThere is no corresponding record for this reference.
- 307Khaliullin, R. Z.; Eshet, H.; Kühne, T. D.; Behler, J.; Parrinello, M. Graphite-diamond phase coexistence study employing a neural-network mapping of the ab initio potential energy surface. Phys. Rev. B 2010, 81, 100103, DOI: 10.1103/PhysRevB.81.100103Google ScholarThere is no corresponding record for this reference.
- 308Khaliullin, R. Z.; Eshet, H.; Kühne, T. D.; Behler, J.; Parrinello, M. Nucleation mechanism for the direct graphite-to-diamond phase transition. Nat. Mater. 2011, 10, 693– 697, DOI: 10.1038/nmat3078Google ScholarThere is no corresponding record for this reference.
- 309Omranpour, A.; Montero De Hijes, P.; Behler, J.; Dellago, C. Perspective: Atomistic simulations of water and aqueous systems with machine learning potentials. J. Chem. Phys. 2024, 160, 170901, DOI: 10.1063/5.0201241Google ScholarThere is no corresponding record for this reference.
- 310Artrith, N.; Morawietz, T.; Behler, J. High-dimensional neural-network potentials for multicomponent systems: Applications to zinc oxide. Phys. Rev. B 2011, 83, 153101, DOI: 10.1103/PhysRevB.83.153101Google ScholarThere is no corresponding record for this reference.
- 311Ko, T. W.; Finkler, J. A.; Goedecker, S.; Behler, J. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun. 2021, 12, 398, DOI: 10.1038/s41467-020-20427-2Google ScholarThere is no corresponding record for this reference.
- 312Khajehpasha, E. R.; Finkler, J. A.; Kühne, T. D.; Ghasemi, S. A. CENT2: Improved charge equilibration via neural network technique. Phys. Rev. B 2022, 105, 144106, DOI: 10.1103/PhysRevB.105.144106Google ScholarThere is no corresponding record for this reference.
- 313Thiemann, F. L.; O’Neill, N.; Kapil, V.; Michaelides, A.; Schran, C. Introduction to machine learning potentials for atomistic simulations. J. Phys.: Condens. Matter 2025, 37, 073002, DOI: 10.1088/1361-648X/ad9657Google ScholarThere is no corresponding record for this reference.
- 314Behler, J. Neural network potential-energy surfaces in chemistry: a tool for large-scale simulations. Phys. Chem. Chem. Phys. 2011, 13, 17930– 17955, DOI: 10.1039/c1cp21668fGoogle ScholarThere is no corresponding record for this reference.
- 315Behler, J. Constructing high-dimensional neural network potentials: A tutorial review. Int. J. Quantum Chem. 2015, 115, 1032– 1050, DOI: 10.1002/qua.24890Google ScholarThere is no corresponding record for this reference.
- 316Brieuc, F.; Schran, C.; Forbert, H.; Marx, D. RubNNet4MD: Ruhr-Universitat Bochum Neural Networks for Molecular Dynamics Simulations. Software Package 2020–2025. DOI: 10.17877/RESOLV-2025-MDN6B2J4 .Google ScholarThere is no corresponding record for this reference.
- 317Drautz, R. Atomic cluster expansion for accurate and transferable interatomic potentials. Phys. Rev. B 2019, 99, 014104, DOI: 10.1103/PhysRevB.99.014104Google ScholarThere is no corresponding record for this reference.
- 318(a) Bochkarev, A.; Lysogorskiy, Y.; Drautz, R. Graph Atomic Cluster Expansion for Semilocal Interactions beyond Equivariant Message Passing. Phys. Rev. X 2024, 14, 021036, DOI: 10.1103/PhysRevX.14.021036Google ScholarThere is no corresponding record for this reference.https://github.com/ICAMS/lammps-user-pace.Google ScholarThere is no corresponding record for this reference.
- 319Zeng, J.; Zhang, D.; Lu, D.; Mo, P.; Li, Z.; Chen, Y.; Rynik, M.; Huang, L.; Li, Z.; Shi, S. DeePMD-kit v2: A software package for deep potential models. J. Chem. Phys. 2023, 159, 054801, DOI: 10.1063/5.0155600Google ScholarThere is no corresponding record for this reference.
- 320Batzner, S.; Musaelian, A.; Sun, L.; Geiger, M.; Mailoa, J. P.; Kornbluth, M.; Molinari, N.; Smidt, T. E.; Kozinsky, B. E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 2022, 13, 2453, DOI: 10.1038/s41467-022-29939-5Google ScholarThere is no corresponding record for this reference.
- 321Musaelian, A.; Batzner, S.; Johansson, A.; Sun, L.; Owen, C. J.; Kornbluth, M.; Kozinsky, B. Learning local equivariant representations for large-scale atomistic dynamics. Nat. Commun. 2023, 14, 579, DOI: 10.1038/s41467-023-36329-yGoogle ScholarThere is no corresponding record for this reference.
- 322Schran, C.; Brezina, K.; Marsalek, O. Committee neural network potentials control generalization errors and enable active learning. J. Chem. Phys. 2020, 153, 104105, DOI: 10.1063/5.0016004Google ScholarThere is no corresponding record for this reference.
- 323Imbalzano, G.; Zhuang, Y.; Kapil, V.; Rossi, K.; Engel, E. A.; Grasselli, F.; Ceriotti, M. Uncertainty estimation for molecular dynamics and sampling. J. Chem. Phys. 2021, 154, 074102, DOI: 10.1063/5.0036522Google ScholarThere is no corresponding record for this reference.
- 324Gastegger, M.; Behler, J.; Marquetand, P. Machine learning molecular dynamics for the simulation of infrared spectra. Chem. Sci. 2017, 8, 6924– 6935, DOI: 10.1039/C7SC02267KGoogle ScholarThere is no corresponding record for this reference.
- 325Podryabinkin, E. V.; Shapeev, A. V. Active learning of linearly parametrized interatomic potentials. Comput. Mater. Sci. 2017, 140, 171– 180, DOI: 10.1016/j.commatsci.2017.08.031Google ScholarThere is no corresponding record for this reference.
- 326Smith, J. S.; Nebgen, B.; Lubbers, N.; Isayev, O.; Roitberg, A. E. Less is more: Sampling chemical space with active learning. J. Chem. Phys. 2018, 148, 241733, DOI: 10.1063/1.5023802Google ScholarThere is no corresponding record for this reference.
- 327Schran, C.; Thiemann, F. L.; Rowe, P.; Müller, E. A.; Marsalek, O.; Michaelides, A. Machine learning potentials for complex aqueous systems made simple. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2110077118 DOI: 10.1073/pnas.2110077118Google ScholarThere is no corresponding record for this reference.
- 328Stolte, N.; Daru, J.; Forbert, H.; Marx, D.; Behler, J. Random sampling versus active learning algorithms for machine learning potentials of quantum liquid water. J. Chem. Theory Comput. 2025, 21, 886– 899, DOI: 10.1021/acs.jctc.4c01382Google ScholarThere is no corresponding record for this reference.
- 329Marx, D.; Tuckerman, M. E.; Hutter, J.; Parrinello, M. The nature of the hydrated excess proton in water. Nature 1999, 397, 601– 604, DOI: 10.1038/17579Google ScholarThere is no corresponding record for this reference.
- 330Marx, D.. In Computer Simulations in Condensed Matter: From Materials to Chemical Biology (Lecture Notes in Physics 704); Ferrario, M., Binder, K., Ciccotti, G., Eds.; Springer, 2006; Chapter 2, pp 507– 539.Google ScholarThere is no corresponding record for this reference.
- 331Marx, D. Proton Transfer 200 Years after von Grotthuss: Insights from Ab Initio Simulations. ChemPhysChem 2006, 7, 1848– 1870, DOI: 10.1002/cphc.200600128Google ScholarThere is no corresponding record for this reference.
- 332Li, X.-Z.; Walker, B.; Michaelides, A. Quantum nature of the hydrogen bond. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6369– 6373, DOI: 10.1073/pnas.1016653108Google ScholarThere is no corresponding record for this reference.
- 333Singh, R.; Azadi, S.; Kühne, T. D. Anharmonicity and finite-temperature effects on the structure, stability, and vibrational spectrum of phase III of solid molecular hydrogen. Phys. Rev. B 2014, 90, 014110, DOI: 10.1103/PhysRevB.90.014110Google ScholarThere is no corresponding record for this reference.
- 334Spura, T.; Elgabarty, H.; Kühne, T. D. “On-the-fly” coupled cluster path-integral molecular dynamics: impact of nuclear quantum effects on the protonated water dimer. Phys. Chem. Chem. Phys. 2015, 17, 14355– 14359, DOI: 10.1039/C4CP05192KGoogle ScholarThere is no corresponding record for this reference.
- 335Kessler, J.; Elgabarty, H.; Spura, T.; Karhan, K.; Partovi-Azar, P.; Hassanali, A. A.; Kühne, T. D. Structure and dynamics of the instantaneous water/vapor interface revisited by path-integral and ab initio molecular dynamics simulations. J. Phys. Chem. B 2015, 119, 10079– 10086, DOI: 10.1021/acs.jpcb.5b04185Google ScholarThere is no corresponding record for this reference.
- 336Marsalek, O.; Markland, T. E. Quantum Dynamics and Spectroscopy of Ab Initio Liquid Water: The Interplay of Nuclear and Electronic Quantum Effects. J. Phys. Chem. Lett. 2017, 8, 1545– 1551, DOI: 10.1021/acs.jpclett.7b00391Google ScholarThere is no corresponding record for this reference.
- 337Azadi, S.; Singh, R.; Kühne, T. D. Nuclear quantum effects induce metallization of dense solid molecular hydrogen. J. Comput. Chem. 2018, 39, 262– 268, DOI: 10.1002/jcc.25104Google ScholarThere is no corresponding record for this reference.
- 338Ojha, D.; Henao, A.; Kühne, T. D. Nuclear quantum effects on the vibrational dynamics of liquid water. J. Chem. Phys. 2018, 148, 102328, DOI: 10.1063/1.5005500Google ScholarThere is no corresponding record for this reference.
- 339Clark, T.; Heske, J.; Kühne, T. D. Opposing electronic and nuclear quantum effects on hydrogen bonds in H2O and D2O. ChemPhysChem 2019, 20, 2461– 2465, DOI: 10.1002/cphc.201900839Google ScholarThere is no corresponding record for this reference.
- 340Schran, C.; Marx, D. Quantum nature of the hydrogen bond from ambient conditions down to ultra-low temperatures. Phys. Chem. Chem. Phys. 2019, 21, 24967– 24975, DOI: 10.1039/C9CP04795FGoogle ScholarThere is no corresponding record for this reference.
- 341Ohto, T.; Dodia, M.; Xu, J.; Imoto, S.; Tang, F.; Zysk, F.; Kühne, T. D.; Shigeta, Y.; Bonn, M.; Wu, X. Accessing the accuracy of density functional theory through structure and dynamics of the water–air interface. J. Phys. Chem. Lett. 2019, 10, 4914– 4919, DOI: 10.1021/acs.jpclett.9b01983Google ScholarThere is no corresponding record for this reference.
- 342Kaliannan, N. K.; Henao Aristizabal, A.; Wiebeler, H.; Zysk, F.; Ohto, T.; Nagata, Y.; Kühne, T. D. Impact of intermolecular vibrational coupling effects on the sum-frequency generation spectra of the water/air interface. Mol. Phys. 2020, 118, 1620358, DOI: 10.1080/00268976.2019.1620358Google ScholarThere is no corresponding record for this reference.
- 343Ojha, D.; Henao, A.; Zysk, F.; Kühne, T. D. Nuclear quantum effects on the vibrational dynamics of the water–air interface. J. Chem. Phys. 2024, 160, 204114, DOI: 10.1063/5.0204071Google ScholarThere is no corresponding record for this reference.
- 344Kac, M. On Distributions of Certain Wiener Functionals. Trans. Am. Math. Soc. 1949, 65, 1– 13, DOI: 10.1090/S0002-9947-1949-0027960-XGoogle ScholarThere is no corresponding record for this reference.
- 345Feynman, R. P.; Hibbs, A. R. Quantum Mechanics and Path Integrals; McGraw-Hill: New York, 1965.Google ScholarThere is no corresponding record for this reference.
- 346Feynman, R. P. Statistical Mechanics; Addison-Wesley: Redwood City, 1972.Google ScholarThere is no corresponding record for this reference.
- 347Barker, J. A. A quantum–statistical Monte Carlo method; path integrals with boundary conditions. J. Chem. Phys. 1979, 70, 2914– 2918, DOI: 10.1063/1.437829Google ScholarThere is no corresponding record for this reference.
- 348Chandler, D.; Wolynes, P. G. Exploiting the isomorphism between quantum-theory and classical statistical-mechanics of polyatomic fluids. J. Chem. Phys. 1981, 74, 4078– 4095, DOI: 10.1063/1.441588Google ScholarThere is no corresponding record for this reference.
- 349Ceperley, D. M. Path integrals in the theory of condensed helium. Rev. Mod. Phys. 1995, 67, 279– 355, DOI: 10.1103/RevModPhys.67.279Google ScholarThere is no corresponding record for this reference.
- 350Markland, T. E.; Ceriotti, M. Nuclear quantum effects enter the mainstream. Nat. Rev. Chem. 2018, 2, 0109, DOI: 10.1038/s41570-017-0109Google ScholarThere is no corresponding record for this reference.
- 351Callaway, D. J. E.; Rahman, A. Micro-canonical ensemble formulation of lattice gauge-theory. Phys. Rev. Lett. 1982, 49, 613– 616, DOI: 10.1103/PhysRevLett.49.613Google ScholarThere is no corresponding record for this reference.
- 352Parrinello, M.; Rahman, A. Study of an F–center in molten KCl. J. Chem. Phys. 1984, 80, 860– 867, DOI: 10.1063/1.446740Google ScholarThere is no corresponding record for this reference.
- 353Coalson, R. D. On the connection between Fourier coefficient and discretized cartesian path integration. J. Chem. Phys. 1986, 85, 926– 936, DOI: 10.1063/1.451248Google ScholarThere is no corresponding record for this reference.
- 354Martyna, G. J.; Tuckerman, M. E.; Tobias, D. J.; Klein, M. L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996, 87, 1117– 1157, DOI: 10.1080/00268979600100761Google ScholarThere is no corresponding record for this reference.
- 355Ceriotti, M.; Parrinello, M.; Markland, T. E.; Manolopoulos, D. E. Efficient stochastic thermostatting of path integral molecular dynamics. J. Chem. Phys. 2010, 133, 124104, DOI: 10.1063/1.3489925Google ScholarThere is no corresponding record for this reference.
- 356Martyna, G. J.; Klein, M. L.; Tuckerman, M. Nosé-Hoover chains - The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635– 2643, DOI: 10.1063/1.463940Google ScholarThere is no corresponding record for this reference.
- 357Tuckerman, M. E.; Marx, D.; Klein, M. L.; Parrinello, M. Efficient and general algorithms for path integral Car–Parrinello molecular dynamics. J. Chem. Phys. 1996, 104, 5579– 5588, DOI: 10.1063/1.471771Google ScholarThere is no corresponding record for this reference.
- 358Kästner, J.; Thiel, W. Bridging the gap between thermodynamic integration and umbrella sampling provides a novel analysis method: “Umbrella integration”. J. Chem. Phys. 2005, 123, 144104, DOI: 10.1063/1.2052648Google ScholarThere is no corresponding record for this reference.
- 359Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 932– 942, DOI: 10.1002/wcms.66Google ScholarThere is no corresponding record for this reference.
- 360Schran, C.; Brieuc, F.; Marx, D. Converged Colored Noise Path Integral Molecular Dynamics Study of the Zundel Cation Down to Ultralow Temperatures at Coupled Cluster Accuracy. J. Chem. Theory Comput. 2018, 14, 5068– 5078, DOI: 10.1021/acs.jctc.8b00705Google ScholarThere is no corresponding record for this reference.
- 361John, C.; Spura, T.; Habershon, S.; Kühne, T. D. Quantum ring-polymer contraction method: Including nuclear quantum effects at no additional computational cost in comparison to ab initio molecular dynamics. Phys. Rev. E 2016, 93, 043305, DOI: 10.1103/PhysRevE.93.043305Google ScholarThere is no corresponding record for this reference.
- 362Ceriotti, M.; Manolopoulos, D. E.; Parrinello, M. Accelerating the convergence of path integral dynamics with a generalized Langevin equation. J. Chem. Phys. 2011, 134, 084104, DOI: 10.1063/1.3556661Google ScholarThere is no corresponding record for this reference.
- 363Ceriotti, M.; Manolopoulos, D. E. Efficient First-Principles Calculation of the Quantum Kinetic Energy and Momentum Distribution of Nuclei. Phys. Rev. Lett. 2012, 109, 100604, DOI: 10.1103/PhysRevLett.109.100604Google ScholarThere is no corresponding record for this reference.
- 364Brieuc, F.; Dammak, H.; Hayoun, M. Quantum Thermal Bath for Path Integral Molecular Dynamics Simulation. J. Chem. Theory Comput. 2016, 12, 1351– 1359, DOI: 10.1021/acs.jctc.5b01146Google ScholarThere is no corresponding record for this reference.
- 365Uhl, F.; Marx, D.; Ceriotti, M. Accelerated path integral methods for atomistic simulations at ultra-low temperatures. J. Chem. Phys. 2016, 145, 054101, DOI: 10.1063/1.4959602Google ScholarThere is no corresponding record for this reference.
- 366Cao, J. S.; Voth, G. A. A new perspective on quantum time-correlation functions. J. Chem. Phys. 1993, 99, 10070– 10073, DOI: 10.1063/1.465512Google ScholarThere is no corresponding record for this reference.
- 367Craig, I. R.; Manolopoulos, D. E. Quantum statistics and classical mechanics: Real time correlation functions from ring polymer molecular dynamics. J. Chem. Phys. 2004, 121, 3368– 3373, DOI: 10.1063/1.1777575Google ScholarThere is no corresponding record for this reference.
- 368Hele, T. J.; Willatt, M. J.; Muolo, A.; Althorpe, S. C. Communication: Relation of centroid molecular dynamics and ring-polymer molecular dynamics to exact quantum dynamics. J. Chem. Phys. 2015, 142, 191101, DOI: 10.1063/1.4921234Google ScholarThere is no corresponding record for this reference.
- 369Voth, G. A. Path–integral centroid methods in quantum statistical mechanics and dynamics. Adv. Chem. Phys. 1996, 93, 135– 218, DOI: 10.1002/9780470141526.ch4Google ScholarThere is no corresponding record for this reference.
- 370Althorpe, S. C. Path-integral approximations to quantum dynamics. Eur. Phys. J. B 2021, 94, 155, DOI: 10.1140/epjb/s10051-021-00155-2Google ScholarThere is no corresponding record for this reference.
- 371Althorpe, S. C. Path Integral Simulations of Condensed-Phase Vibrational Spectroscopy. Annu. Rev. Phys. Chem. 2024, 75, 397– 420, DOI: 10.1146/annurev-physchem-090722-124705Google ScholarThere is no corresponding record for this reference.
- 372Witt, A.; Ivanov, S. D.; Shiga, M.; Forbert, H.; Marx, D. On the applicability of centroid and ring polymer path integral molecular dynamics for vibrational spectroscopy. J. Chem. Phys. 2009, 130, 194510, DOI: 10.1063/1.3125009Google ScholarThere is no corresponding record for this reference.
- 373Rossi, M.; Ceriotti, M.; Manolopoulos, D. E. How to remove the spurious resonances from ring polymer molecular dynamics. J. Chem. Phys. 2014, 140, 234116, DOI: 10.1063/1.4883861Google ScholarThere is no corresponding record for this reference.
- 374Shiga, M. Path integral Brownian chain molecular dynamics: A simple approximation of quantum vibrational dynamics. J. Comput. Chem. 2022, 43, 1864– 1879, DOI: 10.1002/jcc.26989Google ScholarThere is no corresponding record for this reference.
- 375Tuckerman, M. E.; Berne, B. J.; Martyna, G. J.; Klein, M. L. Efficient molecular-dynamics and hybrid Monte-Carlo algorithms for path-integrals. J. Chem. Phys. 1993, 99, 2796– 2808, DOI: 10.1063/1.465188Google ScholarThere is no corresponding record for this reference.
- 376Spura, T.; John, C.; Habershon, S.; Kühne, T. D. Nuclear quantum effects in liquid water from path-integral simulations using an ab initio force-matching approach. Mol. Phys. 2015, 113, 808– 822, DOI: 10.1080/00268976.2014.981231Google ScholarThere is no corresponding record for this reference.
- 377Stolte, N.; Daru, J.; Forbert, H.; Behler, J.; Marx, D. Nuclear Quantum Effects in Liquid Water Are Marginal for Its Average Structure but Significant for Dynamics. J. Phys. Chem. Lett. 2024, 15, 12144– 12150, DOI: 10.1021/acs.jpclett.4c02925Google ScholarThere is no corresponding record for this reference.
- 378Malik, R.; Stolte, N.; Forbert, H.; Chandra, A.; Marx, D. Accurate Determination of Isotope Effects on the Dynamics of H-Bond Breaking and Making in Liquid Water. J. Phys. Chem. Lett. 2025, 16, 3727– 3733, DOI: 10.1021/acs.jpclett.5c00210Google ScholarThere is no corresponding record for this reference.
- 379Feynman, R. P. Atomic Theory of the λ Transition in Helium. Phys. Rev. 1953, 91, 1291– 1301, DOI: 10.1103/PhysRev.91.1291Google ScholarThere is no corresponding record for this reference.
- 380Troyer, M.; Wiese, U.-J. Computational Complexity and Fundamental Limitations to Fermionic Quantum Monte Carlo Simulations. Phys. Rev. Lett. 2005, 94, 170201, DOI: 10.1103/PhysRevLett.94.170201Google ScholarThere is no corresponding record for this reference.
- 381Calcavecchia, F.; Pederiva, F.; Kalos, M. H.; Kühne, T. D. Sign problem of the fermionic shadow wave function. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2014, 90, 053304, DOI: 10.1103/PhysRevE.90.053304Google ScholarThere is no corresponding record for this reference.
- 382Ceperley, D. M.; Pollock, E. L. Path-Integral Computation of the Low-Temperature Properties of Liquid 4He. Phys. Rev. Lett. 1986, 56, 351– 354, DOI: 10.1103/PhysRevLett.56.351Google ScholarThere is no corresponding record for this reference.
- 383Pollock, E. L.; Ceperley, D. M. Path-integral computation of superfluid densities. Phys. Rev. B 1987, 36, 8343– 8352, DOI: 10.1103/PhysRevB.36.8343Google ScholarThere is no corresponding record for this reference.
- 384Boninsegni, M.; Prokof’ev, N.; Svistunov, B. Worm Algorithm for Continuous-Space Path Integral Monte Carlo Simulations. Phys. Rev. Lett. 2006, 96, 070601, DOI: 10.1103/PhysRevLett.96.070601Google ScholarThere is no corresponding record for this reference.
- 385Boninsegni, M.; Prokof’ev, N. V.; Svistunov, B. V. Worm algorithm and diagrammatic Monte Carlo: A new approach to continuous-space path integral Monte Carlo simulations. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2006, 74, 036701, DOI: 10.1103/PhysRevE.74.036701Google ScholarThere is no corresponding record for this reference.
- 386Brieuc, F.; Schran, C.; Uhl, F.; Forbert, H.; Marx, D. Converged quantum simulations of reactive solutes in superfluid helium: The Bochum perspective. J. Chem. Phys. 2020, 152, 210901, DOI: 10.1063/5.0008309Google ScholarThere is no corresponding record for this reference.
- 387Walewski, L.; Forbert, H.; Marx, D. Reactive path integral quantum simulations of molecules solvated in superfluid helium. Comput. Phys. Commun. 2014, 185, 884– 899, DOI: 10.1016/j.cpc.2013.12.011Google ScholarThere is no corresponding record for this reference.
- 388Pollock, E. L.; Ceperley, D. M. Simulation of quantum many-body systems by path-integral methods. Phys. Rev. B 1984, 30, 2555– 2568, DOI: 10.1103/PhysRevB.30.2555Google ScholarThere is no corresponding record for this reference.
- 389Aziz, R. A.; Janzen, A. R.; Moldover, M. R. Ab Initio Calculations for Helium: A Standard for Transport Property Measurements. Phys. Rev. Lett. 1995, 74, 1586– 1589, DOI: 10.1103/PhysRevLett.74.1586Google ScholarThere is no corresponding record for this reference.
- 390Pollock, E. L.; Runge, K. J. Finite-size-scaling analysis of a simulation of the 4He superfluid transition. Phys. Rev. B 1992, 46, 3535– 3539, DOI: 10.1103/PhysRevB.46.3535Google ScholarThere is no corresponding record for this reference.
- 391Keesom, W. H.; Keesom, A. P. New measurements on the specific heat of liquid helium. Physica 1935, 2, 557– 572, DOI: 10.1016/S0031-8914(35)90128-8Google ScholarThere is no corresponding record for this reference.
- 392Klein, M. L.; Venables, J. A. Rare Gas Solids Vol. II; Academic Press: London, New York, San Francisco, 1977.Google ScholarThere is no corresponding record for this reference.
- 393Sindzingre, P.; Klein, M. L.; Ceperley, D. M. Path-integral Monte Carlo study of low-temperature 4He clusters. Phys. Rev. Lett. 1989, 63, 1601– 1604, DOI: 10.1103/PhysRevLett.63.1601Google ScholarThere is no corresponding record for this reference.
- 394Zeng, T.; Guillon, G.; Cantin, J. T.; Roy, P.-N. Probing the Superfluid Response of para-Hydrogen with a Sulfur Dioxide Dopant. J. Phys. Chem. Lett. 2013, 4, 2391– 2396, DOI: 10.1021/jz401188jGoogle ScholarThere is no corresponding record for this reference.
- 395Zeng, T.; Roy, P. N. Microscopic molecular superfluid response: Theory and simulations. Rep. Prog. Phys. 2014, 77, 046601, DOI: 10.1088/0034-4885/77/4/046601Google ScholarThere is no corresponding record for this reference.
- 396Brieuc, F.; Schran, C.; Marx, D. Manifestations of local supersolidity of 4He around a charged molecular impurity. Phys. Rev. Res. 2023, 5, 043083, DOI: 10.1103/PhysRevResearch.5.043083Google ScholarThere is no corresponding record for this reference.
- 397Walewski, Ł.; Forbert, H.; Marx, D. Solvation of molecules in superfluid helium enhances the “interaction induced localization” effect. J. Chem. Phys. 2014, 140, 144305, DOI: 10.1063/1.4870595Google ScholarThere is no corresponding record for this reference.
- 398Duran Caballero, L.; Schran, C.; Brieuc, F.; Marx, D. Neural network interaction potentials for para-hydrogen with flexible molecules. J. Chem. Phys. 2022, 157, 074302, DOI: 10.1063/5.0100953Google ScholarThere is no corresponding record for this reference.
- 399Marx, D.; Parrinello, M. Ab initio path–integral molecular dynamics. Z. Phys. B 1994, 95, 143– 144, DOI: 10.1007/BF01312185Google ScholarThere is no corresponding record for this reference.
- 400Marx, D.; Parrinello, M. Ab initio path integral molecular dynamics: Basic ideas. J. Chem. Phys. 1996, 104, 4077– 4082, DOI: 10.1063/1.471221Google ScholarThere is no corresponding record for this reference.
- 401Boese, A. D.; Forbert, H.; Masia, M.; Tekin, A.; Marx, D.; Jansen, G. Constructing simple yet accurate potentials for describing the solvation of HCl/water clusters in bulk helium and nanodroplets. Phys. Chem. Chem. Phys. 2011, 13, 14550– 14564, DOI: 10.1039/c1cp20991dGoogle ScholarThere is no corresponding record for this reference.
- 402Kuchenbecker, D.; Uhl, F.; Forbert, H.; Jansen, G.; Marx, D. Constructing accurate interaction potentials to describe the microsolvation of protonated methane by helium atoms. Phys. Chem. Chem. Phys. 2017, 19, 8307– 8321, DOI: 10.1039/C7CP00652GGoogle ScholarThere is no corresponding record for this reference.
- 403Uhl, F.; Marx, D. Helium Tagging of Protonated Methane in Messenger Spectroscopy: Does It Interfere with the Fluxionality of CH5+?. Angew. Chem., Int. Ed. 2018, 57, 14792– 14795, DOI: 10.1002/anie.201808531Google ScholarThere is no corresponding record for this reference.
- 404Uhl, F.; Marx, D. Quantum Microsolvation of Protonated Methane with 4He: Large-Amplitude Motion Heavily Influences Bosonic Exchange. Phys. Rev. Lett. 2019, 123, 123002, DOI: 10.1103/PhysRevLett.123.123002Google ScholarThere is no corresponding record for this reference.
- 405Schran, C.; Uhl, F.; Behler, J.; Marx, D. High-dimensional neural network potentials for solvation: The case of protonated water clusters in helium. J. Chem. Phys. 2018, 148, 102310, DOI: 10.1063/1.4996819Google ScholarThere is no corresponding record for this reference.
- 406Werner, H.-J.; Knowles, P. J.; Manby, F. R.; Black, J. A.; Doll, K.; Heßelmann, A.; Kats, D.; Köhn, A.; Korona, T.; Kreplin, D. A. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107, DOI: 10.1063/5.0005081Google ScholarThere is no corresponding record for this reference.
- 407Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108, DOI: 10.1063/5.0004608Google ScholarThere is no corresponding record for this reference.
- 408Topolnicki, R.; Brieuc, F.; Schran, C.; Marx, D. Deciphering High-Order Structural Correlations within Fluxional Molecules from Classical and Quantum Configurational Entropy. J. Chem. Theory Comput. 2020, 16, 6785– 6794, DOI: 10.1021/acs.jctc.0c00642Google ScholarThere is no corresponding record for this reference.
- 409Schran, C.; Brieuc, F.; Marx, D. Transferability of machine learning potentials: Protonated water neural network potential applied to the protonated water hexamer. J. Chem. Phys. 2021, 154, 051101, DOI: 10.1063/5.0035438Google ScholarThere is no corresponding record for this reference.
- 410Larsson, H. R.; Schröder, M.; Beckmann, R.; Brieuc, F.; Schran, C.; Marx, D.; Vendrell, O. State-resolved infrared spectrum of the protonated water dimer: revisiting the characteristic proton transfer doublet peak. Chem. Sci. 2022, 13, 11119– 11125, DOI: 10.1039/D2SC03189BGoogle ScholarThere is no corresponding record for this reference.
- 411Beckmann, R.; Topolnicki, R.; Marx, D. Deciphering the Impact of Helium Tagging on Flexible Molecules: Probing Microsolvation Effects of Protonated Acetylene by Quantum Configurational Entrop. J. Phys. Chem. A 2023, 127, 2460– 2471, DOI: 10.1021/acs.jpca.2c08967Google ScholarThere is no corresponding record for this reference.
- 412Simko, I.; Schran, C.; Brieuc, F.; Fabri, C.; Asvany, O.; Schlemmer, S.; Marx, D.; Császár, A. G. Quantum Nuclear Delocalization and its Rovibrational Fingerprints. Angew. Chem., Int. Ed. 2023, 62, e202306744 DOI: 10.1002/anie.202306744Google ScholarThere is no corresponding record for this reference.
- 413Davies, J. A.; Schran, C.; Brieuc, F.; Marx, D.; Ellis, A. M. Onset of Rotational Decoupling for a Molecular Ion Solvated in Helium: From Tags to Rings and Shells. Phys. Rev. Lett. 2023, 130, 083001, DOI: 10.1103/PhysRevLett.130.083001Google ScholarThere is no corresponding record for this reference.
- 414Beckmann, R.; Schran, C.; Brieuc, F.; Marx, D. Theoretical infrared spectroscopy of protonated methane isotopologues. Phys. Chem. Chem. Phys. 2024, 26, 22846– 22852, DOI: 10.1039/D4CP02295EGoogle ScholarThere is no corresponding record for this reference.
- 415Beckmann, R.; Brieuc, F.; Schran, C.; Marx, D. Infrared Spectra at Coupled Cluster Accuracy from Neural Network Representations. J. Chem. Theory Comput. 2022, 18, 5492– 5501, DOI: 10.1021/acs.jctc.2c00511Google ScholarThere is no corresponding record for this reference.
- 416Larsen, A. H.; Mortensen, J. J.; Blomqvist, J.; Castelli, I. E.; Christensen, R.; Dulak, M.; Friis, J.; Groves, M. N.; Hammer, B.; Hargus, C. The atomic simulation environment - a Python library for working with atoms. J. Phys.: Condens. Matter 2017, 29, 273002, DOI: 10.1088/1361-648X/aa680eGoogle ScholarThere is no corresponding record for this reference.
- 417Brehm, M.; Kirchner, B. TRAVIS – A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51 (8), 2007– 2023, DOI: 10.1021/ci200217wGoogle ScholarThere is no corresponding record for this reference.
- 418Brehm, M.; Thomas, M.; Gehrke, S.; Kirchner, B. TRAVIS – A Free Analyzer for Trajectories from Molecular Simulation. J. Chem. Phys. 2020, 152 (16), 164105, DOI: 10.1063/5.0005078Google ScholarThere is no corresponding record for this reference.
- 419Lindner, J.; Cringus, D.; Pshenichnikov, M. S.; Vöhringer, P. Anharmonic Bend–Stretch Coupling in Neat Liquid Water. Chem. Phys. 2007, 341, 326– 335, DOI: 10.1016/j.chemphys.2007.07.051Google ScholarThere is no corresponding record for this reference.
- 420Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient. J. Chem. Soc., Perkin Trans. 2 1993, 2, 799– 805, DOI: 10.1039/P29930000799Google ScholarThere is no corresponding record for this reference.
- 421Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506– 10517, DOI: 10.1021/jp971959kGoogle ScholarThere is no corresponding record for this reference.
- 422Gordon, R. G. Molecular motion in infrared and raman spectra. J. Chem. Phys. 1965, 43, 1307– 1312, DOI: 10.1063/1.1696920Google ScholarThere is no corresponding record for this reference.
- 423Shimizu, H. Time-Correlation Function of Molecular Random Motion and Shape of Spectral Bands. J. Chem. Phys. 1965, 43, 2453– 2465, DOI: 10.1063/1.1697145Google ScholarThere is no corresponding record for this reference.
- 424Kubo, R.; Toda, M.; Hashitsume, N. Statistical Physics II; Springer: Berlin, 1991.Google ScholarThere is no corresponding record for this reference.
- 425Spaldin, N. A. A beginner’s guide to the modern theory of polarization. J. Solid State Chem. 2012, 195, 2– 10, DOI: 10.1016/j.jssc.2012.05.010Google ScholarThere is no corresponding record for this reference.
- 426King-Smith, R. D.; Vanderbilt, D. Theory of Polarization of Crystalline Solids. Phys. Rev. B 1993, 47, 1651– 1654, DOI: 10.1103/PhysRevB.47.1651Google ScholarThere is no corresponding record for this reference.
- 427Resta, R. Macroscopic Electric Polarization As a Geometric Quantum Phase. Europhys. Lett. 1993, 22, 133– 138, DOI: 10.1209/0295-5075/22/2/010Google ScholarThere is no corresponding record for this reference.
- 428Resta, R. Macroscopic polarization in crystalline dielectrics: the geometric phase approach. Rev. Mod. Phys. 1994, 66, 899– 915, DOI: 10.1103/RevModPhys.66.899Google ScholarThere is no corresponding record for this reference.
- 429Resta, R. Quantum-Mechanical Position Operator in Extended Systems. Phys. Rev. Lett. 1998, 80, 1800– 1803, DOI: 10.1103/PhysRevLett.80.1800Google ScholarThere is no corresponding record for this reference.
- 430Berry, M. V. Quantal phase factors accompanying adiabatic changes. Proc. R. Soc. London, Ser. A 1984, 392, 45– 57, DOI: 10.1098/rspa.1984.0023Google ScholarThere is no corresponding record for this reference.
- 431Mead, C. A. The geometric phase in molecular systems. Rev. Mod. Phys. 1992, 64, 51, DOI: 10.1103/RevModPhys.64.51Google ScholarThere is no corresponding record for this reference.
- 432Kirchner, B.; Hutter, J. Solvent effects on electronic properties from Wannier functions in a dimethyl sulfoxide/water mixture. J. Chem. Phys. 2004, 121, 5133– 5142, DOI: 10.1063/1.1785780Google ScholarThere is no corresponding record for this reference.
- 433Silvestrelli, P. L.; Parrinello, M. Water Molecule Dipole in the Gas and in the Liquid Phase ─ Erratum. Phys. Rev. Lett. 1999, 82, 5415, DOI: 10.1103/PhysRevLett.82.5415Google ScholarThere is no corresponding record for this reference.
- 434Silvestrelli, P. L.; Parrinello, M. Structural, electronic, and bonding properties of liquid water from first principles. J. Chem. Phys. 1999, 111, 3572– 3580, DOI: 10.1063/1.479638Google ScholarThere is no corresponding record for this reference.
- 435Boys, S. F. Construction of Some Molecular Orbitals to be Approximately Invariant for Changes from One Molecule to Another. Rev. Mod. Phys. 1960, 32, 296– 299, DOI: 10.1103/RevModPhys.32.296Google ScholarThere is no corresponding record for this reference.
- 436Pipek, J.; Mezey, P. G. A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave functions. J. Chem. Phys. 1989, 90, 4916– 4926, DOI: 10.1063/1.456588Google ScholarThere is no corresponding record for this reference.
- 437Wannier, G. H. The Structure of Electronic Excitation Levels in Insulating Crystals. Phys. Rev. 1937, 52, 191– 197, DOI: 10.1103/PhysRev.52.191Google ScholarThere is no corresponding record for this reference.
- 438Marzari, N.; Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 1997, 56, 12847– 12865, DOI: 10.1103/PhysRevB.56.12847Google ScholarThere is no corresponding record for this reference.
- 439Silvestrelli, P. L.; Marzari, N.; Vanderbilt, D.; Parrinello, M. Maximally-localized Wannier functions for disordered systems: Application to amorphous silicon. Solid State Commun. 1998, 107, 7– 11, DOI: 10.1016/S0038-1098(98)00175-6Google ScholarThere is no corresponding record for this reference.
- 440Silvestrelli, P. L. Maximally localized Wannier functions for simulations with supercells of general symmetry. Phys. Rev. B 1999, 59, 9703– 9706, DOI: 10.1103/PhysRevB.59.9703Google ScholarThere is no corresponding record for this reference.
- 441Marzari, N.; Mostofi, A. A.; Yates, J. R.; Souza, I.; Vanderbilt, D. Maximally localized Wannier functions: Theory and applications. Rev. Mod. Phys. 2012, 84, 1419– 1475, DOI: 10.1103/RevModPhys.84.1419Google ScholarThere is no corresponding record for this reference.
- 442Ambrosetti, A.; Silvestrelli, P. L. Introduction to maximally localized Wannier functions. Rev. Comput. Chem. 2016, 29, 327– 368, DOI: 10.1002/9781119148739.ch6Google ScholarThere is no corresponding record for this reference.
- 443Berghold, G.; Mundy, C. J.; Romero, A. H.; Hutter, J.; Parrinello, M. General and efficient algorithms for obtaining maximally localized Wannier functions. Phys. Rev. B 2000, 61, 10040– 10048, DOI: 10.1103/PhysRevB.61.10040Google ScholarThere is no corresponding record for this reference.
- 444Resta, R.; Sorella, S. Electron Localization in the Insulating State. Phys. Rev. Lett. 1999, 82, 370– 373, DOI: 10.1103/PhysRevLett.82.370Google ScholarThere is no corresponding record for this reference.
- 445Edmiston, C.; Ruedenberg, K. Localized Atomic and Molecular Orbitals. Rev. Mod. Phys. 1963, 35, 457– 464, DOI: 10.1103/RevModPhys.35.457Google ScholarThere is no corresponding record for this reference.
- 446Jacobi, C. Über ein Leichtes Verfahren die in der Theorie der Säcularstörungen Vorkommenden Gleichungen Numerisch Aufzulösen. J. für die Reine Angewandte Math. 1846, 30, 51– 94Google ScholarThere is no corresponding record for this reference.
- 447Iftimie, R.; Minary, P.; Tuckerman, M. E. Ab initio molecular dynamics: Concepts, recent developments, and future trends. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 6654– 6659, DOI: 10.1073/pnas.0500193102Google ScholarThere is no corresponding record for this reference.
- 448Ojha, D.; Kühne, T. D. “On-the-fly” calculation of the Vibrational Sum-frequency Generation Spectrum at the Air-water Interface. Molecules 2020, 25, 3939, DOI: 10.3390/molecules25173939Google ScholarThere is no corresponding record for this reference.
- 449Partovi-Azar, P.; Kühne, T. D. Full Assignment of ab-initio Raman Spectra at Finite Temperatures Using Wannier Polarizabilities: Application to Cyclohexane Molecule in Gas Phase. Micromachines 2021, 12, 1212, DOI: 10.3390/mi12101212Google ScholarThere is no corresponding record for this reference.
- 450Partovi-Azar, P.; Kühne, T. D. Efficient “On-the-Fly” calculation of Raman Spectra from Ab-Initio molecular dynamics: Application to hydrophobic/hydrophilic solutes in bulk water. J. Comput. Chem. 2015, 36, 2188– 2192, DOI: 10.1002/jcc.24198Google ScholarThere is no corresponding record for this reference.
- 451Partovi-Azar, P.; Kühne, T. D.; Kaghazchi, P. Evidence for the existence of Li 2 S 2 clusters in lithium–sulfur batteries: ab initio Raman spectroscopy simulation. Phys. Chem. Chem. Phys. 2015, 17, 22009– 22014, DOI: 10.1039/C5CP02781KGoogle ScholarThere is no corresponding record for this reference.
- 452Partovi-Azar, P. Efficient method for estimating the dynamics of the full polarizability tensor during ab initio molecular dynamics simulations. Phys. Rev. B 2023, 108, 235157, DOI: 10.1103/PhysRevB.108.235157Google ScholarThere is no corresponding record for this reference.
- 453Thomas, M.; Brehm, M.; Kirchner, B. Voronoi Dipole Moments for the Simulation of Bulk Phase Vibrational Spectra. Phys. Chem. Chem. Phys. 2015, 17, 3207– 3213, DOI: 10.1039/C4CP05272BGoogle ScholarThere is no corresponding record for this reference.
- 454Brehm, M.; Thomas, M. Optimized Atomic Partial Charges and Radii Defined by Radical Voronoi Tessellation of Bulk Phase Simulations. Molecules 2021, 26 (7), 1875, DOI: 10.3390/molecules26071875Google ScholarThere is no corresponding record for this reference.
- 455Voronoi, G. Nouvelles Applications Des Paramètres Continus à La Théorie Des Formes Quadratiques. Deuxième Mémoire. Recherches Sur Les Parallélloèdres Primitifs. J. für die Reine Angewandte Math. 1908, 1908, 198– 287, DOI: 10.1515/crll.1908.134.198Google ScholarThere is no corresponding record for this reference.
- 456Medvedev, N. N. The Algorithm for Three-Dimensional Voronoi Polyhedra. J. Comput. Phys. 1986, 67, 223– 229, DOI: 10.1016/0021-9991(86)90123-3Google ScholarThere is no corresponding record for this reference.
- 457Pollak, M.; Rein, R. Molecular-Orbital Studies of Intermolecular Interaction Energies. II. Approximations Concerned with Coulomb Interactions and Comparison of the Two London Schemes. J. Chem. Phys. 1967, 47, 2045– 2052, DOI: 10.1063/1.1712236Google ScholarThere is no corresponding record for this reference.
- 458Becke, A. D. A Multicenter Numerical Integration Scheme for Polyatomic Molecules. J. Chem. Phys. 1988, 88, 2547– 2553, DOI: 10.1063/1.454033Google ScholarThere is no corresponding record for this reference.
- 459Gellatly, B. J.; Finney, J. L. Calculation of Protein Volumes: An Alternative to the Voronoi Procedure. J. Mol. Biol. 1982, 161, 305– 322, DOI: 10.1016/0022-2836(82)90155-3Google ScholarThere is no corresponding record for this reference.
- 460Richards, F. M. The Interpretation of Protein Structures: Total Volume, Group Volume Distributions and Packing Density. J. Mol. Biol. 1974, 82, 1– 14, DOI: 10.1016/0022-2836(74)90570-1Google ScholarThere is no corresponding record for this reference.
- 461Brehm, M.; Thomas, M. Computing Bulk Phase Raman Optical Activity Spectra from ab initio Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2017, 8 (14), 3409– 3414, DOI: 10.1021/acs.jpclett.7b01616Google ScholarThere is no corresponding record for this reference.
- 462Brehm, M.; Thomas, M. Computing Bulk Phase Resonance Raman Spectra from ab initio Molecular Dynamics and Real-Time TDDFT. J. Chem. Theory Comput. 2019, 15 (7), 3901– 3905, DOI: 10.1021/acs.jctc.9b00512Google ScholarThere is no corresponding record for this reference.
- 463Yang, Y.; Cheramy, J.; Brehm, M.; Xu, Y. Raman Optical Activity of N-Acetyl-l-Cysteine in Water and in Methanol: The “Clusters-in-a-Liquid” Model and ab initio Molecular Dynamics Simulations. ChemPhysChem 2022, 23, e202200161 DOI: 10.1002/cphc.202200161Google ScholarThere is no corresponding record for this reference.
- 464Brehm, M. libvori: Voronoi Integration for Electrostatic Properties from Electron Density, 2025. https://brehm-research.de/libvori.php (accessed 13 09.Google ScholarThere is no corresponding record for this reference.
- 465Rycroft, C. H. VORO++: A three-dimensional Voronoi cell library in C++. Chaos 2009, 19, 041111, DOI: 10.1063/1.3215722Google ScholarThere is no corresponding record for this reference.
- 466Cordero, B.; Gómez, V.; Platero-Prats, A. E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 2832, 2832, DOI: 10.1039/b801115jGoogle ScholarThere is no corresponding record for this reference.
- 467Brehm, M.; Kirchner, B. TRAVIS - A free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007– 2023, DOI: 10.1021/ci200217wGoogle ScholarThere is no corresponding record for this reference.
- 468Ewald, P. P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253– 287, DOI: 10.1002/andp.19213690304Google ScholarThere is no corresponding record for this reference.
- 469Heaton, R. J.; Madden, P. A.; Clark, S. J.; Jahn, S. Condensed Phase Ionic Polarizabilities from Plane Wave Density Functional Theory Calculations. J. Chem. Phys. 2006, 125, 144104, DOI: 10.1063/1.2357151Google ScholarThere is no corresponding record for this reference.
- 470Salanne, M.; Vuilleumier, R.; Madden, P. A.; Simon, C.; Turq, P.; Guillot, B. Polarizabilities of Individual Molecules and Ions in Liquids from First Principles. J. Phys.: Condens. Matter 2008, 20, 494207, DOI: 10.1088/0953-8984/20/49/494207Google ScholarThere is no corresponding record for this reference.
- 471Wan, Q.; Spanu, L.; Galli, G. A.; Gygi, F. Raman Spectra of Liquid Water from ab initio Molecular Dynamics: Vibrational Signatures of Charge Fluctuations in the Hydrogen Bond Network. J. Chem. Theory Comput. 2013, 9, 4124– 4130, DOI: 10.1021/ct4005307Google ScholarThere is no corresponding record for this reference.
- 472Luber, S.; Iannuzzi, M.; Hutter, J. Raman spectra from ab initio molecular dynamics and its application to liquid S-methyloxirane. J. Chem. Phys. 2014, 141, 094503, DOI: 10.1063/1.4894425Google ScholarThere is no corresponding record for this reference.
- 473Albrecht, A. C. On the Theory of Raman Intensities. J. Chem. Phys. 1961, 34, 1476– 1484, DOI: 10.1063/1.1701032Google ScholarThere is no corresponding record for this reference.
- 474Tang, J.; Albrecht, A. C. Studies in Raman Intensity Theory. J. Chem. Phys. 1968, 49, 1144– 1154, DOI: 10.1063/1.1670202Google ScholarThere is no corresponding record for this reference.
- 475Albrecht, A. C.; Hutley, M. C. On the Dependence of Vibrational Raman Intensity on the Wavelength of Incident Light. J. Chem. Phys. 1971, 55, 4438– 4443, DOI: 10.1063/1.1676771Google ScholarThere is no corresponding record for this reference.
- 476Lee, S.-Y.; Heller, E. J. Time-Dependent Theory of Raman Scattering. J. Chem. Phys. 1979, 71, 4777– 4788, DOI: 10.1063/1.438316Google ScholarThere is no corresponding record for this reference.
- 477Heller, E. J. The Semiclassical Way to Molecular Spectroscopy. Acc. Chem. Res. 1981, 14, 368– 375, DOI: 10.1021/ar00072a002Google ScholarThere is no corresponding record for this reference.
- 478Heller, E. J.; Sundberg, R.; Tannor, D. J. Simple Aspects of Raman Scattering. J. Phys. Chem. 1982, 86, 1822– 1833, DOI: 10.1021/j100207a018Google ScholarThere is no corresponding record for this reference.
- 479Jensen, L.; Zhao, L. L.; Autschbach, J.; Schatz, G. C. Theory and Method for Calculating Resonance Raman Scattering from Resonance Polarizability Derivatives. J. Chem. Phys. 2005, 123, 174110, DOI: 10.1063/1.2046670Google ScholarThere is no corresponding record for this reference.
- 480Jensen, L.; Autschbach, J.; Schatz, G. C. Finite Lifetime Effects on the Polarizability within Time-Dependent Density-Functional Theory. J. Chem. Phys. 2005, 122, 224115, DOI: 10.1063/1.1929740Google ScholarThere is no corresponding record for this reference.
- 481Andermatt, S.; Cha, J.; Schiffmann, F.; VandeVondele, J. Combining Linear-Scaling DFT with Subsystem DFT in Born–Oppenheimer and Ehrenfest Molecular Dynamics Simulations: From Molecules to a Virus in Solution. J. Chem. Theory Comput. 2016, 12, 3214– 3227, DOI: 10.1021/acs.jctc.6b00398Google ScholarThere is no corresponding record for this reference.
- 482Provorse, M. R.; Isborn, C. M. Electron Dynamics with Real-Time Time-Dependent Density Functional Theory. Int. J. Quantum Chem. 2016, 116, 739– 749, DOI: 10.1002/qua.25096Google ScholarThere is no corresponding record for this reference.
- 483Goings, J. J.; Lestrange, P. J.; Li, X. Real-Time Time-Dependent Electronic Structure Theory. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1341 DOI: 10.1002/wcms.1341Google ScholarThere is no corresponding record for this reference.
- 484Chen, H.; Mcmahon, J. M.; Ratner, M. A.; Schatz, G. C. Classical Electrodynamics Coupled to Quantum Mechanics for Calculation of Molecular Optical Properties: A RT-TDDFT/FDTD Approach. J. Phys. Chem. C 2010, 114, 14384– 14392, DOI: 10.1021/jp1043392Google ScholarThere is no corresponding record for this reference.
- 485Ibaceta-Jaña, J.; Chugh, M.; Novikov, A. S.; Mirhosseini, H.; Kühne, T. D.; Szyszka, B.; Wagner, M. R.; Muydinov, R. Do lead halide hybrid perovskites have hydrogen bonds?. J. Phys. Chem. C 2022, 126, 16215– 16226, DOI: 10.1021/acs.jpcc.2c02984Google ScholarThere is no corresponding record for this reference.
- 486Brehm, M.; Thomas, M. An Efficient Lossless Compression Algorithm for Trajectories of Atom Positions and Volumetric Data. J. Chem. Inf. Model. 2018, 58 (10), 2092– 2107, DOI: 10.1021/acs.jcim.8b00501Google ScholarThere is no corresponding record for this reference.
- 487Zhang, C.; Khaliullin, R. Z.; Bovi, D.; Guidoni, L.; Kühne, T. D. Vibrational Signature of Water Molecules in Asymmetric Hydrogen Bonding Environments. J. Phys. Chem. Lett. 2013, 4, 3245– 3250, DOI: 10.1021/jz401321xGoogle ScholarThere is no corresponding record for this reference.
- 488Zhang, C.; Guidoni, L.; Kühne, T. D. Competing factors on the frequency separation between the OH stretching modes in water. J. Mol. Liq. 2015, 205, 42– 45, DOI: 10.1016/j.molliq.2014.09.049Google ScholarThere is no corresponding record for this reference.
- 489Nonella, M.; Mathias, G.; Tavan, P. Infrared spectrum of p–benzoquinone in water obtained from a QM/MM hybrid molecular dynamics simulation. J. Phys. Chem. A 2003, 107, 8638– 8647, DOI: 10.1021/jp027747rGoogle ScholarThere is no corresponding record for this reference.
- 490Schmitz, M.; Tavan, P. Vibrational spectra from atomic fluctuations in dynamics simulations I. Theory, limitations and a sample application. J. Chem. Phys. 2004, 121, 12233– 12246, DOI: 10.1063/1.1822914Google ScholarThere is no corresponding record for this reference.
- 491Pejov, L.; Spångberg, D.; Hermansson, K. Using MD Snapshots in ab initio and DFT Calculations: OH Vibrations in the First Hydration Shell around Li+(aq). J. Phys. Chem. A 2005, 109, 5144– 5152, DOI: 10.1021/jp047395jGoogle ScholarThere is no corresponding record for this reference.
- 492Wheeler, R. A.; Dong, H.; Boesch, S. E. Quasiharmonic Vibrations of Water, Water Dimer, and Liquid Water from Principal Component Analysis of Quantum or QM/MM Trajectories. ChemPhysChem 2003, 4, 382– 384, DOI: 10.1002/cphc.200390066Google ScholarThere is no corresponding record for this reference.
- 493Gaigeot, M.-P.; Sprik, M. Ab initio molecular dynamics computation of the infrared spectrum of aqueous uracil. J. Phys. Chem. B 2003, 107, 10344– 10358, DOI: 10.1021/jp034788uGoogle ScholarThere is no corresponding record for this reference.
- 494Mathias, G.; Baer, M. D. Generalized Normal Coordinates for the Vibrational Analysis of Molecular Dynamics Simulations. J. Chem. Theory Comput. 2011, 7, 2028– 2039, DOI: 10.1021/ct2001304Google ScholarThere is no corresponding record for this reference.
- 495Mathias, G.; Ivanov, S. D.; Witt, A.; Baer, M. D.; Marx, D. Infrared Spectroscopy of Fluxional Molecules from (ab initio) Molecular Dynamics: Resolving Large-Amplitude Motion, Multiple Conformations, and Permutational Symmetries. J. Chem. Theory Comput. 2012, 8, 224– 234, DOI: 10.1021/ct2006665Google ScholarThere is no corresponding record for this reference.
- 496Eckart, C. Some Studies Concerning Rotating Axes and Polyatomic Molecules. Phys. Rev. 1935, 47, 552– 558, DOI: 10.1103/PhysRev.47.552Google ScholarThere is no corresponding record for this reference.
- 497Thomas, M.; Brehm, M.; Hollóczki, O.; Kelemen, Z.; Nyulászi, L.; Pasinszki, T.; Kirchner, B. Simulating the Vibrational Spectra of Ionic Liquid Systems: 1-Ethyl-3-Methylimidazolium Acetate and its Mixtures. J. Chem. Phys. 2014, 141, 024510, DOI: 10.1063/1.4887082Google ScholarThere is no corresponding record for this reference.
- 498CP2K Dashboard. https://dashboard.cp2k.org/index.html, 2024; (accessed, 2025 10 18).Google ScholarThere is no corresponding record for this reference.
- 499(a) Frigo, M.; Johnson, S. G. The design and implementation of FFTW3. Proc. IEEE 2005, 93, 216– 231, DOI: 10.1109/JPROC.2004.840301Google ScholarThere is no corresponding record for this reference.See. http://www.fftw.org.Google ScholarThere is no corresponding record for this reference.
- 500Ramaswami, A.; Kenter, T.; Kühne, T. D.; Plessl, C. Efficient ab-initio molecular dynamic simulations by offloading fast Fourier transformations to FPGAs. In 2020 30th International Conference on Field-Programmable Logic and Applications (FPL); Institute of Electrical and Electronics Engineers, 2020, pp 353– 354.Google ScholarThere is no corresponding record for this reference.
- 501Ramaswami, A.; Kenter, T.; Kühne, T. D.; Plessl, C. Evaluating the design space for offloading 3D FFT calculations to an FPGA for high-performance computing. In International Symposium on Applied Reconfigurable Computing; Springer Nature, 2021, pp 285– 294.Google ScholarThere is no corresponding record for this reference.
- 502Wu, X.; Kenter, T.; Schade, R.; Kühne, T. D.; Plessl, C. Computing and compressing electron repulsion integrals on FPGAs. In 2023 IEEE 31st Annual International Symposium on Field-Programmable Custom Computing Machines (FCCM); Institute of Electrical and Electronics Engineers, 2023, pp 162– 173.Google ScholarThere is no corresponding record for this reference.
- 503FFTFPGA: An OpenCL based library for Fast Fourier Transformations for FPGAs. https://github.com/pc2/fft3d-fpga, 2024; (accessed, 2025 10 01).Google ScholarThere is no corresponding record for this reference.
- 504Heinecke, A.; Henry, G.; Hutchinson, M.; Pabst, H. LIBXSMM: Accelerating Small Matrix Multiplications by Runtime Code Generation. In SC16: International Conference for High Performance Computing, Networking, Storage and Analysis; Institute of Electrical and Electronics Engineers: Piscataway, NJ, USA, 2016.Google ScholarThere is no corresponding record for this reference.
- 505Marek, A.; Blum, V.; Johanni, R.; Havu, V.; Lang, B.; Auckenthaler, T.; Heinecke, A.; Bungartz, H.-J.; Lederer, H. The ELPA library: scalable parallel eigenvalue solutions for electronic structure theory and computational science. J. Phys.: Condens. Matter 2014, 26, 213201, DOI: 10.1088/0953-8984/26/21/213201Google ScholarThere is no corresponding record for this reference.
- 506cuSOLVERMp: A High-Performance CUDA Library for Distributed Dense Linear Algebra. https://docs.nvidia.com/cuda/cusolvermp, 2024; (accessed, 2025 10 19).Google ScholarThere is no corresponding record for this reference.
- 507(a) Solcà, R.; Simberg, M.; Meli, R.; Invernizzi, A.; Reverdell, A.; Biddiscombe, J. DLA-Future: A Task-Based Linear Algebra Library Which Provides a GPU-Enabled Distributed Eigensolver. In Asynchronous Many-Task Systems and Applications; Springer Nature: Cham, 2024; pp 135– 141.Google ScholarThere is no corresponding record for this reference.See https://github.com/eth-cscs/DLA-Future.Google ScholarThere is no corresponding record for this reference.
- 508Kozhevnikov, A.; Taillefumier, M.; Frasch, S.; Pintarelli, V. J., Simon; ; Schulthess, T. Domain specific library for electronic structure calculations. 2019; https://github.com/electronic-structure/SIRIUS, (accessed: 2025 10 02).Google ScholarThere is no corresponding record for this reference.
- 509(a) Caldeweyher, E.; Mewes, J.-M.; Ehlert, S.; Grimme, S. Extension and evaluation of the D4 London-dispersion model for periodic systems. Phys. Chem. Chem. Phys. 2020, 22, 8499– 8512, DOI: 10.1039/D0CP00502AGoogle ScholarThere is no corresponding record for this reference.See. https://github.com/dftd4/dftd4.Google ScholarThere is no corresponding record for this reference.
- 510(a) Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB─An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652– 1671, DOI: 10.1021/acs.jctc.8b01176Google ScholarThere is no corresponding record for this reference.See. https://github.com/tblite/tblite.Google ScholarThere is no corresponding record for this reference.
- 511(a) Oleynichenko, A. V.; Zaitsevskii, A.; Mosyagin, N. S.; Petrov, A. N.; Eliav, E.; Titov, A. V. LIBGRPP: A library for the evaluation of molecular integrals of the generalized relativistic pseudopotential operator over Gaussian functions. Symmetry 2023, 15, 197, DOI: 10.3390/sym15010197Google ScholarThere is no corresponding record for this reference.See. https://github.com/aoleynichenko/libgrpp.Google ScholarThere is no corresponding record for this reference.
- 512Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R. A.; Parrinello, M. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961– 1972, DOI: 10.1016/j.cpc.2009.05.011Google ScholarThere is no corresponding record for this reference.
- 513Togo, A.; Tanaka, I. Spglib: a software library for crystal symmetry search, 2018. https://github.com/spglib/spglib (accessed 18 10 2025).Google ScholarThere is no corresponding record for this reference.
- 514(a) Rocha, A. R.; Garcia-Suarez, V. M.; Bailey, S. W.; Lambert, C. J.; Ferrer, J.; Sanvito, S. Towards molecular spintronics. Nat. Mater. 2005, 4, 335– 339, DOI: 10.1038/nmat1349Google ScholarThere is no corresponding record for this reference.See. https://github.com/StefanoSanvitoGroup/libsmeagol.Google ScholarThere is no corresponding record for this reference.
- 515(a) Posenitskiy, E.; Chilkuri, V. G.; Ammar, A.; Hapka, M.; Pernal, K.; Shinde, R.; Landinez Borda, E. J.; Filippi, C.; Nakano, K.; Kohulák, O. TREXIO: A file format and library for quantum chemistry. J. Chem. Phys. 2023, 158, 174801, DOI: 10.1063/5.0148161Google ScholarThere is no corresponding record for this reference.See. https://github.com/trex-coe/trexio.Google ScholarThere is no corresponding record for this reference.
- 516Azizi, M.; Wilhelm, J.; Golze, D.; Delesma, F. A.; Panadés-Barrueta, R. L.; Rinke, P.; Giantomassi, M.; Gonze, X. Validation of the GreenX library time-frequency component for efficient GW and RPA calculations. Phys. Rev. B 2024, 109, 245101, DOI: 10.1103/PhysRevB.109.245101Google ScholarThere is no corresponding record for this reference.
- 517Gamblin, T.; LeGendre, M.; Collette, M. R.; Lee, G. L.; Moody, A.; de Supinski, B. R.; Futral, S. The Spack package manager: bringing order to HPC software chaos. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; Association for Computing Machinery, 2015, pp 1– 12.Google ScholarThere is no corresponding record for this reference.
- 518Holmberg, N.; Laasonen, K. Efficient constrained density functional theory implementation for simulation of condensed phase electron transfer reactions. J. Chem. Theory Comput. 2017, 13, 587– 601, DOI: 10.1021/acs.jctc.6b01085Google ScholarThere is no corresponding record for this reference.
- 519Holmberg, N.; Laasonen, K. Diabatic model for electrochemical hydrogen evolution based on constrained DFT configuration interaction. J. Chem. Phys. 2018, 149, 104702, DOI: 10.1063/1.5038959Google ScholarThere is no corresponding record for this reference.
- 520Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R. A. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961– 1972, DOI: 10.1016/j.cpc.2009.05.011Google ScholarThere is no corresponding record for this reference.
- 521Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1– 5, DOI: 10.1016/j.scriptamat.2015.07.021Google ScholarThere is no corresponding record for this reference.
- 522Akimov, A. V. Libra: an open-source “methodology discovery” library for quantum and classical dynamics simulations. J. Comput. Chem. 2016, 37, 1626– 1649, DOI: 10.1002/jcc.24367Google ScholarThere is no corresponding record for this reference.
- 523Lervik, A.; Riccardi, E.; van Erp, T. S. PyRETIS: A well-done, medium-sized python library for rare events. J. Comput. Chem. 2017, 38, 2439– 2451, DOI: 10.1002/jcc.24900Google ScholarThere is no corresponding record for this reference.
- 524Riccardi, E.; Lervik, A.; Roet, S.; Aarøen, O.; van Erp, T. S. PyRETIS 2: an improbability drive for rare events. J. Comput. Chem. 2020, 41, 370– 377, DOI: 10.1002/jcc.26112Google ScholarThere is no corresponding record for this reference.
- 525Vervust, W.; Zhang, D. T.; Ghysels, A.; Roet, S.; van Erp, T. S.; Riccardi, E. PyRETIS 3: Conquering rare and slow events without boundaries. J. Comput. Chem. 2024, 45, 1224– 1234, DOI: 10.1002/jcc.27319Google ScholarThere is no corresponding record for this reference.
- 526Pizzi, G.; Vitale, V.; Arita, R.; Bluegel, S.; Freimuth, F.; Géranton, G.; Gibertini, M.; Gresch, D.; Johnson, C.; Koretsune, T. Wannier90 as a Community Code: New Features and Applications. J. Phys.: Condens. Matter 2020, 32, 165902, DOI: 10.1088/1361-648X/ab51ffGoogle ScholarThere is no corresponding record for this reference.
- 527Vogt, J.-R.; Schulz, M.; Mattos, R. S.; Barbatti, M.; Persico, M.; Granucci, G.; Hutter, J.; Hehn, A.-S. A mixed density functional theory and semi-empirical framework for trajectory surface hopping on extended systems. ChemRxiv 2025, 2025-ddxzg, DOI: 10.26434/chemrxiv-2025-ddxzgGoogle ScholarThere is no corresponding record for this reference.
- 528Zeng, J.; Zhang, D.; Peng, A.; Zhang, X.; He, S.; Wang, Y.; Liu, X.; Bi, H.; Li, Y.; Cai, C. DeePMD-kit v3: a multiple-backend framework for machine learning potentials. J. Chem. Theory Comput. 2025, 21, 4375– 4385, DOI: 10.1021/acs.jctc.5c00340Google ScholarThere is no corresponding record for this reference.
- 529Antalík, A.; Levy, A.; Johnson, S. K.; Olsen, J. M. H.; Rothlisberger, U. Making Puzzle Pieces Fit or Reshaping MiMiC for Multiscale Simulations with CP2K and More. J. Chem. Inf. Model. 2025, 65, 4994– 5005, DOI: 10.1021/acs.jcim.5c00409Google ScholarThere is no corresponding record for this reference.
- 530Larsen, A. H.; Mortensen, J. J.; Blomqvist, J.; Castelli, I. E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M. N.; Hammer, B.; Hargus, C. The atomic simulation environment─a Python library for working with atoms. J. Phys.: Condens. Matter 2017, 29, 273002, DOI: 10.1088/1361-648X/aa680eGoogle ScholarThere is no corresponding record for this reference.
- 531Ceriotti, M.; More, J.; Manolopoulos, D. E. i-PI: A Python interface for ab initio path integral molecular dynamics simulations. Comput. Phys. Commun. 2014, 185, 1019– 1026, DOI: 10.1016/j.cpc.2013.10.027Google ScholarThere is no corresponding record for this reference.
- 532Kapil, V.; Rossi, M.; Marsalek, O.; Petraglia, R.; Litman, Y.; Spura, T.; Cheng, B.; Cuzzocrea, A.; Meißner, R. H.; Wilkins, D. M. i-PI 2.0: A universal force engine for advanced molecular simulations. Comput. Phys. Commun. 2019, 236, 214– 223, DOI: 10.1016/j.cpc.2018.09.020Google ScholarThere is no corresponding record for this reference.
- 533Seeber, P.; Seidenath, S.; Steinmetzer, J.; Gräfe, S. Growing Spicy ONIOMs: Extending and generalizing concepts of ONIOM and many body expansions. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2023, 13, e1644 DOI: 10.1002/wcms.1644Google ScholarThere is no corresponding record for this reference.
- 534Verstraelen, T.; Van Houteghem, M.; Van Speybroeck, V.; Waroquier, M. MD-TRACKS: A Productive Solution for the Advanced Analysis of Molecular Dynamics and Monte Carlo simulations. J. Chem. Inf. Model. 2008, 48, 2414– 2424, DOI: 10.1021/ci800233yGoogle ScholarThere is no corresponding record for this reference.
- 535Verstraelen, T.; Van Speybroeck, V.; Waroquier, M. ZEOBUILDER: A GUI Toolkit for the Construction of Complex Molecular Structures on the Nanoscale with Building Blocks. J. Chem. Inf. Model. 2008, 48, 1530– 1541, DOI: 10.1021/ci8000748Google ScholarThere is no corresponding record for this reference.
- 536Ghysels, A.; Verstraelen, T.; Hemelsoet, K.; Waroquier, M.; Van Speybroeck, V. TAMkin: a versatile package for vibrational analysis and chemical kinetics. J. Chem. Inf. Model. 2010, 50, 1736– 1750, DOI: 10.1021/ci100099gGoogle ScholarThere is no corresponding record for this reference.
- 537Verstraelen, T.; Adams, W.; Pujal, L.; Tehrani, A.; Kelly, B. D.; Macaya, L.; Meng, F.; Richer, M.; Hernández-Esparza, R.; Yang, X. D. IOData: A python library for reading, writing, and converting computational chemistry file formats and generating input files. J. Comput. Chem. 2021, 42, 458– 464, DOI: 10.1002/jcc.26468Google ScholarThere is no corresponding record for this reference.
- 538Poeschel, F.; Juncheng, E.; Godoy, W. F.; Podhorszki, N.; Klasky, S.; Eisenhauer, G.; Davis, P. E.; Wan, L.; Gainaru, A.; Gu, J.; Transitioning from file-based HPC workflows to streaming data pipelines with openPMD and ADIOS2. In Smoky Mountains Computational Sciences and Engineering Conference; Springer Nature, 2021, pp 99– 118.Google ScholarThere is no corresponding record for this reference.
- 539Gu, J.; Davis, P.; Eisenhauer, G.; Godoy, W.; Huebl, A.; Klasky, S.; Parashar, M.; Podhorszki, N.; Poeschel, F.; Vay, J. Organizing large data sets for efficient analyses on HPC systems. J. Phys.: Conf. Ser. 2022, 2224, 012042, DOI: 10.1088/1742-6596/2224/1/012042Google ScholarThere is no corresponding record for this reference.
- 540Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn. J. Chem. Phys. 2024, 161, 082503, DOI: 10.1063/5.0216272Google ScholarThere is no corresponding record for this reference.
- 541Los, J. H.; Kühne, T. D. Inverse simulated annealing for the determination of amorphous structures. Phys. Rev. B 2013, 87, 214202, DOI: 10.1103/PhysRevB.87.214202Google ScholarThere is no corresponding record for this reference.
- 542Los, J. H.; Gabardi, S.; Bernasconi, M.; Kühne, T. D. Inverse simulated annealing: Improvements and application to amorphous InSb. Comput. Mater. Sci. 2016, 117, 7– 14, DOI: 10.1016/j.commatsci.2016.01.017Google ScholarThere is no corresponding record for this reference.
- 543Thomsen, B.; Shiga, M. Ab initio study of nuclear quantum effects on sub-and supercritical water. J. Chem. Phys. 2021, 155, 194107, DOI: 10.1063/5.0071857Google ScholarThere is no corresponding record for this reference.
- 544Thomsen, B.; Shiga, M. Structures of liquid and aqueous water isotopologues at ambient temperature from ab initio path integral simulations. Phys. Chem. Chem. Phys. 2022, 24, 10851– 10859, DOI: 10.1039/D2CP00499BGoogle ScholarThere is no corresponding record for this reference.
- 545Togo, A.; Chaput, L.; Tadano, T.; Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys.: Condens. Matter 2023, 35, 353001, DOI: 10.1088/1361-648X/acd831Google ScholarThere is no corresponding record for this reference.
- 546Schreder, L.; Luber, S. Implementation of frozen density embedding in CP2K and OpenMolcas: CASSCF wavefunctions embedded in a Gaussian and plane wave DFT environment. J. Chem. Phys. 2024, 161, 144110, DOI: 10.1063/5.0222409Google ScholarThere is no corresponding record for this reference.
- 547Battaglia, S.; Rossmannek, M.; Rybkin, V. V.; Tavernelli, I.; Hutter, J. A general framework for active space embedding methods with applications in quantum computing. npj Comput. Mater. 2024, 10, 297, DOI: 10.1038/s41524-024-01477-2Google ScholarThere is no corresponding record for this reference.
- 548Johnson, B. A.; Bu, S.; Mundy, C. J.; Ananth, N. MixPI: Mixed-Time Slicing Path Integral Software for Quantized Molecular Dynamics Simulations. arXiv 2024, 11988, DOI: 10.48550/arXiv.2411.11988Google ScholarThere is no corresponding record for this reference.
- 549CP2K Benchmark Suite, 2024. https://www.cp2k.org/performance (accessed, 2025 10 19).Google ScholarThere is no corresponding record for this reference.
Cited By
This article is cited by 1 publications.
- Yann Pouillon, Bill Clintone Oyomo, James Sifuna, María Camarasa-Gómez, Xinming Qin, Carlos Beltrán, Fernando Gómez-Ortiz, Honghui Shang, Javier Junquera. Implementation of the hybrid exchange-correlation functionals in the siesta code. Computer Physics Communications 2026, 323 , 110086. https://doi.org/10.1016/j.cpc.2026.110086

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The Journal of Physical Chemistry B
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
Figure 1

Figure 1. Values of the comparison metric ε for CP2K/Quickstep using the “UZH protocol” with respect to all-electron FP-LAPW calculations using the CP2K/SIRIUS code. For each element up to Rn four monoelemental cubic crystals were considered. (55)
Figure 2

Figure 2. Comparison of NNP and AIMD results for liquid water at 300 K as obtained from CP2K. (a) Time per MD step for 64, 512, and 4096 water molecules with the C-NNP model (solid lines with circles) and 64 water molecules with the revPBE0-D3 hybrid functional (dashed line with crosses) reported on a logarithmic scale. (b) Radial distribution functions and (c) vibrational density of states of the hydrogen atoms. Data in panel (b,c) are partially reused from ref (322). Copyright 2020, American Institute of Physics.
Figure 3

Figure 3. Impact of NQEs on the structure and dynamics of hexagonal ice at 250 K. (a) Bead convergence of the virial kinetic energy estimator using the PILE and PIQTB thermostat. (b) Radial distribution functions with classical and quantum nuclei and (b) vibrational density of states obtained with classical MD, RPMD and TRPMD, respectively. All simulations rely on using the C-NNP trained with revPBE0-D3 reference data, (322) as available in the CP2K data repository (see also Section 10.1).
Figure 4
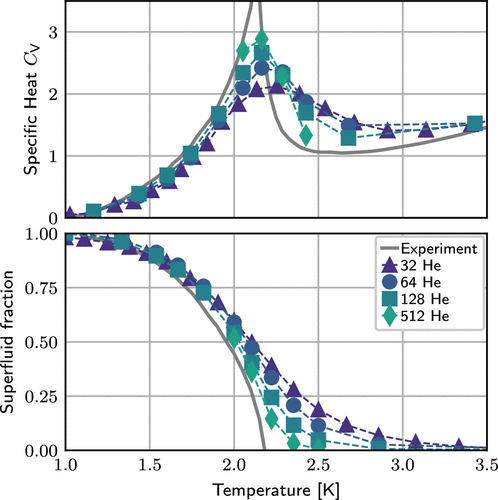
Figure 4. Temperature dependence of (a) the heat capacity and (b) the superfluid fraction of bulk 4He obtained using the canonical worm algorithm to sample bosonic exchange and the winding number estimator to analyze superfluidity (see text). The numerical pair density matrix is the one provided with the CP2K package and has been computed at 80 K using the He···He potential from ref (389), thus P × T = 80 K is kept constant for the temperature scan. The present bosonic PIMC data were obtained from only 32 atoms (in a truncated octahedral supercell according to the sample input provided in the text) and are compared to published results of ref (386) generated from 64, 128, and 512 atoms in the corresponding periodic truncated octahedron using CP2K and the same methodology. For reference, the respective experimental data (391,392) are shown by gray lines; note that deviations of the computed properties from experiment are mainly due to finite-size effects close to the superfluid phase transition given such small system sizes. (390)
Figure 5

Figure 5. For an illustration of the HPIMD/MC method, as implemented in CP2K. (386) The fully flexible and reactive solute species are propagated using PIMD techniques (such as PIQTB in conjunction with the one-body density matrix), while the bosonic solvent (for instance many 4He atoms either forming a finite cluster as illustrated here, or hosted within a periodic supercell) is sampled using bosonic PIMC (for instance based on the canonical worm algorithm in conjunction with the numerical pair density matrix) at a common temperature T (see text). The interactions can be described using high-dimensional NNPs with CCSD(T) accuracy, as described in Section 10.1.
Figure 6

Figure 6. Potential energy Umol along one Trotter replica of quantum PIMD trajectories of (from top to bottom) CH4, CH5+, H3O+, H5O2+, respectively, all at 1.67 K using NNPs trained with CCSD(T*)-F12a/aug-cc-pVTZ reference energies obtained from Molpro. The corresponding CC single-point data were obtained by recomputing the energies at each and every step of the depicted NNP trajectories and are shown as red dotted lines. All energies are reported relative to the equilibrium structures of the respective global minima. Reproduced from Figure 6 of ref (386). Copyright 2020, American Institute of Physics.
Figure 7

Figure 7. Spatial distributions functions stemming from all N helium atoms around a fixed molecular impurity (i.e., X·HeN), as sampled from PIMC simulations (without bosonic exchange) at 1.67 K. The solute···helium interaction energies Uint used in the PIMC sampling of the helium atoms are interpolated from values that have been precomputed on a grid using either the NNP (left column) or single-point CC calculations (right column) on the identical grid both based on counterpoise-corrected CCSD(T*)-F12a/aug-cc-pVTZ energy differences obtained from Molpro. Reproduced from Figure 7 of ref (386). Copyright 2020, American Institute of Physics.
Figure 8
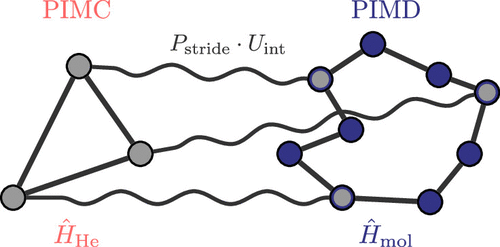
Figure 8. Illustration of PI striding for bosonic HPIMD/MC simulations, as implemented in CP2K: coupling of the PI representation of one helium atom (left) discretized with three Trotter replica (in practice sampled using PIMC based on the pair density matrix representation) to the PI representation of the solute molecule (right) discretized with nine beads (sampled using PIMD based on the one-body density matrix in conjunction with PIQTB thermostatting, as detailed in Section 10.3), thus corresponding to a striding length of three, Pstride = 3. Reproduced from Figure 3 of ref (386). Copyright 2020, American Institute of Physics.
Figure 9

Figure 9. Distance distributions functions of H3O+·4He8 at 1 K with BE sampling of the helium microsolvation environment. The intramolecular O–H, as well as the intermolecular O–He and H–He distance distributions are presented. The inset depicts the beads of the hydronium ion in terms of balls (red/gray for O/H), while all 4He atoms involved in a bosonic exchange cycle are visualized by green strings and all others by green balls.
References
This article references 549 other publications.
- 1Dirac, P. A. M. Quantum mechanics of many-electron systems. Math. Proc. Cambridge Philos. Soc. 1929, 123, 714– 733, DOI: 10.1098/rspa.1929.0094There is no corresponding record for this reference.
- 2Billeter, S. R.; Curioni, A.; Andreoni, W. Efficient linear scaling geometry optimization and transition-state search for direct wavefunction optimization schemes in density functional theory using a plane-wave basis. Comput. Mater. Sci. 2003, 27, 437– 445, DOI: 10.1016/S0927-0256(03)00043-0There is no corresponding record for this reference.
- 3Schlegel, H. B. Geometry Optimization. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 790– 809, DOI: 10.1002/wcms.34There is no corresponding record for this reference.
- 4Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087– 1092, DOI: 10.1063/1.1699114There is no corresponding record for this reference.
- 5Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12562– 12566, DOI: 10.1073/pnas.202427399There is no corresponding record for this reference.
- 6Rahman, A. Correlations in the Motion of Atoms in Liquid Argon. Phys. Rev. 1964, 136, A405– A411, DOI: 10.1103/PhysRev.136.A405There is no corresponding record for this reference.
- 7Tuckerman, M. E. Statistical Mechanics: Theory and Molecular Simulation; Oxford University Press, 2010.There is no corresponding record for this reference.
- 8Frenkel, D.; Smit, B. Understanding Molecular Simulation From Algorithms to Applications; Academic Press, 2023.There is no corresponding record for this reference.
- 9Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press, 2009.There is no corresponding record for this reference.
- 10Hutter, J. Car-Parrinello molecular dynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 604– 612, DOI: 10.1002/wcms.90There is no corresponding record for this reference.
- 11Kühne, T. D.; Krack, M.; Mohamed, F. R.; Parrinello, M. Efficient and accurate Car-Parrinello-like approach to Born-Oppenheimer molecular dynamics. Phys. Rev. Lett. 2007, 98, 066401, DOI: 10.1103/PhysRevLett.98.066401There is no corresponding record for this reference.
- 12Kühne, T. D.; Prodan, E. Disordered crystals from first principles I: Quantifying the configuration space. Ann. Phys. 2018, 391, 120– 149, DOI: 10.1016/j.aop.2018.01.016There is no corresponding record for this reference.
- 13Kühne, T. D. Second generation Car-Parrinello molecular dynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 391– 406, DOI: 10.1002/wcms.1176There is no corresponding record for this reference.
- 14Kühne, T. D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V. V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R. Z.; Schütt, O.; Schiffmann, F. CP2K: An electronic structure and molecular dynamics software package-Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103, DOI: 10.1063/5.0007045There is no corresponding record for this reference.
- 15Hutter, J.; Iannuzzi, M.; Kühne, T. D.. In Comprehensive Computational Chemistry; Yáñez, M., Boyed, R. J., Eds.; Elsevier, 2024; pp 493– 517.There is no corresponding record for this reference.
- 16Kohn, W. Nobel Lecture: Electronic structure of matter─wave functions and density functionals. Rev. Mod. Phys. 1999, 71, 1253– 1266, DOI: 10.1103/RevModPhys.71.1253There is no corresponding record for this reference.
- 17Pople, J. A. Nobel Lecture: Quantum chemical models. Rev. Mod. Phys. 1999, 71, 1267– 1274, DOI: 10.1103/RevModPhys.71.1267There is no corresponding record for this reference.
- 18Ihm, J.; Zunger, A.; Cohen, M. L. Momentum-space formalism for the total energy of solids. J. Phys. C: Solid State Phys. 1979, 12, 4409– 4422, DOI: 10.1088/0022-3719/12/21/009There is no corresponding record for this reference.
- 19Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953– 17979, DOI: 10.1103/PhysRevB.50.17953There is no corresponding record for this reference.
- 20Andersen, O. K. Linear methods in band theory. Phys. Rev. B 1975, 12, 3060– 3083, DOI: 10.1103/PhysRevB.12.3060There is no corresponding record for this reference.
- 21Aryasetiawan, F.; Gunnarsson, O. The GW method. Rep. Prog. Phys. 1998, 61, 237, DOI: 10.1088/0034-4885/61/3/002There is no corresponding record for this reference.
- 22Krack, M.; Parrinello, M. High Performance Computing in Chemistry; NIC Series; NIC-Directors, 2004; Vol. 25, pp 29– 51.There is no corresponding record for this reference.
- 23VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103– 128, DOI: 10.1016/j.cpc.2004.12.014There is no corresponding record for this reference.
- 24Lippert, G.; Hutter, J.; Parrinello, M. A hybrid Gaussian and plane wave density functional scheme. Mol. Phys. 1997, 92, 477– 488, DOI: 10.1080/00268979709482119There is no corresponding record for this reference.
- 25Lippert, G.; Hutter, J.; Parrinello, M. The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations. Theor. Chem. Acc. 1999, 103, 124– 140, DOI: 10.1007/s002140050523There is no corresponding record for this reference.
- 26Slater, J. C. A simplification of the Hartree-Fock method. Phys. Rev. 1951, 81, 385, DOI: 10.1103/PhysRev.81.385There is no corresponding record for this reference.
- 27Møller, C.; Plesset, M. S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618, DOI: 10.1103/PhysRev.46.618There is no corresponding record for this reference.
- 28Bohm, D.; Pines, D. A. collective description of electron interactions: III. Coulomb interactions in a degenerate electron gas. Phys. Rev. 1953, 92, 609, DOI: 10.1103/PhysRev.92.609There is no corresponding record for this reference.
- 29Gell-Mann, M.; Brueckner, K. A. Correlation energy of an electron gas at high density. Phys. Rev. 1957, 106, 364, DOI: 10.1103/PhysRev.106.364There is no corresponding record for this reference.
- 30VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105, DOI: 10.1063/1.2770708There is no corresponding record for this reference.
- 31Goedecker, S.; Teter, M.; Hutter, J. Separable dual–space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703– 1710, DOI: 10.1103/PhysRevB.54.1703There is no corresponding record for this reference.
- 32Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641– 3662, DOI: 10.1103/PhysRevB.58.3641There is no corresponding record for this reference.
- 33Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 2005, 114, 145– 152, DOI: 10.1007/s00214-005-0655-yThere is no corresponding record for this reference.
- 34Reiher, M. Relativistic douglas–kroll–hess theory. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 139– 149, DOI: 10.1002/wcms.67There is no corresponding record for this reference.
- 35Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865– 3868, DOI: 10.1103/PhysRevLett.77.3865There is no corresponding record for this reference.
- 36Sun, J.; Ruzsinszky, A.; Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 2015, 115, 036402, DOI: 10.1103/PhysRevLett.115.036402There is no corresponding record for this reference.
- 37Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158– 6170, DOI: 10.1063/1.478522There is no corresponding record for this reference.
- 38Lu, J.-B.; Cantu, D. C.; Nguyen, M.-T.; Li, J.; Glezakou, V.-A.; Rousseau, R. Norm-conserving pseudopotentials and basis sets to explore lanthanide chemistry in complex environments. J. Chem. Theory Comput. 2019, 15, 5987– 5997, DOI: 10.1021/acs.jctc.9b00553There is no corresponding record for this reference.
- 39Lu, J.-B.; Cantu, D. C.; Xu, C.-Q.; Nguyen, M.-T.; Hu, H.-S.; Glezakou, V.-A.; Rousseau, R.; Li, J. Norm-conserving pseudopotentials and basis sets to explore actinide chemistry in complex environments. J. Chem. Theory Comput. 2021, 17, 3360– 3371, DOI: 10.1021/acs.jctc.1c00026There is no corresponding record for this reference.
- 40Lu, J.-B.; Jiang, X.-L.; Hu, H.-S.; Li, J. Norm-conserving 4f-in-core pseudopotentials and basis sets optimized for trivalent lanthanides (Ln= Ce–Lu). J. Chem. Theory Comput. 2023, 19, 82– 96, DOI: 10.1021/acs.jctc.2c00922There is no corresponding record for this reference.
- 41Lu, J.-B.; Zhang, Y.-Y.; Liu, J.-B.; Li, J. Norm-Conserving 5f-in-Core Pseudopotentials and Gaussian Basis Sets Optimized for Tri-and Tetra-Valent Actinides (An= Pa–Lr). J. Chem. Theory Comput. 2025, 21, 170– 182, DOI: 10.1021/acs.jctc.4c01189There is no corresponding record for this reference.
- 42Willand, A.; Kvashnin, Y. O.; Genovese, L.; Vázquez-Mayagoitia, Á.; Deb, A. K.; Sadeghi, A.; Deutsch, T.; Goedecker, S. Norm-conserving pseudopotentials with chemical accuracy compared to all-electron calculations. J. Chem. Phys. 2013, 138, 104109, DOI: 10.1063/1.4793260There is no corresponding record for this reference.
- 43Oleynichenko, A. V.; Zaitsevskii, A.; Mosyagin, N. S.; Petrov, A. N.; Eliav, E.; Titov, A. V. LIBGRPP: A library for the evaluation of molecular integrals of the generalized relativistic pseudopotential operator over Gaussian functions. Symmetry 2023, 15, 197, DOI: 10.3390/sym15010197There is no corresponding record for this reference.
- 44Pritchard, B. P.; Altarawy, D.; Didier, B.; Gibson, T. D.; Windus, T. L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814– 4820, DOI: 10.1021/acs.jcim.9b00725There is no corresponding record for this reference.
- 45Zijlstra, E. S.; Huntemann, N.; Kalitsov, A.; Garcia, M. E.; Von Barth, U. Optimized Gaussian basis sets for Goedecker–Teter–Hutter pseudopotentials. Model. Simul. Mater. Sci. Eng. 2009, 17, 015009, DOI: 10.1088/0965-0393/17/1/015009There is no corresponding record for this reference.
- 46Peintinger, M. F.; Oliveira, D. V.; Bredow, T. Consistent Gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2013, 34, 451– 459, DOI: 10.1002/jcc.23153There is no corresponding record for this reference.
- 47Vilela Oliveira, D.; Laun, J.; Peintinger, M. F.; Bredow, T. BSSE-correction scheme for consistent gaussian basis sets of double-and triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2019, 40, 2364– 2376, DOI: 10.1002/jcc.26013There is no corresponding record for this reference.
- 48Laun, J.; Bredow, T. BSSE-corrected consistent Gaussian basis sets of triple-zeta valence with polarization quality of the sixth period for solid-state calculations. J. Comput. Chem. 2021, 42, 1064– 1072, DOI: 10.1002/jcc.26521There is no corresponding record for this reference.
- 49Laun, J.; Bredow, T. BSSE-corrected consistent Gaussian basis sets of triple-zeta valence with polarization quality of the fifth period for solid-state calculations. J. Comput. Chem. 2022, 43, 839– 846, DOI: 10.1002/jcc.26839There is no corresponding record for this reference.
- 50Guidon, M.; Hutter, J.; VandeVondele, J. Auxiliary Density Matrix Methods for Hartree-Fock Exchange Calculations. J. Chem. Theory Comput. 2010, 6, 2348– 2364, DOI: 10.1021/ct1002225There is no corresponding record for this reference.
- 51Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 1965, 139, A796– A823, DOI: 10.1103/PhysRev.139.A796There is no corresponding record for this reference.
- 52Aryasetiawan, F.; Gunnarsson, O. The GW method. Rep. Prog. Phys. 1998, 61, 237– 312, DOI: 10.1088/0034-4885/61/3/002There is no corresponding record for this reference.
- 53Whitten, J. L. Coulombic potential energy integrals and approximations. J. Chem. Phys. 1973, 58, 4496– 4501, DOI: 10.1063/1.1679012There is no corresponding record for this reference.
- 54Dunlap, B. I.; Connolly, J.; Sabin, J. On some approximations in applications of Xα theory. J. Chem. Phys. 1979, 71, 3396– 3402, DOI: 10.1063/1.438728There is no corresponding record for this reference.
- 55Bosoni, E.; Beal, L.; Bercx, M.; Blaha, P.; Blügel, S.; Bröder, J.; Callsen, M.; Cottenier, S.; Degomme, A.; Dikan, V. How to verify the precision of density-functional-theory implementations via reproducible and universal workflows. Nat. Rev. Phys. 2024, 6, 45– 58, DOI: 10.1038/s42254-023-00655-3There is no corresponding record for this reference.
- 56Krack, M.; Parrinello, M. All-electron ab-initio molecular dynamics. Phys. Chem. Chem. Phys. 2000, 2, 2105– 2112, DOI: 10.1039/b001167nThere is no corresponding record for this reference.
- 57Dorado, B.; Amadon, B.; Freyss, M.; Bertolus, M. DFT + U calculations of the ground state and metastable states of uranium dioxide. Phys. Rev. B 2009, 79, 235125, DOI: 10.1103/PhysRevB.79.235125There is no corresponding record for this reference.
- 58Krack, M. On the ground state electronic structure of uranium dioxide. Phys. Scr. 2015, 90, 094014, DOI: 10.1088/0031-8949/90/9/094014There is no corresponding record for this reference.
- 59Krack, M.; Gambirasio, A.; Parrinello, M. Ab initio X-ray scattering of liquid water. J. Chem. Phys. 2002, 117, 9409– 9412, DOI: 10.1063/1.1517040There is no corresponding record for this reference.
- 60Müller, P.; Karhan, K.; Krack, M.; Gerstmann, U.; Schmidt, W. G.; Bauer, M.; Kühne, T. D. Impact of finite-temperature and condensed-phase effects on theoretical X-ray absorption spectra of transition metal complexes. J. Comput. Chem. 2019, 40, 712– 716, DOI: 10.1002/jcc.25641There is no corresponding record for this reference.
- 61Krack, M.; Köster, A. M. An adaptive numerical integrator for molecular integrals. J. Chem. Phys. 1998, 108, 3226– 3234, DOI: 10.1063/1.475719There is no corresponding record for this reference.
- 62VandeVondele, J.; Hutter, J. An efficient orbital transformation method for electronic structure calculations. J. Chem. Phys. 2003, 118, 4365– 4369, DOI: 10.1063/1.1543154There is no corresponding record for this reference.
- 63Kerker, G. Efficient iteration scheme for self-consistent pseudopotential calculations. Phys. Rev. B 1981, 23, 3082, DOI: 10.1103/PhysRevB.23.3082There is no corresponding record for this reference.
- 64Pulay, P. Convergence acceleration of iterative sequences. the case of scf iteration. Chem. Phys. Lett. 1980, 73, 393– 398, DOI: 10.1016/0009-2614(80)80396-4There is no corresponding record for this reference.
- 65Marks, L. D.; Luke, D. Robust mixing for ab initio quantum mechanical calculations. Phys. Rev. B 2008, 78, 075114, DOI: 10.1103/PhysRevB.78.075114There is no corresponding record for this reference.
- 66Broyden, C. G. A Class of Methods for Solving Nonlinear Simultaneous Equations. Math. Comput. 1965, 19, 577, DOI: 10.1090/s0025-5718-1965-0198670-6There is no corresponding record for this reference.
- 67Mermin, N. D. Thermal Properties of the Inhomogeneous Electron Gas. Phys. Rev. 1965, 137, A1441– A1443, DOI: 10.1103/PhysRev.137.A1441There is no corresponding record for this reference.
- 68Hutter, J.; Parrinello, M.; Vogel, S. Exponential transformation of molecular orbitals. J. Chem. Phys. 1994, 101, 3862– 3865, DOI: 10.1063/1.467504There is no corresponding record for this reference.
- 69Gan, C.; Haynes, P.; Payne, M. Preconditioned conjugate gradient method for the sparse generalized eigenvalue problem in electronic structure calculations. Comput. Phys. Commun. 2001, 134, 33– 40, DOI: 10.1016/S0010-4655(00)00188-0There is no corresponding record for this reference.
- 70Monkhorst, H. J.; Pack, J. D. Special points for Brillouin–zone integrations. Phys. Rev. B 1976, 13, 5188– 5192, DOI: 10.1103/PhysRevB.13.5188There is no corresponding record for this reference.
- 71Guidon, M.; Hutter, J.; VandeVondele, J. Robust Periodic Hartree-Fock Exchange for Large-Scale Simulations Using Gaussian Basis Sets. J. Chem. Theory Comput. 2009, 5, 3010– 3021, DOI: 10.1021/ct900494gThere is no corresponding record for this reference.
- 72Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098– 3100, DOI: 10.1103/PhysRevA.38.3098There is no corresponding record for this reference.
- 73Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785– 789, DOI: 10.1103/PhysRevB.37.785There is no corresponding record for this reference.
- 74Merlot, P.; Izsák, R.; Borgoo, A.; Kjærgaard, T.; Helgaker, T.; Reine, S. Charge-constrained auxiliary-density-matrix methods for the Hartree–Fock exchange contribution. J. Chem. Phys. 2014, 141, 094104, DOI: 10.1063/1.4894267There is no corresponding record for this reference.
- 75Jensen, F. Unifying general and segmented contracted basis sets. Segmented polarization consistent basis sets. J. Chem. Theory Comput. 2014, 10, 1074– 1085, DOI: 10.1021/ct401026aThere is no corresponding record for this reference.
- 76Kumar, C.; Fliegl, H.; Jensen, F.; Teale, A. M.; Reine, S.; Kjærgaard, T. Accelerating Kohn-Sham response theory using density fitting and the auxiliary-density-matrix method. Int. J. Quantum Chem. 2018, 118, e25639 DOI: 10.1002/qua.25639There is no corresponding record for this reference.
- 77Gavini, V.; Baroni, S.; Blum, V.; Bowler, D. R.; Buccheri, A.; Chelikowsky, J. R.; Das, S.; Dawson, W.; Delugas, P.; Dogan, M. Roadmap on electronic structure codes in the exascale era. Model. Simul. Mater. Sci. Eng. 2023, 31, 063301, DOI: 10.1088/1361-651X/acdf06There is no corresponding record for this reference.
- 78Bussy, A.; Schütt, O.; Hutter, J. Sparse tensor based nuclear gradients for periodic Hartree–Fock and low-scaling correlated wave function methods in the CP2K software package: A massively parallel and GPU accelerated implementation. J. Chem. Phys. 2023, 158, 164109, DOI: 10.1063/5.0144493There is no corresponding record for this reference.
- 79Vahtras, O.; Almlöf, J.; Feyereisen, M. Integral approximations for LCAO-SCF calculations. Chem. Phys. Lett. 1993, 213, 514– 518, DOI: 10.1016/0009-2614(93)89151-7There is no corresponding record for this reference.
- 80Weigend, F. A fully direct RI-HF algorithm: Implementation, optimized auxiliary basis sets, demonstration of accuracy and efficiency. Phys. Chem. Chem. Phys. 2002, 4, 4285– 4291, DOI: 10.1039/b204199pThere is no corresponding record for this reference.
- 81Stoychev, G. L.; Auer, A. A.; Neese, F. Automatic generation of auxiliary basis sets. J. Chem. Theory Comput. 2017, 13, 554– 562, DOI: 10.1021/acs.jctc.6b01041There is no corresponding record for this reference.
- 82Bussy, A.; Hutter, J. Efficient periodic resolution-of-the-identity Hartree–Fock exchange method with k-point sampling and Gaussian basis sets. J. Chem. Phys. 2024, 160, 064116, DOI: 10.1063/5.0189659There is no corresponding record for this reference.
- 83Del Ben, M.; Hutter, J.; VandeVondele, J. Second-Order Møller–Plesset Perturbation Theory in the Condensed Phase: An Efficient and Massively Parallel Gaussian and Plane Waves Approach. J. Chem. Theory Comput. 2012, 8, 4177– 4188, DOI: 10.1021/ct300531wThere is no corresponding record for this reference.
- 84Del Ben, M.; Hutter, J.; VandeVondele, J. Electron Correlation in the Condensed Phase from a Resolution of Identity Approach Based on the Gaussian and Plane Waves Scheme. J. Chem. Theory Comput. 2013, 9, 2654– 2671, DOI: 10.1021/ct4002202There is no corresponding record for this reference.
- 85Weigend, F.; Häser, M. RI-MP2: first derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331– 340, DOI: 10.1007/s002140050269There is no corresponding record for this reference.
- 86Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143– 152, DOI: 10.1016/S0009-2614(98)00862-8There is no corresponding record for this reference.
- 87Del Ben, M.; Hutter, J.; VandeVondele, J. Forces and stress in second order Møller-Plesset perturbation theory for condensed phase systems within the resolution-of-identity Gaussian and plane waves approach. J. Chem. Phys. 2015, 143, 102803, DOI: 10.1063/1.4919238There is no corresponding record for this reference.
- 88Rybkin, V. V.; VandeVondele, J. Spin-Unrestricted Second-Order Møller–Plesset (MP2) Forces for the Condensed Phase: From Molecular Radicals to F-Centers in Solids. J. Chem. Theory Comput. 2016, 12, 2214– 2223, DOI: 10.1021/acs.jctc.6b00015There is no corresponding record for this reference.
- 89Schwartz, C. Importance of Angular Correlations between Atomic Electrons. Phys. Rev. 1962, 126, 1015– 1019, DOI: 10.1103/PhysRev.126.1015There is no corresponding record for this reference.
- 90Borštnik, U.; VandeVondele, J.; Weber, V.; Hutter, J. Sparse matrix multiplication: The distributed block-compressed sparse row library. Parallel Comput. 2014, 40, 47– 58, DOI: 10.1016/j.parco.2014.03.012There is no corresponding record for this reference.
- 91SPLA: Specialized Parallel Linear Algebra. https://github.com/eth-cscs/spla, (accessed, 2025 05 29).There is no corresponding record for this reference.
- 92Stein, F.; Hutter, J. Double-hybrid density functionals for the condensed phase: Gradients, stress tensor, and auxiliary-density matrix method acceleration. J. Chem. Phys. 2022, 156, 074107, DOI: 10.1063/5.0082327There is no corresponding record for this reference.
- 93Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108, DOI: 10.1063/1.2148954There is no corresponding record for this reference.
- 94(a) Lehtola, S.; Steigemann, C.; Oliveira, M. J. T.; Marques, M. A. L. Recent developments in libxc ─ A comprehensive library of functionals for density functional theory. SoftwareX 2018, 7, 1– 5, DOI: 10.1016/j.softx.2017.11.002There is no corresponding record for this reference.See. https://gitlab.com/libxc/libxc.There is no corresponding record for this reference.
- 95Stein, F.; Hutter, J.; Rybkin, V. V. Double-hybrid DFT functionals for the condensed phase: Gaussian and plane waves implementation and evaluation. Molecules 2020, 25, 5174, DOI: 10.3390/molecules25215174There is no corresponding record for this reference.
- 96Furche, F. Developing the random phase approximation into a practical post-Kohn–Sham correlation model. J. Chem. Phys. 2008, 129, 114105, DOI: 10.1063/1.2977789There is no corresponding record for this reference.
- 97Boyd, J. P. Exponentially convergent Fourier-Chebshev quadrature schemes on bounded and infinite intervals. J. Sci. Comput. 1987, 2, 99– 109, DOI: 10.1007/BF01061480There is no corresponding record for this reference.
- 98Braess, D.; Hackbusch, W. Approximation of 1/x by exponential sums in [1, ∞). IMA J. Numer. Anal. 2005, 25, 685– 697, DOI: 10.1093/imanum/dri015There is no corresponding record for this reference.
- 99Ren, X.; Rinke, P.; Scuseria, G. E.; Scheffler, M. Renormalized second-order perturbation theory for the electron correlation energy: Concept, implementation, and benchmarks. Phys. Rev. B 2013, 88, 035120, DOI: 10.1103/PhysRevB.88.035120There is no corresponding record for this reference.
- 100Bates, J. E.; Furche, F. Communication: Random phase approximation renormalized many-body perturbation theory. J. Chem. Phys. 2013, 139, 171103, DOI: 10.1063/1.4827254There is no corresponding record for this reference.
- 101Freeman, D. L. Coupled-cluster expansion applied to the electron gas: Inclusion of ring and exchange effects. Phys. Rev. B 1977, 15, 5512– 5521, DOI: 10.1103/PhysRevB.15.5512There is no corresponding record for this reference.
- 102Stein, F.; Hutter, J. Massively parallel implementation of gradients within the random phase approximation: Application to the polymorphs of benzene. J. Chem. Phys. 2024, 160, 024120, DOI: 10.1063/5.0180704There is no corresponding record for this reference.
- 103Kwasniewski, G.; Kabić, M.; Besta, M.; VandeVondele, J.; Solcà, R.; Hoefler, T. Red-blue pebbling revisited: Near optimal parallel matrix-matrix multiplication. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; Association for Computing Machinery, 2019; pp 1– 22.There is no corresponding record for this reference.
- 104Wilhelm, J.; Seewald, P.; Del Ben, M.; Hutter, J. Large-scale cubic-scaling random phase approximation correlation energy calculations using a Gaussian basis. J. Chem. Theory Comput. 2016, 12, 5851– 5859, DOI: 10.1021/acs.jctc.6b00840There is no corresponding record for this reference.
- 105Valeev, E. F. Libint: A library for the evaluation of molecular integrals of many-body operators over Gaussian functions. http://libint.valeyev.net/, 2024; (accessed, 2025 05 29).There is no corresponding record for this reference.
- 106Anisimov, V. I.; Zaanen, J.; Andersen, O. K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 1991, 44, 943– 954, DOI: 10.1103/PhysRevB.44.943There is no corresponding record for this reference.
- 107Dudarev, S. L.; Manh, D. N.; Sutton, A. P. Effect of Mott-Hubbard correlations on the electronic structure and structural stability of uranium dioxide. Philos. Mag. B 1997, 75, 613– 628, DOI: 10.1080/13642819708202343There is no corresponding record for this reference.
- 108Dudarev, S. L.; Botton, G. A.; Savrasov, S. Y.; Humphreys, C. J.; Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505– 1509, DOI: 10.1103/PhysRevB.57.1505There is no corresponding record for this reference.
- 109Mulliken, R. S. Electronic Population Analysis on LCAO-MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833– 1840, DOI: 10.1063/1.1740588There is no corresponding record for this reference.
- 110Löwdin, P.-O. On the Non-Orthogonality Problem Connected with the Use of Atomic Wave Functions in the Theory of Molecules and Crystals. J. Chem. Phys. 1950, 18, 365– 375, DOI: 10.1063/1.1747632There is no corresponding record for this reference.
- 111Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G. K. H.; Marks, L. D. WIEN2k: An APW+lo program for calculating the properties of solids. J. Chem. Phys. 2020, 152, 074101, DOI: 10.1063/1.5143061There is no corresponding record for this reference.
- 112The FLEUR project. https://www.flapw.de/, (accessed: 2025 10 17).There is no corresponding record for this reference.
- 113The Elk Code. https://elk.sourceforge.io/, (accessed, 2025 10 17).There is no corresponding record for this reference.
- 114Gulans, A.; Kontur, S.; Meisenbichler, C.; Nabok, D.; Pavone, P.; Rigamonti, S.; Sagmeister, S.; Werner, U.; Draxl, C. exciting: a full-potential all-electron package implementing density-functional theory and many-body perturbation theory. J. Phys.: Condens. Matter 2014, 26, 363202, DOI: 10.1088/0953-8984/26/36/363202There is no corresponding record for this reference.
- 115Hamann, D. R.; Schlüter, M.; Chiang, C. Norm-Conserving Pseudopotentials. Phys. Rev. Lett. 1979, 43, 1494– 1497, DOI: 10.1103/PhysRevLett.43.1494There is no corresponding record for this reference.
- 116Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892– 7895, DOI: 10.1103/PhysRevB.41.7892There is no corresponding record for this reference.
- 117Slater, J. C. Wave functions in a periodic potential. Phys. Rev. 1937, 51, 846, DOI: 10.1103/PhysRev.51.846There is no corresponding record for this reference.
- 118Ceperley, D. M.; Alder, B. J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566, DOI: 10.1103/PhysRevLett.45.566There is no corresponding record for this reference.
- 119Golze, D.; Dvorak, M.; Rinke, P. The GW compendium: A practical guide to theoretical photoemission spectroscopy. Front. Chem. 2019, 7, 377, DOI: 10.3389/fchem.2019.00377There is no corresponding record for this reference.
- 120Capelle, K. A. bird’s-eye view of density-functional theory. Braz. J. Phys. 2006, 36, 1318– 1343, DOI: 10.1590/S0103-97332006000700035There is no corresponding record for this reference.
- 121Caravati, S.; Bernasconi, M.; Kühne, T.; Krack, M.; Parrinello, M. First-principles study of crystalline and amorphousGe2Sb2Te5 and the effects of stoichiometric defects. J. Phys.: Condens. Matter 2009, 21, 255501, DOI: 10.1088/0953-8984/21/25/255501There is no corresponding record for this reference.
- 122Caravati, S.; Colleoni, D.; Mazzarello, R.; Kühne, T.; Krack, M.; Bernasconi, M.; Parrinello, M. First-principles study of nitrogen doping in cubic and amorphousGe2Sb2Te5. J. Phys.: Condens. Matter 2011, 23, 265801, DOI: 10.1088/0953-8984/23/26/265801There is no corresponding record for this reference.
- 123Los, J. H.; Kühne, T. D.; Gabardi, S.; Bernasconi, M. First-principles study of the amorphous In 3 SbTe 2 phase change compound. Phys. Rev. B 2013, 88, 174203, DOI: 10.1103/PhysRevB.88.174203There is no corresponding record for this reference.
- 124Gabardi, S.; Caravati, S.; Los, J. H.; Kühne, T. D.; Bernasconi, M. Influence of the exchange and correlation functional on the structure of amorphous InSb and In3SbTe2 compounds. J. Chem. Phys. 2016, 144, 204508, DOI: 10.1063/1.4950817There is no corresponding record for this reference.
- 125Schambeck, M.; Golze, D.; Wilhelm, J. Solving multipole challenges in the GW100 benchmark enables precise low-scaling GW calculations. Phys. Rev. B 2024, 110, 125146, DOI: 10.1103/PhysRevB.110.125146There is no corresponding record for this reference.
- 126Pasquier, R.; Camarasa-Gómez, M.; Hehn, A.-S.; Hernangómez-Pérez, D.; Wilhelm, J. Efficient GW band structure calculations using Gaussian basis functions and application to atomically thin transition-metal dichalcogenides. Phys. Rev. B 2025, 112, 205103, DOI: 10.1103/v4zv-1pf9There is no corresponding record for this reference.
- 127Graml, M.; Zollner, K.; Hernangómez-Pérez, D.; Faria Junior, P. E.; Wilhelm, J. Low-Scaling GW Algorithm Applied to Twisted Transition-Metal Dichalcogenide Heterobilayers. J. Chem. Theory Comput. 2024, 20, 2202– 2208, DOI: 10.1021/acs.jctc.3c01230There is no corresponding record for this reference.
- 128van Setten, M. J.; Caruso, F.; Sharifzadeh, S.; Ren, X.; Scheffler, M.; Liu, F.; Lischner, J.; Lin, L.; Deslippe, J. R.; Louie, S. G. GW100: Benchmarking G0W0 for Molecular Systems. J. Chem. Theory Comput. 2015, 11, 5665– 5687, DOI: 10.1021/acs.jctc.5b00453There is no corresponding record for this reference.
- 129Setyawan, W.; Curtarolo, S. High-throughput electronic band structure calculations: Challenges and tools. Comput. Mater. Sci. 2010, 49, 299– 312, DOI: 10.1016/j.commatsci.2010.05.010There is no corresponding record for this reference.
- 130Schusteritsch, G.; Kühne, T. D.; Guo, Z. X.; Kaxiras, E. The effect of Ag, Pb and Bi impurities on grain boundary sliding and intergranular decohesion in Copper. Philos. Mag. 2016, 96, 2868– 2886, DOI: 10.1080/14786435.2016.1217360There is no corresponding record for this reference.
- 131Praprotnik, M.; Site, L. D.; Kremer, K. Multiscale simulation of soft matter: From scale bridging to adaptive resolution. Annu. Rev. Phys. Chem. 2008, 59, 545– 571, DOI: 10.1146/annurev.physchem.59.032607.093707There is no corresponding record for this reference.
- 132Cortes-Huerto, R.; Praprotnik, M.; Kremer, K.; Delle Site, L. From adaptive resolution to molecular dynamics of open systems. Eur. Phys. J. B 2021, 94, 189, DOI: 10.1140/epjb/s10051-021-00193-wThere is no corresponding record for this reference.
- 133Panahian Jand, S.; Kühne, T. D.; Delle Site, L. On the physical consistency of an open quantum region with a classical reservoir in molecular simulation. Adv. Theory Simul. 2024, 7, 2400833, DOI: 10.1002/adts.202400833There is no corresponding record for this reference.
- 134Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999– 3094, DOI: 10.1021/cr9904009There is no corresponding record for this reference.
- 135Fattebert, J.-L.; Gygi, F. Density functional theory for efficient ab initio molecular dynamics simulations in solution. J. Comput. Chem. 2002, 23, 662– 666, DOI: 10.1002/jcc.10069There is no corresponding record for this reference.
- 136Fattebert, J.-L.; Gygi, F. First-principles molecular dynamics simulations in a continuum solvent. Int. J. Quantum Chem. 2003, 93, 139– 147, DOI: 10.1002/qua.10548There is no corresponding record for this reference.
- 137Andreussi, O.; Dabo, I.; Marzari, N. Revised self-consistent continuum solvation in electronic-structure calculations. J. Chem. Phys. 2012, 136, 064102, DOI: 10.1063/1.3676407There is no corresponding record for this reference.
- 138Yin, W.-J.; Krack, M.; Li, X.; Chen, L.-Z.; Liu, L.-M. Periodic continuum solvation model integrated with first-principles calculations for solid surfaces. Prog. Nat. Sci. 2017, 27, 283– 288, DOI: 10.1016/j.pnsc.2017.03.003There is no corresponding record for this reference.
- 139Yin, W.-J.; Krack, M.; Wen, B.; Ma, S.-Y.; Liu, L.-M. CO2 Capture and Conversion on Rutile TiO2(110) in the Water Environment: Insight by First-Principles Calculations. J. Phys. Chem. Lett. 2015, 6, 2538– 2545, DOI: 10.1021/acs.jpclett.5b00798There is no corresponding record for this reference.
- 140Dupont, C.; Andreussi, O.; Marzari, N. Self-consistent continuum solvation (SCCS): The case of charged systems. J. Chem. Phys. 2013, 139, 214110, DOI: 10.1063/1.4832475There is no corresponding record for this reference.
- 141Martyna, G. J.; Tuckerman, M. E. A reciprocal space based method for treating long range interactions inab initioand force-field-based calculations in clusters. J. Chem. Phys. 1999, 110, 2810– 2821, DOI: 10.1063/1.477923There is no corresponding record for this reference.
- 142Cococcioni, M.; Mauri, F.; Ceder, G.; Marzari, N. Electronic-Enthalpy Functional for Finite Systems Under Pressure. Phys. Rev. Lett. 2005, 94, 145501, DOI: 10.1103/physrevlett.94.145501There is no corresponding record for this reference.
- 143Warshel, A.; Levitt, M. Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme. J. Mol. Biol. 1976, 103, 227– 249, DOI: 10.1016/0022-2836(76)90311-9There is no corresponding record for this reference.
- 144Stewart, J. J. P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173– 1213, DOI: 10.1007/s00894-007-0233-4There is no corresponding record for this reference.
- 145Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260– 7268, DOI: 10.1103/PhysRevB.58.7260There is no corresponding record for this reference.
- 146Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989– 2009, DOI: 10.1021/acs.jctc.7b00118There is no corresponding record for this reference.
- 147Abraham, M. J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J. C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19– 25, DOI: 10.1016/j.softx.2015.06.001There is no corresponding record for this reference.
- 148QM/MM Documentation - GROMACS 2022-beta1. https://manual.gromacs.org/documentation/2022-beta1/reference-manual/special/qmmm.html, 2022; (accessed, 2024 10 29).There is no corresponding record for this reference.
- 149Senn, H. M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed. 2009, 48, 1198– 1229, DOI: 10.1002/anie.200802019There is no corresponding record for this reference.
- 150Zhang, Y.; Lee, T.-S.; Yang, W. A pseudobond approach to combining quantum mechanical and molecular mechanical methods. J. Chem. Phys. 1999, 110, 46– 54, DOI: 10.1063/1.478083There is no corresponding record for this reference.
- 151Ihrig, A. C.; Schiffmann, C.; Sebastiani, D. Specific quantum mechanical/molecular mechanical capping-potentials for biomolecular functional groups. J. Chem. Phys. 2011, 135, 214107, DOI: 10.1063/1.3664300There is no corresponding record for this reference.
- 152Schiffmann, C.; Sebastiani, D. Artificial bee colony optimization of capping potentials for hybrid quantum mechanical/molecular mechanical calculations. J. Chem. Theory Comput. 2011, 7, 1307– 1315, DOI: 10.1021/ct1007108There is no corresponding record for this reference.
- 153Laino, T.; Mohamed, F.; Laio, A.; Parrinello, M. An efficient real space multigrid QM/MM electrostatic coupling. J. Chem. Theory Comput. 2005, 1, 1176– 1184, DOI: 10.1021/ct050123fThere is no corresponding record for this reference.
- 154Laino, T.; Mohamed, F.; Laio, A.; Parrinello, M. An Efficient Linear-Scaling Electrostatic Coupling for treating periodic boundary conditions in QM/MM Simulations. J. Chem. Theory Comput. 2006, 2, 1370– 1378, DOI: 10.1021/ct6001169There is no corresponding record for this reference.
- 155Golze, D.; Iannuzzi, M.; Nguyen, M.-T.; Passerone, D.; Hutter, J. Simulation of Adsorption Processes at Metallic Interfaces: An Image Charge Augmented QM/MM Approach. J. Chem. Theory Comput. 2013, 9, 5086– 5097, DOI: 10.1021/ct400698yThere is no corresponding record for this reference.
- 156Devynck, F.; Iannuzzi, M.; Krack, M. Frenkel pair recombinations in UO2: Importance of explicit description of polarizability in core-shell molecular dynamics simulations. Phys. Rev. B 2012, 85, 184103, DOI: 10.1103/PhysRevB.85.184103There is no corresponding record for this reference.
- 157Blöchl, P. E. Electrostatic decoupling of periodic images of plane-wave-expanded densities and derived atomic point charges. J. Chem. Phys. 1995, 103, 7422– 7428, DOI: 10.1063/1.470314There is no corresponding record for this reference.
- 158Laino, T.; Mohamed, F.; Laio, A.; Parrinello, M. An efficient linear-scaling electrostatic coupling for treating periodic boundary conditions in QM/MM simulations. J. Chem. Theory Comput. 2006, 2, 1370– 1378, DOI: 10.1021/ct6001169There is no corresponding record for this reference.
- 159Image Charges. https://manual.cp2k.org/trunk/methods/qm_mm/image_charges.html, 2015; (accessed, 2025 02 05).There is no corresponding record for this reference.
- 160Bayly, C. I.; Cieplak, P.; Cornell, W. D.; Kollman, P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269– 10280, DOI: 10.1021/j100142a004There is no corresponding record for this reference.
- 161Golze, D.; Hutter, J.; Iannuzzi, M. Wetting of water on hexagonal boron nitrideRh(111): a QM/MM model based on atomic charges derived for nano-structured substrates. Phys. Chem. Chem. Phys. 2015, 17, 14307– 14316, DOI: 10.1039/C4CP04638BThere is no corresponding record for this reference.
- 162https://www.cp2k.org/howto:resp, 2015; (accessed, 2025 06 13).There is no corresponding record for this reference.
- 163Campañá, C.; Mussard, B.; Woo, T. K. Electrostatic Potential Derived Atomic Charges for Periodic Systems Using a Modified Error Functional. J. Chem. Theory Comput. 2009, 5, 2866– 2878, DOI: 10.1021/ct9003405There is no corresponding record for this reference.
- 164Huang, C.; Pavone, M.; Carter, E. A. Quantum mechanical embedding theory based on a unique embedding potential. J. Chem. Phys. 2011, 134, 154110, DOI: 10.1063/1.3577516There is no corresponding record for this reference.
- 165Wu, Q.; Yang, W. A direct optimization method for calculating density functionals and exchange-correlation potentials from electron densities. J. Chem. Phys. 2003, 118, 2498– 2509, DOI: 10.1063/1.1535422There is no corresponding record for this reference.
- 166Van Leeuwen, R.; Baerends, E. Exchange-correlation potential with correct asymptotic behavior. Phys. Rev. A 1994, 49, 2421, DOI: 10.1103/PhysRevA.49.2421There is no corresponding record for this reference.
- 167Putrino, A.; Sebastiani, D.; Parrinello, M. Generalized variational density functional perturbation theory. J. Chem. Phys. 2000, 113, 7102– 7109, DOI: 10.1063/1.1312830There is no corresponding record for this reference.
- 168Sebastiani, D.; Parrinello, M. A New ab-Initio Approach for NMR Chemical Shifts in Periodic Systems. J. Phys. Chem. A 2001, 105, 1951– 1958, DOI: 10.1021/jp002807jThere is no corresponding record for this reference.
- 169Sebastiani, D. Ab-initio calculations of NMR parameters in condensed phases. Mod. Phys. Lett. B 2003, 17, 1301– 1319, DOI: 10.1142/S0217984903006372There is no corresponding record for this reference.
- 170Gonze, X. Adiabatic density-functional perturbation theory. Phys. Rev. A 1995, 52, 1096– 1114, DOI: 10.1103/PhysRevA.52.1096There is no corresponding record for this reference.
- 171Sebastiani, D. Current Densities and Nucleus-Independent Chemical Shift Maps from Reciprocal-Space Density Functional Perturbation Theory Calculations. ChemPhysChem 2006, 7, 164– 175, DOI: 10.1002/cphc.200500438There is no corresponding record for this reference.
- 172Weber, V.; Iannuzzi, M.; Giani, S.; Hutter, J.; Declerck, R.; Waroquier, M. Magnetic linear response properties calculations with the Gaussian and augmented-plane-wave method. J. Chem. Phys. 2009, 131, 014106, DOI: 10.1063/1.3156803There is no corresponding record for this reference.
- 173Putrino, A.; Parrinello, M. Anharmonic Raman spectra in high-pressure ice from ab initio simulations. Phys. Rev. Lett. 2002, 88, 176401, DOI: 10.1103/physrevlett.88.176401There is no corresponding record for this reference.
- 174Scherrer, A.; Vuilleumier, R.; Sebastiani, D. Vibrational circular dichroism from ab initio molecular dynamics and nuclear velocity perturbation theory in the liquid phase. J. Chem. Phys. 2016, 145, 084101, DOI: 10.1063/1.4960653There is no corresponding record for this reference.
- 175Resta, R. Why are insulators insulating and metals conducting?. J. Phys.: Condens. Matter 2002, 14, R625– R656, DOI: 10.1088/0953-8984/14/20/201There is no corresponding record for this reference.
- 176Mauri, F.; Louie, S. Magnetic Susceptibility of Insulators from First Principles. Phys. Rev. Lett. 1996, 76, 4246– 4249, DOI: 10.1103/PhysRevLett.76.4246There is no corresponding record for this reference.
- 177Keith, T. A.; Bader, R. F. W. Calculation of magnetic response properties using atoms in molecules. Chem. Phys. Lett. 1992, 194, 1– 8, DOI: 10.1016/0009-2614(92)85733-QThere is no corresponding record for this reference.
- 178Keith, T. A.; Bader, R. F. W. Calculation of magnetic response properties using a continuous set of gauge transformations. Chem. Phys. Lett. 1993, 210, 223– 231, DOI: 10.1016/0009-2614(93)89127-4There is no corresponding record for this reference.
- 179van Wüllen, C. Calculation of NMR and EPR Parameters: Theory and Applications; Wiley, 2004; pp 83– 100.There is no corresponding record for this reference.
- 180Cowan, B. P. Nuclear Magnetic Resonance and Relaxation, online-ausg ed.; Cambridge University Press: Cambridge, 1997.There is no corresponding record for this reference.
- 181Mondal, A.; Gaultois, M. W.; Pell, A. J.; Iannuzzi, M.; Grey, C. P.; Hutter, J.; Kaupp, M. Large-scale computation of nuclear magnetic resonance shifts for paramagnetic solids using CP2K. J. Chem. Theory Comput. 2018, 14, 377– 394, DOI: 10.1021/acs.jctc.7b00991There is no corresponding record for this reference.
- 182Schreckenbach, G.; Ziegler, T. Calculation of NMR shielding tensors based on density functional theory and a scalar relativistic Pauli-type Hamiltonian. The application to transition metal complexes. Int. J. Quantum Chem. 1997, 61, 899– 918, DOI: 10.1002/(SICI)1097-461X(1997)61:6<899::AID-QUA3>3.0.CO;2-RThere is no corresponding record for this reference.
- 183Pickard, C.; Mauri, F. First-Principles Theory of the EPR G Tensor in Solids: Defects in Quartz. Phys. Rev. Lett. 2002, 88, 086403, DOI: 10.1103/physrevlett.88.086403There is no corresponding record for this reference.
- 184Van Yperen-De Deyne, A.; Pauwels, E.; Van Speybroeck, V.; Waroquier, M. Accurate Spin–orbit and Spin–other-orbit Contributions to the G-tensor for Transition Metal Containing Systems. Phys. Chem. Chem. Phys. 2012, 14, 10690– 10704, DOI: 10.1039/c2cp41086aThere is no corresponding record for this reference.
- 185Neese, F. Efficient and Accurate Approximations to the Molecular Spin-orbit Coupling Operator and Their Use in Molecular G-tensor Calculations. J. Chem. Phys. 2005, 122, 034107, DOI: 10.1063/1.1829047There is no corresponding record for this reference.
- 186Malkina, O. L.; Vaara, J.; Schimmelpfennig, B.; Munzarova, M.; Malkin, V. G.; Kaupp, M. Density Functional Calculations of Electronic G-tensors Using Spin-orbit Pseudopotentials and Mean-field All-electron Spin-orbit Operators. J. Am. Chem. Soc. 2000, 122, 9206– 9218, DOI: 10.1021/ja000984sThere is no corresponding record for this reference.
- 187Pigliapochi, R.; Pell, A. J.; Seymour, I. D.; Grey, C. P.; Ceresoli, D.; Kaupp, M. DFT investigation of the effect of spin-orbit coupling on the NMR shifts in paramagnetic solids. Phys. Rev. B 2017, 95, 054412, DOI: 10.1103/PhysRevB.95.054412There is no corresponding record for this reference.
- 188Declerck, R.; Pauwels, E.; Van Speybroeck, V.; Waroquier, M. First-principles Calculations of Hyperfine Parameters with the Gaussian and Augmented-plane-wave Method: Application to Radicals Embedded in a Crystalline Environment. Phys. Rev. B 2006, 74, 245103, DOI: 10.1103/physrevb.74.245103There is no corresponding record for this reference.
- 189Blase, X.; Duchemin, I.; Jacquemin, D. The Bethe–Salpeter equation in chemistry: relations with TD-DFT, applications and challenges. Chem. Soc. Rev. 2018, 47, 1022– 1043, DOI: 10.1039/C7CS00049AThere is no corresponding record for this reference.
- 190Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291, DOI: 10.1016/S0009-2614(99)01149-5There is no corresponding record for this reference.
- 191Hutter, J. Excited state nuclear forces from the Tamm–Dancoff approximation to time–dependent density functional theory within the plane wave basis set framework. J. Chem. Phys. 2003, 118, 3928– 3934, DOI: 10.1063/1.1540109There is no corresponding record for this reference.
- 192Grimme, S. A simplified Tamm-Dancoff density functional approach for the electronic excitation spectra of very large molecules. J. Chem. Phys. 2013, 138, 244104, DOI: 10.1063/1.4811331There is no corresponding record for this reference.
- 193Hehn, A.-S.; Sertcan, B.; Belleflamme, F.; Chulkov, S. K.; Watkins, M. B.; Hutter, J. Excited-State Properties for Extended Systems: Efficient Hybrid Density Functional Methods. J. Chem. Theory Comput. 2022, 18, 4186– 4202, DOI: 10.1021/acs.jctc.2c00144There is no corresponding record for this reference.
- 194Sertcan Gokmen, B.; Hutter, J.; Hehn, A.-S. Excited-State Forces with the Gaussian and Augmented Plane Wave Method for the Tamm–Dancoff Approximation of Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2024, 20, 8494– 8504, DOI: 10.1021/acs.jctc.4c00614There is no corresponding record for this reference.
- 195Vogt, J.-R.; Wilhelm, J.; Hehn, A.-S. Perturbative spin–orbit couplings for the simulation of extended framework materials. J. Chem. Phys. 2025, 162, 082502, DOI: 10.1063/5.0242940There is no corresponding record for this reference.
- 196van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392– 399, DOI: 10.1063/1.476576There is no corresponding record for this reference.
- 197Bussy, A.; Hutter, J. Efficient and low-scaling linear-response time-dependent density functional theory implementation for core-level spectroscopy of large and periodic systems. Phys. Chem. Chem. Phys. 2021, 23, 4736– 4746, DOI: 10.1039/D0CP06164FThere is no corresponding record for this reference.
- 198Ljungberg, M. P.; Koval, P.; Ferrari, F.; Foerster, D.; Sánchez-Portal, D. Cubic-Scaling Iterative Solution of the Bethe-Salpeter Equation for Finite Systems. Phys. Rev. B 2015, 92, 075422, DOI: 10.1103/PhysRevB.92.075422There is no corresponding record for this reference.
- 199Jacquemin, D.; Duchemin, I.; Blondel, A.; Blase, X. Assessment of the Accuracy of the Bethe–Salpeter (BSE/GW) Oscillator Strengths. J. Chem. Theory Comput. 2016, 12, 3969– 3981, DOI: 10.1021/acs.jctc.6b00419There is no corresponding record for this reference.
- 200Bruneval, F.; Hamed, S. M.; Neaton, J. B. A systematic benchmark of the ab initio Bethe-Salpeter equation approach for low-lying optical excitations of small organic molecules. J. Chem. Phys. 2015, 142, 244101, DOI: 10.1063/1.4922489There is no corresponding record for this reference.
- 201Sander, T.; Maggio, E.; Kresse, G. Beyond the Tamm-Dancoff approximation for extended systems using exact diagonalization. Phys. Rev. B 2015, 92, 045209, DOI: 10.1103/PhysRevB.92.045209There is no corresponding record for this reference.
- 202Liu, C.; Kloppenburg, J.; Yao, Y.; Ren, X.; Appel, H.; Kanai, Y.; Blum, V. All-electron ab initio Bethe-Salpeter equation approach to neutral excitations in molecules with numeric atom-centered orbitals. J. Chem. Phys. 2020, 152, 044105, DOI: 10.1063/1.5123290There is no corresponding record for this reference.
- 203Blase, X.; Duchemin, I.; Jacquemin, D.; Loos, P.-F. The Bethe–Salpeter Equation Formalism: From Physics to Chemistry. J. Phys. Chem. Lett. 2020, 11, 7371– 7382, DOI: 10.1021/acs.jpclett.0c01875There is no corresponding record for this reference.
- 204Bechstedt, F. Many-Body Approach to Electronic Excitations: Concepts and Applications; Springer Series in Solid-State Sciences; Springer-Verlag: Berlin, Heidelberg, 2015; Vol. 181.There is no corresponding record for this reference.
- 205Mewes, S. A.; Plasser, F.; Krylov, A.; Dreuw, A. Benchmarking Excited-State Calculations Using Exciton Properties. J. Chem. Theory Comput. 2018, 14, 710– 725, DOI: 10.1021/acs.jctc.7b01145There is no corresponding record for this reference.
- 206Ullrich, C. A. Time-Dependent Density-Functional Theory: Concepts and Applications; Oxford University Press: New York, 2012.There is no corresponding record for this reference.
- 207Bruneval, F.; Rangel, T.; Hamed, S. M.; Shao, M.; Yang, C.; Neaton, J. B. Molgw 1: Many-body Perturbation Theory Software for Atoms, Molecules, and Clusters. Comput. Phys. Commun. 2016, 208, 149– 161, DOI: 10.1016/j.cpc.2016.06.019There is no corresponding record for this reference.
- 208Knysh, I.; Lipparini, F.; Blondel, A.; Duchemin, I.; Blase, X.; Loos, P.-F.; Jacquemin, D. Reference CC3 Excitation Energies for Organic Chromophores: Benchmarking TD-DFT, BSE/GW, and Wave Function Methods. J. Chem. Theory Comput. 2024, 20, 8152– 8174, DOI: 10.1021/acs.jctc.4c00906There is no corresponding record for this reference.
- 209Kendall, R. A.; Dunning, T. H.; Harrison, R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796– 6806, DOI: 10.1063/1.462569There is no corresponding record for this reference.
- 210Schreiber, M.; Silva-Junior, M. R.; Sauer, S. P. A.; Thiel, W. Benchmarks for Electronically Excited States: CASPT2, CC2, CCSD, and CC3. J. Chem. Phys. 2008, 128, 134110, DOI: 10.1063/1.2889385There is no corresponding record for this reference.
- 211Kunert, T.; Schmidt, R. Non-adiabatic quantum molecular dynamics: General formalism and case study H2+ in strong laser fields. Eur. Phys. J. D 2003, 25, 15– 24, DOI: 10.1140/epjd/e2003-00086-8There is no corresponding record for this reference.
- 212Castro, A.; Marques, M. A. L.; Rubio, A. Propagators for the time-dependent Kohn-Sham equations. J. Chem. Phys. 2004, 121, 3425– 3433, DOI: 10.1063/1.1774980There is no corresponding record for this reference.
- 213Moler, C.; Loan, C. V. Nineteen Dubious Ways to Compute the Exponential of a Matrix, Twenty-Five Years Later. SIAM Rev. 2003, 45, 3– 49, DOI: 10.1137/s00361445024180There is no corresponding record for this reference.
- 214Andermatt, S.; Bani-Hashemian, M. H.; Brück, S.; VandeVondele, J.; Luisier, M. Transport simulations with density-matrix-based real-time time-dependant density functional theory. In 2017 International Conference on Simulation of Semiconductor Processes and Devices (SISPAD); Institute of Electrical and Electronics Engineers, 2017, pp 177– 180.There is no corresponding record for this reference.
- 215Bertsch, G. F.; Iwata, J.-I.; Rubio, A.; Yabana, K. Real-space, real-time method for the dielectric function. Phys. Rev. B 2000, 62, 7998– 8002, DOI: 10.1103/PhysRevB.62.7998There is no corresponding record for this reference.
- 216Yabana, K.; Sugiyama, T.; Shinohara, Y.; Otobe, T.; Bertsch, G. F. Time-dependent density functional theory for strong electromagnetic fields in crystalline solids. Phys. Rev. B 2012, 85, 045134, DOI: 10.1103/physrevb.85.045134There is no corresponding record for this reference.
- 217Pemmaraju, C.; Vila, F.; Kas, J.; Sato, S.; Rehr, J.; Yabana, K.; Prendergast, D. Velocity-gauge real-time TDDFT within a numerical atomic orbital basis set. Comput. Phys. Commun. 2018, 226, 30– 38, DOI: 10.1016/j.cpc.2018.01.013There is no corresponding record for this reference.
- 218Onida, G.; Reining, L.; Rubio, A. Electronic excitations: density-functional versus many-body Green’s-function approaches. Rev. Mod. Phys. 2002, 74, 601– 659, DOI: 10.1103/RevModPhys.74.601There is no corresponding record for this reference.
- 219Marek, S.; Wilhelm, J. Linear and Nonlinear Optical Properties of Molecules from Real-Time Propagation Based on the Bethe-Salpeter Equation. J. Chem. Theory Comput. 2025, 21, 9814– 9822, DOI: 10.1021/acs.jctc.5c01246There is no corresponding record for this reference.
- 220Attaccalite, C.; Grüning, M.; Marini, A. Real-time approach to the optical properties of solids and nanostructures: Time-dependent Bethe-Salpeter equation. Phys. Rev. B 2011, 84, 245110, DOI: 10.1103/PhysRevB.84.245110There is no corresponding record for this reference.
- 221Casida, M. E.; Chong, D. P. Physical interpretation and assessment of the Coulomb-hole and screened-exchange approximation for molecules. Phys. Rev. A 1989, 40, 4837– 4848, DOI: 10.1103/PhysRevA.40.4837There is no corresponding record for this reference.
- 222Farid, B.; Daling, R.; Lenstra, D.; van Haeringen, W. GW approach to the calculation of electron self-energies in semiconductors. Phys. Rev. B 1988, 38, 7530– 7534, DOI: 10.1103/PhysRevB.38.7530There is no corresponding record for this reference.
- 223Betzinger, M.; Friedrich, C.; Görling, A.; Blügel, S. Precise all-electron dynamical response functions: Application to COHSEX and the RPA correlation energy. Phys. Rev. B 2015, 92, 245101, DOI: 10.1103/PhysRevB.92.245101There is no corresponding record for this reference.
- 224O’Rourke, C.; Bowler, D. R. Linear scaling density matrix real time TDDFT: Propagator unitarity and matrix truncation. J. Chem. Phys. 2015, 143, 102801, DOI: 10.1063/1.4919128There is no corresponding record for this reference.
- 225Mattiat, J.; Luber, S. Comparison of Length, Velocity, and Symmetric Gauges for the Calculation of Absorption and Electric Circular Dichroism Spectra with Real-Time Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2022, 18, 5513– 5526, DOI: 10.1021/acs.jctc.2c00644There is no corresponding record for this reference.
- 226Müller, C.; Sharma, M.; Sierka, M. Real-time time-dependent density functional theory using density fitting and the continuous fast multipole method. J. Comput. Chem. 2020, 41, 2573– 2582, DOI: 10.1002/jcc.26412There is no corresponding record for this reference.
- 227Yabana, K.; Nakatsukasa, T.; Iwata, J.-I.; Bertsch, G. F. Real-time, real-space implementation of the linear response time-dependent density-functional theory. Phys. Status Solidi B 2006, 243, 1121– 1138, DOI: 10.1002/pssb.200642005There is no corresponding record for this reference.
- 228Li, X.; Govind, N.; Isborn, C.; DePrince, A. E., III; Lopata, K. Real-time time-dependent electronic structure theory. Chem. Rev. 2020, 120, 9951– 9993, DOI: 10.1021/acs.chemrev.0c00223There is no corresponding record for this reference.
- 229Mattiat, J.; Luber, S. Efficient calculation of (resonance) Raman spectra and excitation profiles with real-time propagation. J. Chem. Phys. 2018, 149, 174108, DOI: 10.1063/1.5051250There is no corresponding record for this reference.
- 230Bruner, A.; LaMaster, D.; Lopata, K. Accelerated Broadband Spectra Using Transition Dipole Decomposition and Padé Approximants. J. Chem. Theory Comput. 2016, 12, 3741– 3750, DOI: 10.1021/acs.jctc.6b00511There is no corresponding record for this reference.
- 231Leucke, M.; Panadés-Barrueta, R. L.; Bas, E. E.; Golze, D. Analytic continuation component of the GreenX library: robust Padé approximants with symmetry constraints. J. Open Source Softw. 2025, 10, 7859, DOI: 10.21105/joss.07859There is no corresponding record for this reference.
- 232Barbatti, M.; Bondanza, M.; Crespo-Otero, R.; Demoulin, B.; Dral, P. O.; Granucci, G.; Kossoski, F.; Lischka, H.; Mennucci, B.; Mukherjee, S. Newton-X Platform: New Software Developments for Surface Hopping and Nuclear Ensembles. J. Chem. Theory Comput. 2022, 18, 6851– 6865, DOI: 10.1021/acs.jctc.2c00804There is no corresponding record for this reference.
- 233Barbatti, M.; Ruckenbauer, M.; Plasser, F.; Pittner, J.; Granucci, G.; Persico, M.; Lischka, H. Newton-X: a surface-hopping program for nonadiabatic molecular dynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 26– 33, DOI: 10.1002/wcms.1158There is no corresponding record for this reference.
- 234Barbatti, M.; Granucci, G.; Persico, M.; Ruckenbauer, M.; Vazdar, M.; Eckert-Maksić, M.; Lischka, H. The on-the-fly surface-hopping program system Newton-X: Application to ab initio simulation of the nonadiabatic photodynamics of benchmark systems. J. Photochem. Photobiol., A 2007, 190, 228– 240, DOI: 10.1016/j.jphotochem.2006.12.008There is no corresponding record for this reference.
- 235Cederbaum, L. S.; Domcke, W.; Schirmer, J. Many-body theory of core holes. Phys. Rev. A 1980, 22, 206, DOI: 10.1103/PhysRevA.22.206There is no corresponding record for this reference.
- 236DeBeer George, S.; Petrenko, T.; Neese, F. Time-dependent density functional calculations of ligand K-edge X-ray absorption spectra. Inorg. Chim. Acta 2008, 361, 965– 972, DOI: 10.1016/j.ica.2007.05.046There is no corresponding record for this reference.
- 237Müller, P.; Karhan, K.; Krack, M.; Gerstmann, U.; Schmidt, W. G.; Bauer, M.; Kühne, T. D. Impact of finite-temperature and condensed-phase effects on theoretical X-ray absorption spectra of transition metal complexes. J. Comput. Chem. 2019, 40, 712– 716, DOI: 10.1002/jcc.25641There is no corresponding record for this reference.
- 238Franco de Carvalho, F.; Curchod, B. F.; Penfold, T. J.; Tavernelli, I. Derivation of spin-orbit couplings in collinear linear-response TDDFT: A rigorous formulation. J. Chem. Phys. 2014, 140, 144103, DOI: 10.1063/1.4870010There is no corresponding record for this reference.
- 239Bussy, A.; Hutter, J. Data for: Efficient and low-scaling linear-response time-dependent density functional theory implementation for core-level spectroscopy of large and periodic systems, 2021. accessed: 2025–08–23. DOI: 10.24435/materialscloud:js-me .There is no corresponding record for this reference.
- 240Lopata, K.; Van Kuiken, B.; Khalil, M.; Govind, N. Linear-response and real-time time-dependent density functional theory studies of core-level near-edge X-ray absorption. J. Chem. Theory Comput. 2012, 8, 3284– 3292, DOI: 10.1021/ct3005613There is no corresponding record for this reference.
- 241Khaliullin, R. Z.; Cobar, E. A.; Lochan, R. C.; Bell, A. T.; Head-Gordon, M. Unravelling the Origin of Intermolecular Interactions Using Absolutely Localized Molecular Orbitals. J. Phys. Chem. A 2007, 111, 8753– 8765, DOI: 10.1021/jp073685zThere is no corresponding record for this reference.
- 242Khaliullin, R. Z.; Kühne, T. D. Microscopic properties of liquid water from combined ab initio molecular dynamics and energy decomposition studies. Phys. Chem. Chem. Phys. 2013, 15, 15746– 15766, DOI: 10.1039/c3cp51039eThere is no corresponding record for this reference.
- 243Mo, Y.; Gao, J.; Peyerimhoff, S. D. Energy decomposition analysis of intermolecular interactions using a block-localized wave function approach. J. Chem. Phys. 2000, 112, 5530– 5538, DOI: 10.1063/1.481185There is no corresponding record for this reference.
- 244Azar, R. J.; Horn, P. R.; Sundstrom, E. J.; Head-Gordon, M. Useful lower limits to polarization contributions to intermolecular interactions using a minimal basis of localized orthogonal orbitals: Theory and analysis of the water dimer. J. Chem. Phys. 2013, 138, 084102, DOI: 10.1063/1.4792434There is no corresponding record for this reference.
- 245Stoll, H.; Wagenblast, G.; Preuß, H. On the use of local basis sets for localized molecular orbitals. Theor. Chim. Acta 1980, 57, 169– 178, DOI: 10.1007/BF00574903There is no corresponding record for this reference.
- 246Khaliullin, R. Z.; Head-Gordon, M.; Bell, A. T. An efficient self-consistent field method for large systems of weakly interacting components. J. Chem. Phys. 2006, 124, 204105, DOI: 10.1063/1.2191500There is no corresponding record for this reference.
- 247Khaliullin, R. Z.; Bell, A. T.; Head-Gordon, M. Analysis of charge transfer effects in molecular complexes based on absolutely localized molecular orbitals. J. Chem. Phys. 2008, 128, 184112, DOI: 10.1063/1.2912041There is no corresponding record for this reference.
- 248Khaliullin, R. Z.; VandeVondele, J.; Hutter, J. Efficient Linear-Scaling Density Functional Theory for Molecular Systems. J. Chem. Theory Comput. 2013, 9, 4421– 4427, DOI: 10.1021/ct400595kThere is no corresponding record for this reference.
- 249Staub, R.; Iannuzzi, M.; Khaliullin, R. Z.; Steinmann, S. N. Energy Decomposition Analysis for Metal Surface,ÄìAdsorbate Interactions by Block Localized Wave Functions. J. Chem. Theory Comput. 2019, 15, 265– 275, DOI: 10.1021/acs.jctc.8b00957There is no corresponding record for this reference.
- 250Shi, Y.; Scheiber, H.; Khaliullin, R. Z. Contribution of the Covalent Component of the Hydrogen-Bond Network to the Properties of Liquid Water. J. Phys. Chem. A 2018, 122, 7482– 7490, DOI: 10.1021/acs.jpca.8b06857There is no corresponding record for this reference.
- 251Scheiber, H.; Shi, Y.; Khaliullin, R. Z. Communication: Compact orbitals enable low-cost linear-scaling ab initio molecular dynamics for weakly-interacting systems. J. Chem. Phys. 2018, 148, 231103, DOI: 10.1063/1.5029939There is no corresponding record for this reference.
- 252Luo, Z.; Friscic, T.; Khaliullin, R. Z. Why pregnenolone and progesterone, two structurally similar steroids, exhibit remarkably different cocrystallization with aromatic molecules. Phys. Chem. Chem. Phys. 2018, 20, 898– 904, DOI: 10.1039/C7CP06828JThere is no corresponding record for this reference.
- 253Ojha, D.; Kaliannan, N. K.; Kühne, T. D. Time-dependent vibrational sum-frequency generation spectroscopy of the air-water interface. Commun. Chem. 2019, 2, 116, DOI: 10.1038/s42004-019-0220-6There is no corresponding record for this reference.
- 254Ojha, D.; Kühne, T. D. Hydrogen bond dynamics of interfacial water molecules revealed from two-dimensional vibrational sum-frequency generation spectroscopy. Sci. Rep. 2021, 11, 2456, DOI: 10.1038/s41598-021-81635-4There is no corresponding record for this reference.
- 255Kühne, T. D.; Khaliullin, R. Z. Electronic signature of the instantaneous asymmetry in the first coordination shell of liquid water. Nat. Commun. 2013, 4, 1450, DOI: 10.1038/ncomms2459There is no corresponding record for this reference.
- 256Elgabarty, H.; Khaliullin, R. Z.; Kühne, T. D. Covalency of hydrogen bonds in liquid water can be probed by proton nuclear magnetic resonance experiments. Nat. Commun. 2015, 6, 8318, DOI: 10.1038/ncomms9318There is no corresponding record for this reference.
- 257Elgabarty, H.; Kühne, T. D. Tumbling with a limp: local asymmetry in water’s hydrogen bond network and its consequences. Phys. Chem. Chem. Phys. 2020, 22, 10397– 10411, DOI: 10.1039/C9CP06960GThere is no corresponding record for this reference.
- 258Kühne, T. D.; Khaliullin, R. Z. Nature of the Asymmetry in the Hydrogen-Bond Networks of Hexagonal Ice and Liquid Water. J. Am. Chem. Soc. 2014, 136, 3395– 3399, DOI: 10.1021/ja411161aThere is no corresponding record for this reference.
- 259Yun, Y.; Khaliullin, R. Z.; Jung, Y. Low-Dimensional Confined Ice Has the Electronic Signature of Liquid Water. J. Phys. Chem. Lett. 2019, 10, 2008– 2016, DOI: 10.1021/acs.jpclett.9b00921There is no corresponding record for this reference.
- 260Salem, M. A.; Kühne, T. D. Insight from energy decomposition analysis on a hydrogen-bond-mediated mechanism for on-water catalysis. Mol. Phys. 2020, 118, e1797920 DOI: 10.1080/00268976.2020.1797920There is no corresponding record for this reference.
- 261Heske, J.; Kühne, T. D.; Antonietti, M. Water in PHI nanopores: modeling adsorption, solvent structure, and thermodynamics. ACS Omega 2023, 8, 26526– 26532, DOI: 10.1021/acsomega.3c03308There is no corresponding record for this reference.
- 262Ramos-Cordoba, E.; Lambrecht, D. S.; Head-Gordon, M. Charge-transfer and the hydrogen bond: Spectroscopic and structural implications from electronic structure calculations. Faraday Discuss. 2011, 150, 345– 362, DOI: 10.1039/c1fd00004gThere is no corresponding record for this reference.
- 263Fransson, T.; Harada, Y.; Kosugi, N.; Besley, N. A.; Winter, B.; Rehr, J. J.; Pettersson, L. G. M.; Nilsson, A. X-ray and Electron Spectroscopy of Water. Chem. Rev. 2016, 116, 7551– 7569, DOI: 10.1021/acs.chemrev.5b00672There is no corresponding record for this reference.
- 264Lenz, A.; Ojamäe, L. Theoretical IR Spectra for Water Clusters (H2O)n(n= 6–22, 28, 30) and Identification of Spectral Contributions from Different H-Bond Conformations in Gaseous and Liquid Water. J. Phys. Chem. A 2006, 110, 13388– 13393, DOI: 10.1021/jp066372xThere is no corresponding record for this reference.
- 265Ojha, D.; Karhan, K.; Kühne, T. D. On the hydrogen bond strength and vibrational spectroscopy of liquid water. Sci. Rep. 2018, 8, 16888, DOI: 10.1038/s41598-018-35357-9There is no corresponding record for this reference.
- 266Elgabarty, H.; Kaliannan, N. K.; Kühne, T. D. Enhancement of the local asymmetry in the hydrogen bond network of liquid water by an ultrafast electric field pulse. Sci. Rep. 2019, 9, 10002, DOI: 10.1038/s41598-019-46449-5There is no corresponding record for this reference.
- 267Yun, Y.; Khaliullin, R. Z.; Jung, Y. Correlated Local Fluctuations in the Hydrogen Bond Network of Liquid Water. J. Am. Chem. Soc. 2022, 144, 13127– 13136, DOI: 10.1021/jacs.2c02362There is no corresponding record for this reference.
- 268Balos, V.; Kaliannan, N. K.; Elgabarty, H.; Wolf, M.; Kühne, T. D.; Sajadi, M. Time-resolved terahertz–Raman spectroscopy reveals that cations and anions distinctly modify intermolecular interactions of water. Nat. Chem. 2022, 14, 1031– 1037, DOI: 10.1038/s41557-022-00977-2There is no corresponding record for this reference.
- 269McGrath, M. J.; Siepmann, J. I.; Kuo, I.-F. W.; Mundy, C. J.; VandeVondele, J.; Sprik, M.; Hutter, J.; Mohamed, F.; Krack, M.; Parrinello, M. Toward a Monte Carlo program for simulating vapor–liquid phase equilibria from first principles. Comput. Phys. Commun. 2005, 169, 289– 294, DOI: 10.1016/j.cpc.2005.03.065There is no corresponding record for this reference.
- 270McGrath, M. J.; Siepmann, J. I.; Kuo, I.-F. W.; Mundy, C. J.; VandeVondele, J.; Hutter, J.; Mohamed, F.; Krack, M. Isobaric–isothermal Monte Carlo simulations from first principles: application to liquid water at ambient conditions. ChemPhysChem 2005, 6, 1894– 1901, DOI: 10.1002/cphc.200400580There is no corresponding record for this reference.
- 271McGrath, M. J.; Siepmann, J. I.; Kuo, I.-F. W.; Mundy, C. J.; VandeVondele, J.; Hutter, J.; Mohamed, F.; Krack, M. Simulating fluid-phase equilibria of water from first principles. J. Phys. Chem. A 2006, 110, 640– 646, DOI: 10.1021/jp0535947There is no corresponding record for this reference.
- 272Kuo, I. W.; Mundy, C. J.; Eggimann, B. L.; McGrath, M. J.; Siepmann, J. I.; Chen, B.; Vieceli, J.; Tobias, D. J. Structure and Dynamics of the Aqueous Liquid- Vapor Interface: A Comprehensive Particle-Based Simulation Study. J. Phys. Chem. B 2006, 110, 3738– 3746, DOI: 10.1021/jp056330tThere is no corresponding record for this reference.
- 273Iannuzzi, M.; Laio, A.; Parrinello, M. Efficient Exploration of Reactive Potential Energy Surfaces Using Car–Parrinello Molecular Dynamics. Phys. Rev. Lett. 2003, 90, 238302, DOI: 10.1103/PhysRevLett.90.238302There is no corresponding record for this reference.
- 274Schade, R.; Kenter, T.; Elgabarty, H.; Lass, M.; Schütt, O.; Lazzaro, A.; Pabst, H.; Mohr, S.; Hutter, J.; Kühne, T. D. Towards electronic structure-based ab-initio molecular dynamics simulations with hundreds of millions of atoms. Parallel Comput. 2022, 111, 102920, DOI: 10.1016/j.parco.2022.102920There is no corresponding record for this reference.
- 275Schade, R.; Kenter, T.; Elgabarty, H.; Lass, M.; Kühne, T. D.; Plessl, C. Breaking the exascale barrier for the electronic structure problem in ab-initio molecular dynamics. Int. J. High Perform. Comput. Appl. 2023, 37, 530– 538, DOI: 10.1177/10943420231177631There is no corresponding record for this reference.
- 276Arias, T. A.; Payne, M. C.; Joannopoulos, J. D. Ab initio molecular dynamics: Analytically continued energy functionals and insights into iterative solutions. Phys. Rev. Lett. 1992, 69, 1077– 1080, DOI: 10.1103/PhysRevLett.69.1077There is no corresponding record for this reference.
- 277Arias, T. A.; Payne, M. C.; Joannopoulos, J. D. Ab initio molecular-dynamics techniques extended to large-length-scale systems. Phys. Rev. B 1992, 45, 1538– 1549, DOI: 10.1103/PhysRevB.45.1538There is no corresponding record for this reference.
- 278Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558, DOI: 10.1103/PhysRevB.47.558There is no corresponding record for this reference.
- 279Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251, DOI: 10.1103/PhysRevB.49.14251There is no corresponding record for this reference.
- 280Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471– 2474, DOI: 10.1103/PhysRevLett.55.2471There is no corresponding record for this reference.
- 281Caravati, S.; Bernasconi, M.; Kühne, T.; Krack, M.; Parrinello, M. Coexistence of tetrahedral-and octahedral-like sites in amorphous phase change materials. Appl. Phys. Lett. 2007, 91, 171906, DOI: 10.1063/1.2801626There is no corresponding record for this reference.
- 282Kühne, T. D.; Krack, M.; Parrinello, M. Static and Dynamical Properties of Liquid Water from First Principles by a Novel Car-Parrinello-like Approach. J. Chem. Theory Comput. 2009, 5, 235– 241, DOI: 10.1021/ct800417qThere is no corresponding record for this reference.
- 283Cucinotta, C. S.; Miceli, G.; Raiteri, P.; Krack, M.; Kühne, T. D.; Bernasconi, M.; Parrinello, M. Superionic Conduction in Substoichiometric LiAl Alloy: An Ab Initio Study. Phys. Rev. Lett. 2009, 103, 125901, DOI: 10.1103/PhysRevLett.103.125901There is no corresponding record for this reference.
- 284Caravati, S.; Bernasconi, M.; Kühne, T. D.; Krack, M.; Parrinello, M. Unravelling the Mechanism of Pressure Induced Amorphization of Phase Change Materials. Phys. Rev. Lett. 2009, 102, 205502, DOI: 10.1103/PhysRevLett.102.205502There is no corresponding record for this reference.
- 285Luduena, G. A.; Kühne, T. D.; Sebastiani, D. Mixed Grotthuss and Vehicle Transport Mechanism in Proton Conducting Polymers from Ab initio Molecular Dynamics Simulations. Chem. Mater. 2011, 23, 1424– 1429, DOI: 10.1021/cm102674uThere is no corresponding record for this reference.
- 286Kühne, T. D.; Pascal, T. A.; Kaxiras, E.; Jung, Y. New Insights into the Structure of the Vapor/Water Interface from Large-Scale First-Principles Simulations. J. Phys. Chem. Lett. 2011, 2, 105– 113, DOI: 10.1021/jz101391rThere is no corresponding record for this reference.
- 287Behler, J. Perspective: Machine learning potentials for atomistic simulations. J. Chem. Phys. 2016, 145, 170901, DOI: 10.1063/1.4966192There is no corresponding record for this reference.
- 288Behler, J. First Principles Neural Network Potentials for Reactive Simulations of Large Molecular and Condensed Systems. Angew. Chem., Int. Ed. 2017, 56, 12828– 12840, DOI: 10.1002/anie.201703114There is no corresponding record for this reference.
- 289Bartók, A. P.; De, S.; Poelking, C.; Bernstein, N.; Kermode, J. R.; Csányi, G.; Ceriotti, M. Machine learning unifies the modeling of materials and molecules. Sci. Adv. 2017, 3, e1701816 DOI: 10.1126/sciadv.1701816There is no corresponding record for this reference.
- 290Butler, K. T.; Davies, D. W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547– 555, DOI: 10.1038/s41586-018-0337-2There is no corresponding record for this reference.
- 291Deringer, V. L.; Caro, M. A.; Csányi, G. Machine Learning Interatomic Potentials as Emerging Tools for Materials Science. Adv. Mater. 2019, 31, 1902765, DOI: 10.1002/adma.201902765There is no corresponding record for this reference.
- 292Kang, P. L.; Shang, C.; Liu, Z. P. Large-Scale Atomic Simulation via Machine Learning Potentials Constructed by Global Potential Energy Surface Exploration. Acc. Chem. Res. 2020, 53, 2119– 2129, DOI: 10.1021/acs.accounts.0c00472There is no corresponding record for this reference.
- 293Behler, J. Four Generations of High-Dimensional Neural Network Potentials. Chem. Rev. 2021, 121, 10037– 10072, DOI: 10.1021/acs.chemrev.0c00868There is no corresponding record for this reference.
- 294Fiedler, L.; Shah, K.; Bussmann, M.; Cangi, A. Deep dive into machine learning density functional theory for materials science and chemistry. Phys. Rev. Mater. 2022, 6, 040301, DOI: 10.1103/PhysRevMaterials.6.040301There is no corresponding record for this reference.
- 295Behler, J.; Parrinello, M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 2007, 98, 146401, DOI: 10.1103/PhysRevLett.98.146401There is no corresponding record for this reference.
- 296Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 2011, 134, 074106, DOI: 10.1063/1.3553717There is no corresponding record for this reference.
- 297Natarajan, S. K.; Morawietz, T.; Behler, J. Representing the potential-energy surface of protonated water clusters by high-dimensional neural network potentials. Phys. Chem. Chem. Phys. 2015, 17, 8356– 8371, DOI: 10.1039/c4cp04751fThere is no corresponding record for this reference.
- 298Schran, C.; Behler, J.; Marx, D. Automated Fitting of Neural Network Potentials at Coupled Cluster Accuracy: Protonated Water Clusters as Testing Ground. J. Chem. Theory Comput. 2020, 16, 88– 99, DOI: 10.1021/acs.jctc.9b00805There is no corresponding record for this reference.
- 299Morawietz, T.; Singraber, A.; Dellago, C.; Behler, J. How van der Waals interactions determine the unique properties of water. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 8368– 8373, DOI: 10.1073/pnas.1602375113There is no corresponding record for this reference.
- 300Daru, J.; Forbert, H.; Behler, J.; Marx, D. Coupled Cluster Molecular Dynamics of Condensed Phase Systems Enabled by Machine Learning Potentials: Liquid Water Benchmark. Phys. Rev. Lett. 2022, 129, 226001, DOI: 10.1103/PhysRevLett.129.226001There is no corresponding record for this reference.
- 301Quaranta, V.; Hellström, M.; Behler, J. Proton-Transfer Mechanisms at the Water-ZnO Interface: The Role of Presolvation. J. Phys. Chem. Lett. 2017, 8, 1476– 1483, DOI: 10.1021/acs.jpclett.7b00358There is no corresponding record for this reference.
- 302Behler, J.; Martoňák, R.; Donadio, D.; Parrinello, M. Metadynamics simulations of the high-pressure phases of silicon employing a high-dimensional neural network potential. Phys. Rev. Lett. 2008, 100, 185501, DOI: 10.1103/PhysRevLett.100.185501There is no corresponding record for this reference.
- 303Eshet, H.; Khaliullin, R. Z.; Kühne, T. D.; Behler, J.; Parrinello, M. Ab initio quality neural-network potential for sodium. Phys. Rev. B 2010, 81, 184107, DOI: 10.1103/PhysRevB.81.184107There is no corresponding record for this reference.
- 304Eshet, H.; Khaliullin, R. Z.; Kühne, T. D.; Behler, J.; Parrinello, M. Microscopic origins of the anomalous melting behavior of sodium under high pressure. Phys. Rev. Lett. 2012, 108, 115701, DOI: 10.1103/PhysRevLett.108.115701There is no corresponding record for this reference.
- 305Eckhoff, M.; Behler, J. From Molecular Fragments to the Bulk: Development of a Neural Network Potential for MOF-5. J. Chem. Theory Comput. 2019, 15, 3793– 3809, DOI: 10.1021/acs.jctc.8b01288There is no corresponding record for this reference.
- 306Eckhoff, M.; Behler, J. Insights into lithium manganese oxide-water interfaces using machine learning potentials. J. Chem. Phys. 2021, 155, 244703, DOI: 10.1063/5.0073449There is no corresponding record for this reference.
- 307Khaliullin, R. Z.; Eshet, H.; Kühne, T. D.; Behler, J.; Parrinello, M. Graphite-diamond phase coexistence study employing a neural-network mapping of the ab initio potential energy surface. Phys. Rev. B 2010, 81, 100103, DOI: 10.1103/PhysRevB.81.100103There is no corresponding record for this reference.
- 308Khaliullin, R. Z.; Eshet, H.; Kühne, T. D.; Behler, J.; Parrinello, M. Nucleation mechanism for the direct graphite-to-diamond phase transition. Nat. Mater. 2011, 10, 693– 697, DOI: 10.1038/nmat3078There is no corresponding record for this reference.
- 309Omranpour, A.; Montero De Hijes, P.; Behler, J.; Dellago, C. Perspective: Atomistic simulations of water and aqueous systems with machine learning potentials. J. Chem. Phys. 2024, 160, 170901, DOI: 10.1063/5.0201241There is no corresponding record for this reference.
- 310Artrith, N.; Morawietz, T.; Behler, J. High-dimensional neural-network potentials for multicomponent systems: Applications to zinc oxide. Phys. Rev. B 2011, 83, 153101, DOI: 10.1103/PhysRevB.83.153101There is no corresponding record for this reference.
- 311Ko, T. W.; Finkler, J. A.; Goedecker, S.; Behler, J. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun. 2021, 12, 398, DOI: 10.1038/s41467-020-20427-2There is no corresponding record for this reference.
- 312Khajehpasha, E. R.; Finkler, J. A.; Kühne, T. D.; Ghasemi, S. A. CENT2: Improved charge equilibration via neural network technique. Phys. Rev. B 2022, 105, 144106, DOI: 10.1103/PhysRevB.105.144106There is no corresponding record for this reference.
- 313Thiemann, F. L.; O’Neill, N.; Kapil, V.; Michaelides, A.; Schran, C. Introduction to machine learning potentials for atomistic simulations. J. Phys.: Condens. Matter 2025, 37, 073002, DOI: 10.1088/1361-648X/ad9657There is no corresponding record for this reference.
- 314Behler, J. Neural network potential-energy surfaces in chemistry: a tool for large-scale simulations. Phys. Chem. Chem. Phys. 2011, 13, 17930– 17955, DOI: 10.1039/c1cp21668fThere is no corresponding record for this reference.
- 315Behler, J. Constructing high-dimensional neural network potentials: A tutorial review. Int. J. Quantum Chem. 2015, 115, 1032– 1050, DOI: 10.1002/qua.24890There is no corresponding record for this reference.
- 316Brieuc, F.; Schran, C.; Forbert, H.; Marx, D. RubNNet4MD: Ruhr-Universitat Bochum Neural Networks for Molecular Dynamics Simulations. Software Package 2020–2025. DOI: 10.17877/RESOLV-2025-MDN6B2J4 .There is no corresponding record for this reference.
- 317Drautz, R. Atomic cluster expansion for accurate and transferable interatomic potentials. Phys. Rev. B 2019, 99, 014104, DOI: 10.1103/PhysRevB.99.014104There is no corresponding record for this reference.
- 318(a) Bochkarev, A.; Lysogorskiy, Y.; Drautz, R. Graph Atomic Cluster Expansion for Semilocal Interactions beyond Equivariant Message Passing. Phys. Rev. X 2024, 14, 021036, DOI: 10.1103/PhysRevX.14.021036There is no corresponding record for this reference.https://github.com/ICAMS/lammps-user-pace.There is no corresponding record for this reference.
- 319Zeng, J.; Zhang, D.; Lu, D.; Mo, P.; Li, Z.; Chen, Y.; Rynik, M.; Huang, L.; Li, Z.; Shi, S. DeePMD-kit v2: A software package for deep potential models. J. Chem. Phys. 2023, 159, 054801, DOI: 10.1063/5.0155600There is no corresponding record for this reference.
- 320Batzner, S.; Musaelian, A.; Sun, L.; Geiger, M.; Mailoa, J. P.; Kornbluth, M.; Molinari, N.; Smidt, T. E.; Kozinsky, B. E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 2022, 13, 2453, DOI: 10.1038/s41467-022-29939-5There is no corresponding record for this reference.
- 321Musaelian, A.; Batzner, S.; Johansson, A.; Sun, L.; Owen, C. J.; Kornbluth, M.; Kozinsky, B. Learning local equivariant representations for large-scale atomistic dynamics. Nat. Commun. 2023, 14, 579, DOI: 10.1038/s41467-023-36329-yThere is no corresponding record for this reference.
- 322Schran, C.; Brezina, K.; Marsalek, O. Committee neural network potentials control generalization errors and enable active learning. J. Chem. Phys. 2020, 153, 104105, DOI: 10.1063/5.0016004There is no corresponding record for this reference.
- 323Imbalzano, G.; Zhuang, Y.; Kapil, V.; Rossi, K.; Engel, E. A.; Grasselli, F.; Ceriotti, M. Uncertainty estimation for molecular dynamics and sampling. J. Chem. Phys. 2021, 154, 074102, DOI: 10.1063/5.0036522There is no corresponding record for this reference.
- 324Gastegger, M.; Behler, J.; Marquetand, P. Machine learning molecular dynamics for the simulation of infrared spectra. Chem. Sci. 2017, 8, 6924– 6935, DOI: 10.1039/C7SC02267KThere is no corresponding record for this reference.
- 325Podryabinkin, E. V.; Shapeev, A. V. Active learning of linearly parametrized interatomic potentials. Comput. Mater. Sci. 2017, 140, 171– 180, DOI: 10.1016/j.commatsci.2017.08.031There is no corresponding record for this reference.
- 326Smith, J. S.; Nebgen, B.; Lubbers, N.; Isayev, O.; Roitberg, A. E. Less is more: Sampling chemical space with active learning. J. Chem. Phys. 2018, 148, 241733, DOI: 10.1063/1.5023802There is no corresponding record for this reference.
- 327Schran, C.; Thiemann, F. L.; Rowe, P.; Müller, E. A.; Marsalek, O.; Michaelides, A. Machine learning potentials for complex aqueous systems made simple. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2110077118 DOI: 10.1073/pnas.2110077118There is no corresponding record for this reference.
- 328Stolte, N.; Daru, J.; Forbert, H.; Marx, D.; Behler, J. Random sampling versus active learning algorithms for machine learning potentials of quantum liquid water. J. Chem. Theory Comput. 2025, 21, 886– 899, DOI: 10.1021/acs.jctc.4c01382There is no corresponding record for this reference.
- 329Marx, D.; Tuckerman, M. E.; Hutter, J.; Parrinello, M. The nature of the hydrated excess proton in water. Nature 1999, 397, 601– 604, DOI: 10.1038/17579There is no corresponding record for this reference.
- 330Marx, D.. In Computer Simulations in Condensed Matter: From Materials to Chemical Biology (Lecture Notes in Physics 704); Ferrario, M., Binder, K., Ciccotti, G., Eds.; Springer, 2006; Chapter 2, pp 507– 539.There is no corresponding record for this reference.
- 331Marx, D. Proton Transfer 200 Years after von Grotthuss: Insights from Ab Initio Simulations. ChemPhysChem 2006, 7, 1848– 1870, DOI: 10.1002/cphc.200600128There is no corresponding record for this reference.
- 332Li, X.-Z.; Walker, B.; Michaelides, A. Quantum nature of the hydrogen bond. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6369– 6373, DOI: 10.1073/pnas.1016653108There is no corresponding record for this reference.
- 333Singh, R.; Azadi, S.; Kühne, T. D. Anharmonicity and finite-temperature effects on the structure, stability, and vibrational spectrum of phase III of solid molecular hydrogen. Phys. Rev. B 2014, 90, 014110, DOI: 10.1103/PhysRevB.90.014110There is no corresponding record for this reference.
- 334Spura, T.; Elgabarty, H.; Kühne, T. D. “On-the-fly” coupled cluster path-integral molecular dynamics: impact of nuclear quantum effects on the protonated water dimer. Phys. Chem. Chem. Phys. 2015, 17, 14355– 14359, DOI: 10.1039/C4CP05192KThere is no corresponding record for this reference.
- 335Kessler, J.; Elgabarty, H.; Spura, T.; Karhan, K.; Partovi-Azar, P.; Hassanali, A. A.; Kühne, T. D. Structure and dynamics of the instantaneous water/vapor interface revisited by path-integral and ab initio molecular dynamics simulations. J. Phys. Chem. B 2015, 119, 10079– 10086, DOI: 10.1021/acs.jpcb.5b04185There is no corresponding record for this reference.
- 336Marsalek, O.; Markland, T. E. Quantum Dynamics and Spectroscopy of Ab Initio Liquid Water: The Interplay of Nuclear and Electronic Quantum Effects. J. Phys. Chem. Lett. 2017, 8, 1545– 1551, DOI: 10.1021/acs.jpclett.7b00391There is no corresponding record for this reference.
- 337Azadi, S.; Singh, R.; Kühne, T. D. Nuclear quantum effects induce metallization of dense solid molecular hydrogen. J. Comput. Chem. 2018, 39, 262– 268, DOI: 10.1002/jcc.25104There is no corresponding record for this reference.
- 338Ojha, D.; Henao, A.; Kühne, T. D. Nuclear quantum effects on the vibrational dynamics of liquid water. J. Chem. Phys. 2018, 148, 102328, DOI: 10.1063/1.5005500There is no corresponding record for this reference.
- 339Clark, T.; Heske, J.; Kühne, T. D. Opposing electronic and nuclear quantum effects on hydrogen bonds in H2O and D2O. ChemPhysChem 2019, 20, 2461– 2465, DOI: 10.1002/cphc.201900839There is no corresponding record for this reference.
- 340Schran, C.; Marx, D. Quantum nature of the hydrogen bond from ambient conditions down to ultra-low temperatures. Phys. Chem. Chem. Phys. 2019, 21, 24967– 24975, DOI: 10.1039/C9CP04795FThere is no corresponding record for this reference.
- 341Ohto, T.; Dodia, M.; Xu, J.; Imoto, S.; Tang, F.; Zysk, F.; Kühne, T. D.; Shigeta, Y.; Bonn, M.; Wu, X. Accessing the accuracy of density functional theory through structure and dynamics of the water–air interface. J. Phys. Chem. Lett. 2019, 10, 4914– 4919, DOI: 10.1021/acs.jpclett.9b01983There is no corresponding record for this reference.
- 342Kaliannan, N. K.; Henao Aristizabal, A.; Wiebeler, H.; Zysk, F.; Ohto, T.; Nagata, Y.; Kühne, T. D. Impact of intermolecular vibrational coupling effects on the sum-frequency generation spectra of the water/air interface. Mol. Phys. 2020, 118, 1620358, DOI: 10.1080/00268976.2019.1620358There is no corresponding record for this reference.
- 343Ojha, D.; Henao, A.; Zysk, F.; Kühne, T. D. Nuclear quantum effects on the vibrational dynamics of the water–air interface. J. Chem. Phys. 2024, 160, 204114, DOI: 10.1063/5.0204071There is no corresponding record for this reference.
- 344Kac, M. On Distributions of Certain Wiener Functionals. Trans. Am. Math. Soc. 1949, 65, 1– 13, DOI: 10.1090/S0002-9947-1949-0027960-XThere is no corresponding record for this reference.
- 345Feynman, R. P.; Hibbs, A. R. Quantum Mechanics and Path Integrals; McGraw-Hill: New York, 1965.There is no corresponding record for this reference.
- 346Feynman, R. P. Statistical Mechanics; Addison-Wesley: Redwood City, 1972.There is no corresponding record for this reference.
- 347Barker, J. A. A quantum–statistical Monte Carlo method; path integrals with boundary conditions. J. Chem. Phys. 1979, 70, 2914– 2918, DOI: 10.1063/1.437829There is no corresponding record for this reference.
- 348Chandler, D.; Wolynes, P. G. Exploiting the isomorphism between quantum-theory and classical statistical-mechanics of polyatomic fluids. J. Chem. Phys. 1981, 74, 4078– 4095, DOI: 10.1063/1.441588There is no corresponding record for this reference.
- 349Ceperley, D. M. Path integrals in the theory of condensed helium. Rev. Mod. Phys. 1995, 67, 279– 355, DOI: 10.1103/RevModPhys.67.279There is no corresponding record for this reference.
- 350Markland, T. E.; Ceriotti, M. Nuclear quantum effects enter the mainstream. Nat. Rev. Chem. 2018, 2, 0109, DOI: 10.1038/s41570-017-0109There is no corresponding record for this reference.
- 351Callaway, D. J. E.; Rahman, A. Micro-canonical ensemble formulation of lattice gauge-theory. Phys. Rev. Lett. 1982, 49, 613– 616, DOI: 10.1103/PhysRevLett.49.613There is no corresponding record for this reference.
- 352Parrinello, M.; Rahman, A. Study of an F–center in molten KCl. J. Chem. Phys. 1984, 80, 860– 867, DOI: 10.1063/1.446740There is no corresponding record for this reference.
- 353Coalson, R. D. On the connection between Fourier coefficient and discretized cartesian path integration. J. Chem. Phys. 1986, 85, 926– 936, DOI: 10.1063/1.451248There is no corresponding record for this reference.
- 354Martyna, G. J.; Tuckerman, M. E.; Tobias, D. J.; Klein, M. L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996, 87, 1117– 1157, DOI: 10.1080/00268979600100761There is no corresponding record for this reference.
- 355Ceriotti, M.; Parrinello, M.; Markland, T. E.; Manolopoulos, D. E. Efficient stochastic thermostatting of path integral molecular dynamics. J. Chem. Phys. 2010, 133, 124104, DOI: 10.1063/1.3489925There is no corresponding record for this reference.
- 356Martyna, G. J.; Klein, M. L.; Tuckerman, M. Nosé-Hoover chains - The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635– 2643, DOI: 10.1063/1.463940There is no corresponding record for this reference.
- 357Tuckerman, M. E.; Marx, D.; Klein, M. L.; Parrinello, M. Efficient and general algorithms for path integral Car–Parrinello molecular dynamics. J. Chem. Phys. 1996, 104, 5579– 5588, DOI: 10.1063/1.471771There is no corresponding record for this reference.
- 358Kästner, J.; Thiel, W. Bridging the gap between thermodynamic integration and umbrella sampling provides a novel analysis method: “Umbrella integration”. J. Chem. Phys. 2005, 123, 144104, DOI: 10.1063/1.2052648There is no corresponding record for this reference.
- 359Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 932– 942, DOI: 10.1002/wcms.66There is no corresponding record for this reference.
- 360Schran, C.; Brieuc, F.; Marx, D. Converged Colored Noise Path Integral Molecular Dynamics Study of the Zundel Cation Down to Ultralow Temperatures at Coupled Cluster Accuracy. J. Chem. Theory Comput. 2018, 14, 5068– 5078, DOI: 10.1021/acs.jctc.8b00705There is no corresponding record for this reference.
- 361John, C.; Spura, T.; Habershon, S.; Kühne, T. D. Quantum ring-polymer contraction method: Including nuclear quantum effects at no additional computational cost in comparison to ab initio molecular dynamics. Phys. Rev. E 2016, 93, 043305, DOI: 10.1103/PhysRevE.93.043305There is no corresponding record for this reference.
- 362Ceriotti, M.; Manolopoulos, D. E.; Parrinello, M. Accelerating the convergence of path integral dynamics with a generalized Langevin equation. J. Chem. Phys. 2011, 134, 084104, DOI: 10.1063/1.3556661There is no corresponding record for this reference.
- 363Ceriotti, M.; Manolopoulos, D. E. Efficient First-Principles Calculation of the Quantum Kinetic Energy and Momentum Distribution of Nuclei. Phys. Rev. Lett. 2012, 109, 100604, DOI: 10.1103/PhysRevLett.109.100604There is no corresponding record for this reference.
- 364Brieuc, F.; Dammak, H.; Hayoun, M. Quantum Thermal Bath for Path Integral Molecular Dynamics Simulation. J. Chem. Theory Comput. 2016, 12, 1351– 1359, DOI: 10.1021/acs.jctc.5b01146There is no corresponding record for this reference.
- 365Uhl, F.; Marx, D.; Ceriotti, M. Accelerated path integral methods for atomistic simulations at ultra-low temperatures. J. Chem. Phys. 2016, 145, 054101, DOI: 10.1063/1.4959602There is no corresponding record for this reference.
- 366Cao, J. S.; Voth, G. A. A new perspective on quantum time-correlation functions. J. Chem. Phys. 1993, 99, 10070– 10073, DOI: 10.1063/1.465512There is no corresponding record for this reference.
- 367Craig, I. R.; Manolopoulos, D. E. Quantum statistics and classical mechanics: Real time correlation functions from ring polymer molecular dynamics. J. Chem. Phys. 2004, 121, 3368– 3373, DOI: 10.1063/1.1777575There is no corresponding record for this reference.
- 368Hele, T. J.; Willatt, M. J.; Muolo, A.; Althorpe, S. C. Communication: Relation of centroid molecular dynamics and ring-polymer molecular dynamics to exact quantum dynamics. J. Chem. Phys. 2015, 142, 191101, DOI: 10.1063/1.4921234There is no corresponding record for this reference.
- 369Voth, G. A. Path–integral centroid methods in quantum statistical mechanics and dynamics. Adv. Chem. Phys. 1996, 93, 135– 218, DOI: 10.1002/9780470141526.ch4There is no corresponding record for this reference.
- 370Althorpe, S. C. Path-integral approximations to quantum dynamics. Eur. Phys. J. B 2021, 94, 155, DOI: 10.1140/epjb/s10051-021-00155-2There is no corresponding record for this reference.
- 371Althorpe, S. C. Path Integral Simulations of Condensed-Phase Vibrational Spectroscopy. Annu. Rev. Phys. Chem. 2024, 75, 397– 420, DOI: 10.1146/annurev-physchem-090722-124705There is no corresponding record for this reference.
- 372Witt, A.; Ivanov, S. D.; Shiga, M.; Forbert, H.; Marx, D. On the applicability of centroid and ring polymer path integral molecular dynamics for vibrational spectroscopy. J. Chem. Phys. 2009, 130, 194510, DOI: 10.1063/1.3125009There is no corresponding record for this reference.
- 373Rossi, M.; Ceriotti, M.; Manolopoulos, D. E. How to remove the spurious resonances from ring polymer molecular dynamics. J. Chem. Phys. 2014, 140, 234116, DOI: 10.1063/1.4883861There is no corresponding record for this reference.
- 374Shiga, M. Path integral Brownian chain molecular dynamics: A simple approximation of quantum vibrational dynamics. J. Comput. Chem. 2022, 43, 1864– 1879, DOI: 10.1002/jcc.26989There is no corresponding record for this reference.
- 375Tuckerman, M. E.; Berne, B. J.; Martyna, G. J.; Klein, M. L. Efficient molecular-dynamics and hybrid Monte-Carlo algorithms for path-integrals. J. Chem. Phys. 1993, 99, 2796– 2808, DOI: 10.1063/1.465188There is no corresponding record for this reference.
- 376Spura, T.; John, C.; Habershon, S.; Kühne, T. D. Nuclear quantum effects in liquid water from path-integral simulations using an ab initio force-matching approach. Mol. Phys. 2015, 113, 808– 822, DOI: 10.1080/00268976.2014.981231There is no corresponding record for this reference.
- 377Stolte, N.; Daru, J.; Forbert, H.; Behler, J.; Marx, D. Nuclear Quantum Effects in Liquid Water Are Marginal for Its Average Structure but Significant for Dynamics. J. Phys. Chem. Lett. 2024, 15, 12144– 12150, DOI: 10.1021/acs.jpclett.4c02925There is no corresponding record for this reference.
- 378Malik, R.; Stolte, N.; Forbert, H.; Chandra, A.; Marx, D. Accurate Determination of Isotope Effects on the Dynamics of H-Bond Breaking and Making in Liquid Water. J. Phys. Chem. Lett. 2025, 16, 3727– 3733, DOI: 10.1021/acs.jpclett.5c00210There is no corresponding record for this reference.
- 379Feynman, R. P. Atomic Theory of the λ Transition in Helium. Phys. Rev. 1953, 91, 1291– 1301, DOI: 10.1103/PhysRev.91.1291There is no corresponding record for this reference.
- 380Troyer, M.; Wiese, U.-J. Computational Complexity and Fundamental Limitations to Fermionic Quantum Monte Carlo Simulations. Phys. Rev. Lett. 2005, 94, 170201, DOI: 10.1103/PhysRevLett.94.170201There is no corresponding record for this reference.
- 381Calcavecchia, F.; Pederiva, F.; Kalos, M. H.; Kühne, T. D. Sign problem of the fermionic shadow wave function. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2014, 90, 053304, DOI: 10.1103/PhysRevE.90.053304There is no corresponding record for this reference.
- 382Ceperley, D. M.; Pollock, E. L. Path-Integral Computation of the Low-Temperature Properties of Liquid 4He. Phys. Rev. Lett. 1986, 56, 351– 354, DOI: 10.1103/PhysRevLett.56.351There is no corresponding record for this reference.
- 383Pollock, E. L.; Ceperley, D. M. Path-integral computation of superfluid densities. Phys. Rev. B 1987, 36, 8343– 8352, DOI: 10.1103/PhysRevB.36.8343There is no corresponding record for this reference.
- 384Boninsegni, M.; Prokof’ev, N.; Svistunov, B. Worm Algorithm for Continuous-Space Path Integral Monte Carlo Simulations. Phys. Rev. Lett. 2006, 96, 070601, DOI: 10.1103/PhysRevLett.96.070601There is no corresponding record for this reference.
- 385Boninsegni, M.; Prokof’ev, N. V.; Svistunov, B. V. Worm algorithm and diagrammatic Monte Carlo: A new approach to continuous-space path integral Monte Carlo simulations. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2006, 74, 036701, DOI: 10.1103/PhysRevE.74.036701There is no corresponding record for this reference.
- 386Brieuc, F.; Schran, C.; Uhl, F.; Forbert, H.; Marx, D. Converged quantum simulations of reactive solutes in superfluid helium: The Bochum perspective. J. Chem. Phys. 2020, 152, 210901, DOI: 10.1063/5.0008309There is no corresponding record for this reference.
- 387Walewski, L.; Forbert, H.; Marx, D. Reactive path integral quantum simulations of molecules solvated in superfluid helium. Comput. Phys. Commun. 2014, 185, 884– 899, DOI: 10.1016/j.cpc.2013.12.011There is no corresponding record for this reference.
- 388Pollock, E. L.; Ceperley, D. M. Simulation of quantum many-body systems by path-integral methods. Phys. Rev. B 1984, 30, 2555– 2568, DOI: 10.1103/PhysRevB.30.2555There is no corresponding record for this reference.
- 389Aziz, R. A.; Janzen, A. R.; Moldover, M. R. Ab Initio Calculations for Helium: A Standard for Transport Property Measurements. Phys. Rev. Lett. 1995, 74, 1586– 1589, DOI: 10.1103/PhysRevLett.74.1586There is no corresponding record for this reference.
- 390Pollock, E. L.; Runge, K. J. Finite-size-scaling analysis of a simulation of the 4He superfluid transition. Phys. Rev. B 1992, 46, 3535– 3539, DOI: 10.1103/PhysRevB.46.3535There is no corresponding record for this reference.
- 391Keesom, W. H.; Keesom, A. P. New measurements on the specific heat of liquid helium. Physica 1935, 2, 557– 572, DOI: 10.1016/S0031-8914(35)90128-8There is no corresponding record for this reference.
- 392Klein, M. L.; Venables, J. A. Rare Gas Solids Vol. II; Academic Press: London, New York, San Francisco, 1977.There is no corresponding record for this reference.
- 393Sindzingre, P.; Klein, M. L.; Ceperley, D. M. Path-integral Monte Carlo study of low-temperature 4He clusters. Phys. Rev. Lett. 1989, 63, 1601– 1604, DOI: 10.1103/PhysRevLett.63.1601There is no corresponding record for this reference.
- 394Zeng, T.; Guillon, G.; Cantin, J. T.; Roy, P.-N. Probing the Superfluid Response of para-Hydrogen with a Sulfur Dioxide Dopant. J. Phys. Chem. Lett. 2013, 4, 2391– 2396, DOI: 10.1021/jz401188jThere is no corresponding record for this reference.
- 395Zeng, T.; Roy, P. N. Microscopic molecular superfluid response: Theory and simulations. Rep. Prog. Phys. 2014, 77, 046601, DOI: 10.1088/0034-4885/77/4/046601There is no corresponding record for this reference.
- 396Brieuc, F.; Schran, C.; Marx, D. Manifestations of local supersolidity of 4He around a charged molecular impurity. Phys. Rev. Res. 2023, 5, 043083, DOI: 10.1103/PhysRevResearch.5.043083There is no corresponding record for this reference.
- 397Walewski, Ł.; Forbert, H.; Marx, D. Solvation of molecules in superfluid helium enhances the “interaction induced localization” effect. J. Chem. Phys. 2014, 140, 144305, DOI: 10.1063/1.4870595There is no corresponding record for this reference.
- 398Duran Caballero, L.; Schran, C.; Brieuc, F.; Marx, D. Neural network interaction potentials for para-hydrogen with flexible molecules. J. Chem. Phys. 2022, 157, 074302, DOI: 10.1063/5.0100953There is no corresponding record for this reference.
- 399Marx, D.; Parrinello, M. Ab initio path–integral molecular dynamics. Z. Phys. B 1994, 95, 143– 144, DOI: 10.1007/BF01312185There is no corresponding record for this reference.
- 400Marx, D.; Parrinello, M. Ab initio path integral molecular dynamics: Basic ideas. J. Chem. Phys. 1996, 104, 4077– 4082, DOI: 10.1063/1.471221There is no corresponding record for this reference.
- 401Boese, A. D.; Forbert, H.; Masia, M.; Tekin, A.; Marx, D.; Jansen, G. Constructing simple yet accurate potentials for describing the solvation of HCl/water clusters in bulk helium and nanodroplets. Phys. Chem. Chem. Phys. 2011, 13, 14550– 14564, DOI: 10.1039/c1cp20991dThere is no corresponding record for this reference.
- 402Kuchenbecker, D.; Uhl, F.; Forbert, H.; Jansen, G.; Marx, D. Constructing accurate interaction potentials to describe the microsolvation of protonated methane by helium atoms. Phys. Chem. Chem. Phys. 2017, 19, 8307– 8321, DOI: 10.1039/C7CP00652GThere is no corresponding record for this reference.
- 403Uhl, F.; Marx, D. Helium Tagging of Protonated Methane in Messenger Spectroscopy: Does It Interfere with the Fluxionality of CH5+?. Angew. Chem., Int. Ed. 2018, 57, 14792– 14795, DOI: 10.1002/anie.201808531There is no corresponding record for this reference.
- 404Uhl, F.; Marx, D. Quantum Microsolvation of Protonated Methane with 4He: Large-Amplitude Motion Heavily Influences Bosonic Exchange. Phys. Rev. Lett. 2019, 123, 123002, DOI: 10.1103/PhysRevLett.123.123002There is no corresponding record for this reference.
- 405Schran, C.; Uhl, F.; Behler, J.; Marx, D. High-dimensional neural network potentials for solvation: The case of protonated water clusters in helium. J. Chem. Phys. 2018, 148, 102310, DOI: 10.1063/1.4996819There is no corresponding record for this reference.
- 406Werner, H.-J.; Knowles, P. J.; Manby, F. R.; Black, J. A.; Doll, K.; Heßelmann, A.; Kats, D.; Köhn, A.; Korona, T.; Kreplin, D. A. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107, DOI: 10.1063/5.0005081There is no corresponding record for this reference.
- 407Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108, DOI: 10.1063/5.0004608There is no corresponding record for this reference.
- 408Topolnicki, R.; Brieuc, F.; Schran, C.; Marx, D. Deciphering High-Order Structural Correlations within Fluxional Molecules from Classical and Quantum Configurational Entropy. J. Chem. Theory Comput. 2020, 16, 6785– 6794, DOI: 10.1021/acs.jctc.0c00642There is no corresponding record for this reference.
- 409Schran, C.; Brieuc, F.; Marx, D. Transferability of machine learning potentials: Protonated water neural network potential applied to the protonated water hexamer. J. Chem. Phys. 2021, 154, 051101, DOI: 10.1063/5.0035438There is no corresponding record for this reference.
- 410Larsson, H. R.; Schröder, M.; Beckmann, R.; Brieuc, F.; Schran, C.; Marx, D.; Vendrell, O. State-resolved infrared spectrum of the protonated water dimer: revisiting the characteristic proton transfer doublet peak. Chem. Sci. 2022, 13, 11119– 11125, DOI: 10.1039/D2SC03189BThere is no corresponding record for this reference.
- 411Beckmann, R.; Topolnicki, R.; Marx, D. Deciphering the Impact of Helium Tagging on Flexible Molecules: Probing Microsolvation Effects of Protonated Acetylene by Quantum Configurational Entrop. J. Phys. Chem. A 2023, 127, 2460– 2471, DOI: 10.1021/acs.jpca.2c08967There is no corresponding record for this reference.
- 412Simko, I.; Schran, C.; Brieuc, F.; Fabri, C.; Asvany, O.; Schlemmer, S.; Marx, D.; Császár, A. G. Quantum Nuclear Delocalization and its Rovibrational Fingerprints. Angew. Chem., Int. Ed. 2023, 62, e202306744 DOI: 10.1002/anie.202306744There is no corresponding record for this reference.
- 413Davies, J. A.; Schran, C.; Brieuc, F.; Marx, D.; Ellis, A. M. Onset of Rotational Decoupling for a Molecular Ion Solvated in Helium: From Tags to Rings and Shells. Phys. Rev. Lett. 2023, 130, 083001, DOI: 10.1103/PhysRevLett.130.083001There is no corresponding record for this reference.
- 414Beckmann, R.; Schran, C.; Brieuc, F.; Marx, D. Theoretical infrared spectroscopy of protonated methane isotopologues. Phys. Chem. Chem. Phys. 2024, 26, 22846– 22852, DOI: 10.1039/D4CP02295EThere is no corresponding record for this reference.
- 415Beckmann, R.; Brieuc, F.; Schran, C.; Marx, D. Infrared Spectra at Coupled Cluster Accuracy from Neural Network Representations. J. Chem. Theory Comput. 2022, 18, 5492– 5501, DOI: 10.1021/acs.jctc.2c00511There is no corresponding record for this reference.
- 416Larsen, A. H.; Mortensen, J. J.; Blomqvist, J.; Castelli, I. E.; Christensen, R.; Dulak, M.; Friis, J.; Groves, M. N.; Hammer, B.; Hargus, C. The atomic simulation environment - a Python library for working with atoms. J. Phys.: Condens. Matter 2017, 29, 273002, DOI: 10.1088/1361-648X/aa680eThere is no corresponding record for this reference.
- 417Brehm, M.; Kirchner, B. TRAVIS – A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51 (8), 2007– 2023, DOI: 10.1021/ci200217wThere is no corresponding record for this reference.
- 418Brehm, M.; Thomas, M.; Gehrke, S.; Kirchner, B. TRAVIS – A Free Analyzer for Trajectories from Molecular Simulation. J. Chem. Phys. 2020, 152 (16), 164105, DOI: 10.1063/5.0005078There is no corresponding record for this reference.
- 419Lindner, J.; Cringus, D.; Pshenichnikov, M. S.; Vöhringer, P. Anharmonic Bend–Stretch Coupling in Neat Liquid Water. Chem. Phys. 2007, 341, 326– 335, DOI: 10.1016/j.chemphys.2007.07.051There is no corresponding record for this reference.
- 420Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient. J. Chem. Soc., Perkin Trans. 2 1993, 2, 799– 805, DOI: 10.1039/P29930000799There is no corresponding record for this reference.
- 421Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506– 10517, DOI: 10.1021/jp971959kThere is no corresponding record for this reference.
- 422Gordon, R. G. Molecular motion in infrared and raman spectra. J. Chem. Phys. 1965, 43, 1307– 1312, DOI: 10.1063/1.1696920There is no corresponding record for this reference.
- 423Shimizu, H. Time-Correlation Function of Molecular Random Motion and Shape of Spectral Bands. J. Chem. Phys. 1965, 43, 2453– 2465, DOI: 10.1063/1.1697145There is no corresponding record for this reference.
- 424Kubo, R.; Toda, M.; Hashitsume, N. Statistical Physics II; Springer: Berlin, 1991.There is no corresponding record for this reference.
- 425Spaldin, N. A. A beginner’s guide to the modern theory of polarization. J. Solid State Chem. 2012, 195, 2– 10, DOI: 10.1016/j.jssc.2012.05.010There is no corresponding record for this reference.
- 426King-Smith, R. D.; Vanderbilt, D. Theory of Polarization of Crystalline Solids. Phys. Rev. B 1993, 47, 1651– 1654, DOI: 10.1103/PhysRevB.47.1651There is no corresponding record for this reference.
- 427Resta, R. Macroscopic Electric Polarization As a Geometric Quantum Phase. Europhys. Lett. 1993, 22, 133– 138, DOI: 10.1209/0295-5075/22/2/010There is no corresponding record for this reference.
- 428Resta, R. Macroscopic polarization in crystalline dielectrics: the geometric phase approach. Rev. Mod. Phys. 1994, 66, 899– 915, DOI: 10.1103/RevModPhys.66.899There is no corresponding record for this reference.
- 429Resta, R. Quantum-Mechanical Position Operator in Extended Systems. Phys. Rev. Lett. 1998, 80, 1800– 1803, DOI: 10.1103/PhysRevLett.80.1800There is no corresponding record for this reference.
- 430Berry, M. V. Quantal phase factors accompanying adiabatic changes. Proc. R. Soc. London, Ser. A 1984, 392, 45– 57, DOI: 10.1098/rspa.1984.0023There is no corresponding record for this reference.
- 431Mead, C. A. The geometric phase in molecular systems. Rev. Mod. Phys. 1992, 64, 51, DOI: 10.1103/RevModPhys.64.51There is no corresponding record for this reference.
- 432Kirchner, B.; Hutter, J. Solvent effects on electronic properties from Wannier functions in a dimethyl sulfoxide/water mixture. J. Chem. Phys. 2004, 121, 5133– 5142, DOI: 10.1063/1.1785780There is no corresponding record for this reference.
- 433Silvestrelli, P. L.; Parrinello, M. Water Molecule Dipole in the Gas and in the Liquid Phase ─ Erratum. Phys. Rev. Lett. 1999, 82, 5415, DOI: 10.1103/PhysRevLett.82.5415There is no corresponding record for this reference.
- 434Silvestrelli, P. L.; Parrinello, M. Structural, electronic, and bonding properties of liquid water from first principles. J. Chem. Phys. 1999, 111, 3572– 3580, DOI: 10.1063/1.479638There is no corresponding record for this reference.
- 435Boys, S. F. Construction of Some Molecular Orbitals to be Approximately Invariant for Changes from One Molecule to Another. Rev. Mod. Phys. 1960, 32, 296– 299, DOI: 10.1103/RevModPhys.32.296There is no corresponding record for this reference.
- 436Pipek, J.; Mezey, P. G. A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave functions. J. Chem. Phys. 1989, 90, 4916– 4926, DOI: 10.1063/1.456588There is no corresponding record for this reference.
- 437Wannier, G. H. The Structure of Electronic Excitation Levels in Insulating Crystals. Phys. Rev. 1937, 52, 191– 197, DOI: 10.1103/PhysRev.52.191There is no corresponding record for this reference.
- 438Marzari, N.; Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 1997, 56, 12847– 12865, DOI: 10.1103/PhysRevB.56.12847There is no corresponding record for this reference.
- 439Silvestrelli, P. L.; Marzari, N.; Vanderbilt, D.; Parrinello, M. Maximally-localized Wannier functions for disordered systems: Application to amorphous silicon. Solid State Commun. 1998, 107, 7– 11, DOI: 10.1016/S0038-1098(98)00175-6There is no corresponding record for this reference.
- 440Silvestrelli, P. L. Maximally localized Wannier functions for simulations with supercells of general symmetry. Phys. Rev. B 1999, 59, 9703– 9706, DOI: 10.1103/PhysRevB.59.9703There is no corresponding record for this reference.
- 441Marzari, N.; Mostofi, A. A.; Yates, J. R.; Souza, I.; Vanderbilt, D. Maximally localized Wannier functions: Theory and applications. Rev. Mod. Phys. 2012, 84, 1419– 1475, DOI: 10.1103/RevModPhys.84.1419There is no corresponding record for this reference.
- 442Ambrosetti, A.; Silvestrelli, P. L. Introduction to maximally localized Wannier functions. Rev. Comput. Chem. 2016, 29, 327– 368, DOI: 10.1002/9781119148739.ch6There is no corresponding record for this reference.
- 443Berghold, G.; Mundy, C. J.; Romero, A. H.; Hutter, J.; Parrinello, M. General and efficient algorithms for obtaining maximally localized Wannier functions. Phys. Rev. B 2000, 61, 10040– 10048, DOI: 10.1103/PhysRevB.61.10040There is no corresponding record for this reference.
- 444Resta, R.; Sorella, S. Electron Localization in the Insulating State. Phys. Rev. Lett. 1999, 82, 370– 373, DOI: 10.1103/PhysRevLett.82.370There is no corresponding record for this reference.
- 445Edmiston, C.; Ruedenberg, K. Localized Atomic and Molecular Orbitals. Rev. Mod. Phys. 1963, 35, 457– 464, DOI: 10.1103/RevModPhys.35.457There is no corresponding record for this reference.
- 446Jacobi, C. Über ein Leichtes Verfahren die in der Theorie der Säcularstörungen Vorkommenden Gleichungen Numerisch Aufzulösen. J. für die Reine Angewandte Math. 1846, 30, 51– 94There is no corresponding record for this reference.
- 447Iftimie, R.; Minary, P.; Tuckerman, M. E. Ab initio molecular dynamics: Concepts, recent developments, and future trends. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 6654– 6659, DOI: 10.1073/pnas.0500193102There is no corresponding record for this reference.
- 448Ojha, D.; Kühne, T. D. “On-the-fly” calculation of the Vibrational Sum-frequency Generation Spectrum at the Air-water Interface. Molecules 2020, 25, 3939, DOI: 10.3390/molecules25173939There is no corresponding record for this reference.
- 449Partovi-Azar, P.; Kühne, T. D. Full Assignment of ab-initio Raman Spectra at Finite Temperatures Using Wannier Polarizabilities: Application to Cyclohexane Molecule in Gas Phase. Micromachines 2021, 12, 1212, DOI: 10.3390/mi12101212There is no corresponding record for this reference.
- 450Partovi-Azar, P.; Kühne, T. D. Efficient “On-the-Fly” calculation of Raman Spectra from Ab-Initio molecular dynamics: Application to hydrophobic/hydrophilic solutes in bulk water. J. Comput. Chem. 2015, 36, 2188– 2192, DOI: 10.1002/jcc.24198There is no corresponding record for this reference.
- 451Partovi-Azar, P.; Kühne, T. D.; Kaghazchi, P. Evidence for the existence of Li 2 S 2 clusters in lithium–sulfur batteries: ab initio Raman spectroscopy simulation. Phys. Chem. Chem. Phys. 2015, 17, 22009– 22014, DOI: 10.1039/C5CP02781KThere is no corresponding record for this reference.
- 452Partovi-Azar, P. Efficient method for estimating the dynamics of the full polarizability tensor during ab initio molecular dynamics simulations. Phys. Rev. B 2023, 108, 235157, DOI: 10.1103/PhysRevB.108.235157There is no corresponding record for this reference.
- 453Thomas, M.; Brehm, M.; Kirchner, B. Voronoi Dipole Moments for the Simulation of Bulk Phase Vibrational Spectra. Phys. Chem. Chem. Phys. 2015, 17, 3207– 3213, DOI: 10.1039/C4CP05272BThere is no corresponding record for this reference.
- 454Brehm, M.; Thomas, M. Optimized Atomic Partial Charges and Radii Defined by Radical Voronoi Tessellation of Bulk Phase Simulations. Molecules 2021, 26 (7), 1875, DOI: 10.3390/molecules26071875There is no corresponding record for this reference.
- 455Voronoi, G. Nouvelles Applications Des Paramètres Continus à La Théorie Des Formes Quadratiques. Deuxième Mémoire. Recherches Sur Les Parallélloèdres Primitifs. J. für die Reine Angewandte Math. 1908, 1908, 198– 287, DOI: 10.1515/crll.1908.134.198There is no corresponding record for this reference.
- 456Medvedev, N. N. The Algorithm for Three-Dimensional Voronoi Polyhedra. J. Comput. Phys. 1986, 67, 223– 229, DOI: 10.1016/0021-9991(86)90123-3There is no corresponding record for this reference.
- 457Pollak, M.; Rein, R. Molecular-Orbital Studies of Intermolecular Interaction Energies. II. Approximations Concerned with Coulomb Interactions and Comparison of the Two London Schemes. J. Chem. Phys. 1967, 47, 2045– 2052, DOI: 10.1063/1.1712236There is no corresponding record for this reference.
- 458Becke, A. D. A Multicenter Numerical Integration Scheme for Polyatomic Molecules. J. Chem. Phys. 1988, 88, 2547– 2553, DOI: 10.1063/1.454033There is no corresponding record for this reference.
- 459Gellatly, B. J.; Finney, J. L. Calculation of Protein Volumes: An Alternative to the Voronoi Procedure. J. Mol. Biol. 1982, 161, 305– 322, DOI: 10.1016/0022-2836(82)90155-3There is no corresponding record for this reference.
- 460Richards, F. M. The Interpretation of Protein Structures: Total Volume, Group Volume Distributions and Packing Density. J. Mol. Biol. 1974, 82, 1– 14, DOI: 10.1016/0022-2836(74)90570-1There is no corresponding record for this reference.
- 461Brehm, M.; Thomas, M. Computing Bulk Phase Raman Optical Activity Spectra from ab initio Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2017, 8 (14), 3409– 3414, DOI: 10.1021/acs.jpclett.7b01616There is no corresponding record for this reference.
- 462Brehm, M.; Thomas, M. Computing Bulk Phase Resonance Raman Spectra from ab initio Molecular Dynamics and Real-Time TDDFT. J. Chem. Theory Comput. 2019, 15 (7), 3901– 3905, DOI: 10.1021/acs.jctc.9b00512There is no corresponding record for this reference.
- 463Yang, Y.; Cheramy, J.; Brehm, M.; Xu, Y. Raman Optical Activity of N-Acetyl-l-Cysteine in Water and in Methanol: The “Clusters-in-a-Liquid” Model and ab initio Molecular Dynamics Simulations. ChemPhysChem 2022, 23, e202200161 DOI: 10.1002/cphc.202200161There is no corresponding record for this reference.
- 464Brehm, M. libvori: Voronoi Integration for Electrostatic Properties from Electron Density, 2025. https://brehm-research.de/libvori.php (accessed 13 09.There is no corresponding record for this reference.
- 465Rycroft, C. H. VORO++: A three-dimensional Voronoi cell library in C++. Chaos 2009, 19, 041111, DOI: 10.1063/1.3215722There is no corresponding record for this reference.
- 466Cordero, B.; Gómez, V.; Platero-Prats, A. E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 2832, 2832, DOI: 10.1039/b801115jThere is no corresponding record for this reference.
- 467Brehm, M.; Kirchner, B. TRAVIS - A free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007– 2023, DOI: 10.1021/ci200217wThere is no corresponding record for this reference.
- 468Ewald, P. P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253– 287, DOI: 10.1002/andp.19213690304There is no corresponding record for this reference.
- 469Heaton, R. J.; Madden, P. A.; Clark, S. J.; Jahn, S. Condensed Phase Ionic Polarizabilities from Plane Wave Density Functional Theory Calculations. J. Chem. Phys. 2006, 125, 144104, DOI: 10.1063/1.2357151There is no corresponding record for this reference.
- 470Salanne, M.; Vuilleumier, R.; Madden, P. A.; Simon, C.; Turq, P.; Guillot, B. Polarizabilities of Individual Molecules and Ions in Liquids from First Principles. J. Phys.: Condens. Matter 2008, 20, 494207, DOI: 10.1088/0953-8984/20/49/494207There is no corresponding record for this reference.
- 471Wan, Q.; Spanu, L.; Galli, G. A.; Gygi, F. Raman Spectra of Liquid Water from ab initio Molecular Dynamics: Vibrational Signatures of Charge Fluctuations in the Hydrogen Bond Network. J. Chem. Theory Comput. 2013, 9, 4124– 4130, DOI: 10.1021/ct4005307There is no corresponding record for this reference.
- 472Luber, S.; Iannuzzi, M.; Hutter, J. Raman spectra from ab initio molecular dynamics and its application to liquid S-methyloxirane. J. Chem. Phys. 2014, 141, 094503, DOI: 10.1063/1.4894425There is no corresponding record for this reference.
- 473Albrecht, A. C. On the Theory of Raman Intensities. J. Chem. Phys. 1961, 34, 1476– 1484, DOI: 10.1063/1.1701032There is no corresponding record for this reference.
- 474Tang, J.; Albrecht, A. C. Studies in Raman Intensity Theory. J. Chem. Phys. 1968, 49, 1144– 1154, DOI: 10.1063/1.1670202There is no corresponding record for this reference.
- 475Albrecht, A. C.; Hutley, M. C. On the Dependence of Vibrational Raman Intensity on the Wavelength of Incident Light. J. Chem. Phys. 1971, 55, 4438– 4443, DOI: 10.1063/1.1676771There is no corresponding record for this reference.
- 476Lee, S.-Y.; Heller, E. J. Time-Dependent Theory of Raman Scattering. J. Chem. Phys. 1979, 71, 4777– 4788, DOI: 10.1063/1.438316There is no corresponding record for this reference.
- 477Heller, E. J. The Semiclassical Way to Molecular Spectroscopy. Acc. Chem. Res. 1981, 14, 368– 375, DOI: 10.1021/ar00072a002There is no corresponding record for this reference.
- 478Heller, E. J.; Sundberg, R.; Tannor, D. J. Simple Aspects of Raman Scattering. J. Phys. Chem. 1982, 86, 1822– 1833, DOI: 10.1021/j100207a018There is no corresponding record for this reference.
- 479Jensen, L.; Zhao, L. L.; Autschbach, J.; Schatz, G. C. Theory and Method for Calculating Resonance Raman Scattering from Resonance Polarizability Derivatives. J. Chem. Phys. 2005, 123, 174110, DOI: 10.1063/1.2046670There is no corresponding record for this reference.
- 480Jensen, L.; Autschbach, J.; Schatz, G. C. Finite Lifetime Effects on the Polarizability within Time-Dependent Density-Functional Theory. J. Chem. Phys. 2005, 122, 224115, DOI: 10.1063/1.1929740There is no corresponding record for this reference.
- 481Andermatt, S.; Cha, J.; Schiffmann, F.; VandeVondele, J. Combining Linear-Scaling DFT with Subsystem DFT in Born–Oppenheimer and Ehrenfest Molecular Dynamics Simulations: From Molecules to a Virus in Solution. J. Chem. Theory Comput. 2016, 12, 3214– 3227, DOI: 10.1021/acs.jctc.6b00398There is no corresponding record for this reference.
- 482Provorse, M. R.; Isborn, C. M. Electron Dynamics with Real-Time Time-Dependent Density Functional Theory. Int. J. Quantum Chem. 2016, 116, 739– 749, DOI: 10.1002/qua.25096There is no corresponding record for this reference.
- 483Goings, J. J.; Lestrange, P. J.; Li, X. Real-Time Time-Dependent Electronic Structure Theory. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1341 DOI: 10.1002/wcms.1341There is no corresponding record for this reference.
- 484Chen, H.; Mcmahon, J. M.; Ratner, M. A.; Schatz, G. C. Classical Electrodynamics Coupled to Quantum Mechanics for Calculation of Molecular Optical Properties: A RT-TDDFT/FDTD Approach. J. Phys. Chem. C 2010, 114, 14384– 14392, DOI: 10.1021/jp1043392There is no corresponding record for this reference.
- 485Ibaceta-Jaña, J.; Chugh, M.; Novikov, A. S.; Mirhosseini, H.; Kühne, T. D.; Szyszka, B.; Wagner, M. R.; Muydinov, R. Do lead halide hybrid perovskites have hydrogen bonds?. J. Phys. Chem. C 2022, 126, 16215– 16226, DOI: 10.1021/acs.jpcc.2c02984There is no corresponding record for this reference.
- 486Brehm, M.; Thomas, M. An Efficient Lossless Compression Algorithm for Trajectories of Atom Positions and Volumetric Data. J. Chem. Inf. Model. 2018, 58 (10), 2092– 2107, DOI: 10.1021/acs.jcim.8b00501There is no corresponding record for this reference.
- 487Zhang, C.; Khaliullin, R. Z.; Bovi, D.; Guidoni, L.; Kühne, T. D. Vibrational Signature of Water Molecules in Asymmetric Hydrogen Bonding Environments. J. Phys. Chem. Lett. 2013, 4, 3245– 3250, DOI: 10.1021/jz401321xThere is no corresponding record for this reference.
- 488Zhang, C.; Guidoni, L.; Kühne, T. D. Competing factors on the frequency separation between the OH stretching modes in water. J. Mol. Liq. 2015, 205, 42– 45, DOI: 10.1016/j.molliq.2014.09.049There is no corresponding record for this reference.
- 489Nonella, M.; Mathias, G.; Tavan, P. Infrared spectrum of p–benzoquinone in water obtained from a QM/MM hybrid molecular dynamics simulation. J. Phys. Chem. A 2003, 107, 8638– 8647, DOI: 10.1021/jp027747rThere is no corresponding record for this reference.
- 490Schmitz, M.; Tavan, P. Vibrational spectra from atomic fluctuations in dynamics simulations I. Theory, limitations and a sample application. J. Chem. Phys. 2004, 121, 12233– 12246, DOI: 10.1063/1.1822914There is no corresponding record for this reference.
- 491Pejov, L.; Spångberg, D.; Hermansson, K. Using MD Snapshots in ab initio and DFT Calculations: OH Vibrations in the First Hydration Shell around Li+(aq). J. Phys. Chem. A 2005, 109, 5144– 5152, DOI: 10.1021/jp047395jThere is no corresponding record for this reference.
- 492Wheeler, R. A.; Dong, H.; Boesch, S. E. Quasiharmonic Vibrations of Water, Water Dimer, and Liquid Water from Principal Component Analysis of Quantum or QM/MM Trajectories. ChemPhysChem 2003, 4, 382– 384, DOI: 10.1002/cphc.200390066There is no corresponding record for this reference.
- 493Gaigeot, M.-P.; Sprik, M. Ab initio molecular dynamics computation of the infrared spectrum of aqueous uracil. J. Phys. Chem. B 2003, 107, 10344– 10358, DOI: 10.1021/jp034788uThere is no corresponding record for this reference.
- 494Mathias, G.; Baer, M. D. Generalized Normal Coordinates for the Vibrational Analysis of Molecular Dynamics Simulations. J. Chem. Theory Comput. 2011, 7, 2028– 2039, DOI: 10.1021/ct2001304There is no corresponding record for this reference.
- 495Mathias, G.; Ivanov, S. D.; Witt, A.; Baer, M. D.; Marx, D. Infrared Spectroscopy of Fluxional Molecules from (ab initio) Molecular Dynamics: Resolving Large-Amplitude Motion, Multiple Conformations, and Permutational Symmetries. J. Chem. Theory Comput. 2012, 8, 224– 234, DOI: 10.1021/ct2006665There is no corresponding record for this reference.
- 496Eckart, C. Some Studies Concerning Rotating Axes and Polyatomic Molecules. Phys. Rev. 1935, 47, 552– 558, DOI: 10.1103/PhysRev.47.552There is no corresponding record for this reference.
- 497Thomas, M.; Brehm, M.; Hollóczki, O.; Kelemen, Z.; Nyulászi, L.; Pasinszki, T.; Kirchner, B. Simulating the Vibrational Spectra of Ionic Liquid Systems: 1-Ethyl-3-Methylimidazolium Acetate and its Mixtures. J. Chem. Phys. 2014, 141, 024510, DOI: 10.1063/1.4887082There is no corresponding record for this reference.
- 498CP2K Dashboard. https://dashboard.cp2k.org/index.html, 2024; (accessed, 2025 10 18).There is no corresponding record for this reference.
- 499(a) Frigo, M.; Johnson, S. G. The design and implementation of FFTW3. Proc. IEEE 2005, 93, 216– 231, DOI: 10.1109/JPROC.2004.840301There is no corresponding record for this reference.See. http://www.fftw.org.There is no corresponding record for this reference.
- 500Ramaswami, A.; Kenter, T.; Kühne, T. D.; Plessl, C. Efficient ab-initio molecular dynamic simulations by offloading fast Fourier transformations to FPGAs. In 2020 30th International Conference on Field-Programmable Logic and Applications (FPL); Institute of Electrical and Electronics Engineers, 2020, pp 353– 354.There is no corresponding record for this reference.
- 501Ramaswami, A.; Kenter, T.; Kühne, T. D.; Plessl, C. Evaluating the design space for offloading 3D FFT calculations to an FPGA for high-performance computing. In International Symposium on Applied Reconfigurable Computing; Springer Nature, 2021, pp 285– 294.There is no corresponding record for this reference.
- 502Wu, X.; Kenter, T.; Schade, R.; Kühne, T. D.; Plessl, C. Computing and compressing electron repulsion integrals on FPGAs. In 2023 IEEE 31st Annual International Symposium on Field-Programmable Custom Computing Machines (FCCM); Institute of Electrical and Electronics Engineers, 2023, pp 162– 173.There is no corresponding record for this reference.
- 503FFTFPGA: An OpenCL based library for Fast Fourier Transformations for FPGAs. https://github.com/pc2/fft3d-fpga, 2024; (accessed, 2025 10 01).There is no corresponding record for this reference.
- 504Heinecke, A.; Henry, G.; Hutchinson, M.; Pabst, H. LIBXSMM: Accelerating Small Matrix Multiplications by Runtime Code Generation. In SC16: International Conference for High Performance Computing, Networking, Storage and Analysis; Institute of Electrical and Electronics Engineers: Piscataway, NJ, USA, 2016.There is no corresponding record for this reference.
- 505Marek, A.; Blum, V.; Johanni, R.; Havu, V.; Lang, B.; Auckenthaler, T.; Heinecke, A.; Bungartz, H.-J.; Lederer, H. The ELPA library: scalable parallel eigenvalue solutions for electronic structure theory and computational science. J. Phys.: Condens. Matter 2014, 26, 213201, DOI: 10.1088/0953-8984/26/21/213201There is no corresponding record for this reference.
- 506cuSOLVERMp: A High-Performance CUDA Library for Distributed Dense Linear Algebra. https://docs.nvidia.com/cuda/cusolvermp, 2024; (accessed, 2025 10 19).There is no corresponding record for this reference.
- 507(a) Solcà, R.; Simberg, M.; Meli, R.; Invernizzi, A.; Reverdell, A.; Biddiscombe, J. DLA-Future: A Task-Based Linear Algebra Library Which Provides a GPU-Enabled Distributed Eigensolver. In Asynchronous Many-Task Systems and Applications; Springer Nature: Cham, 2024; pp 135– 141.There is no corresponding record for this reference.See https://github.com/eth-cscs/DLA-Future.There is no corresponding record for this reference.
- 508Kozhevnikov, A.; Taillefumier, M.; Frasch, S.; Pintarelli, V. J., Simon; ; Schulthess, T. Domain specific library for electronic structure calculations. 2019; https://github.com/electronic-structure/SIRIUS, (accessed: 2025 10 02).There is no corresponding record for this reference.
- 509(a) Caldeweyher, E.; Mewes, J.-M.; Ehlert, S.; Grimme, S. Extension and evaluation of the D4 London-dispersion model for periodic systems. Phys. Chem. Chem. Phys. 2020, 22, 8499– 8512, DOI: 10.1039/D0CP00502AThere is no corresponding record for this reference.See. https://github.com/dftd4/dftd4.There is no corresponding record for this reference.
- 510(a) Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB─An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652– 1671, DOI: 10.1021/acs.jctc.8b01176There is no corresponding record for this reference.See. https://github.com/tblite/tblite.There is no corresponding record for this reference.
- 511(a) Oleynichenko, A. V.; Zaitsevskii, A.; Mosyagin, N. S.; Petrov, A. N.; Eliav, E.; Titov, A. V. LIBGRPP: A library for the evaluation of molecular integrals of the generalized relativistic pseudopotential operator over Gaussian functions. Symmetry 2023, 15, 197, DOI: 10.3390/sym15010197There is no corresponding record for this reference.See. https://github.com/aoleynichenko/libgrpp.There is no corresponding record for this reference.
- 512Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R. A.; Parrinello, M. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961– 1972, DOI: 10.1016/j.cpc.2009.05.011There is no corresponding record for this reference.
- 513Togo, A.; Tanaka, I. Spglib: a software library for crystal symmetry search, 2018. https://github.com/spglib/spglib (accessed 18 10 2025).There is no corresponding record for this reference.
- 514(a) Rocha, A. R.; Garcia-Suarez, V. M.; Bailey, S. W.; Lambert, C. J.; Ferrer, J.; Sanvito, S. Towards molecular spintronics. Nat. Mater. 2005, 4, 335– 339, DOI: 10.1038/nmat1349There is no corresponding record for this reference.See. https://github.com/StefanoSanvitoGroup/libsmeagol.There is no corresponding record for this reference.
- 515(a) Posenitskiy, E.; Chilkuri, V. G.; Ammar, A.; Hapka, M.; Pernal, K.; Shinde, R.; Landinez Borda, E. J.; Filippi, C.; Nakano, K.; Kohulák, O. TREXIO: A file format and library for quantum chemistry. J. Chem. Phys. 2023, 158, 174801, DOI: 10.1063/5.0148161There is no corresponding record for this reference.See. https://github.com/trex-coe/trexio.There is no corresponding record for this reference.
- 516Azizi, M.; Wilhelm, J.; Golze, D.; Delesma, F. A.; Panadés-Barrueta, R. L.; Rinke, P.; Giantomassi, M.; Gonze, X. Validation of the GreenX library time-frequency component for efficient GW and RPA calculations. Phys. Rev. B 2024, 109, 245101, DOI: 10.1103/PhysRevB.109.245101There is no corresponding record for this reference.
- 517Gamblin, T.; LeGendre, M.; Collette, M. R.; Lee, G. L.; Moody, A.; de Supinski, B. R.; Futral, S. The Spack package manager: bringing order to HPC software chaos. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; Association for Computing Machinery, 2015, pp 1– 12.There is no corresponding record for this reference.
- 518Holmberg, N.; Laasonen, K. Efficient constrained density functional theory implementation for simulation of condensed phase electron transfer reactions. J. Chem. Theory Comput. 2017, 13, 587– 601, DOI: 10.1021/acs.jctc.6b01085There is no corresponding record for this reference.
- 519Holmberg, N.; Laasonen, K. Diabatic model for electrochemical hydrogen evolution based on constrained DFT configuration interaction. J. Chem. Phys. 2018, 149, 104702, DOI: 10.1063/1.5038959There is no corresponding record for this reference.
- 520Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R. A. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961– 1972, DOI: 10.1016/j.cpc.2009.05.011There is no corresponding record for this reference.
- 521Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1– 5, DOI: 10.1016/j.scriptamat.2015.07.021There is no corresponding record for this reference.
- 522Akimov, A. V. Libra: an open-source “methodology discovery” library for quantum and classical dynamics simulations. J. Comput. Chem. 2016, 37, 1626– 1649, DOI: 10.1002/jcc.24367There is no corresponding record for this reference.
- 523Lervik, A.; Riccardi, E.; van Erp, T. S. PyRETIS: A well-done, medium-sized python library for rare events. J. Comput. Chem. 2017, 38, 2439– 2451, DOI: 10.1002/jcc.24900There is no corresponding record for this reference.
- 524Riccardi, E.; Lervik, A.; Roet, S.; Aarøen, O.; van Erp, T. S. PyRETIS 2: an improbability drive for rare events. J. Comput. Chem. 2020, 41, 370– 377, DOI: 10.1002/jcc.26112There is no corresponding record for this reference.
- 525Vervust, W.; Zhang, D. T.; Ghysels, A.; Roet, S.; van Erp, T. S.; Riccardi, E. PyRETIS 3: Conquering rare and slow events without boundaries. J. Comput. Chem. 2024, 45, 1224– 1234, DOI: 10.1002/jcc.27319There is no corresponding record for this reference.
- 526Pizzi, G.; Vitale, V.; Arita, R.; Bluegel, S.; Freimuth, F.; Géranton, G.; Gibertini, M.; Gresch, D.; Johnson, C.; Koretsune, T. Wannier90 as a Community Code: New Features and Applications. J. Phys.: Condens. Matter 2020, 32, 165902, DOI: 10.1088/1361-648X/ab51ffThere is no corresponding record for this reference.
- 527Vogt, J.-R.; Schulz, M.; Mattos, R. S.; Barbatti, M.; Persico, M.; Granucci, G.; Hutter, J.; Hehn, A.-S. A mixed density functional theory and semi-empirical framework for trajectory surface hopping on extended systems. ChemRxiv 2025, 2025-ddxzg, DOI: 10.26434/chemrxiv-2025-ddxzgThere is no corresponding record for this reference.
- 528Zeng, J.; Zhang, D.; Peng, A.; Zhang, X.; He, S.; Wang, Y.; Liu, X.; Bi, H.; Li, Y.; Cai, C. DeePMD-kit v3: a multiple-backend framework for machine learning potentials. J. Chem. Theory Comput. 2025, 21, 4375– 4385, DOI: 10.1021/acs.jctc.5c00340There is no corresponding record for this reference.
- 529Antalík, A.; Levy, A.; Johnson, S. K.; Olsen, J. M. H.; Rothlisberger, U. Making Puzzle Pieces Fit or Reshaping MiMiC for Multiscale Simulations with CP2K and More. J. Chem. Inf. Model. 2025, 65, 4994– 5005, DOI: 10.1021/acs.jcim.5c00409There is no corresponding record for this reference.
- 530Larsen, A. H.; Mortensen, J. J.; Blomqvist, J.; Castelli, I. E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M. N.; Hammer, B.; Hargus, C. The atomic simulation environment─a Python library for working with atoms. J. Phys.: Condens. Matter 2017, 29, 273002, DOI: 10.1088/1361-648X/aa680eThere is no corresponding record for this reference.
- 531Ceriotti, M.; More, J.; Manolopoulos, D. E. i-PI: A Python interface for ab initio path integral molecular dynamics simulations. Comput. Phys. Commun. 2014, 185, 1019– 1026, DOI: 10.1016/j.cpc.2013.10.027There is no corresponding record for this reference.
- 532Kapil, V.; Rossi, M.; Marsalek, O.; Petraglia, R.; Litman, Y.; Spura, T.; Cheng, B.; Cuzzocrea, A.; Meißner, R. H.; Wilkins, D. M. i-PI 2.0: A universal force engine for advanced molecular simulations. Comput. Phys. Commun. 2019, 236, 214– 223, DOI: 10.1016/j.cpc.2018.09.020There is no corresponding record for this reference.
- 533Seeber, P.; Seidenath, S.; Steinmetzer, J.; Gräfe, S. Growing Spicy ONIOMs: Extending and generalizing concepts of ONIOM and many body expansions. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2023, 13, e1644 DOI: 10.1002/wcms.1644There is no corresponding record for this reference.
- 534Verstraelen, T.; Van Houteghem, M.; Van Speybroeck, V.; Waroquier, M. MD-TRACKS: A Productive Solution for the Advanced Analysis of Molecular Dynamics and Monte Carlo simulations. J. Chem. Inf. Model. 2008, 48, 2414– 2424, DOI: 10.1021/ci800233yThere is no corresponding record for this reference.
- 535Verstraelen, T.; Van Speybroeck, V.; Waroquier, M. ZEOBUILDER: A GUI Toolkit for the Construction of Complex Molecular Structures on the Nanoscale with Building Blocks. J. Chem. Inf. Model. 2008, 48, 1530– 1541, DOI: 10.1021/ci8000748There is no corresponding record for this reference.
- 536Ghysels, A.; Verstraelen, T.; Hemelsoet, K.; Waroquier, M.; Van Speybroeck, V. TAMkin: a versatile package for vibrational analysis and chemical kinetics. J. Chem. Inf. Model. 2010, 50, 1736– 1750, DOI: 10.1021/ci100099gThere is no corresponding record for this reference.
- 537Verstraelen, T.; Adams, W.; Pujal, L.; Tehrani, A.; Kelly, B. D.; Macaya, L.; Meng, F.; Richer, M.; Hernández-Esparza, R.; Yang, X. D. IOData: A python library for reading, writing, and converting computational chemistry file formats and generating input files. J. Comput. Chem. 2021, 42, 458– 464, DOI: 10.1002/jcc.26468There is no corresponding record for this reference.
- 538Poeschel, F.; Juncheng, E.; Godoy, W. F.; Podhorszki, N.; Klasky, S.; Eisenhauer, G.; Davis, P. E.; Wan, L.; Gainaru, A.; Gu, J.; Transitioning from file-based HPC workflows to streaming data pipelines with openPMD and ADIOS2. In Smoky Mountains Computational Sciences and Engineering Conference; Springer Nature, 2021, pp 99– 118.There is no corresponding record for this reference.
- 539Gu, J.; Davis, P.; Eisenhauer, G.; Godoy, W.; Huebl, A.; Klasky, S.; Parashar, M.; Podhorszki, N.; Poeschel, F.; Vay, J. Organizing large data sets for efficient analyses on HPC systems. J. Phys.: Conf. Ser. 2022, 2224, 012042, DOI: 10.1088/1742-6596/2224/1/012042There is no corresponding record for this reference.
- 540Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn. J. Chem. Phys. 2024, 161, 082503, DOI: 10.1063/5.0216272There is no corresponding record for this reference.
- 541Los, J. H.; Kühne, T. D. Inverse simulated annealing for the determination of amorphous structures. Phys. Rev. B 2013, 87, 214202, DOI: 10.1103/PhysRevB.87.214202There is no corresponding record for this reference.
- 542Los, J. H.; Gabardi, S.; Bernasconi, M.; Kühne, T. D. Inverse simulated annealing: Improvements and application to amorphous InSb. Comput. Mater. Sci. 2016, 117, 7– 14, DOI: 10.1016/j.commatsci.2016.01.017There is no corresponding record for this reference.
- 543Thomsen, B.; Shiga, M. Ab initio study of nuclear quantum effects on sub-and supercritical water. J. Chem. Phys. 2021, 155, 194107, DOI: 10.1063/5.0071857There is no corresponding record for this reference.
- 544Thomsen, B.; Shiga, M. Structures of liquid and aqueous water isotopologues at ambient temperature from ab initio path integral simulations. Phys. Chem. Chem. Phys. 2022, 24, 10851– 10859, DOI: 10.1039/D2CP00499BThere is no corresponding record for this reference.
- 545Togo, A.; Chaput, L.; Tadano, T.; Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys.: Condens. Matter 2023, 35, 353001, DOI: 10.1088/1361-648X/acd831There is no corresponding record for this reference.
- 546Schreder, L.; Luber, S. Implementation of frozen density embedding in CP2K and OpenMolcas: CASSCF wavefunctions embedded in a Gaussian and plane wave DFT environment. J. Chem. Phys. 2024, 161, 144110, DOI: 10.1063/5.0222409There is no corresponding record for this reference.
- 547Battaglia, S.; Rossmannek, M.; Rybkin, V. V.; Tavernelli, I.; Hutter, J. A general framework for active space embedding methods with applications in quantum computing. npj Comput. Mater. 2024, 10, 297, DOI: 10.1038/s41524-024-01477-2There is no corresponding record for this reference.
- 548Johnson, B. A.; Bu, S.; Mundy, C. J.; Ananth, N. MixPI: Mixed-Time Slicing Path Integral Software for Quantized Molecular Dynamics Simulations. arXiv 2024, 11988, DOI: 10.48550/arXiv.2411.11988There is no corresponding record for this reference.
- 549CP2K Benchmark Suite, 2024. https://www.cp2k.org/performance (accessed, 2025 10 19).There is no corresponding record for this reference.