



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
A New Set of Combining Rules for Mie (λ, 6) PotentialClick to copy article linkArticle link copied!
- Nguyen Van PhuocNguyen Van PhuocFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City 70000, VietnamMore by Nguyen Van Phuoc
- Thanh Doanh LeThanh Doanh LeFaculty of Electrical Engineering, Electric Power University, Ministry of Industry and Trade, Hanoi 10000, VietnamMore by Thanh Doanh Le
- Van Hoa Nguyen*Van Hoa Nguyen*Email: [email protected]Institute of Fundamental and Applied Sciences, Duy Tan University, Tran Nhat Duat Street, Ho Chi Minh City 70000, VietnamFaculty of Environmental and Natural Sciences, Duy Tan University, 03 Quang Trung Street, Da Nang 50000, VietnamMore by Van Hoa Nguyen
- Suresh AlapatiSuresh AlapatiDepartment of Mechatronics Engineering, Kyungsung University, 309, Suyeong-ro (Daeyeon-dong), Nam-gu, Busan 48434, KoreaMore by Suresh Alapati
- Stéphanie Delage SantacreuStéphanie Delage SantacreuLaboratoire de Mathématiques et de leurs Applications de Pau, UMR5142, CNRS, Université de Pau et des Pays de l’Adour, Pau 64000, FranceMore by Stéphanie Delage Santacreu
- Guillaume GalliéroGuillaume GalliéroLaboratoire des Fluides Complexes et leurs Réservoirs, Université de Pau et des Pays de l’Adour, E2S UPPA, CNRS, Pau 64000, FranceMore by Guillaume Galliéro
- Hai Hoang*Hai Hoang*Email: [email protected]Institute of Fundamental and Applied Sciences, Duy Tan University, Tran Nhat Duat Street, Ho Chi Minh City 70000, VietnamFaculty of Environmental and Natural Sciences, Duy Tan University, 03 Quang Trung Street, Da Nang 50000, VietnamMore by Hai Hoang
Abstract
Force fields based on the Mie (λ, 6) potential, combined with theoretical methods and molecular simulations, offer a promising framework for predicting the thermophysical properties of fluids. Despite this potential, the availability of reliable combining rules for unlike interaction parameters in mixtures remains limited, thereby constraining the broader application of Mie (λ, 6)-based force fields. In this study, a new set of combining rules for the Mie (λ, 6) potential is proposed, derived by using a distortion model for the repulsive interaction and a geometric mean approximation for the attractive interaction, combined with first-order mathematical approximations. The capability of the new combining rules was first evaluated for noble gas pairs modeled with the Lennard–Jones potential, a specific case of Mie (λ, 6) potential with λ = 12, for which experimentally derived data on unlike interaction parameters are available. The results showed noticeably better agreement with experimentally derived values than those obtained using the two commonly used combining rules. Further assessment was carried out through the evaluation of Henry’s law constants, phase diagrams, and excess molar volumes, which are highly sensitive to cross-interactions, for various binary mixtures modeled using the Mie chain coarse-grained force field, obtained from NVT-GEMC, NPT-GEMC, and NPT-MC simulations, respectively. For mixtures with similar Mie (λ, 6) potential parameters for the components, all of the combining rules, including the new ones, yielded comparable predictions. In contrast, for asymmetric systems with significant force field parameter disparities, the new rules yielded substantially improved accuracy relative to experimental data for all considered thermodynamic properties, whereas the commonly used combining rules exhibited poor performance with markedly larger deviations. These findings highlight the improved robustness and broader applicability of the proposed combining rules for extending Mie (λ, 6)-based force fields to complex fluid mixtures.
This publication is licensed for personal use by The American Chemical Society.
1. Introduction
2. Combining Rules for Mie Potentials
3. Molecular Simulations
4. Results and Discussion
4.1. Validation of the New Set of Combining Rules
| noble gas | ε/kB [K] | σ [Å] |
|---|---|---|
| He | 24.80 | 2.366 |
| Ne | 43.00 | 2.730 |
| Ar | 135.00 | 3.360 |
| Kr | 193.00 | 3.570 |
| Xe | 256.00 | 3.920 |
| pair | classical combining rules | Lafitte et al. combining rules | Kong combining rules | new combining rules | data deduced from experiments |
|---|---|---|---|---|---|
| ε/kB [K] | |||||
| He–Ar | 57.86 | 55.27 | 41.50 | 42.32 | 40.00 ± 3.00 |
| He–Xe | 79.68 | 72.49 | 40.94 | 39.88 | 49.00 ± 5.00 |
| Ne–Ar | 76.19 | 74.97 | 67.12 | 69.17 | 64.50 ± 4.00 |
| Ne–Kr | 91.10 | 88.68 | 73.61 | 75.57 | 71.50 ± 3.50 |
| Ne–Xe | 104.92 | 99.92 | 73.38 | 74.49 | 73.00 ± 4.00 |
| Ar–Kr | 161.42 | 161.19 | 159.60 | 160.79 | 148.00 ± 7.00 |
| Ar–Xe | 185.90 | 184.26 | 175.16 | 177.94 | 178.00 ± 6.00 |
| σ [Å] | |||||
| He–Ar | 2.863 | 2.863 | 2.980 | 2.970 | 2.980 ± 0.020 |
| He–Xe | 3.143 | 3.143 | 3.403 | 3.418 | 3.360 ± 0.030 |
| Ne–Ar | 3.045 | 3.045 | 3.093 | 3.078 | 3.090 ± 0.030 |
| Ne–Kr | 3.150 | 3.150 | 3.235 | 3.221 | 3.220 ± 0.030 |
| Ne–Xe | 3.325 | 3.325 | 3.472 | 3.464 | 3.460 ± 0.030 |
| Ar–Kr | 3.465 | 3.465 | 3.470 | 3.466 | 3.510 ± 0.030 |
| Ar–Xe | 3.640 | 3.640 | 3.665 | 3.656 | 3.650 ± 0.030 |
4.2. Tests of the New Set of Combining Rules
4.2.1. Lennard–Jones Parameters of Unlike Interactions
4.2.2. Thermodynamic Properties of Binary Mixtures
| species | Nc | λ | ε/kB [K] | σ [Å] |
|---|---|---|---|---|
| neon | 1 | 12.510 | 34.912 | 2.813 |
| argon | 1 | 13.926 | 125.571 | 3.407 |
| krypton | 1 | 13.428 | 171.149 | 3.634 |
| xenon | 1 | 14.223 | 244.131 | 3.962 |
| methane | 1 | 14.000 | 161.000 | 3.740 |
| carbon dioxide | 2 | 16.933 | 211.539 | 2.861 |
| n-heptane | 3 | 14.034 | 294.293 | 4.049 |
| n-decane | 4 | 16.025 | 336.756 | 4.111 |
| toluene | 3 | 12.27 | 293.247 | 3.658 |
| pair | classical combining rules | Lafitte et al. combining rules | new combining rules |
|---|---|---|---|
| repulsive exponent | |||
| argon/krypton | 13.68 | 13.67 | 13.66 |
| krypton/xenon | 13.83 | 13.82 | 13.83 |
| methane/xenon | 14.11 | 14.11 | 14.12 |
| krypton/n-heptane | 13.73 | 13.73 | 13.74 |
| methane/n-heptane | 14.02 | 14.02 | 14.02 |
| neon/n-heptane | 13.27 | 13.24 | 13.40 |
| argon/n-heptane | 13.98 | 13.98 | 13.99 |
| carbon dioxide/n-heptane | 15.48 | 15.40 | 15.11 |
| methane/n-decane | 15.01 | 14.97 | 15.01 |
| carbon dioxide/n-decane | 16.48 | 16.47 | 16.38 |
| methane/toluene | 13.13 | 13.10 | 13.07 |
| carbon dioxide/toluene | 14.60 | 14.36 | 13.95 |
| ε/kB [K] | |||
| argon/krypton | 146.60 | 146.37 | 145.38 |
| krypton/xenon | 204.41 | 203.84 | 203.32 |
| methane/xenon | 198.25 | 198.01 | 198.23 |
| krypton/n-heptane | 224.43 | 223.45 | 222.15 |
| methane/n-heptane | 217.67 | 217.16 | 216.69 |
| neon/n-heptane | 101.36 | 96.47 | 76.15 |
| argon/n-heptane | 192.24 | 190.10 | 183.43 |
| carbon dioxide/n-heptane | 249.51 | 238.53 | 207.18 |
| methane/n-decane | 232.85 | 232.07 | 235.69 |
| carbon dioxide/n-decane | 266.9 | 254.14 | 223.62 |
| methane/toluene | 217.29 | 217.25 | 217.24 |
| carbon dioxide/toluene | 249.07 | 243.50 | 217.07 |
| σ [Å] | |||
| argon/krypton | 3.521 | 3.521 | 3.523 |
| krypton/xenon | 3.798 | 3.798 | 3.799 |
| methane/xenon | 3.851 | 3.851 | 3.850 |
| krypton/n-heptane | 3.841 | 3.841 | 3.844 |
| methane/n-heptane | 3.895 | 3.895 | 3.894 |
| neon/n-heptane | 3.431 | 3.431 | 3.549 |
| argon/n-heptane | 3.728 | 3.728 | 3.744 |
| carbon dioxide/n-heptane | 3.455 | 3.455 | 3.502 |
| methane/n-decane | 3.925 | 3.925 | 3.917 |
| carbon dioxide/n-decane | 3.486 | 3.486 | 3.530 |
| methane–toluene | 3.699 | 3.699 | 3.699 |
| carbon dioxide–toluene | 3.260 | 3.260 | 3.300 |
4.2.2.1. Henry’s Law Constant
Figure 1
Figure 1. Comparison of Henry’s law constants obtained from the NVT-GEMC simulations using different combining rules and experiment for seven binary mixtures for which the Mie (λ, 6) potential parameters of the components are relatively similar. (a) Argon/Krypton mixture. (b) Krypton/Xenon mixture. (c) Methane/Xenon mixture. (d) Krypton/n-Heptane mixture. (e) Methane/n-Heptane mixture. (f) Methane/n-Decane mixture. (g) Methane–Toluene mixture. Solid circles (red) correspond to the experimental data. Open squares (green) correspond to the simulation data by using the classical combining rules. Open deltas (blue) correspond to the simulation data using Lafitte et al. combining rules. Open right triangles (red) correspond to the simulation data by using the new combining rules. Lines serve as a guide to the eye for the experimental data.
Figure 2
Figure 2. Comparison of Henry’s law constants obtained from the NVT-GEMC simulations using different combining rules and experiment for five binary mixtures for which the Mie (λ, 6) potential parameters of the components are significantly different. (a) Neon/n-heptane mixture. (b) Argon/n-heptane mixture. (c) Carbon dioxide/n-heptane mixture. (d) Carbon dioxide/n-decane mixture. (e) Carbon dioxide/toluene mixture. The legend is identical to that in Figure 1.
4.2.2.2. Phase Diagram of a Binary Mixture
Figure 3
Figure 3. Comparison of pressure–composition phase diagrams of binary mixtures obtained from the NPT-GEMC simulations using different combining rules and experiment for six binary mixtures for which the Mie (λ, 6) potential parameters of the components are relatively similar. (a) Argon/krypton mixture at T = 177.38 K. (b) Krypton/xenon mixture at T = 190.03 K. (c) Methane/xenon mixture at T = 189.78 K. (d) Methane/n-heptane mixture at T = 310.93 K. (e) Methane/n-decane mixture at T = 373.15 K. (f) Methane/toluene mixture at T = 422.45 K. The legend is identical to that in Figure 1.
Figure 4
Figure 4. Comparison of pressure–composition phase diagrams of binary mixtures obtained from the NPT-GEMC simulations using different combining rules and experiment for three binary mixtures for which the Mie (λ, 6) potential parameters of the species are significantly different. (a) Carbon dioxide/n-heptane mixture at T = 310.65 K. (b) Carbon dioxide/n-decane mixture at T = 377.59 K. (c) Carbon dioxide/toluene mixture at T = 308.16 K. The legend is identical to that in Figure 1.
4.2.2.3. Excess Molar Volume of a Binary Mixture
Figure 5
Figure 5. Comparison of excess molar volumes of binary mixtures obtained from the NPT-MC simulations using different combining rules and experiment for three binary mixtures for which the Mie (λ, 6) potential parameters of the components are relatively similar. (a) Methane/n-heptane mixture at T = 303.15 K and P = 30 MPa. (b) Methane/n-decane mixture at T = 373.15 K and P = 40 MPa. (c) Methane/toluene mixture at T = 333.15 K and P = 45 MPa. The legend is identical to that in Figure 1.
Figure 6
Figure 6. Comparison of excess molar volumes of binary mixtures obtained from the NPT-MC simulations using different combining rules and experiment for three binary mixtures for which the Mie (λ, 6) potential parameters of the components are significantly different. (a) Carbon dioxide/n-heptane mixture at T = 313.15 K and P = 10 MPa. (b) Carbon dioxide/n-decane mixture at T = 318.15 K and P = 15 MPa. (c) Carbon dioxide/toluene mixture at T = 313.15 K and P = 10 MPa. The legend is identical to that in Figure 1.
5. Conclusions
Author Information
- Hai Hoang - Institute of Fundamental and Applied Sciences, Duy Tan University, Tran Nhat Duat Street, Ho Chi Minh City 70000, Vietnam; Faculty of Environmental and Natural Sciences, Duy Tan University, 03 Quang Trung Street, Da Nang 50000, Vietnam;
 https://orcid.org/0000-0003-2992-0866;
https://orcid.org/0000-0003-2992-0866;
- Guillaume Galliéro - Laboratoire des Fluides Complexes et leurs Réservoirs, Université de Pau et des Pays de l’Adour, E2S UPPA, CNRS, Pau 64000, France;https://orcid.org/0000-0001-6393-8387
N.V.P.: data curation; methodology; investigation; formal analysis; software; visualization; writing─original draft; and writing─review and editing. T.D.L.: investigation; formal analysis; software; visualization; and writing─review and editing. V.H.N.: data curation; methodology; investigation; formal analysis; software; visualization; and writing─review and editing. S.A.: formal analysis; validation; visualization; and writing─review and editing. S.D.S.: formal analysis; software; validation; and writing─review and editing. G.G.: conceptualization; supervision; writing─original draft; and writing─review and editing. H.H.: conceptualization; data curation; methodology; supervision; validation; writing─original draft; and writing─review and editing.
Acknowledgments
The authors gratefully acknowledge the support of the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 103.01-2025.42. Computational resources were provided by the University of Pau and Pays de l’Adour (UPPA) and the MCIA (Mésocentre de Calcul Intensif Aquitain). Hai Hoang also acknowledges financial support from the Centre National de la Recherche Scientifique (CNRS), France. The authors thank Dr. Dung Chinh Nguyen for valuable discussions on the mathematical derivations.
Appendix: Derivation of the New Set of Combining Rules for the Mie (λ, 6) Potential
References
This article references 60 other publications.
- 1Dohrn, R.; Pfohl, O. Thermophysical properties─industrial directions. Fluid Phase Equilib. 2002, 194–197, 15– 29, DOI: 10.1016/S0378-3812(01)00791-9Google ScholarThere is no corresponding record for this reference.
- 2Gupta, S.; Olson, J. D. Industrial needs in physical properties. Ind. Eng. Chem. Res. 2003, 42, 6359– 6374, DOI: 10.1021/ie030170vGoogle ScholarThere is no corresponding record for this reference.
- 3Hendriks, E.; Kontogeorgis, G. M.; Dohrn, R.; Hemptinne, J.-C. de.; Economou, I. G.; Žilnik, L. F.; Vesovic, V. Industrial requirements for thermodynamics and transport properties. Ind. Eng. Chem. Res. 2010, 49, 11131– 11141, DOI: 10.1021/ie101231bGoogle ScholarThere is no corresponding record for this reference.
- 4Kontogeorgis, G. M.; Dohrn, R.; Economou, I. G.; Hemptinne, J.-C. de.; Kate, A. Ten.; Kuitunen, S.; Mooijer, M.; Žilnik, L. F.; Vesovic, V. Industrial requirements for thermodynamic and transport properties: 2020. Ind. Eng. Chem. Res. 2021, 60, 4987– 5013, DOI: 10.1021/acs.iecr.0c05356Google ScholarThere is no corresponding record for this reference.
- 5de Hemptinne, J.-C.; Kontogeorgis, G. M.; Dohrn, R.; Economou, I. G.; Ten Kate, A.; Kuitunen, S.; Fele Žilnik, L. F.; De Angelis, M. G.; Vesovic, V. A view on the future of applied thermodynamics. Ind. Eng. Chem. Res. 2022, 61, 14664– 14680, DOI: 10.1021/acs.iecr.2c01906Google ScholarThere is no corresponding record for this reference.
- 6Gupta, S.; Elliott, J. R.; Anderko, A.; Crosthwaite, J.; Chapman, W. G.; Lira, C. T. Current practices and continuing needs in thermophysical properties for the chemical industry. Ind. Eng. Chem. Res. 2023, 62, 3394– 3427, DOI: 10.1021/acs.iecr.2c03153Google ScholarThere is no corresponding record for this reference.
- 7Poling, B. E.; Prausnitz, M.; O’Connell, J. P. The Properties of Gases and Liquids; McGraw-Hill: New York, 2004.Google ScholarThere is no corresponding record for this reference.
- 8Goodwin, A. R.; Sengers, J.; Peters, C. J. Applied Thermodynamics of Fluids; Royal Society of Chemistry: Cambridge, 2010.Google ScholarThere is no corresponding record for this reference.
- 9Assael, M. J.; Goodwin, A. R. H.; Vesovic, V.; Wakeham, W. A. Experimental Thermodynamics Volume IX: Advances in Transport Properties of Fluids; Royal Society of Chemistry: London, 2014.Google ScholarThere is no corresponding record for this reference.
- 10Tillotson, M. J.; Diamantonis, N. I.; Buda, C.; Bolton, L. W.; Müller, E. A. Molecular modelling of the thermophysical properties of fluids: expectations, limitations, gaps and opportunities. Phys. Chem. Chem. Phys. 2023, 25, 12607– 12628, DOI: 10.1039/D2CP05423JGoogle ScholarThere is no corresponding record for this reference.
- 11Mejía, A.; Herdes, C.; Müller, E. A. Force fields for coarse-grained molecular simulations from a corresponding states correlation. Ind. Eng. Chem. Res. 2014, 53, 4131– 4141, DOI: 10.1021/ie404247eGoogle ScholarThere is no corresponding record for this reference.
- 12Herdes, C.; Totton, T. S.; Müller, E. A. Coarse grained force field for the molecular simulation of natural gases and condensates. Fluid Phase Equilib. 2015, 406, 91– 100, DOI: 10.1016/j.fluid.2015.07.014Google ScholarThere is no corresponding record for this reference.
- 13Mick, J. R.; Barhaghi, M. S.; Jackman, B.; Rushaidat, K.; Schwiebert, L.; Potoff, J. J. Optimized Mie potentials for phase equilibria: Application to noble gases and their mixtures with n-alkanes. J. Chem. Phys. 2015, 143, 114304 DOI: 10.1063/1.4930138Google ScholarThere is no corresponding record for this reference.
- 14Hoang, H.; Delage-Santacreu, S.; Galliero, G. Simultaneous description of equilibrium, interfacial, and transport properties of fluids using a Mie chain coarse-grained force field. Ind. Eng. Chem. Res. 2017, 56, 9213– 9226, DOI: 10.1021/acs.iecr.7b01397Google ScholarThere is no corresponding record for this reference.
- 15Jervell, V. G.; Wilhelmsen, Ø. Revised Enskog theory for Mie fluids: Prediction of diffusion coefficients, thermal diffusion coefficients, viscosities, and thermal conductivities. J. Chem. Phys. 2023, 158, 224103 DOI: 10.1063/5.0149865Google ScholarThere is no corresponding record for this reference.
- 16Chaparro, G.; Müller, E. A. Simulation and data-driven modeling of the transport properties of the Mie fluid. J. Phys. Chem. B 2024, 128, 551– 566, DOI: 10.1021/acs.jpcb.3c06813Google ScholarThere is no corresponding record for this reference.
- 17Ervik, Å.; Serratos, G. J.; Müller, E. A. raaSAFT: A framework enabling coarse-grained molecular dynamics simulations based on the SAFT-γ Mie force field. Comput. Phys. Commun. 2017, 212, 161– 179, DOI: 10.1016/j.cpc.2016.07.035Google ScholarThere is no corresponding record for this reference.
- 18Calvin, D. W.; Reed III, T. M. Mixture rules for the Mie (n, 6) intermolecular pair potential and the Dymond–Alder pair potential. J. Chem. Phys. 1971, 54, 3733– 3738, DOI: 10.1063/1.1675422Google ScholarThere is no corresponding record for this reference.
- 19Lafitte, T.; Apostolakou, A.; Avendaño, C.; Galindo, A.; Adjiman, C. S.; Müller, E. A.; Jackson, G. Accurate statistical associating fluid theory for chain molecules formed from Mie segments. J. Chem. Phys. 2013, 139, 154504 DOI: 10.1063/1.4819786Google ScholarThere is no corresponding record for this reference.
- 20Lorentz, H. A. Ueber die Anwendung des Satzes vom Virial in der kinetischen Theorie der Gase. Ann. Phys. 1881, 248, 127– 136, DOI: 10.1002/andp.18812480110Google ScholarThere is no corresponding record for this reference.
- 21Berthelot, D. Sur le mélange des gaz. Compt. Rendus 1898, 126, 15Google ScholarThere is no corresponding record for this reference.
- 22Hamani, A. W. S.; Bazile, J. P.; Hoang, H.; Luc, H. T.; Daridon, J. L.; Galliero, G. Thermophysical properties of simple molecular liquid mixtures: On the limitations of some force fields. J. Mol. Liq. 2020, 303, 112663 DOI: 10.1016/j.molliq.2020.112663Google ScholarThere is no corresponding record for this reference.
- 23Hamani, A. W. S.; Hoang, H.; Viet, T. Q. Q.; Daridon, J. L.; Galliero, G. Excess volume, isothermal compressibility, isentropic compressibility and speed of sound of carbon dioxide+ n-heptane binary mixture under pressure up to 70 MPa. II. Molecular simulations. J. Supercrit. Fluids 2020, 164, 104890 DOI: 10.1016/j.supflu.2020.104890Google ScholarThere is no corresponding record for this reference.
- 24Delhommelle, J.; Millié, P. Inadequacy of the Lorentz–Berthelot combining rules for accurate predictions of equilibrium properties by molecular simulation. Mol. Phys. 2001, 99, 619– 625, DOI: 10.1080/00268970010020041Google ScholarThere is no corresponding record for this reference.
- 25Desgranges, C.; Delhommelle, J. Evaluation of the grand-canonical partition function using expanded Wang–Landau simulations. III. Impact of combining rules on mixtures properties. J. Chem. Phys. 2014, 140, 104109 DOI: 10.1063/1.4867498Google ScholarThere is no corresponding record for this reference.
- 26Hoang, H.; Ho, K. H.; Battani, A.; Scott, J. A.; Collell, J.; Pujol, M.; Galliero, G. Modeling solubility induced elemental fractionation of noble gases in oils. Geochim. Cosmochim. Acta 2025, 388, 127– 142, DOI: 10.1016/j.gca.2024.09.004Google ScholarThere is no corresponding record for this reference.
- 27Smith, F. T. Atomic distortion and the combining rule for repulsive potentials. Phys. Rev. A 1972, 5, 1708 DOI: 10.1103/PhysRevA.5.1708Google ScholarThere is no corresponding record for this reference.
- 28Waldman, M.; Hagler, A. T. New combining rules for rare gas van der Waals parameters. J. Comput. Chem. 1993, 14, 1077– 1084, DOI: 10.1002/jcc.540140909Google ScholarThere is no corresponding record for this reference.
- 29Dauber-Osguthorpe, P.; Hagler, A. T. Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there?. J. Comput.-Aided Mol. Des. 2019, 33, 133– 203, DOI: 10.1007/s10822-018-0111-4Google ScholarThere is no corresponding record for this reference.
- 30Kong, C. L. Combining rules for intermolecular potential parameters. II. Rules for the Lennard-Jones (12–6) potential and the Morse potential. J. Chem. Phys. 1973, 59, 2464– 2467, DOI: 10.1063/1.1680358Google ScholarThere is no corresponding record for this reference.
- 31Kong, C. L.; Chakrabarty, M. R. Combining rules for intermolecular potential parameters. III. Application to the Exp-6 potential. J. Phys. Chem. A 1973, 77, 2668– 2670, DOI: 10.1021/j100640a019Google ScholarThere is no corresponding record for this reference.
- 32Prausnitz, J. M.; Lichtenthaler, R. N.; De Azevedo, E. G. Molecular Thermodynamics of Fluid-Phase Equilibria; Pearson Education: London, 1998.Google ScholarThere is no corresponding record for this reference.
- 33Panagiotopoulos, A. Z. Monte Carlo methods for phase equilibria of fluids. J. Phys.: Condens. Matter 2000, 12, R25, DOI: 10.1088/0953-8984/12/3/201Google ScholarThere is no corresponding record for this reference.
- 34Shing, K. S.; Gubbins, K. E.; Lucas, K. Henry constants in non-ideal fluid mixtures: Computer simulation and theory. Mol. Phys. 1988, 65, 1235– 1252, DOI: 10.1080/00268978800101731Google ScholarThere is no corresponding record for this reference.
- 35Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 3rd ed.; Elsevier: Amsterdam, 2023.Google ScholarThere is no corresponding record for this reference.
- 36Widom, B. Some topics in the theory of fluids. J. Chem. Phys. 1963, 39, 2808– 2812, DOI: 10.1063/1.1734110Google ScholarThere is no corresponding record for this reference.
- 37Ungerer, P.; Tavitian, B.; Boutin, A. Applications of Molecular Simulation in the Oil and Gas Industry; Technip: Paris, 2005.Google ScholarThere is no corresponding record for this reference.
- 38Hogervorst, W. Transport and equilibrium properties of simple gases and forces between like and unlike atoms. Physica 1971, 51, 77– 89, DOI: 10.1016/0031-8914(71)90138-8Google ScholarThere is no corresponding record for this reference.
- 39Schouten, J. A.; Deerenberg, A.; Trappeniers, N. J. Vapour-liquid and gas-gas equilibria in simple systems: IV. The system argon-krypton. Physica A 1975, 81, 151– 160, DOI: 10.1016/0378-4371(75)90042-4Google ScholarThere is no corresponding record for this reference.
- 40Calado, J. C.; Chang, E.; Streett, W. B. Vapour-liquid equilibrium in the krypton-xenon system. Physica A 1983, 117, 127– 138, DOI: 10.1016/0378-4371(83)90025-0Google ScholarThere is no corresponding record for this reference.
- 41Dias, L. M. B.; Filipe, E. J.; McCabe, C.; Calado, J. C. Thermodynamics of liquid (xenon + methane) mixtures. J. Phys. Chem. B 2004, 108, 7377– 7381, DOI: 10.1021/jp037070nGoogle ScholarThere is no corresponding record for this reference.
- 42Clever, H. L. Krypton, Xenon, and Radon─Gas Solubilities; Elsevier: Oxford, UK, 1979b; Vol. 2.Google ScholarThere is no corresponding record for this reference.
- 43Reamer, H. H.; Sage, B. H.; Lacey, W. N. Phase equilibria in hydrocarbon systems. Volumetric and phase behavior of the methane-n-heptane system. Ind. Eng. Chem. Chem. Eng. Data Ser. 1956, 1, 29– 42, DOI: 10.1021/i460001a007Google ScholarThere is no corresponding record for this reference.
- 44Beaudoin, J. M.; Kohn, J. P. Multiphase and volumetric equilibria of the methane-n-decane binary system at temperatures between – 36° and 150°. J. Chem. Eng. Data 1967, 12, 189– 191, DOI: 10.1021/je60033a007Google ScholarThere is no corresponding record for this reference.
- 45Lin, Y. N.; Hwang, S. C.; Kobayashi, R. Vapor-liquid equilibrium of the methane-toluene system at low temperatures. J. Chem. Eng. Data 1978, 23, 231– 234, DOI: 10.1021/je60078a007Google ScholarThere is no corresponding record for this reference.
- 46Elbishlawi, M.; Spencer, J. R. Equilibrium relations of two methane-aromatic binary systems at 150°F. Ind. Eng. Chem. 1951, 43, 1811– 1815, DOI: 10.1021/ie50500a036Google ScholarThere is no corresponding record for this reference.
- 47Lin, H. M.; Sebastian, H. M.; Simnick, J. J.; Chao, K. C. Gas-liquid equilibrium in binary mixtures of methane with n-decane, benzene, and toluene. J. Chem. Eng. Data 1979, 24, 146– 149, DOI: 10.1021/je60081a004Google ScholarThere is no corresponding record for this reference.
- 48Sander, R.; Acree, W. E., Jr; Visscher, A. De.; Schwartz, S. E.; Wallington, T. J. Henry’s law constants (IUPAC Recommendations 2021). Pure Appl. Chem. 2022, 94, 71– 85, DOI: 10.1515/pac-2020-0302Google ScholarThere is no corresponding record for this reference.
- 49Clever, H. L. Helium and Neon─Gas Solubilities; Elsevier: Oxford, UK, 1979a; Vol. 1.Google ScholarThere is no corresponding record for this reference.
- 50Clever, H. L. Argon─Gas Solubilities; Elsevier: Oxford, UK, 1980; Vol. 4.Google ScholarThere is no corresponding record for this reference.
- 51Kalra, H.; Kubota, H.; Robinson, D. B.; Ng, H. J. Equilibrium phase properties of the carbon dioxide-n-heptane system. J. Chem. Eng. Data 1978, 23, 317– 321, DOI: 10.1021/je60079a016Google ScholarThere is no corresponding record for this reference.
- 52Reamer, H. H.; Sage, B. H. Phase equilibria in hydrocarbon systems. Volumetric and phase behavior of the n-decane–CO2 system. J. Chem. Eng. Data 1963, 8, 508– 513, DOI: 10.1021/je60019a010Google ScholarThere is no corresponding record for this reference.
- 53Nagarajan, N.; Robinson, R. L., Jr. Equilibrium phase compositions, phase densities, and interfacial tensions for carbon dioxide + hydrocarbon systems. 2. Carbon dioxide + n-decane. J. Chem. Eng. Data 1986, 31, 168– 171, DOI: 10.1021/je00044a012Google ScholarThere is no corresponding record for this reference.
- 54Fink, S. D.; Hershey, H. C. Modeling the vapor-liquid equilibria of 1,1,1-trichloroethane + carbon dioxide and toluene + carbon dioxide at 308, 323, and 353 K. Ind. Eng. Chem. Res. 1990, 29, 295– 306, DOI: 10.1021/ie00098a022Google ScholarThere is no corresponding record for this reference.
- 55Bazile, J.-P.; Nasri, D.; Hoang, H.; Galliero, G.; Daridon, J.-L. Density, speed of sound, compressibility and related excess properties of methane + n-heptane at T = 303.15 K and p = 10 to 70 MPa. Int. J. Thermophys. 2020, 41, 115 DOI: 10.1007/s10765-020-02694-9Google ScholarThere is no corresponding record for this reference.
- 56Regueira, T.; Pantelide, G.; Yan, W.; Stenby, E. H. Density and phase equilibrium of the binary system methane + n-decane under high temperatures and pressures. Fluid Phase Equilib. 2016, 428, 48– 61, DOI: 10.1016/j.fluid.2016.08.004Google ScholarThere is no corresponding record for this reference.
- 57Baylaucq, A.; Boned, C.; Canet, X.; Zéberg-Mikkelsen, C. K. High-pressure (up to 140 MPa) dynamic viscosity of the methane and toluene system: Measurements and comparative study of some representative models. Int. J. Thermophys. 2003, 24, 621– 638, DOI: 10.1023/A:1024023913165Google ScholarThere is no corresponding record for this reference.
- 58Bazile, J.-P.; Nasri, D.; Hamani, A. W. S.; Galliero, G.; Daridon, J.-L. Excess volume, isothermal compressibility, isentropic compressibility and speed of sound of carbon dioxide + n-heptane binary mixture under pressure up to 70 MPa. I. Experimental measurements. J. Supercrit. Fluids 2018, 140, 218– 232, DOI: 10.1016/j.supflu.2018.05.028Google ScholarThere is no corresponding record for this reference.
- 59Chacon Valero, A. M.; Feitosa, F. X.; de Sant’Ana, H. B. Density and volumetric behavior of binary CO2 + n-decane and ternary CO2 + n-decane + naphthalene systems at high pressure and high temperature. J. Chem. Eng. Data 2020, 65, 3499– 3509, DOI: 10.1021/acs.jced.0c00090Google ScholarThere is no corresponding record for this reference.
- 60Matsukawa, H.; Tsuji, T.; Otake, K. Measurement of the density of carbon dioxide/toluene homogeneous mixtures and correlation with equations of state. J. Chem. Thermodyn. 2022, 164, 106618 DOI: 10.1016/j.jct.2021.106618Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
Figure 1

Figure 1. Comparison of Henry’s law constants obtained from the NVT-GEMC simulations using different combining rules and experiment for seven binary mixtures for which the Mie (λ, 6) potential parameters of the components are relatively similar. (a) Argon/Krypton mixture. (b) Krypton/Xenon mixture. (c) Methane/Xenon mixture. (d) Krypton/n-Heptane mixture. (e) Methane/n-Heptane mixture. (f) Methane/n-Decane mixture. (g) Methane–Toluene mixture. Solid circles (red) correspond to the experimental data. Open squares (green) correspond to the simulation data by using the classical combining rules. Open deltas (blue) correspond to the simulation data using Lafitte et al. combining rules. Open right triangles (red) correspond to the simulation data by using the new combining rules. Lines serve as a guide to the eye for the experimental data.
Figure 2
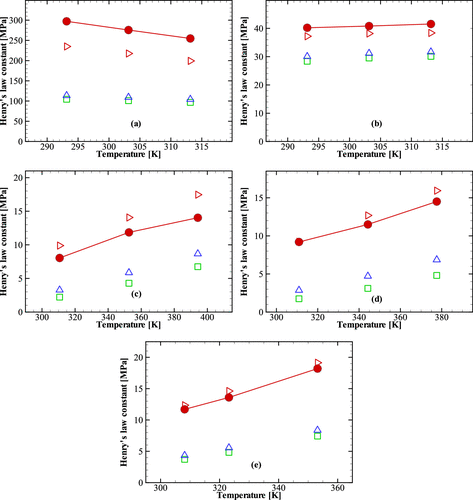
Figure 2. Comparison of Henry’s law constants obtained from the NVT-GEMC simulations using different combining rules and experiment for five binary mixtures for which the Mie (λ, 6) potential parameters of the components are significantly different. (a) Neon/n-heptane mixture. (b) Argon/n-heptane mixture. (c) Carbon dioxide/n-heptane mixture. (d) Carbon dioxide/n-decane mixture. (e) Carbon dioxide/toluene mixture. The legend is identical to that in Figure 1.
Figure 3
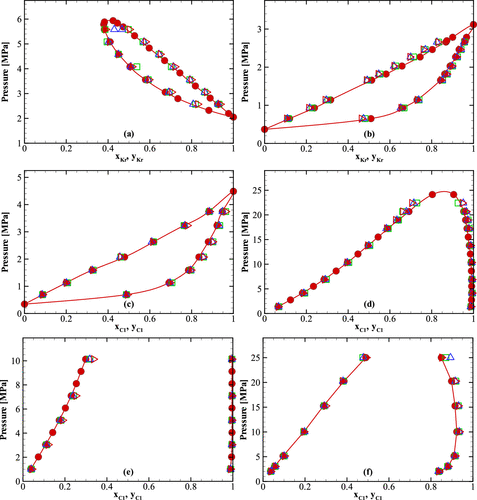
Figure 3. Comparison of pressure–composition phase diagrams of binary mixtures obtained from the NPT-GEMC simulations using different combining rules and experiment for six binary mixtures for which the Mie (λ, 6) potential parameters of the components are relatively similar. (a) Argon/krypton mixture at T = 177.38 K. (b) Krypton/xenon mixture at T = 190.03 K. (c) Methane/xenon mixture at T = 189.78 K. (d) Methane/n-heptane mixture at T = 310.93 K. (e) Methane/n-decane mixture at T = 373.15 K. (f) Methane/toluene mixture at T = 422.45 K. The legend is identical to that in Figure 1.
Figure 4
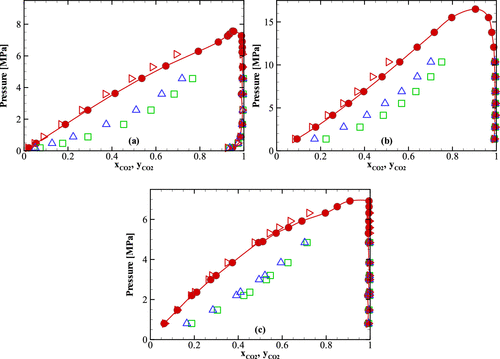
Figure 4. Comparison of pressure–composition phase diagrams of binary mixtures obtained from the NPT-GEMC simulations using different combining rules and experiment for three binary mixtures for which the Mie (λ, 6) potential parameters of the species are significantly different. (a) Carbon dioxide/n-heptane mixture at T = 310.65 K. (b) Carbon dioxide/n-decane mixture at T = 377.59 K. (c) Carbon dioxide/toluene mixture at T = 308.16 K. The legend is identical to that in Figure 1.
Figure 5

Figure 5. Comparison of excess molar volumes of binary mixtures obtained from the NPT-MC simulations using different combining rules and experiment for three binary mixtures for which the Mie (λ, 6) potential parameters of the components are relatively similar. (a) Methane/n-heptane mixture at T = 303.15 K and P = 30 MPa. (b) Methane/n-decane mixture at T = 373.15 K and P = 40 MPa. (c) Methane/toluene mixture at T = 333.15 K and P = 45 MPa. The legend is identical to that in Figure 1.
Figure 6

Figure 6. Comparison of excess molar volumes of binary mixtures obtained from the NPT-MC simulations using different combining rules and experiment for three binary mixtures for which the Mie (λ, 6) potential parameters of the components are significantly different. (a) Carbon dioxide/n-heptane mixture at T = 313.15 K and P = 10 MPa. (b) Carbon dioxide/n-decane mixture at T = 318.15 K and P = 15 MPa. (c) Carbon dioxide/toluene mixture at T = 313.15 K and P = 10 MPa. The legend is identical to that in Figure 1.
References
This article references 60 other publications.
- 1Dohrn, R.; Pfohl, O. Thermophysical properties─industrial directions. Fluid Phase Equilib. 2002, 194–197, 15– 29, DOI: 10.1016/S0378-3812(01)00791-9There is no corresponding record for this reference.
- 2Gupta, S.; Olson, J. D. Industrial needs in physical properties. Ind. Eng. Chem. Res. 2003, 42, 6359– 6374, DOI: 10.1021/ie030170vThere is no corresponding record for this reference.
- 3Hendriks, E.; Kontogeorgis, G. M.; Dohrn, R.; Hemptinne, J.-C. de.; Economou, I. G.; Žilnik, L. F.; Vesovic, V. Industrial requirements for thermodynamics and transport properties. Ind. Eng. Chem. Res. 2010, 49, 11131– 11141, DOI: 10.1021/ie101231bThere is no corresponding record for this reference.
- 4Kontogeorgis, G. M.; Dohrn, R.; Economou, I. G.; Hemptinne, J.-C. de.; Kate, A. Ten.; Kuitunen, S.; Mooijer, M.; Žilnik, L. F.; Vesovic, V. Industrial requirements for thermodynamic and transport properties: 2020. Ind. Eng. Chem. Res. 2021, 60, 4987– 5013, DOI: 10.1021/acs.iecr.0c05356There is no corresponding record for this reference.
- 5de Hemptinne, J.-C.; Kontogeorgis, G. M.; Dohrn, R.; Economou, I. G.; Ten Kate, A.; Kuitunen, S.; Fele Žilnik, L. F.; De Angelis, M. G.; Vesovic, V. A view on the future of applied thermodynamics. Ind. Eng. Chem. Res. 2022, 61, 14664– 14680, DOI: 10.1021/acs.iecr.2c01906There is no corresponding record for this reference.
- 6Gupta, S.; Elliott, J. R.; Anderko, A.; Crosthwaite, J.; Chapman, W. G.; Lira, C. T. Current practices and continuing needs in thermophysical properties for the chemical industry. Ind. Eng. Chem. Res. 2023, 62, 3394– 3427, DOI: 10.1021/acs.iecr.2c03153There is no corresponding record for this reference.
- 7Poling, B. E.; Prausnitz, M.; O’Connell, J. P. The Properties of Gases and Liquids; McGraw-Hill: New York, 2004.There is no corresponding record for this reference.
- 8Goodwin, A. R.; Sengers, J.; Peters, C. J. Applied Thermodynamics of Fluids; Royal Society of Chemistry: Cambridge, 2010.There is no corresponding record for this reference.
- 9Assael, M. J.; Goodwin, A. R. H.; Vesovic, V.; Wakeham, W. A. Experimental Thermodynamics Volume IX: Advances in Transport Properties of Fluids; Royal Society of Chemistry: London, 2014.There is no corresponding record for this reference.
- 10Tillotson, M. J.; Diamantonis, N. I.; Buda, C.; Bolton, L. W.; Müller, E. A. Molecular modelling of the thermophysical properties of fluids: expectations, limitations, gaps and opportunities. Phys. Chem. Chem. Phys. 2023, 25, 12607– 12628, DOI: 10.1039/D2CP05423JThere is no corresponding record for this reference.
- 11Mejía, A.; Herdes, C.; Müller, E. A. Force fields for coarse-grained molecular simulations from a corresponding states correlation. Ind. Eng. Chem. Res. 2014, 53, 4131– 4141, DOI: 10.1021/ie404247eThere is no corresponding record for this reference.
- 12Herdes, C.; Totton, T. S.; Müller, E. A. Coarse grained force field for the molecular simulation of natural gases and condensates. Fluid Phase Equilib. 2015, 406, 91– 100, DOI: 10.1016/j.fluid.2015.07.014There is no corresponding record for this reference.
- 13Mick, J. R.; Barhaghi, M. S.; Jackman, B.; Rushaidat, K.; Schwiebert, L.; Potoff, J. J. Optimized Mie potentials for phase equilibria: Application to noble gases and their mixtures with n-alkanes. J. Chem. Phys. 2015, 143, 114304 DOI: 10.1063/1.4930138There is no corresponding record for this reference.
- 14Hoang, H.; Delage-Santacreu, S.; Galliero, G. Simultaneous description of equilibrium, interfacial, and transport properties of fluids using a Mie chain coarse-grained force field. Ind. Eng. Chem. Res. 2017, 56, 9213– 9226, DOI: 10.1021/acs.iecr.7b01397There is no corresponding record for this reference.
- 15Jervell, V. G.; Wilhelmsen, Ø. Revised Enskog theory for Mie fluids: Prediction of diffusion coefficients, thermal diffusion coefficients, viscosities, and thermal conductivities. J. Chem. Phys. 2023, 158, 224103 DOI: 10.1063/5.0149865There is no corresponding record for this reference.
- 16Chaparro, G.; Müller, E. A. Simulation and data-driven modeling of the transport properties of the Mie fluid. J. Phys. Chem. B 2024, 128, 551– 566, DOI: 10.1021/acs.jpcb.3c06813There is no corresponding record for this reference.
- 17Ervik, Å.; Serratos, G. J.; Müller, E. A. raaSAFT: A framework enabling coarse-grained molecular dynamics simulations based on the SAFT-γ Mie force field. Comput. Phys. Commun. 2017, 212, 161– 179, DOI: 10.1016/j.cpc.2016.07.035There is no corresponding record for this reference.
- 18Calvin, D. W.; Reed III, T. M. Mixture rules for the Mie (n, 6) intermolecular pair potential and the Dymond–Alder pair potential. J. Chem. Phys. 1971, 54, 3733– 3738, DOI: 10.1063/1.1675422There is no corresponding record for this reference.
- 19Lafitte, T.; Apostolakou, A.; Avendaño, C.; Galindo, A.; Adjiman, C. S.; Müller, E. A.; Jackson, G. Accurate statistical associating fluid theory for chain molecules formed from Mie segments. J. Chem. Phys. 2013, 139, 154504 DOI: 10.1063/1.4819786There is no corresponding record for this reference.
- 20Lorentz, H. A. Ueber die Anwendung des Satzes vom Virial in der kinetischen Theorie der Gase. Ann. Phys. 1881, 248, 127– 136, DOI: 10.1002/andp.18812480110There is no corresponding record for this reference.
- 21Berthelot, D. Sur le mélange des gaz. Compt. Rendus 1898, 126, 15There is no corresponding record for this reference.
- 22Hamani, A. W. S.; Bazile, J. P.; Hoang, H.; Luc, H. T.; Daridon, J. L.; Galliero, G. Thermophysical properties of simple molecular liquid mixtures: On the limitations of some force fields. J. Mol. Liq. 2020, 303, 112663 DOI: 10.1016/j.molliq.2020.112663There is no corresponding record for this reference.
- 23Hamani, A. W. S.; Hoang, H.; Viet, T. Q. Q.; Daridon, J. L.; Galliero, G. Excess volume, isothermal compressibility, isentropic compressibility and speed of sound of carbon dioxide+ n-heptane binary mixture under pressure up to 70 MPa. II. Molecular simulations. J. Supercrit. Fluids 2020, 164, 104890 DOI: 10.1016/j.supflu.2020.104890There is no corresponding record for this reference.
- 24Delhommelle, J.; Millié, P. Inadequacy of the Lorentz–Berthelot combining rules for accurate predictions of equilibrium properties by molecular simulation. Mol. Phys. 2001, 99, 619– 625, DOI: 10.1080/00268970010020041There is no corresponding record for this reference.
- 25Desgranges, C.; Delhommelle, J. Evaluation of the grand-canonical partition function using expanded Wang–Landau simulations. III. Impact of combining rules on mixtures properties. J. Chem. Phys. 2014, 140, 104109 DOI: 10.1063/1.4867498There is no corresponding record for this reference.
- 26Hoang, H.; Ho, K. H.; Battani, A.; Scott, J. A.; Collell, J.; Pujol, M.; Galliero, G. Modeling solubility induced elemental fractionation of noble gases in oils. Geochim. Cosmochim. Acta 2025, 388, 127– 142, DOI: 10.1016/j.gca.2024.09.004There is no corresponding record for this reference.
- 27Smith, F. T. Atomic distortion and the combining rule for repulsive potentials. Phys. Rev. A 1972, 5, 1708 DOI: 10.1103/PhysRevA.5.1708There is no corresponding record for this reference.
- 28Waldman, M.; Hagler, A. T. New combining rules for rare gas van der Waals parameters. J. Comput. Chem. 1993, 14, 1077– 1084, DOI: 10.1002/jcc.540140909There is no corresponding record for this reference.
- 29Dauber-Osguthorpe, P.; Hagler, A. T. Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there?. J. Comput.-Aided Mol. Des. 2019, 33, 133– 203, DOI: 10.1007/s10822-018-0111-4There is no corresponding record for this reference.
- 30Kong, C. L. Combining rules for intermolecular potential parameters. II. Rules for the Lennard-Jones (12–6) potential and the Morse potential. J. Chem. Phys. 1973, 59, 2464– 2467, DOI: 10.1063/1.1680358There is no corresponding record for this reference.
- 31Kong, C. L.; Chakrabarty, M. R. Combining rules for intermolecular potential parameters. III. Application to the Exp-6 potential. J. Phys. Chem. A 1973, 77, 2668– 2670, DOI: 10.1021/j100640a019There is no corresponding record for this reference.
- 32Prausnitz, J. M.; Lichtenthaler, R. N.; De Azevedo, E. G. Molecular Thermodynamics of Fluid-Phase Equilibria; Pearson Education: London, 1998.There is no corresponding record for this reference.
- 33Panagiotopoulos, A. Z. Monte Carlo methods for phase equilibria of fluids. J. Phys.: Condens. Matter 2000, 12, R25, DOI: 10.1088/0953-8984/12/3/201There is no corresponding record for this reference.
- 34Shing, K. S.; Gubbins, K. E.; Lucas, K. Henry constants in non-ideal fluid mixtures: Computer simulation and theory. Mol. Phys. 1988, 65, 1235– 1252, DOI: 10.1080/00268978800101731There is no corresponding record for this reference.
- 35Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 3rd ed.; Elsevier: Amsterdam, 2023.There is no corresponding record for this reference.
- 36Widom, B. Some topics in the theory of fluids. J. Chem. Phys. 1963, 39, 2808– 2812, DOI: 10.1063/1.1734110There is no corresponding record for this reference.
- 37Ungerer, P.; Tavitian, B.; Boutin, A. Applications of Molecular Simulation in the Oil and Gas Industry; Technip: Paris, 2005.There is no corresponding record for this reference.
- 38Hogervorst, W. Transport and equilibrium properties of simple gases and forces between like and unlike atoms. Physica 1971, 51, 77– 89, DOI: 10.1016/0031-8914(71)90138-8There is no corresponding record for this reference.
- 39Schouten, J. A.; Deerenberg, A.; Trappeniers, N. J. Vapour-liquid and gas-gas equilibria in simple systems: IV. The system argon-krypton. Physica A 1975, 81, 151– 160, DOI: 10.1016/0378-4371(75)90042-4There is no corresponding record for this reference.
- 40Calado, J. C.; Chang, E.; Streett, W. B. Vapour-liquid equilibrium in the krypton-xenon system. Physica A 1983, 117, 127– 138, DOI: 10.1016/0378-4371(83)90025-0There is no corresponding record for this reference.
- 41Dias, L. M. B.; Filipe, E. J.; McCabe, C.; Calado, J. C. Thermodynamics of liquid (xenon + methane) mixtures. J. Phys. Chem. B 2004, 108, 7377– 7381, DOI: 10.1021/jp037070nThere is no corresponding record for this reference.
- 42Clever, H. L. Krypton, Xenon, and Radon─Gas Solubilities; Elsevier: Oxford, UK, 1979b; Vol. 2.There is no corresponding record for this reference.
- 43Reamer, H. H.; Sage, B. H.; Lacey, W. N. Phase equilibria in hydrocarbon systems. Volumetric and phase behavior of the methane-n-heptane system. Ind. Eng. Chem. Chem. Eng. Data Ser. 1956, 1, 29– 42, DOI: 10.1021/i460001a007There is no corresponding record for this reference.
- 44Beaudoin, J. M.; Kohn, J. P. Multiphase and volumetric equilibria of the methane-n-decane binary system at temperatures between – 36° and 150°. J. Chem. Eng. Data 1967, 12, 189– 191, DOI: 10.1021/je60033a007There is no corresponding record for this reference.
- 45Lin, Y. N.; Hwang, S. C.; Kobayashi, R. Vapor-liquid equilibrium of the methane-toluene system at low temperatures. J. Chem. Eng. Data 1978, 23, 231– 234, DOI: 10.1021/je60078a007There is no corresponding record for this reference.
- 46Elbishlawi, M.; Spencer, J. R. Equilibrium relations of two methane-aromatic binary systems at 150°F. Ind. Eng. Chem. 1951, 43, 1811– 1815, DOI: 10.1021/ie50500a036There is no corresponding record for this reference.
- 47Lin, H. M.; Sebastian, H. M.; Simnick, J. J.; Chao, K. C. Gas-liquid equilibrium in binary mixtures of methane with n-decane, benzene, and toluene. J. Chem. Eng. Data 1979, 24, 146– 149, DOI: 10.1021/je60081a004There is no corresponding record for this reference.
- 48Sander, R.; Acree, W. E., Jr; Visscher, A. De.; Schwartz, S. E.; Wallington, T. J. Henry’s law constants (IUPAC Recommendations 2021). Pure Appl. Chem. 2022, 94, 71– 85, DOI: 10.1515/pac-2020-0302There is no corresponding record for this reference.
- 49Clever, H. L. Helium and Neon─Gas Solubilities; Elsevier: Oxford, UK, 1979a; Vol. 1.There is no corresponding record for this reference.
- 50Clever, H. L. Argon─Gas Solubilities; Elsevier: Oxford, UK, 1980; Vol. 4.There is no corresponding record for this reference.
- 51Kalra, H.; Kubota, H.; Robinson, D. B.; Ng, H. J. Equilibrium phase properties of the carbon dioxide-n-heptane system. J. Chem. Eng. Data 1978, 23, 317– 321, DOI: 10.1021/je60079a016There is no corresponding record for this reference.
- 52Reamer, H. H.; Sage, B. H. Phase equilibria in hydrocarbon systems. Volumetric and phase behavior of the n-decane–CO2 system. J. Chem. Eng. Data 1963, 8, 508– 513, DOI: 10.1021/je60019a010There is no corresponding record for this reference.
- 53Nagarajan, N.; Robinson, R. L., Jr. Equilibrium phase compositions, phase densities, and interfacial tensions for carbon dioxide + hydrocarbon systems. 2. Carbon dioxide + n-decane. J. Chem. Eng. Data 1986, 31, 168– 171, DOI: 10.1021/je00044a012There is no corresponding record for this reference.
- 54Fink, S. D.; Hershey, H. C. Modeling the vapor-liquid equilibria of 1,1,1-trichloroethane + carbon dioxide and toluene + carbon dioxide at 308, 323, and 353 K. Ind. Eng. Chem. Res. 1990, 29, 295– 306, DOI: 10.1021/ie00098a022There is no corresponding record for this reference.
- 55Bazile, J.-P.; Nasri, D.; Hoang, H.; Galliero, G.; Daridon, J.-L. Density, speed of sound, compressibility and related excess properties of methane + n-heptane at T = 303.15 K and p = 10 to 70 MPa. Int. J. Thermophys. 2020, 41, 115 DOI: 10.1007/s10765-020-02694-9There is no corresponding record for this reference.
- 56Regueira, T.; Pantelide, G.; Yan, W.; Stenby, E. H. Density and phase equilibrium of the binary system methane + n-decane under high temperatures and pressures. Fluid Phase Equilib. 2016, 428, 48– 61, DOI: 10.1016/j.fluid.2016.08.004There is no corresponding record for this reference.
- 57Baylaucq, A.; Boned, C.; Canet, X.; Zéberg-Mikkelsen, C. K. High-pressure (up to 140 MPa) dynamic viscosity of the methane and toluene system: Measurements and comparative study of some representative models. Int. J. Thermophys. 2003, 24, 621– 638, DOI: 10.1023/A:1024023913165There is no corresponding record for this reference.
- 58Bazile, J.-P.; Nasri, D.; Hamani, A. W. S.; Galliero, G.; Daridon, J.-L. Excess volume, isothermal compressibility, isentropic compressibility and speed of sound of carbon dioxide + n-heptane binary mixture under pressure up to 70 MPa. I. Experimental measurements. J. Supercrit. Fluids 2018, 140, 218– 232, DOI: 10.1016/j.supflu.2018.05.028There is no corresponding record for this reference.
- 59Chacon Valero, A. M.; Feitosa, F. X.; de Sant’Ana, H. B. Density and volumetric behavior of binary CO2 + n-decane and ternary CO2 + n-decane + naphthalene systems at high pressure and high temperature. J. Chem. Eng. Data 2020, 65, 3499– 3509, DOI: 10.1021/acs.jced.0c00090There is no corresponding record for this reference.
- 60Matsukawa, H.; Tsuji, T.; Otake, K. Measurement of the density of carbon dioxide/toluene homogeneous mixtures and correlation with equations of state. J. Chem. Thermodyn. 2022, 164, 106618 DOI: 10.1016/j.jct.2021.106618There is no corresponding record for this reference.