



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Decoding the Structural Controversy of Li- and Mn-Rich Cathodes through Synthesis-Dependent Domain EvolutionClick to copy article linkArticle link copied!
- Minji KimMinji KimDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of KoreaMore by Minji Kim
- Dohyun KimDohyun KimDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of KoreaMore by Dohyun Kim
- Hyeona JoHyeona JoDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of KoreaMore by Hyeona Jo
- Keun Chan YooKeun Chan YooDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of KoreaMore by Keun Chan Yoo
- Hyeokjun Park*Hyeokjun Park*Email: [email protected]Department of Materials Science and Engineering, Korea University, Seoul 02841, Republic of KoreaMore by Hyeokjun Park
Abstract
Li- and Mn-rich layered oxides (LMR) have attracted significant research interest because of their potential merits of low cost and high capacity that exceed current Ni-rich cathodes. Despite nearly two decades of intensive investigations, however, the structural nature of LMRs, particularly with respect to Li2MnO3-like domains has remained under debate. Early structure models described LMRs as an intergrowth of monoclinic Li2MnO3 (C2/m) and rhombohedral LiTMO2 (R3̅m) phases. In contrast, subsequent studies revealed that these structural signatures can also arise from local Li–Mn ordering within a single solid-solution framework. These conflicting interpretations have complicated a clear understanding of the structure–property relationships and impeded progress toward the commercial implementation of LMRs. In this perspective, we present a unified view showing that the structural state of LMRs is determined not solely by bulk composition but critically by synthesis conditions. By integrating evidence related to precursor chemistry and calcination parameters, we elaborate on how cation homogeneity, the distribution of excess Li, Mn oxidation states, and diffusion kinetics collectively dictate whether Li2MnO3 forms extended slabs or nanoscale, delocalized structural motifs. Through a comparative assessment of synthesis routes, we review how LMRs span a continuous structural spectrum rather than conforming to a simple binary phase model. Finally, we outline synthesis-driven design principles for controlling the evolution of Li2MnO3 domains, providing practical guidelines to develop structurally robust, high-energy LMR cathodes.
This publication is licensed for personal use by The American Chemical Society.
Introduction
Main
Brief Historical Overview of the Structural Controversy in LMRs
Figure 1
Figure 1. Structural signatures and diagnostic characterization of Li2MnO3 domains in Li- and Mn-rich layered oxides. (a) Schematic illustration comparing the two structural interpretations of Li-rich layered oxides: a two-phase intergrowth model consisting of monoclinic Li2MnO3 (C2/m) and rhombohedral LiTMO2 (R3̅m) domains, and a solid-solution model in which Li–Mn ordering is embedded as local motifs within a single layered lattice. (b) Powder XRD patterns for Li-rich layered oxides with increasing Li2MnO3 content, showing the emergence and growth of monoclinic superstructure reflections. Reproduced from ref (22) under CC BY-NC-ND 4.0 license. Copyright 2019, The Electrochemical Society. (c) HAADF-STEM images and simulated atomic configurations demonstrating Li/Mn honeycomb ordering and the contrast differences between Li columns and TM columns, used to distinguish Li2MnO3-like local ordering from fully random TM distributions. Reproduced with permission from ref (23). Copyright 2012, American Chemical Society.
Figure 2
Figure 2. Chronological evolution of structural interpretations of Li2MnO3domain in LMRs. (a) 6Li MAS NMR spectra of Li2MnO3 samples prepared at 1000 °C (blue) and 850 °C (black). Reproduced with permission from ref (25). Copyright 2005, Elsevier. (b) Convergent-beam electron diffraction (CBED) patterns comparing LiMn0.5Ni0.5O2 and Li1.048[Mn0.333Ni0.333Co0.333]0.952O2. Reproduced with permission from ref (26). Copyright 2006, Elsevier. (c) HAADF-STEM imaging identifying localized excess-Li regions responsible for Li2MnO3-like contrast in LMR particles. Reproduced with permission from ref (17). Copyright 2020, Wiley. (d) X-ray diffraction superlattice peaks (20–25°) comparing DS-LMR (delocalized excess-Li) and LS-LMR (localized excess-Li). Reproduced with permission from ref (18). Copyright 2021, Wiley. (e) Scanning electron microscopy and XRD analysis correlating superstructure evolution and stacking faults with precursor-dependent synthesis pathways (Solid state and Sol–gel method). Reproduced from ref (21) under CC BY 4.0 license. Copyright 2021, American Chemical Society. (f) In situ STEM tracking of sol–gel precursors during heating, revealing atomic-level TM mixing. Reproduced with permission from ref (27). Copyright 2022, Elsevier.
Synthesis-Driven Transformation of Li2MnO3-Like Domains
Figure 3
Figure 3. Synthesis-dependent evolution of Li2MnO3-like domains in LMRs. (a) STEM–EDS elemental maps comparing cathodes prepared from solid state, and sol–gel precursors. Reproduced from ref (21) under CC BY 4.0 license. Copyright 2021, American Chemical Society. (b) Schematic illustration of the different synthesis route and FAULTS-refined XRD patterns of LMR samples annealed precursor at 900 °C for 3 h prepared by SG (Sol–gel) and CP (Coprecipitation) method. Reproduced from ref (28) under CC BY 3.0 license. Copyright 2022, Royal Society of Chemistry. (c) In situ HT XRD patterns for carbonate-derived (CO3-LRLO) and hydroxide-derived (OH-LRLO) Li-rich layered oxides. Reproduced with permission from ref (29). Copyright 2025, Wiley. (d) In situ Raman spectra showing distinct phase evolution pathways during calcination: lithium carbonate precursors rapidly develop C2/m Li2MnO3-like modes, whereas lithium hydroxide precursors progress through spinel-like intermediates prior to layered ordering. Reproduced with permission from ref (30). Copyright 2024, Royal Society of Chemistry. (e) EDS mapping of samples heated at low-temperature (∼700 °C, left) and high-temperature (∼1100 °C, right), showing the transition from well-defined Li2MnO3 domains to fused, single-solution-like lattices at elevated temperatures. Reproduced with permission from ref (19). Copyright 2024, Royal Society of Chemistry. (f) Effect of oxygen partial pressure during calcination, where high pO2 promotes extended Li2MnO3-like ordering and faulted monoclinic slabs, while reduced pO2 collapses ordering and yields more homogeneous monoclinic solid-solutions. Reproduced with permission from ref (20). Copyright 2023, American Chemical Society.
Influence of Synthesis-Driven Li2MnO3 Domains on Electrochemical Behavior
Figure 4
Figure 4. Synthesis-dependent electrochemical manifestations associated with Li2MnO3 domain regulation in LMR cathodes. (a) First-cycle charge–discharge profiles of LMNCO cathodes synthesized via sol–gel (SG) and solid state (SS) precursor routes. Reproduced from ref (21) under CC BY 4.0 license. Copyright 2021, American Chemical Society. (b) Charge–discharge curves and corresponding dQ/dV profiles of carbonate-derived (CO3-LRLO) and hydroxide-derived (OH-LRLO) Li-rich layered oxides collected at selected cycles (1st–300th) at 25 °C within a voltage window of 2.0–4.8 V. Reproduced with permission from ref (29). Copyright 2025, Wiley. (c) Voltage–capacity profiles of LLO cathodes prepared with lithium carbonate (LLO-LC) and lithium hydroxide monohydrate (LLO-LHM) during cycling following rate capability tests. Reproduced with permission from ref (30). Copyright 2024, Royal Society of Chemistry. (d) Differential capacity (dQ/dV) plots of LS-LMR and DS-LMR cathodes at the 1st, 50th, and 100th cycles. Reproduced with permission from ref (18). Copyright 2021, Wiley. (e) Voltage–capacity profiles of LMR cathodes calcined at 750 and 1000 °C, respectively, together with their cycling performance evaluated in the voltage range of 2.0–4.5 V after electrochemical activation. Reproduced with permission from ref (19). Copyright 2024, Royal Society of Chemistry. (f) Electrochemical behavior of LNMO-X cathodes calcined under different oxygen partial pressures (X = 1, 0.1, and 0.01) and air atmosphere (X = 20). Reproduced with permission from ref (20). Copyright 2023, American Chemical Society.
Conclusions and Outlook
Figure 5
Figure 5. Schematic illustration of synthesis-driven control over Li2MnO3 domain structures in LMRs.
Author Information
- Hyeokjun Park - Department of Materials Science and Engineering, Korea University, Seoul 02841, Republic of Korea;
 https://orcid.org/0000-0003-3367-8881;
https://orcid.org/0000-0003-3367-8881;
- Minji Kim - Department of Materials Science and Engineering, Korea University, Seoul 02841, Republic of Korea;https://orcid.org/0000-0002-9136-0866
M.K., D.K., H.J., K.C.Y., and H.P. conceived the idea and wrote the manuscript. H.P. supervised the work. All authors approved the final version of the manuscript.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT; Nos. RS-2021-NR062019).
References
This article references 36 other publications.
- 1Nitta, N.; Wu, F.; Lee, J. T.; Yushin, G. Li-Ion Battery Materials: Present and Future. Mater. Today 2015, 18 (5), 252– 264, DOI: 10.1016/j.mattod.2014.10.040Google ScholarThere is no corresponding record for this reference.
- 2Grey, C. P.; Tarascon, J. M. Sustainability and in Situ Monitoring in Battery Development. Nat. Mater. 2017, 16 (1), 45– 56, DOI: 10.1038/nmat4777Google ScholarThere is no corresponding record for this reference.
- 3Dixit, M.; Witherspoon, B.; Muralidharan, N.; Mench, M. M.; Kweon, C.-B. M.; Sun, Y.-K.; Belharouak, I. Insights into the Critical Materials Supply Chain of the Battery Market for Enhanced Energy Security. ACS Energy Lett. 2024, 9 (8), 3780– 3789, DOI: 10.1021/acsenergylett.4c01300Google ScholarThere is no corresponding record for this reference.
- 4Thackeray, M. M.; Kang, S.-H.; Johnson, C. S.; Vaughey, J. T.; Benedek, R.; Hackney, S. A. Li2MnO3-Stabilized LiMO2 (M = Mn, Ni, Co) Electrodes for Lithium-Ion Batteries. J. Mater. Chem. 2007, 17 (30), 3112, DOI: 10.1039/b702425hGoogle ScholarThere is no corresponding record for this reference.
- 5Zhao, J.; Su, Y.; Dong, J.; Shi, Q.; Lu, Y.; Li, N.; Wang, H.; Fang, Y.; Li, W.; Hao, J.; Wu, Y.; Qi, Q.; Wu, F.; Chen, L. Regulating Anionic Redox Reversibility in Li-Rich Layered Cathodes via Diffusion-Induced Entropy-Assisted Surface Engineering. Energy Storage Materials 2024, 70, 103550 DOI: 10.1016/j.ensm.2024.103550Google ScholarThere is no corresponding record for this reference.
- 6Assat, G.; Tarascon, J.-M. Fundamental Understanding and Practical Challenges of Anionic Redox Activity in Li-Ion Batteries. Nat. Energy 2018, 3 (5), 373– 386, DOI: 10.1038/s41560-018-0097-0Google ScholarThere is no corresponding record for this reference.
- 7Assat, G.; Delacourt, C.; Corte, D. A. D.; Tarascon, J.-M. Editors’ Choice─Practical Assessment of Anionic Redox in Li-Rich Layered Oxide Cathodes: A Mixed Blessing for High Energy Li-Ion Batteries. J. Electrochem. Soc. 2016, 163 (14), A2965– A2976, DOI: 10.1149/2.0531614jesGoogle ScholarThere is no corresponding record for this reference.
- 8Gallagher, K. G.; Croy, J. R.; Balasubramanian, M.; Bettge, M.; Abraham, D. P.; Burrell, A. K.; Thackeray, M. M. Correlating Hysteresis and Voltage Fade in Lithium- and Manganese-Rich Layered Transition-Metal Oxide Electrodes. Electrochem. Commun. 2013, 33, 96– 98, DOI: 10.1016/j.elecom.2013.04.022Google ScholarThere is no corresponding record for this reference.
- 9Assat, G.; Foix, D.; Delacourt, C.; Iadecola, A.; Dedryvère, R.; Tarascon, J.-M. Fundamental Interplay between Anionic/Cationic Redox Governing the Kinetics and Thermodynamics of Lithium-Rich Cathodes. Nat. Commun. 2017, 8 (1), 2219, DOI: 10.1038/s41467-017-02291-9Google ScholarThere is no corresponding record for this reference.
- 10Croy, J. R.; Gallagher, K. G.; Balasubramanian, M.; Chen, Z.; Ren, Y.; Kim, D.; Kang, S.-H.; Dees, D. W.; Thackeray, M. M. Examining Hysteresis in Composite x Li2 MnO3 ·(1– x)LiMO2 Cathode Structures. J. Phys. Chem. C 2013, 117 (13), 6525– 6536, DOI: 10.1021/jp312658qGoogle ScholarThere is no corresponding record for this reference.
- 11Armstrong, A. R.; Holzapfel, M.; Novák, P.; Johnson, C. S.; Kang, S.-H.; Thackeray, M. M.; Bruce, P. G. Demonstrating Oxygen Loss and Associated Structural Reorganization in the Lithium Battery Cathode Li[Ni0.2 Li0.2 Mn0.6 ]O2. J. Am. Chem. Soc. 2006, 128 (26), 8694– 8698, DOI: 10.1021/ja062027+Google ScholarThere is no corresponding record for this reference.
- 12Zhao, J.; Su, Y.; Dong, J.; Hao, J.; Che, H.; Lu, Y.; Li, N.; Zhang, B.; Zhang, P.; Wu, F.; Chen, L. Boosting Reaction Homogeneity through Reverse Gradient Fluorination for High-Performance Li-Rich Layered Cathodes. Energy Storage Materials 2025, 76, 104137 DOI: 10.1016/j.ensm.2025.104137Google ScholarThere is no corresponding record for this reference.
- 13McCalla, E.; Abakumov, A. M.; Saubanère, M.; Foix, D.; Berg, E. J.; Rousse, G.; Doublet, M.-L.; Gonbeau, D.; Novák, P.; Van Tendeloo, G.; Dominko, R.; Tarascon, J.-M. Visualization of O-O Peroxo-like Dimers in High-Capacity Layered Oxides for Li-Ion Batteries. Science 2015, 350 (6267), 1516– 1521, DOI: 10.1126/science.aac8260Google ScholarThere is no corresponding record for this reference.
- 14Singer, A.; Zhang, M.; Hy, S.; Cela, D.; Fang, C.; Wynn, T. A.; Qiu, B.; Xia, Y.; Liu, Z.; Ulvestad, A.; Hua, N.; Wingert, J.; Liu, H.; Sprung, M.; Zozulya, A. V.; Maxey, E.; Harder, R.; Meng, Y. S.; Shpyrko, O. G. Nucleation of Dislocations and Their Dynamics in Layered Oxide Cathode Materials during Battery Charging. Nat. Energy 2018, 3 (8), 641– 647, DOI: 10.1038/s41560-018-0184-2Google ScholarThere is no corresponding record for this reference.
- 15Kang, S.; Choi, D.; Lee, H.; Choi, B.; Kang, Y. A Mechanistic Insight into the Oxygen Redox of Li-Rich Layered Cathodes and Their Related Electronic/Atomic Behaviors Upon Cycling. Adv. Mater. 2023, 35 (43), 2211965 DOI: 10.1002/adma.202211965Google ScholarThere is no corresponding record for this reference.
- 16Bareño, J.; Lei, C. H.; Wen, J. G.; Kang, S.-H.; Petrov, I.; Abraham, D. P. Local Structure of Layered Oxide Electrode Materials for Lithium-Ion Batteries. Adv. Mater. 2010, 22 (10), 1122– 1127, DOI: 10.1002/adma.200904247Google ScholarThere is no corresponding record for this reference.
- 17Hwang, J.; Myeong, S.; Jin, W.; Jang, H.; Nam, G.; Yoon, M.; Kim, S. H.; Joo, S. H.; Kwak, S. K.; Kim, M. G.; Cho, J. Excess-Li Localization Triggers Chemical Irreversibility in Li- and Mn-Rich Layered Oxides. Adv. Mater. 2020, 32 (34), 2001944 DOI: 10.1002/adma.202001944Google ScholarThere is no corresponding record for this reference.
- 18Hwang, J.; Myeong, S.; Lee, E.; Jang, H.; Yoon, M.; Cha, H.; Sung, J.; Kim, M. G.; Seo, D.; Cho, J. Lattice-Oxygen-Stabilized Li- and Mn-Rich Cathodes with Sub-Micrometer Particles by Modifying the Excess-Li Distribution. Adv. Mater. 2021, 33 (18), 2100352 DOI: 10.1002/adma.202100352Google ScholarThere is no corresponding record for this reference.
- 19Xu, S.; Chen, Z.; Zhao, W.; Ren, W.; Hou, C.; Liu, J.; Wang, W.; Yin, C.; Tan, X.; Lou, X.; Yao, X.; Gao, Z.; Liu, H.; Wang, L.; Yin, Z.; Qiu, B.; Hu, B.; Li, T.; Dong, C.; Pan, F.; Zhang, M. Multi-Angle Tracking Synthetic Kinetics of Phase Evolution in Li-Rich Mn-Based Cathodes. Energy Environ. Sci. 2024, 17 (11), 3807– 3818, DOI: 10.1039/D3EE04199AGoogle ScholarThere is no corresponding record for this reference.
- 20Li, J.; Li, W.; Zhang, C.; Han, C.; Chen, X.; Zhao, H.; Xu, H.; Jia, G.; Li, Z.; Li, J.; Zhang, Y.; Guo, X.; Gao, F.; Liu, J.; Qiu, X. Tuning Li2 MnO3 -Like Domain Size and Surface Structure Enables Highly Stabilized Li-Rich Layered Oxide Cathodes. ACS Nano 2023, 17 (17), 16827– 16839, DOI: 10.1021/acsnano.3c03666Google ScholarThere is no corresponding record for this reference.
- 21Menon, A. S.; Ulusoy, S.; Ojwang, D. O.; Riekehr, L.; Didier, C.; Peterson, V. K.; Salazar-Alvarez, G.; Svedlindh, P.; Edström, K.; Gomez, C. P.; Brant, W. R. Synthetic Pathway Determines the Nonequilibrium Crystallography of Li- and Mn-Rich Layered Oxide Cathode Materials. ACS Appl. Energy Mater. 2021, 4 (2), 1924– 1935, DOI: 10.1021/acsaem.0c03027Google ScholarThere is no corresponding record for this reference.
- 22Redel, K.; Kulka, A.; Plewa, A.; Molenda, J. High-Performance Li-Rich Layered Transition Metal Oxide Cathode Materials for Li-Ion Batteries. J. Electrochem. Soc. 2019, 166 (3), A5333– A5342, DOI: 10.1149/2.0511903jesGoogle ScholarThere is no corresponding record for this reference.
- 23Boulineau, A.; Simonin, L.; Colin, J.-F.; Canévet, E.; Daniel, L.; Patoux, S. Evolutions of Li1.2 Mn0.61 Ni0.18 Mg0.01 O2 during the Initial Charge/Discharge Cycle Studied by Advanced Electron Microscopy. Chem. Mater. 2012, 24 (18), 3558– 3566, DOI: 10.1021/cm301140gGoogle ScholarThere is no corresponding record for this reference.
- 24Jarvis, K.; Deng, Z.; Manthiram, A.; Ferreira, P. Understanding the Role of Lithium Content on the Structure and Capacity of Lithium-Rich Layered Oxides by Aberration-Corrected STEM, D-STEM, and EDS. Microsc Microanal 2012, 18 (S2), 1484– 1485, DOI: 10.1017/S1431927612009270Google ScholarThere is no corresponding record for this reference.
- 25Bréger, J.; Jiang, M.; Dupré, N.; Meng, Y. S.; Shao-Horn, Y.; Ceder, G.; Grey, C. P. High-Resolution X-Ray Diffraction, DIFFaX, NMR and First Principles Study of Disorder in the Li2MnO3–Li[Ni1/2Mn1/2]O2 Solid Solution. J. Solid State Chem. 2005, 178 (9), 2575– 2585, DOI: 10.1016/j.jssc.2005.05.027Google ScholarThere is no corresponding record for this reference.
- 26Thackeray, M. M.; Kang, S.-H.; Johnson, C. S.; Vaughey, J. T.; Hackney, S. A. Comments on the Structural Complexity of Lithium-Rich Li1+xM1–xO2 Electrodes (M = Mn, Ni, Co) for Lithium Batteries. Electrochem. Commun. 2006, 8 (9), 1531– 1538, DOI: 10.1016/j.elecom.2006.06.030Google ScholarThere is no corresponding record for this reference.
- 27Li, Y.; Xu, S.; Zhao, W.; Chen, Z.; Chen, Z.; Li, S.; Hu, J.; Cao, B.; Li, J.; Zheng, S.; Chen, Z.; Zhang, T.; Zhang, M.; Pan, F. Correlating the Dispersion of Li@Mn6 Superstructure Units with the Oxygen Activation in Li-Rich Layered Cathode. Energy Storage Materials 2022, 45, 422– 431, DOI: 10.1016/j.ensm.2021.12.003Google ScholarThere is no corresponding record for this reference.
- 28Menon, A. S.; Khalil, S.; Ojwang, D. O.; Edström, K.; Gomez, C. P.; Brant, W. R. Synthesis–Structure Relationships in Li- and Mn-Rich Layered Oxides: Phase Evolution, Superstructure Ordering and Stacking Faults. Dalton Trans. 2022, 51 (11), 4435– 4446, DOI: 10.1039/D2DT00104GGoogle ScholarThere is no corresponding record for this reference.
- 29Huang, Y.; Li, C.; Zhang, K.; Tang, Y.; Tu, W.; Tian, Y.; Wang, J.; Yan, Y.; Chen, Y.; Zou, Y.; Li, L.; Zhang, B.; Bao, J.; Ding, C.; Wang, Y.; Qiu, T.; Sun, X.; Qiao, Y.; Sun, S. How Does the Precursor Influence the Li-Rich Layered Oxide Cathode?. Angew. Chem. Int. Ed 2025, 64, e18277 DOI: 10.1002/anie.202518277Google ScholarThere is no corresponding record for this reference.
- 30Choi, G.; Chang, U.; Lee, J.; Park, K.; Kwon, H.; Lee, H.; Kim, Y.-I.; Seo, J. H.; Park, Y.-C.; Park, I.; Kim, J.; Lee, S.; Choi, J.; Yu, B.; Song, J.-H.; Shin, H.; Baek, S.-W.; Lee, S. K.; Park, H.; Jung, K. Unraveling and Regulating Superstructure Domain Dispersion in Lithium-Rich Layered Oxide Cathodes for High Stability and Reversibility. Energy Environ. Sci. 2024, 17 (13), 4634– 4645, DOI: 10.1039/D4EE00487FGoogle ScholarThere is no corresponding record for this reference.
- 31Park, H.; Park, H.; Song, K.; Song, S. H.; Kang, S.; Ko, K.-H.; Eum, D.; Jeon, Y.; Kim, J.; Seong, W. M.; Kim, H.; Park, J.; Kang, K. In Situ Multiscale Probing of the Synthesis of a Ni-Rich Layered Oxide Cathode Reveals Reaction Heterogeneity Driven by Competing Kinetic Pathways. Nat. Chem. 2022, 14 (6), 614– 622, DOI: 10.1038/s41557-022-00915-2Google ScholarThere is no corresponding record for this reference.
- 32Song, S. H.; Kim, H. S.; Kim, K. S.; Hong, S.; Jeon, H.; Lim, J.; Jung, Y. H.; Ahn, H.; Jang, J. D.; Kim, M.; Seo, J. H.; Kwon, J.; Kim, D.; Lee, Y. J.; Han, Y.; Park, K.; Kim, C.; Yu, S.; Park, H.; Jin, H. M.; Kim, H. Toward a Nanoscale-Defect-Free Ni-Rich Layered Oxide Cathode Through Regulated Pore Evolution for Long-Lifespan Li Rechargeable Batteries. Adv. Funct Materials 2024, 34 (3), 2306654 DOI: 10.1002/adfm.202306654Google ScholarThere is no corresponding record for this reference.
- 33Ko, K.-H.; Park, H.; Yoon, J.; Song, S. H.; Choi, E.; Seo, S.; Kwon, J.; Lee, S.; Heo, J.; Han, S.; Park, J.; Kim, W.; Kim, Y.-T.; Lim, J.; Lee, Y. S.; Kim, H.; Kang, K. Multiscale Defect Regulation of Cobalt-Free Layered Oxides for High-Energy and Long-Lasting Cathodes. ACS Energy Lett. 2025, 10 (4), 1605– 1614, DOI: 10.1021/acsenergylett.5c00207Google ScholarThere is no corresponding record for this reference.
- 34Zuo, W.; Gim, J.; Li, T.; Hou, D.; Gao, Y.; Zhou, S.; Zhao, C.; Jia, X.; Yang, Z.; Liu, Y.; Xu, W.; Xiao, X.; Xu, G.-L.; Amine, K. Microstrain Screening towards Defect-Less Layered Transition Metal Oxide Cathodes. Nat. Nanotechnol. 2024, 19 (11), 1644– 1653, DOI: 10.1038/s41565-024-01734-xGoogle ScholarThere is no corresponding record for this reference.
- 35Zuo, W.; Ren, F.; Barai, P.; Hou, D.; Zhou, S.; Wang, G.; Li, T.; Jia, X.; Qin, Y.; Yang, Z.; Xu, W.; Liu, Y.; Yan, H.; Chu, Y. S.; Yang, Y.; Srinivasan, V.; Xiao, X.; Amine, K.; Xu, G.-L. Gas-Mediated Defect Engineering in Earth-Abundant Mn-Rich Layered Oxides for Non-Aqueous Sodium-Based Batteries. Nat. Nanotechnol. 2025, 20 (11), 1667– 1677, DOI: 10.1038/s41565-025-01998-xGoogle ScholarThere is no corresponding record for this reference.
- 36Qiu, Y.; Liu, Q.; Tao, J.; Yan, P.; Tan, G.; Liu, F.; Wang, D.; Yu, N.; Zhang, N.; Yang, Y.; Wang, W.; Wang, Y.; Cao, K.; Wang, J.; Lun, Z.; Xu, C. Enabling the Synthesis of O3-Type Sodium Anion-Redox Cathodes via Atmosphere Modulation. Nat. Commun. 2025, 16 (1), 2343, DOI: 10.1038/s41467-025-57665-1Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
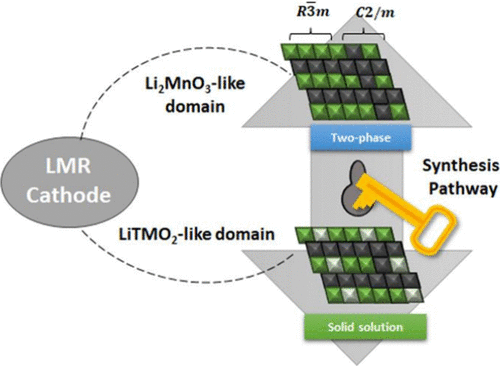
Figure 1
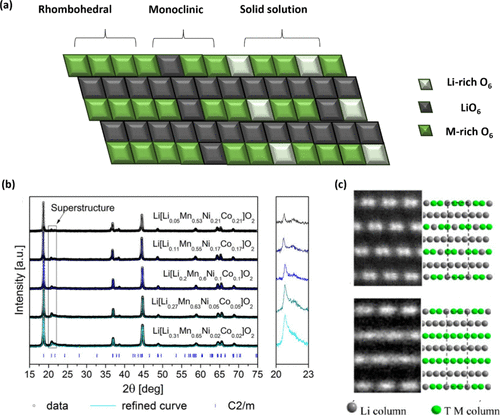
Figure 1. Structural signatures and diagnostic characterization of Li2MnO3 domains in Li- and Mn-rich layered oxides. (a) Schematic illustration comparing the two structural interpretations of Li-rich layered oxides: a two-phase intergrowth model consisting of monoclinic Li2MnO3 (C2/m) and rhombohedral LiTMO2 (R3̅m) domains, and a solid-solution model in which Li–Mn ordering is embedded as local motifs within a single layered lattice. (b) Powder XRD patterns for Li-rich layered oxides with increasing Li2MnO3 content, showing the emergence and growth of monoclinic superstructure reflections. Reproduced from ref (22) under CC BY-NC-ND 4.0 license. Copyright 2019, The Electrochemical Society. (c) HAADF-STEM images and simulated atomic configurations demonstrating Li/Mn honeycomb ordering and the contrast differences between Li columns and TM columns, used to distinguish Li2MnO3-like local ordering from fully random TM distributions. Reproduced with permission from ref (23). Copyright 2012, American Chemical Society.
Figure 2

Figure 2. Chronological evolution of structural interpretations of Li2MnO3domain in LMRs. (a) 6Li MAS NMR spectra of Li2MnO3 samples prepared at 1000 °C (blue) and 850 °C (black). Reproduced with permission from ref (25). Copyright 2005, Elsevier. (b) Convergent-beam electron diffraction (CBED) patterns comparing LiMn0.5Ni0.5O2 and Li1.048[Mn0.333Ni0.333Co0.333]0.952O2. Reproduced with permission from ref (26). Copyright 2006, Elsevier. (c) HAADF-STEM imaging identifying localized excess-Li regions responsible for Li2MnO3-like contrast in LMR particles. Reproduced with permission from ref (17). Copyright 2020, Wiley. (d) X-ray diffraction superlattice peaks (20–25°) comparing DS-LMR (delocalized excess-Li) and LS-LMR (localized excess-Li). Reproduced with permission from ref (18). Copyright 2021, Wiley. (e) Scanning electron microscopy and XRD analysis correlating superstructure evolution and stacking faults with precursor-dependent synthesis pathways (Solid state and Sol–gel method). Reproduced from ref (21) under CC BY 4.0 license. Copyright 2021, American Chemical Society. (f) In situ STEM tracking of sol–gel precursors during heating, revealing atomic-level TM mixing. Reproduced with permission from ref (27). Copyright 2022, Elsevier.
Figure 3

Figure 3. Synthesis-dependent evolution of Li2MnO3-like domains in LMRs. (a) STEM–EDS elemental maps comparing cathodes prepared from solid state, and sol–gel precursors. Reproduced from ref (21) under CC BY 4.0 license. Copyright 2021, American Chemical Society. (b) Schematic illustration of the different synthesis route and FAULTS-refined XRD patterns of LMR samples annealed precursor at 900 °C for 3 h prepared by SG (Sol–gel) and CP (Coprecipitation) method. Reproduced from ref (28) under CC BY 3.0 license. Copyright 2022, Royal Society of Chemistry. (c) In situ HT XRD patterns for carbonate-derived (CO3-LRLO) and hydroxide-derived (OH-LRLO) Li-rich layered oxides. Reproduced with permission from ref (29). Copyright 2025, Wiley. (d) In situ Raman spectra showing distinct phase evolution pathways during calcination: lithium carbonate precursors rapidly develop C2/m Li2MnO3-like modes, whereas lithium hydroxide precursors progress through spinel-like intermediates prior to layered ordering. Reproduced with permission from ref (30). Copyright 2024, Royal Society of Chemistry. (e) EDS mapping of samples heated at low-temperature (∼700 °C, left) and high-temperature (∼1100 °C, right), showing the transition from well-defined Li2MnO3 domains to fused, single-solution-like lattices at elevated temperatures. Reproduced with permission from ref (19). Copyright 2024, Royal Society of Chemistry. (f) Effect of oxygen partial pressure during calcination, where high pO2 promotes extended Li2MnO3-like ordering and faulted monoclinic slabs, while reduced pO2 collapses ordering and yields more homogeneous monoclinic solid-solutions. Reproduced with permission from ref (20). Copyright 2023, American Chemical Society.
Figure 4
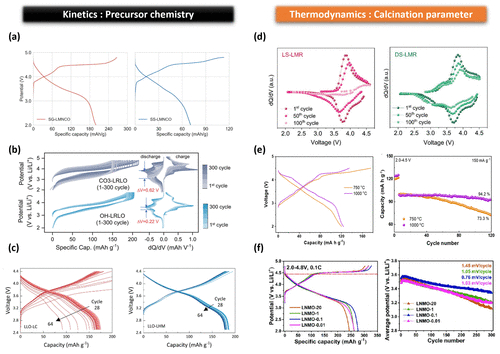
Figure 4. Synthesis-dependent electrochemical manifestations associated with Li2MnO3 domain regulation in LMR cathodes. (a) First-cycle charge–discharge profiles of LMNCO cathodes synthesized via sol–gel (SG) and solid state (SS) precursor routes. Reproduced from ref (21) under CC BY 4.0 license. Copyright 2021, American Chemical Society. (b) Charge–discharge curves and corresponding dQ/dV profiles of carbonate-derived (CO3-LRLO) and hydroxide-derived (OH-LRLO) Li-rich layered oxides collected at selected cycles (1st–300th) at 25 °C within a voltage window of 2.0–4.8 V. Reproduced with permission from ref (29). Copyright 2025, Wiley. (c) Voltage–capacity profiles of LLO cathodes prepared with lithium carbonate (LLO-LC) and lithium hydroxide monohydrate (LLO-LHM) during cycling following rate capability tests. Reproduced with permission from ref (30). Copyright 2024, Royal Society of Chemistry. (d) Differential capacity (dQ/dV) plots of LS-LMR and DS-LMR cathodes at the 1st, 50th, and 100th cycles. Reproduced with permission from ref (18). Copyright 2021, Wiley. (e) Voltage–capacity profiles of LMR cathodes calcined at 750 and 1000 °C, respectively, together with their cycling performance evaluated in the voltage range of 2.0–4.5 V after electrochemical activation. Reproduced with permission from ref (19). Copyright 2024, Royal Society of Chemistry. (f) Electrochemical behavior of LNMO-X cathodes calcined under different oxygen partial pressures (X = 1, 0.1, and 0.01) and air atmosphere (X = 20). Reproduced with permission from ref (20). Copyright 2023, American Chemical Society.
Figure 5
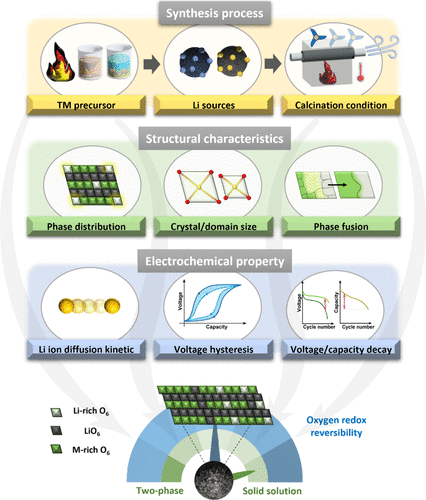
Figure 5. Schematic illustration of synthesis-driven control over Li2MnO3 domain structures in LMRs.
References
This article references 36 other publications.
- 1Nitta, N.; Wu, F.; Lee, J. T.; Yushin, G. Li-Ion Battery Materials: Present and Future. Mater. Today 2015, 18 (5), 252– 264, DOI: 10.1016/j.mattod.2014.10.040There is no corresponding record for this reference.
- 2Grey, C. P.; Tarascon, J. M. Sustainability and in Situ Monitoring in Battery Development. Nat. Mater. 2017, 16 (1), 45– 56, DOI: 10.1038/nmat4777There is no corresponding record for this reference.
- 3Dixit, M.; Witherspoon, B.; Muralidharan, N.; Mench, M. M.; Kweon, C.-B. M.; Sun, Y.-K.; Belharouak, I. Insights into the Critical Materials Supply Chain of the Battery Market for Enhanced Energy Security. ACS Energy Lett. 2024, 9 (8), 3780– 3789, DOI: 10.1021/acsenergylett.4c01300There is no corresponding record for this reference.
- 4Thackeray, M. M.; Kang, S.-H.; Johnson, C. S.; Vaughey, J. T.; Benedek, R.; Hackney, S. A. Li2MnO3-Stabilized LiMO2 (M = Mn, Ni, Co) Electrodes for Lithium-Ion Batteries. J. Mater. Chem. 2007, 17 (30), 3112, DOI: 10.1039/b702425hThere is no corresponding record for this reference.
- 5Zhao, J.; Su, Y.; Dong, J.; Shi, Q.; Lu, Y.; Li, N.; Wang, H.; Fang, Y.; Li, W.; Hao, J.; Wu, Y.; Qi, Q.; Wu, F.; Chen, L. Regulating Anionic Redox Reversibility in Li-Rich Layered Cathodes via Diffusion-Induced Entropy-Assisted Surface Engineering. Energy Storage Materials 2024, 70, 103550 DOI: 10.1016/j.ensm.2024.103550There is no corresponding record for this reference.
- 6Assat, G.; Tarascon, J.-M. Fundamental Understanding and Practical Challenges of Anionic Redox Activity in Li-Ion Batteries. Nat. Energy 2018, 3 (5), 373– 386, DOI: 10.1038/s41560-018-0097-0There is no corresponding record for this reference.
- 7Assat, G.; Delacourt, C.; Corte, D. A. D.; Tarascon, J.-M. Editors’ Choice─Practical Assessment of Anionic Redox in Li-Rich Layered Oxide Cathodes: A Mixed Blessing for High Energy Li-Ion Batteries. J. Electrochem. Soc. 2016, 163 (14), A2965– A2976, DOI: 10.1149/2.0531614jesThere is no corresponding record for this reference.
- 8Gallagher, K. G.; Croy, J. R.; Balasubramanian, M.; Bettge, M.; Abraham, D. P.; Burrell, A. K.; Thackeray, M. M. Correlating Hysteresis and Voltage Fade in Lithium- and Manganese-Rich Layered Transition-Metal Oxide Electrodes. Electrochem. Commun. 2013, 33, 96– 98, DOI: 10.1016/j.elecom.2013.04.022There is no corresponding record for this reference.
- 9Assat, G.; Foix, D.; Delacourt, C.; Iadecola, A.; Dedryvère, R.; Tarascon, J.-M. Fundamental Interplay between Anionic/Cationic Redox Governing the Kinetics and Thermodynamics of Lithium-Rich Cathodes. Nat. Commun. 2017, 8 (1), 2219, DOI: 10.1038/s41467-017-02291-9There is no corresponding record for this reference.
- 10Croy, J. R.; Gallagher, K. G.; Balasubramanian, M.; Chen, Z.; Ren, Y.; Kim, D.; Kang, S.-H.; Dees, D. W.; Thackeray, M. M. Examining Hysteresis in Composite x Li2 MnO3 ·(1– x)LiMO2 Cathode Structures. J. Phys. Chem. C 2013, 117 (13), 6525– 6536, DOI: 10.1021/jp312658qThere is no corresponding record for this reference.
- 11Armstrong, A. R.; Holzapfel, M.; Novák, P.; Johnson, C. S.; Kang, S.-H.; Thackeray, M. M.; Bruce, P. G. Demonstrating Oxygen Loss and Associated Structural Reorganization in the Lithium Battery Cathode Li[Ni0.2 Li0.2 Mn0.6 ]O2. J. Am. Chem. Soc. 2006, 128 (26), 8694– 8698, DOI: 10.1021/ja062027+There is no corresponding record for this reference.
- 12Zhao, J.; Su, Y.; Dong, J.; Hao, J.; Che, H.; Lu, Y.; Li, N.; Zhang, B.; Zhang, P.; Wu, F.; Chen, L. Boosting Reaction Homogeneity through Reverse Gradient Fluorination for High-Performance Li-Rich Layered Cathodes. Energy Storage Materials 2025, 76, 104137 DOI: 10.1016/j.ensm.2025.104137There is no corresponding record for this reference.
- 13McCalla, E.; Abakumov, A. M.; Saubanère, M.; Foix, D.; Berg, E. J.; Rousse, G.; Doublet, M.-L.; Gonbeau, D.; Novák, P.; Van Tendeloo, G.; Dominko, R.; Tarascon, J.-M. Visualization of O-O Peroxo-like Dimers in High-Capacity Layered Oxides for Li-Ion Batteries. Science 2015, 350 (6267), 1516– 1521, DOI: 10.1126/science.aac8260There is no corresponding record for this reference.
- 14Singer, A.; Zhang, M.; Hy, S.; Cela, D.; Fang, C.; Wynn, T. A.; Qiu, B.; Xia, Y.; Liu, Z.; Ulvestad, A.; Hua, N.; Wingert, J.; Liu, H.; Sprung, M.; Zozulya, A. V.; Maxey, E.; Harder, R.; Meng, Y. S.; Shpyrko, O. G. Nucleation of Dislocations and Their Dynamics in Layered Oxide Cathode Materials during Battery Charging. Nat. Energy 2018, 3 (8), 641– 647, DOI: 10.1038/s41560-018-0184-2There is no corresponding record for this reference.
- 15Kang, S.; Choi, D.; Lee, H.; Choi, B.; Kang, Y. A Mechanistic Insight into the Oxygen Redox of Li-Rich Layered Cathodes and Their Related Electronic/Atomic Behaviors Upon Cycling. Adv. Mater. 2023, 35 (43), 2211965 DOI: 10.1002/adma.202211965There is no corresponding record for this reference.
- 16Bareño, J.; Lei, C. H.; Wen, J. G.; Kang, S.-H.; Petrov, I.; Abraham, D. P. Local Structure of Layered Oxide Electrode Materials for Lithium-Ion Batteries. Adv. Mater. 2010, 22 (10), 1122– 1127, DOI: 10.1002/adma.200904247There is no corresponding record for this reference.
- 17Hwang, J.; Myeong, S.; Jin, W.; Jang, H.; Nam, G.; Yoon, M.; Kim, S. H.; Joo, S. H.; Kwak, S. K.; Kim, M. G.; Cho, J. Excess-Li Localization Triggers Chemical Irreversibility in Li- and Mn-Rich Layered Oxides. Adv. Mater. 2020, 32 (34), 2001944 DOI: 10.1002/adma.202001944There is no corresponding record for this reference.
- 18Hwang, J.; Myeong, S.; Lee, E.; Jang, H.; Yoon, M.; Cha, H.; Sung, J.; Kim, M. G.; Seo, D.; Cho, J. Lattice-Oxygen-Stabilized Li- and Mn-Rich Cathodes with Sub-Micrometer Particles by Modifying the Excess-Li Distribution. Adv. Mater. 2021, 33 (18), 2100352 DOI: 10.1002/adma.202100352There is no corresponding record for this reference.
- 19Xu, S.; Chen, Z.; Zhao, W.; Ren, W.; Hou, C.; Liu, J.; Wang, W.; Yin, C.; Tan, X.; Lou, X.; Yao, X.; Gao, Z.; Liu, H.; Wang, L.; Yin, Z.; Qiu, B.; Hu, B.; Li, T.; Dong, C.; Pan, F.; Zhang, M. Multi-Angle Tracking Synthetic Kinetics of Phase Evolution in Li-Rich Mn-Based Cathodes. Energy Environ. Sci. 2024, 17 (11), 3807– 3818, DOI: 10.1039/D3EE04199AThere is no corresponding record for this reference.
- 20Li, J.; Li, W.; Zhang, C.; Han, C.; Chen, X.; Zhao, H.; Xu, H.; Jia, G.; Li, Z.; Li, J.; Zhang, Y.; Guo, X.; Gao, F.; Liu, J.; Qiu, X. Tuning Li2 MnO3 -Like Domain Size and Surface Structure Enables Highly Stabilized Li-Rich Layered Oxide Cathodes. ACS Nano 2023, 17 (17), 16827– 16839, DOI: 10.1021/acsnano.3c03666There is no corresponding record for this reference.
- 21Menon, A. S.; Ulusoy, S.; Ojwang, D. O.; Riekehr, L.; Didier, C.; Peterson, V. K.; Salazar-Alvarez, G.; Svedlindh, P.; Edström, K.; Gomez, C. P.; Brant, W. R. Synthetic Pathway Determines the Nonequilibrium Crystallography of Li- and Mn-Rich Layered Oxide Cathode Materials. ACS Appl. Energy Mater. 2021, 4 (2), 1924– 1935, DOI: 10.1021/acsaem.0c03027There is no corresponding record for this reference.
- 22Redel, K.; Kulka, A.; Plewa, A.; Molenda, J. High-Performance Li-Rich Layered Transition Metal Oxide Cathode Materials for Li-Ion Batteries. J. Electrochem. Soc. 2019, 166 (3), A5333– A5342, DOI: 10.1149/2.0511903jesThere is no corresponding record for this reference.
- 23Boulineau, A.; Simonin, L.; Colin, J.-F.; Canévet, E.; Daniel, L.; Patoux, S. Evolutions of Li1.2 Mn0.61 Ni0.18 Mg0.01 O2 during the Initial Charge/Discharge Cycle Studied by Advanced Electron Microscopy. Chem. Mater. 2012, 24 (18), 3558– 3566, DOI: 10.1021/cm301140gThere is no corresponding record for this reference.
- 24Jarvis, K.; Deng, Z.; Manthiram, A.; Ferreira, P. Understanding the Role of Lithium Content on the Structure and Capacity of Lithium-Rich Layered Oxides by Aberration-Corrected STEM, D-STEM, and EDS. Microsc Microanal 2012, 18 (S2), 1484– 1485, DOI: 10.1017/S1431927612009270There is no corresponding record for this reference.
- 25Bréger, J.; Jiang, M.; Dupré, N.; Meng, Y. S.; Shao-Horn, Y.; Ceder, G.; Grey, C. P. High-Resolution X-Ray Diffraction, DIFFaX, NMR and First Principles Study of Disorder in the Li2MnO3–Li[Ni1/2Mn1/2]O2 Solid Solution. J. Solid State Chem. 2005, 178 (9), 2575– 2585, DOI: 10.1016/j.jssc.2005.05.027There is no corresponding record for this reference.
- 26Thackeray, M. M.; Kang, S.-H.; Johnson, C. S.; Vaughey, J. T.; Hackney, S. A. Comments on the Structural Complexity of Lithium-Rich Li1+xM1–xO2 Electrodes (M = Mn, Ni, Co) for Lithium Batteries. Electrochem. Commun. 2006, 8 (9), 1531– 1538, DOI: 10.1016/j.elecom.2006.06.030There is no corresponding record for this reference.
- 27Li, Y.; Xu, S.; Zhao, W.; Chen, Z.; Chen, Z.; Li, S.; Hu, J.; Cao, B.; Li, J.; Zheng, S.; Chen, Z.; Zhang, T.; Zhang, M.; Pan, F. Correlating the Dispersion of Li@Mn6 Superstructure Units with the Oxygen Activation in Li-Rich Layered Cathode. Energy Storage Materials 2022, 45, 422– 431, DOI: 10.1016/j.ensm.2021.12.003There is no corresponding record for this reference.
- 28Menon, A. S.; Khalil, S.; Ojwang, D. O.; Edström, K.; Gomez, C. P.; Brant, W. R. Synthesis–Structure Relationships in Li- and Mn-Rich Layered Oxides: Phase Evolution, Superstructure Ordering and Stacking Faults. Dalton Trans. 2022, 51 (11), 4435– 4446, DOI: 10.1039/D2DT00104GThere is no corresponding record for this reference.
- 29Huang, Y.; Li, C.; Zhang, K.; Tang, Y.; Tu, W.; Tian, Y.; Wang, J.; Yan, Y.; Chen, Y.; Zou, Y.; Li, L.; Zhang, B.; Bao, J.; Ding, C.; Wang, Y.; Qiu, T.; Sun, X.; Qiao, Y.; Sun, S. How Does the Precursor Influence the Li-Rich Layered Oxide Cathode?. Angew. Chem. Int. Ed 2025, 64, e18277 DOI: 10.1002/anie.202518277There is no corresponding record for this reference.
- 30Choi, G.; Chang, U.; Lee, J.; Park, K.; Kwon, H.; Lee, H.; Kim, Y.-I.; Seo, J. H.; Park, Y.-C.; Park, I.; Kim, J.; Lee, S.; Choi, J.; Yu, B.; Song, J.-H.; Shin, H.; Baek, S.-W.; Lee, S. K.; Park, H.; Jung, K. Unraveling and Regulating Superstructure Domain Dispersion in Lithium-Rich Layered Oxide Cathodes for High Stability and Reversibility. Energy Environ. Sci. 2024, 17 (13), 4634– 4645, DOI: 10.1039/D4EE00487FThere is no corresponding record for this reference.
- 31Park, H.; Park, H.; Song, K.; Song, S. H.; Kang, S.; Ko, K.-H.; Eum, D.; Jeon, Y.; Kim, J.; Seong, W. M.; Kim, H.; Park, J.; Kang, K. In Situ Multiscale Probing of the Synthesis of a Ni-Rich Layered Oxide Cathode Reveals Reaction Heterogeneity Driven by Competing Kinetic Pathways. Nat. Chem. 2022, 14 (6), 614– 622, DOI: 10.1038/s41557-022-00915-2There is no corresponding record for this reference.
- 32Song, S. H.; Kim, H. S.; Kim, K. S.; Hong, S.; Jeon, H.; Lim, J.; Jung, Y. H.; Ahn, H.; Jang, J. D.; Kim, M.; Seo, J. H.; Kwon, J.; Kim, D.; Lee, Y. J.; Han, Y.; Park, K.; Kim, C.; Yu, S.; Park, H.; Jin, H. M.; Kim, H. Toward a Nanoscale-Defect-Free Ni-Rich Layered Oxide Cathode Through Regulated Pore Evolution for Long-Lifespan Li Rechargeable Batteries. Adv. Funct Materials 2024, 34 (3), 2306654 DOI: 10.1002/adfm.202306654There is no corresponding record for this reference.
- 33Ko, K.-H.; Park, H.; Yoon, J.; Song, S. H.; Choi, E.; Seo, S.; Kwon, J.; Lee, S.; Heo, J.; Han, S.; Park, J.; Kim, W.; Kim, Y.-T.; Lim, J.; Lee, Y. S.; Kim, H.; Kang, K. Multiscale Defect Regulation of Cobalt-Free Layered Oxides for High-Energy and Long-Lasting Cathodes. ACS Energy Lett. 2025, 10 (4), 1605– 1614, DOI: 10.1021/acsenergylett.5c00207There is no corresponding record for this reference.
- 34Zuo, W.; Gim, J.; Li, T.; Hou, D.; Gao, Y.; Zhou, S.; Zhao, C.; Jia, X.; Yang, Z.; Liu, Y.; Xu, W.; Xiao, X.; Xu, G.-L.; Amine, K. Microstrain Screening towards Defect-Less Layered Transition Metal Oxide Cathodes. Nat. Nanotechnol. 2024, 19 (11), 1644– 1653, DOI: 10.1038/s41565-024-01734-xThere is no corresponding record for this reference.
- 35Zuo, W.; Ren, F.; Barai, P.; Hou, D.; Zhou, S.; Wang, G.; Li, T.; Jia, X.; Qin, Y.; Yang, Z.; Xu, W.; Liu, Y.; Yan, H.; Chu, Y. S.; Yang, Y.; Srinivasan, V.; Xiao, X.; Amine, K.; Xu, G.-L. Gas-Mediated Defect Engineering in Earth-Abundant Mn-Rich Layered Oxides for Non-Aqueous Sodium-Based Batteries. Nat. Nanotechnol. 2025, 20 (11), 1667– 1677, DOI: 10.1038/s41565-025-01998-xThere is no corresponding record for this reference.
- 36Qiu, Y.; Liu, Q.; Tao, J.; Yan, P.; Tan, G.; Liu, F.; Wang, D.; Yu, N.; Zhang, N.; Yang, Y.; Wang, W.; Wang, Y.; Cao, K.; Wang, J.; Lun, Z.; Xu, C. Enabling the Synthesis of O3-Type Sodium Anion-Redox Cathodes via Atmosphere Modulation. Nat. Commun. 2025, 16 (1), 2343, DOI: 10.1038/s41467-025-57665-1There is no corresponding record for this reference.