



This publication is Open Access under the license indicated. Learn More
Artificial Intelligence in Chemical Engineering: Protein Design from First Principles to Structural PredictionClick to copy article linkArticle link copied!
- Joseph S. Bailey Jr.Joseph S. Bailey, Jr.Department of Chemical and Biomolecular Engineering, The Ohio State University, 151 W Woodruff Avenue, Columbus, Ohio 43210, United StatesMore by Joseph S. Bailey, Jr.
- Søren C. SpinaSøren C. SpinaDepartment of Chemical and Biomolecular Engineering, The Ohio State University, 151 W Woodruff Avenue, Columbus, Ohio 43210, United StatesMore by Søren C. Spina
- Andrew HuAndrew HuCollege of Medicine, The Ohio State University, 460 W 10th Avenue, Columbus, Ohio 43210, United StatesMore by Andrew Hu
- Nathan PhanNathan PhanDepartment of Chemical and Biomolecular Engineering, The Ohio State University, 151 W Woodruff Avenue, Columbus, Ohio 43210, United StatesMore by Nathan Phan
- Rachel B. GetmanRachel B. GetmanDepartment of Chemical and Biomolecular Engineering, The Ohio State University, 151 W Woodruff Avenue, Columbus, Ohio 43210, United StatesMore by Rachel B. Getman
- Blaise R. Kimmel*Blaise R. Kimmel*Email: [email protected]Department of Chemical and Biomolecular Engineering, The Ohio State University, 151 W Woodruff Avenue, Columbus, Ohio 43210, United StatesCenter for Cancer Engineering, Ohio State University Comprehensive Cancer Center, The Ohio State University, 2255 Kenny Road, Columbus, Ohio 43210, United StatesPelotonia Institute for Immuno-Oncology, Ohio State University Comprehensive Cancer Center, The Ohio State University, 2255 Kenny Road, Columbus, Ohio 43210, United StatesMore by Blaise R. Kimmel
ACS Engineering Au
© 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.

Non-Commercial (NC): Only non-commercial uses of the work are permitted.

No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
Machine learning and artificial intelligence are improving the speed and accuracy of every step during the protein design process. Early computational strategies relied on physics-based modeling and energy functions to identify amino acid sequences and desired folds. Recent advances in deep-learning structure prediction, diffusion-based backbone generation, and graph-based sequence design now allow researchers to explore the protein sequence and structural space more efficiently. These developments allow proteins to be used as fundamental systems whose components can be engineered with high precision. Computational predictions still struggle to properly account for conformational dynamics, catalytic environments, external interactions, and the broader chemical diversity present in natural enzymes. This review covers the progression from physics-based methods to deep learning, generative methods, and includes current strategies for evaluating stability and function in silico and experimentally.
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Special Issue
Published as part of ACS Engineering Au special issue “AI and Machine Learning in Chemical Engineering: Breakthroughs and Applications”.
Introduction
Figure 1
Figure 1. From sequence to protein structure and conformational behavior. (A) Biological information transfer follows a deterministic pathway from DNA to RNA to protein, linking the encoded sequence information to the emergent molecular function and dynamics. (B) Input amino acid sequences serve as the basis for predictive modeling frameworks. (C) Sequence-informed A.I./ML frameworks trained on sequence and structural ensemble data learn the mapping between linear sequences and conformation space. (D) The resulting structural ensemble offers a data-driven view of protein flexibility and structural diversity derived directly from the amino acid sequence. Reprinted or adapted with permission under a CC-BY 3.0 License from Ille et al. (14) Copyright 2025 AIP Publishing.
Figure 2
Figure 2. Conceptual view of the protein functional universe. The diagram maps the relationships among sequence, structure, and function spaces. Each circle represents an individual protein defined by its amino acid sequence, 3D folds, and biological activity. The blue circles correspond to proteins accessible through natural evolution or traditional protein engineering, primarily clustered within well-explored regions (yellow). Gray circles indicate proteins that remain uncharacterized and lie within the unexplored sequence–structure–function space. The red circles represent proteins accessible through ML-driven de novo design, which extends exploration beyond natural boundaries into previously inaccessible regions. In this framework, sequence space (top layer) is linked to structure space (middle layer) and ultimately to function space (bottom layer), with A.I. methods systematically probing across all three layers. Reprinted or adapted with permission under a CC-BY 4.0 License from Zhang et al. (22) Copyright 2025 MDPI.
Foundational Structure Prediction and Design Frameworks
ROSETTA and PyRosetta
| package | primary role | integration | reported accuracy/benchmarks | key architecture | limitations |
|---|---|---|---|---|---|
| ROSETTA (1998) | physics-based modeling, de novo folding, docking, enzyme/binder design | (1) modular protocols (RosettaScripts) | (1) ab initio folding within 2–4 Å RMSD for small proteins | (1) Monte Carlo sampling with fragment assembly | (1) computationally expensive |
| (2) integrates with PyRosetta and experimental pipelines | (2) accurate ligand docking (≤2 Å in RosettaLigand); successful in antibody modeling and enzyme design (24−26) | (2) hybrid energy function (physics and knowledge-based potentials) | (2) limited backbone flexibility in fixed-backbone design | ||
| (3) underrepresents entropy and solvent dynamics | |||||
| (4) requires large sampling for success | |||||
| PyRosetta (2010) | scriptable interface for custom design workflows | (1) python API to ROSETTA core | (1) comparable accuracy to ROSETTA protocols | exposes ROSETTA “Pose” object, scoring functions, and movers to Python | (1) requires user scripting; limited scalability without HPC |
| (2) integrates with NumPy/pandas/ML tools | (2) flexible pipelines for alanine scanning, ΔΔG, interface mapping (29,30) | (2) inherits ROSETTA’s scoring function, biases | |||
| (3) not inherently generative | |||||
| AlphaFold2 (2020) | high-accuracy structure prediction from sequence | used in nearly all modern pipelines as a validation filter | (1) CASP14: median GDT_TS > 90 | deep attention networks (Evoformer and structure module) with iterative refinement | (1) deterministic outputs |
| (2) subangstrom accuracy for many folds | (2) limited conformational diversity | ||||
| (3) proteome-scale modeling (34,37,47,48) | (3) no motif conditioning | ||||
| (4) no explicit ligand/cofactor modeling | |||||
| ColabFold (2021) | accessible high-throughput structure prediction | (1) integrates AlphaFold2/RF models | (1) CASP14 free modeling accuracy close to AlphaFold2 | (1) AlphaFold2/RF backbone adapted to Colab notebook | (1) dependent on MSA quality |
| (2) uses MMseq2 for fast MSA generation | (2) ≥40× faster MSA generation; robust on toxin families and multimer predictions (40−42,49) | (2) MMseq2 for sequence search | (2) reduce precision vs AlphaFold2 | ||
| (3) used on Google Colab/local | (3) deterministic | ||||
| (4) limited support for rare folds or novel chemistries | |||||
| RoseTTAFold (2021–2023) | multitrack prediction (RF), nucleic acid complexes (RFNA), all-atom assemblies (RFAA) | (1) extends to motif scaffolding, protein–ligand/nucleic acid complexes | (1) RF: three-track models within 2–3 Å | (1) three-track neural network (RF) | (1) deterministic (RF/RFAA/RFNA) |
| (2) paired with ProteinMPNN/LigandMPNN | (2) RFAA: subangstrom ligand placement | (2) graph-based all-atom encoding (RFAA) | (2) limited dynamics | ||
| (3) RFNA: improved protein–DNA/RNA accuracy (44−46) | (3) sequence and structure alphabet expansion (RFNA) | (3) incomplete coverage of novel chemistries |
AlphaFold: Structure Prediction at Scale
ColabFold: Standardized Prediction
RoseTTAFold: Expansion to All-Atom Modeling
Figure 3
Figure 3. Overview of the current protein design dogma. Traditional protein science is often described as a one-way flow in which (A) amino acid sequences give rise to (B) folded structures, which in turn underpin (C) biological function. Modern de novo design inverts this logic: researchers now begin with the desired function and work backward to identify compatible folds and sequences. Current computational frameworks align with three broad strategies: (1) two-stage design, in which structural generators such as ROSETTA, RoseTTAFold, or PyRosetta first propose candidate protein backbones that are then optimized by sequence design engines; (2) sequence-driven methods, exemplified by AlphaFold2 and ColabFold, which predict protein structures directly from amino acid sequence information and are widely used to validate or filter design candidates; and (3) coguided approaches, including multitrack RoseTTAFold variants (RF, RFNA, RFAA) and diffusion-based models (RFDiffusion), which integrate amino acid sequence and protein structure generation simultaneously. These complementary strategies extend the protein design beyond natural sequence–structure relationships, enabling a function-first exploration of protein space. Reprinted or adapted with permission under a CC-BY 4.0 License from Zhang et al. (22) Copyright 2025 MDPI.
Shared Constraints of Foundational Frameworks
Generative Backbone and Sequence Design
Figure 4
Figure 4. Overview of the A.I.-driven protein design toolbox. According to their functional roles in A.I.-driven generative protein design, the protein design toolbox can be divided into five categories: (A) structure prediction frameworks (e.g., AlphaFold2, RoseTTAFold) that validate fold accuracy; (B) de novo backbone generators (RFDiffusion, RFDiffusionAA) that embed motifs or active sites into novel folds; (C) fixed-backbone sequence designers (LigandMPNN) that optimize sequences against a defined structural context; (D) sequence generation models (ProteinMPNN), which not only perform fixed-backbone optimization but also function as a generative sampler of amino acid sequences; and (E) sequence–structure cogeneration and refinement frameworks (PLACER), which jointly optimize side chains, ligands, and active-site geometry. Reprinted or adapted with permission under a CC-BY 4.0 License from Zhang et al. (22) Copyright 2025 MDPI.
Diffusion: Backbone Construction and Modeling as a Generative Process
| package | primary role | integration | reported accuracy/benchmarks | key architecture | limitations |
|---|---|---|---|---|---|
| RFDiffusion (2023) | De novo protein backbone and functional motif design | generates protein scaffolds for motif/catalyst embedding | (1) diverse novel folds (up to 600 residues) | (1) 3D frame-based denoising diffusion model using RoseTTAFold | (1) high GPU cost |
| (2) RMSD ∼ 2 Å for motif placement; 42–54% success for TIM barrels | (2) supports symmetric design, self-conditioning, and partial motif constraints | (2) sampling variance | |||
| (3) 19% hit rate for binders | (3) limited explicit ligand handling (addressed in RFDiffusionAA) | ||||
| (4) 23/25 success rate in motif scaffolding | (4) sensitive to motif constraints | ||||
| (5) improved interface and side-chain quality in RFDiffusionAA (19) | (5) challenges with polar interfaces | ||||
| (6) stochastic outputs may vary | |||||
| RFDiffusionAA (2024) | active-site-aware protein backbone generation and binder design | used for enzyme pocket design, synthetase-ligand scaffolding, and interface tuning | (1) >20% increase in ΔΔG success | (1) RFDiffusion fine-tuned on active-site data | (1) requires detailed active-site input |
| (2) supports joint active-site and motif design | (2) supports per-residue conditioning, side-chain aware diffusion, and flexible residue input | (2) no end-to-end sequence optimization (must be coupled with ProteinMPNN and LigandMPNN) | |||
| (3) improved hallucination accuracy (19,45) | |||||
| ProteinMPNN (2022) | amino acid sequence design for fixed backbones | follows RF/RFDiffusion scaffold generation | (1) ∼50–55% native sequence recovery overall | message passing graph neural network on protein backbone context | (1) fixed backbone |
| (2) ∼90–95% for buried residues; 200× faster than ROSETTA (57,58) | (2) no ligand/cofactor support | ||||
| (3) no noncanonical AA modeling | |||||
| (4) lacks backbone flexibility | |||||
| LigandMPNN (2025) | sequence optimization in the presence of ligands | pocket-specific redesign postdocking or PLACER-generated poses | (1) 63.3% sequence recovery (small molecules), 50.5% nucleotides, 77.5% metals | (1) dual-graph neural network linking ligand atoms and protein residues | (1) requires accurate initial ligand pose and placement |
| (2) Chi1 recovery ∼86% (59) | (2) ligand-aware autoregressive design and side-chain packing | (2) sparse data for rare chemotypes | |||
| PLACER (2025) | active-site evaluation and pose refinement | filters/optimizes RFDiffusion and LigandMPNN output | (1) RMSD ≈ 1.1 Å for ligand active-site alignment | SE(3)-equivariant graph transformer and denoising-based side-chain and ligand optimization | (1) requires known ligand pose or transition-state geometry |
| (2) improves functional design success by 3–5× in catalytic benchmarks (63,64) | (2) limited support for de novo ligand generation | ||||
| (3) sensitive to backbone geometry errors |
ProteinMPNN: Sequence Design as Geometric Prediction
LigandMPNN: Incorporating Chemical Context into Sequence Design
PLACER: Active-Site Geometry as a Filtering Step
Figure 5
Figure 5. Timeline of major developments in protein structure prediction (black) and design methodologies (red). Following early innovations such as ROSETTA (1998) and PyRosetta (2010), the field saw nearly two decades of incremental progress before the emergence of transformative A.I.-based models such as AlphaFold2 (2020). Since then, breakthroughs in generative frameworks, including ProteinMPNN, RFDiffusion, and LigandMPNN, have rapidly expanded, marking a shift toward integrated prediction-design pipelines.
Protein Large Language Models and Sequence-Space Design
Generative A.I.
Workflows for Model Training and Protein Design
In Silico Evaluation of Designed Proteins
Static Scoring and Foldability Screening
Binding Energetics and Interface Quality
Figure 6
Figure 6. Computational strategies for evaluating amino acid sequence perturbations. (A) Structural stability analysis introduces mutations into a sequence and applies ab initio folding to predict conformational shifts, highlighting favorable and unfavorable perturbations. (B) Binding affinity analysis docks protein constituents, incorporates mutations, and estimates changes in binding free energy (ΔΔG) to evaluate the interaction stability. (C) Interface hotspot probing systematically mutates residues at binding interfaces to pinpoint the positions that are most critical for binding energetics.
Dynamics, Sampling, and the Accuracy-Efficiency Trade-Off
Figure 7
Figure 7. Computational evaluation of the biological and functional properties of proteins. (A) Molecular dynamics and catalysis simulate mutated proteins in solvated environments to capture conformational flexibility and catalytic changes through trajectory analyses. Hybrid pipelines that integrate molecular dynamics (MD) with ROSETTA and directed evolution have yielded efficient de novo and redesigned biocatalysts, such as HG3.17 and BH32.14, whose catalytic power emerges from MD-guided active-site reorganization and solvent shielding. (B) Solubility analysis predicts the effects of amino acid sequence variation on protein solubility by comparing mutant distributions to wild-type benchmarks. CamSol-based workflows enable the rational optimization of both solubility and conformational stability, as demonstrated for six antibodies (including two approved therapeutics), enhancing developability without compromising binding. (137) (C) Aggregation propensity assesses structural and sequence features to identify residues or motifs that drive aggregation, distinguishing soluble variants from aggregation-prone variants. Using Aggrescan3D, researchers computationally minimized aggregation hotspots to engineer green fluorescent protein (GFP) mutants with significantly improved solubility and reduced aggregation, resulting in a fast-folding, aggregation-resistant variant. (138) Together, these approaches extend computational evaluation to capture dynamic solubility and aggregation behaviors that critically influence protein performance in physiological and industrial contexts.
Solubility and Aggregation Behaviors
Limitations of In Silico Evaluation
| metric | purpose | example methods | significance | limitations |
|---|---|---|---|---|
| structural stability | predict foldability | ROSETTA (ref2015), RMSD, AlphaFold, pLDDT (34,35,90,92) | ensures the designed fold is retained postmutation | static models neglect entropy and conformational flexibility |
| binding affinity | assess interaction strength | flex ddG, InterfaceAnalyzer, alanine/proline scanning, PDBbind (98,102,104,105,108) | guides interface design and ligand-binding optimization | sensitive to backbone quality and local packing residues |
| interface hotspot probing | localize key residues | alanine/proline scanning, ncAA probe libraries (e.g., PheCN, Bpa) (103,105,139) | identifies energetic “anchors” and enables targeted mutation design | noncanonical probes may bias geometry or introduce steric clashes |
| molecular dynamics and catalysis | model flexibility and transition states | MetaDynamics, MD, QM/MM, REMD, EVB (117,121,126,132,140) | reveals loop dynamics and allosteric networks for catalytic preorganization | high computational cost: enhanced methods require expertise and tuning |
| solubility | predict aggregation or expression risk | CamSol, PROTOSOLM, GATSol (130,131,133,141) | critical for developability, expression, and therapeutic viability | underperforms for IDPs, membrane proteins, or large multichain assemblies |
| aggregation propensity | identify aggregation-prone regions | Aggrescan3D, β-strand exposure models (132,142−144) | detects amyloid risk, hydrophobic patches | may misclassify functional β-sheets or multimer interfaces |
Directed Evolution as a Complement to De Novo Design
Figure 8
Figure 8. Conceptual framework contrasting traditional and A.I.-assisted directed evolution (DE) workflows. The diagram is divided into two pathways: the upper route represents conventional DE, where (A) natural sequence diversity is explored, (B) mutational libraries are generated, (C) variants are expressed, and (E) high-throughput screening identifies improved candidates through iterative experimental cycles. The lower route introduces A.I./ML-assisted or hybrid methodologies, in which (D) supervised models with uncertainty quantification learn the sequence-fitness landscape and use acquisition functions to propose new variants, balancing exploration (high uncertainty) and exploitation (high predicted fitness). These feedback-driven optimization strategies accelerate variant discovery with a reduced screening effort. Combined approaches, such as active learning-assisted directed evolution (ALDE), have yielded (F) optimized protoglobin-based biocatalysts for nonnative cyclopropanation reactions, enhancing their activity, selectivity, and stability while minimizing experimental costs. (148)
Experimental Validation of AI-Generated Protein Tools
| method | measurement | strengths | limitations | example applications |
|---|---|---|---|---|
| X-ray crystallography | atomic-resolution structural “snapshots” | well-established refinement pipelines | (1) requires crystallization (often challenging/time-consuming) | (1) benchmarking AlphaFold prediction |
| (2) static lattice limits dynamic studies | (2) validation of active sites (158,159) | |||
| Cryo-EM | structural validation of large assemblies and complexes | (1) no crystallization needed | (1) historically lower resolution for small proteins (<100 kDa) | (1) antibody–antigen complexes |
| (2) captures transient or unstable complexes | (2) requires advanced processing software | (2) complement to crystallography | ||
| (3) excels at large proteins, complexes, and membrane proteins | (3) ML refinement of maps (151,155,160) | |||
| NMR spectroscopy | conformational ensembles, loop dynamics, chemical environment | (1) probes protein, motion in solution | (1) limited to smaller proteins | (1) loop dynamics in catalysis |
| (2) reveals catalytic loop mobility and reaction intermediates | (2) requires isotopic labeling; lower spatial resolution than crystals | (2) conformational changes critical for function (152) | ||
| hybrid approaches | integrated models combining experimental and computational restraints | combines ML predictions (AlphaFold/ROSETTA) with sparse restraints (XL-MS, cryo-EM maps, covalent labeling) | requires careful alignment of computational and experimental data sets | refinement of protein–protein interfaces and complexes via XL-MS and AlphaFold/ROSETTA (154,155) |
Figure 9
Figure 9. Experimental–computational pipeline for protein engineering. (A) Protein mutant libraries are generated by introducing sequence variations across the regions of interest. AlphaFold-guided domain-motif design (e.g., FBXO23-STX1B) has revealed novel regulatory interfaces relevant to therapeutic target discovery. (155) (B) Mutants are expressed in Escherichia coli, yeast, or mammalian systems to generate protein ensembles for screening; such expression-labeling pipelines support enzyme and biocatalyst development used in pharmaceuticals and green chemistry. (154) (C) Structural characterization via cryo-EM, NMR, and X-ray crystallography (and hybrid methods) refined with predictive models such as AlphaFold2 or ROSETTA resolves folding and conformational dynamics, as shown in ribosomal complex refinements in NMR-ROSETTA modeling of ubiquitin. (156,157) (D) Functional screening evaluates the activity and binding properties of mutant sets, such as hydroxyl radical footprinting, to identify active-site or interface residues that control activity and stability, as applied to Hsp90-co-chaperone systems and engineered oxidoreductase. (154) (E) Finally, A.I./ML integration combines experimental data with modeling to predict next-generation variants, accelerating industrial enzyme design, antibody optimization, and biosensor development.
Limitations and Future Directions in Computational Protein Design
Author Information
- Blaise R. Kimmel - Department of Chemical and Biomolecular Engineering, The Ohio State University, 151 W Woodruff Avenue, Columbus, Ohio 43210, United States; Center for Cancer Engineering, Ohio State University Comprehensive Cancer Center, The Ohio State University, 2255 Kenny Road, Columbus, Ohio 43210, United States; Pelotonia Institute for Immuno-Oncology, Ohio State University Comprehensive Cancer Center, The Ohio State University, 2255 Kenny Road, Columbus, Ohio 43210, United States;
 https://orcid.org/0000-0002-9582-9887;
https://orcid.org/0000-0002-9582-9887;
- Rachel B. Getman - Department of Chemical and Biomolecular Engineering, The Ohio State University, 151 W Woodruff Avenue, Columbus, Ohio 43210, United States;https://orcid.org/0000-0003-0755-0534
J.S.B. Jr.: Wrote the original draft of the manuscript, generated all figures and graphics for the manuscript, edited, revised, and approved the final version of the manuscript; S.C.S., A.H., and N.P.: supported the generation of graphics and writing for the manuscript; R.G.: edited, revised, and approved the final version of the manuscript; B.R.K.: wrote the original draft of the manuscript, edited, revised, and approved the final version of the manuscript, and acquired funding to support the work. CRediT: Joseph S. Bailey Jr. conceptualization, data curation, formal analysis, investigation, methodology, writing - original draft, writing - review & editing; Søren Spina visualization, writing - original draft, writing - review & editing; Andrew Hu visualization, writing - original draft, writing - review & editing; Nathan Phan visualization, writing - original draft, writing - review & editing; Rachel B. Getman funding acquisition, writing - review & editing; Blaise R. Kimmel conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation, visualization, writing - original draft, writing - review & editing.
We gratefully thank the Ohio State University Comprehensive Cancer Center (OSUCCC), OSUCCC Center for Cancer, and the Department of Chemical and Biomolecular Engineering at The Ohio State University for support of this work. B.R.K. acknowledges financial support from the Prostate Cancer Foundation Young Investigator Award.
Acknowledgments
This work was supported in part by The Ohio State University Center for Cancer Engineering─Curing Cancer Through Research in Engineering and Sciences. B.R.K. acknowledges financial support from the Prostate Cancer Foundation Young Investigator Award. We acknowledge the use of PaperPal and Grammarly as AI tools to modify the grammar, phrasing, and sentence structure while writing this review. Each author takes full responsibility for the manuscript’s content.
References
This article references 160 other publications.
- 1Yu, Y.; Hu, C.; Xia, L.; Wang, J. Artificial Metalloenzyme Design with Unnatural Amino Acids and Non-Native Cofactors. ACS Catal. 2018, 8, 1851– 1863, DOI: 10.1021/acscatal.7b03754Google ScholarThere is no corresponding record for this reference.
- 2Mirts, E. N.; Bhagi-Damodaran, A.; Lu, Y. Understanding and Modulating Metalloenzymes with Unnatural Amino Acids, Non-Native Metal Ions, and Non-Native Metallocofactors. Acc. Chem. Res. 2019, 52, 935– 944, DOI: 10.1021/acs.accounts.9b00011Google ScholarThere is no corresponding record for this reference.
- 3Mann, S. I.; Nayak, A.; Gassner, G. T.; Therien, M. J.; DeGrado, W. F. De Novo Design, Solution Characterization, and Crystallographic Structure of an Abiological Mn–Porphyrin-Binding Protein Capable of Stabilizing a Mn(V) Species. J. Am. Chem. Soc. 2021, 143, 252– 259, DOI: 10.1021/jacs.0c10136Google ScholarThere is no corresponding record for this reference.
- 4Bergman, M. T.; Xiao, X.; Hall, C. K. In Silico Design and Analysis of Plastic-Binding Peptides. J. Phys. Chem. B 2023, 127, 8370– 8381, DOI: 10.1021/acs.jpcb.3c04319Google ScholarThere is no corresponding record for this reference.
- 5García-Moreno, P. J. Recent advances in the production of emulsifying peptides with the aid of proteomics and bioinformatics. Curr. Opin. Food Sci. 2023, 51, 101039 DOI: 10.1016/j.cofs.2023.101039Google ScholarThere is no corresponding record for this reference.
- 6Ndochinwa, G. O.; Wang, Q. Y.; Okoro, N. O. New advances in protein engineering for industrial applications: Key takeaways. Open Life Sci. 2024, 19, 20220856 DOI: 10.1515/biol-2022-0856Google ScholarThere is no corresponding record for this reference.
- 7Marcos, E.; Silva, D. Essentials of de novo protein design: Methods and applications. WIREs Comput. Mol. Sci. 2018, 8 (6), e1374 DOI: 10.1002/wcms.1374Google ScholarThere is no corresponding record for this reference.
- 8Huang, P.-S.; Boyken, S. E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320– 327, DOI: 10.1038/nature19946Google ScholarThere is no corresponding record for this reference.
- 9Woolfson, D. N. A Brief History of De Novo Protein Design: Minimal, Rational, and Computational. J. Mol. Biol. 2021, 433, 167160 DOI: 10.1016/j.jmb.2021.167160Google ScholarThere is no corresponding record for this reference.
- 10Dill, K. A.; Ozkan, S. B.; Shell, M. S.; Weikl, T. R. Protein Folding Problem. Annu. Rev. Biophys. 2008, 37, 289– 316, DOI: 10.1146/annurev.biophys.37.092707.153558Google ScholarThere is no corresponding record for this reference.
- 11Kocher, C. D.; Dill, K. A. Origins of life: The Protein Folding Problem all over again?. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2315000121 DOI: 10.1073/pnas.2315000121Google ScholarThere is no corresponding record for this reference.
- 12Chen, S.-J.; Hassan, M.; Jernigan, R. L. Protein folds vs. protein folding: Differing questions, different challenges. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2214423119 DOI: 10.1073/pnas.2214423119Google ScholarThere is no corresponding record for this reference.
- 13Kiss, G.; Çelebi-Ölçüm, N.; Moretti, R.; Baker, D.; Houk, K. N. Computational Enzyme Design. Angew. Chem., Int. Ed. 2013, 52, 5700– 5725, DOI: 10.1002/anie.201204077Google ScholarThere is no corresponding record for this reference.
- 14Ille, A. M.; Anas, E.; Mathews, M. B.; Burley, S. K. From sequence to protein structure and conformational dynamics with artificial intelligence/machine learning. Struct. Dyn. 2025, 12, 030902 DOI: 10.1063/4.0000765Google ScholarThere is no corresponding record for this reference.
- 15Anfinsen, C. B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223– 230, DOI: 10.1126/science.181.4096.223Google ScholarThere is no corresponding record for this reference.
- 16Voigt, C. A.; Mayo, S. L.; Arnold, F. H.; Wang, Z.-G. Computational method to reduce the search space for directed protein evolution. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 3778– 3783, DOI: 10.1073/pnas.051614498Google ScholarThere is no corresponding record for this reference.
- 17Kuhlman, B.; Baker, D. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10383– 10388, DOI: 10.1073/pnas.97.19.10383Google ScholarThere is no corresponding record for this reference.
- 18Sleator, R. D. Solving the protein folding problem···. FEBS Lett. 2024, 598, 2831– 2835, DOI: 10.1002/1873-3468.15043Google ScholarThere is no corresponding record for this reference.
- 19Watson, J. L.; Juergens, D.; Bennett, N. R. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089– 1100, DOI: 10.1038/s41586-023-06415-8Google ScholarThere is no corresponding record for this reference.
- 20Leveson-Gower, R. B. Designing Enzymatic Reactivity with an Expanded Palette. ChemBioChem 2025, 26, e202500076 DOI: 10.1002/cbic.202500076Google ScholarThere is no corresponding record for this reference.
- 21Hartman, M. C. T. Non-canonical Amino Acid Substrates of Escherichia coli Aminoacyl-tRNA Synthetases. ChemBioChem 2022, 23, e202100299 DOI: 10.1002/cbic.202100299Google ScholarThere is no corresponding record for this reference.
- 22Zhang, G.; Liu, C.; Lu, J.; Zhang, S.; Zhu, L. The Role of AI-Driven De Novo Protein Design in the Exploration of the Protein Functional Universe. Biology 2025, 14, 1268 DOI: 10.3390/biology14091268Google ScholarThere is no corresponding record for this reference.
- 23Rohl, C. A.; Strauss, C. E. M.; Misura, K. M. S.; Baker, D. Protein Structure Prediction Using Rosetta. Methods Enzymol. 2004, 383, 66– 93, DOI: 10.1016/S0076-6879(04)83004-0Google ScholarThere is no corresponding record for this reference.
- 24Simons, K. T.; Kooperberg, C.; Huang, E.; Baker, D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and bayesian scoring functions. J. Mol. Biol. 1997, 268, 209– 225, DOI: 10.1006/jmbi.1997.0959Google ScholarThere is no corresponding record for this reference.
- 25Leman, J. K.; Weitzner, B. D.; Lewis, S. M. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 2020, 17, 665– 680, DOI: 10.1038/s41592-020-0848-2Google ScholarThere is no corresponding record for this reference.
- 26Das, R.; Baker, D. Macromolecular Modeling with Rosetta. Annu. Rev. Biochem. 2008, 77, 363– 382, DOI: 10.1146/annurev.biochem.77.062906.171838Google ScholarThere is no corresponding record for this reference.
- 27Kaufmann, K. W.; Meiler, J. Using RosettaLigand for Small Molecule Docking into Comparative Models. PLoS One 2012, 7, e50769 DOI: 10.1371/journal.pone.0050769Google ScholarThere is no corresponding record for this reference.
- 28Lemmon, G.; Kaufmann, K.; Meiler, J. Prediction of HIV-1 Protease/Inhibitor Affinity using RosettaLigand. Chem. Biol. Drug Des. 2012, 79, 888– 896, DOI: 10.1111/j.1747-0285.2012.01356.xGoogle ScholarThere is no corresponding record for this reference.
- 29Chaudhury, S.; Lyskov, S.; Gray, J. J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 2010, 26, 689– 691, DOI: 10.1093/bioinformatics/btq007Google ScholarThere is no corresponding record for this reference.
- 30Le, K. H.; Adolf-Bryfogle, J.; Klima, J. C. PyRosetta Jupyter Notebooks Teach Biomolecular Structure Prediction and Design. Biophysicist 2021, 2, 108– 122, DOI: 10.35459/tbp.2019.000147Google ScholarThere is no corresponding record for this reference.
- 31Ford, A. S.; Weitzner, B. D.; Bahl, C. D. Integration of the Rosetta suite with the python software stack via reproducible packaging and core programming interfaces for distributed simulation. Protein Sci. 2020, 29, 43– 51, DOI: 10.1002/pro.3721Google ScholarThere is no corresponding record for this reference.
- 32Van Stappen, C.; Deng, Y.; Liu, Y. Designing Artificial Metalloenzymes by Tuning of the Environment beyond the Primary Coordination Sphere. Chem. Rev. 2022, 122, 11974– 12045, DOI: 10.1021/acs.chemrev.2c00106Google ScholarThere is no corresponding record for this reference.
- 33Tivon, B.; Wiese, J.; Müller, M. P. Computational Design of Lysine Targeting Covalent Binders Using Rosetta. J. Chem. Inf. Model. 2025, 65, 5612– 5622, DOI: 10.1021/acs.jcim.5c00212Google ScholarThere is no corresponding record for this reference.
- 34Jumper, J.; Evans, R.; Pritzel, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583– 589, DOI: 10.1038/s41586-021-03819-2Google ScholarThere is no corresponding record for this reference.
- 35Tunyasuvunakool, K.; Adler, J.; Wu, Z. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590– 596, DOI: 10.1038/s41586-021-03828-1Google ScholarThere is no corresponding record for this reference.
- 36Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)─Round XIV. Proteins:Struct., Funct., Bioinf. 2021, 89, 1607– 1617, DOI: 10.1002/prot.26237Google ScholarThere is no corresponding record for this reference.
- 37Kovalevskiy, O.; Mateos-Garcia, J.; Tunyasuvunakool, K. AlphaFold two years on: Validation and impact. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2315002121 DOI: 10.1073/pnas.2315002121Google ScholarThere is no corresponding record for this reference.
- 38Schneider, B.; Sweeney, B. A.; Bateman, A. When will RNA get its AlphaFold moment?. Nucleic Acids Res. 2023, 51, 9522– 9532, DOI: 10.1093/nar/gkad726Google ScholarThere is no corresponding record for this reference.
- 39Terwilliger, T. C.; Liebschner, D.; Croll, T. I. AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nat. Methods 2024, 21, 110– 116, DOI: 10.1038/s41592-023-02087-4Google ScholarThere is no corresponding record for this reference.
- 40Mirdita, M.; Schütze, K.; Moriwaki, Y. ColabFold: making protein folding accessible to all. Nat. Methods 2022, 19, 679– 682, DOI: 10.1038/s41592-022-01488-1Google ScholarThere is no corresponding record for this reference.
- 41Kim, G.; Lee, S.; Levy Karin, E. Easy and accurate protein structure prediction using ColabFold. Nat. Protoc. 2025, 20, 620– 642, DOI: 10.1038/s41596-024-01060-5Google ScholarThere is no corresponding record for this reference.
- 42Kalogeropoulos, K.; Bohn, M. F.; Jenkins, D. E. A comparative study of protein structure prediction tools for challenging targets: Snake venom toxins. Toxicon 2024, 238, 107559 DOI: 10.1016/j.toxicon.2023.107559Google ScholarThere is no corresponding record for this reference.
- 43Baek, M.; DiMaio, F.; Anishchenko, I. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871– 876, DOI: 10.1126/science.abj8754Google ScholarThere is no corresponding record for this reference.
- 44Baek, M.; McHugh, R.; Anishchenko, I. Accurate prediction of protein–nucleic acid complexes using RoseTTAFoldNA. Nat. Methods 2024, 21, 117– 121, DOI: 10.1038/s41592-023-02086-5Google ScholarThere is no corresponding record for this reference.
- 45Krishna, R.; Wang, J.; Ahern, W. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science 2024, 384, eadl2528 DOI: 10.1126/science.adl2528Google ScholarThere is no corresponding record for this reference.
- 46Liu, S.; Wu, K.; Chen, C. Obtaining protein foldability information from computational models of AlphaFold2 and RoseTTAFold. Comput. Struct. Biotechnol. J. 2022, 20, 4481– 4489, DOI: 10.1016/j.csbj.2022.08.034Google ScholarThere is no corresponding record for this reference.
- 47Wayment-Steele, H. K.; Ojoawo, A.; Otten, R. Predicting multiple conformations via sequence clustering and AlphaFold2. Nature 2024, 625, 832– 839, DOI: 10.1038/s41586-023-06832-9Google ScholarThere is no corresponding record for this reference.
- 48Casadevall, G.; Duran, C.; Osuna, S. AlphaFold2 and Deep Learning for Elucidating Enzyme Conformational Flexibility and Its Application for Design. JACS Au 2023, 3, 1554– 1562, DOI: 10.1021/jacsau.3c00188Google ScholarThere is no corresponding record for this reference.
- 49Vallejo, W.; Díaz-Uribe, C.; Fajardo, C. Google Colab and Virtual Simulations: Practical e-Learning Tools to Support the Teaching of Thermodynamics and to Introduce Coding to Students. ACS Omega 2022, 7, 7421– 7429, DOI: 10.1021/acsomega.2c00362Google ScholarThere is no corresponding record for this reference.
- 50Adiyaman, R.; Edmunds, N. S.; Genc, A. G.; Alharbi, S. M. A.; McGuffin, L. J. Improvement of protein tertiary and quaternary structure predictions using the ReFOLD refinement method and the AlphaFold2 recycling process. Bioinforma. Adv. 2023, 3 (1), vbad078 DOI: 10.1093/bioadv/vbad078Google ScholarThere is no corresponding record for this reference.
- 51Ahern, W.; Yim, J.; Tischer, D. Atom level enzyme active site scaffolding using RFdiffusion2. Nat. Methods 2026, 23, 96– 105, DOI: 10.1038/s41592-025-02975-xGoogle ScholarThere is no corresponding record for this reference.
- 52Wang, W.; Feng, C.; Han, R. trRosettaRNA: automated prediction of RNA 3D structure with transformer network. Nat. Commun. 2023, 14, 7266 DOI: 10.1038/s41467-023-42528-4Google ScholarThere is no corresponding record for this reference.
- 53Corso, G.; Stärk, H.; Jing, B.; Barzilay, R.; Jaakkola, T. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking, arXiv:2210.01776. arXiv.org e-Print archive. https://arxiv.org/abs/2210.01776. 2023.Google ScholarThere is no corresponding record for this reference.
- 54Alamdari, S. Protein generation with evolutionary diffusion: sequence is all you need. Preprint at https://doi.org/10.1101/2023.09.11.556673. 2023.Google ScholarThere is no corresponding record for this reference.
- 55Chu, A. E.; Kim, J.; Cheng, L. An all-atom protein generative model. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2311500121 DOI: 10.1073/pnas.2311500121Google ScholarThere is no corresponding record for this reference.
- 56Dauparas, J. Robust deep learning based protein sequence design using ProteinMPNN.Google ScholarThere is no corresponding record for this reference.
- 57Sumida, K. H.; Núñez-Franco, R.; Kalvet, I. Improving Protein Expression, Stability, and Function with ProteinMPNN. J. Am. Chem. Soc. 2024, 146, 2054– 2061, DOI: 10.1021/jacs.3c10941Google ScholarThere is no corresponding record for this reference.
- 58De Haas, R. J.; Brunette, N.; Goodson, A. Rapid and automated design of two-component protein nanomaterials using ProteinMPNN. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2314646121 DOI: 10.1073/pnas.2314646121Google ScholarThere is no corresponding record for this reference.
- 59Dauparas, J.; Lee, G. R.; Pecoraro, R. Atomic context-conditioned protein sequence design using LigandMPNN. Nat. Methods 2025, 22, 717– 723, DOI: 10.1038/s41592-025-02626-1Google ScholarThere is no corresponding record for this reference.
- 60Clark-Elsayed, A. Comparing LigandMPNN and Directed Evolution for Altering the Effector-Binding Site in the RamR Transcription Factor.Google ScholarThere is no corresponding record for this reference.
- 61An, L.; Said, M.; Tran, L. Binding and sensing diverse small molecules using shape-complementary pseudocycles. Science 2024, 385, 276– 282, DOI: 10.1126/science.adn3780Google ScholarThere is no corresponding record for this reference.
- 62Agu, P. C.; Afiukwa, C. A.; Orji, O. U. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 2023, 13, 13398 DOI: 10.1038/s41598-023-40160-2Google ScholarThere is no corresponding record for this reference.
- 63Anishchenko, I. Modeling protein-small molecule conformational ensembles with ChemNet. Preprint at https://doi.org/10.1101/2024.09.25.614868. 2024.Google ScholarThere is no corresponding record for this reference.
- 64Lauko, A.; Pellock, S. J.; Sumida, K. H. Computational design of serine hydrolases. Science 2025, 388, eadu2454 DOI: 10.1126/science.adu2454Google ScholarThere is no corresponding record for this reference.
- 65Park, H.; Zhou, G.; Baek, M.; Baker, D.; DiMaio, F. Force Field Optimization Guided by Small Molecule Crystal Lattice Data Enables Consistent Sub-Angstrom Protein–Ligand Docking. J. Chem. Theory Comput. 2021, 17, 2000– 2010, DOI: 10.1021/acs.jctc.0c01184Google ScholarThere is no corresponding record for this reference.
- 66Garcia, M.; Dixit, S. M.; Rocklin, G. J. Evaluating zero-shot prediction of protein design success by AlphaFold, ESMFold, and ProteinMPNN.Google ScholarThere is no corresponding record for this reference.
- 67Kong, Z. ProtFlow: Flow Matching-based Protein Sequence Design with Comprehensive Protein Semantic Distribution Learning and High-quality Generation.Google ScholarThere is no corresponding record for this reference.
- 68Elnaggar, A.; Heinzinger, M.; Dallago, C. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112– 7127, DOI: 10.1109/TPAMI.2021.3095381Google ScholarThere is no corresponding record for this reference.
- 69Madani, A. ProGen: Language Modeling for Protein Generation, arXiv:2004.03497. arXiv.org e-Print archive. https://arxiv.org/abs/2004.03497. 2020.Google ScholarThere is no corresponding record for this reference.
- 70Nijkamp, E.; Ruffolo, J. A.; Weinstein, E. N.; Naik, N.; Madani, A. ProGen2: Exploring the boundaries of protein language models. Cell Syst. 2023, 14, 968– 978.e3, DOI: 10.1016/j.cels.2023.10.002Google ScholarThere is no corresponding record for this reference.
- 71Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 2022, 13, 4348 DOI: 10.1038/s41467-022-32007-7Google ScholarThere is no corresponding record for this reference.
- 72Nguyen, E.; Poli, M.; Durrant, M. G. Sequence modeling and design from molecular to genome scale with Evo. Science 2024, 386, eado9336 DOI: 10.1126/science.ado9336Google ScholarThere is no corresponding record for this reference.
- 73Dalla-Torre, H.; Gonzalez, L.; Mendoza-Revilla, J. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nat. Methods 2025, 22, 287– 297, DOI: 10.1038/s41592-024-02523-zGoogle ScholarThere is no corresponding record for this reference.
- 74Avsec, Ž.; Latysheva, N.; Cheng, J. Advancing regulatory variant effect prediction with AlphaGenome. Nature 2026, 649, 1206– 1218, DOI: 10.1038/s41586-025-10014-0Google ScholarThere is no corresponding record for this reference.
- 75Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction.Google ScholarThere is no corresponding record for this reference.
- 76Chai Discovery. Chai-1: Decoding the molecular interactions of life. Preprint at https://doi.org/10.1101/2024.10.10.615955. 2024.Google ScholarThere is no corresponding record for this reference.
- 77Ingraham, J. B.; Baranov, M.; Costello, Z. Illuminating protein space with a programmable generative model. Nature 2023, 623, 1070– 1078, DOI: 10.1038/s41586-023-06728-8Google ScholarThere is no corresponding record for this reference.
- 78Mille-Fragoso, L. S. Efficient generation of epitope-targeted de novo antibodies with Germinal.Google ScholarThere is no corresponding record for this reference.
- 79Pacesa, M.; Nickel, L.; Schellhaas, C. One-shot design of functional protein binders with BindCraft. Nature 2025, 646, 483– 492, DOI: 10.1038/s41586-025-09429-6Google ScholarThere is no corresponding record for this reference.
- 80BoltzGen: Toward Universal Binder Design.Google ScholarThere is no corresponding record for this reference.
- 81Zhang, O. ODesign: A World Model for Biomolecular Interaction Design, arXiv:2510.22304. arXiv.org e-Print archive. https://arxiv.org/abs/2510.22304. 2025.Google ScholarThere is no corresponding record for this reference.
- 82Parks, M. Blind Virtual Screening at Scale: A Scalable End-to-End Pipeline for Blind Docking and Affinity Prediction.Google ScholarThere is no corresponding record for this reference.
- 83John, P. S. BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery, arXiv:2411.10548. arXiv.org e-Print archive. https://arxiv.org/abs/2411.10548. 2025.Google ScholarThere is no corresponding record for this reference.
- 84Silke, D.; Iskander, J.; Pan, J. ProteinDJ : A high-performance and modular protein design pipeline. Protein Sci. 2026, 35, e70464 DOI: 10.1002/pro.70464Google ScholarThere is no corresponding record for this reference.
- 85González-Rodríguez, N.; Chacón-Sánchez, C.; Llorca, O.; Fernández-Leiro, R. Automated and modular protein binder design with BinderFlow. PLOS Comput. Biol. 2025, 21, e1013747 DOI: 10.1371/journal.pcbi.1013747Google ScholarThere is no corresponding record for this reference.
- 86Danny, B. Ovo, an Open-Source Ecosystem for De Novo Protein Design.Google ScholarThere is no corresponding record for this reference.
- 87Baker, D. An exciting but challenging road ahead for computational enzyme design. Protein Sci. 2010, 19, 1817– 1819, DOI: 10.1002/pro.481Google ScholarThere is no corresponding record for this reference.
- 88Beadle, B. M.; Shoichet, B. K. Structural Bases of Stability–function Tradeoffs in Enzymes. J. Mol. Biol. 2002, 321, 285– 296, DOI: 10.1016/S0022-2836(02)00599-5Google ScholarThere is no corresponding record for this reference.
- 89Barlow, K. A.; Conchúir, S. Ó.; Thompson, S. Flex ddG: Rosetta Ensemble-Based Estimation of Changes in Protein–Protein Binding Affinity upon Mutation. J. Phys. Chem. B 2018, 122, 5389– 5399, DOI: 10.1021/acs.jpcb.7b11367Google ScholarThere is no corresponding record for this reference.
- 90Shringari, S. R.; Giannakoulias, S.; Ferrie, J. J.; Petersson, E. J. Rosetta custom score functions accurately predict ΔΔG of mutations at protein–protein interfaces using machine learning. Chem. Commun. 2020, 56, 6774– 6777, DOI: 10.1039/D0CC01959CGoogle ScholarThere is no corresponding record for this reference.
- 91Smith, S. T.; Meiler, J. Assessing multiple score functions in Rosetta for drug discovery. PLoS One 2020, 15, e0240450 DOI: 10.1371/journal.pone.0240450Google ScholarThere is no corresponding record for this reference.
- 92Alford, R. F.; Leaver-Fay, A.; Jeliazkov, J. R. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031– 3048, DOI: 10.1021/acs.jctc.7b00125Google ScholarThere is no corresponding record for this reference.
- 93Tyka, M. D.; Keedy, D. A.; André, I. Alternate States of Proteins Revealed by Detailed Energy Landscape Mapping. J. Mol. Biol. 2011, 405, 607– 618, DOI: 10.1016/j.jmb.2010.11.008Google ScholarThere is no corresponding record for this reference.
- 94Planas-Iglesias, J.; Marques, S. M.; Pinto, G. P. Computational design of enzymes for biotechnological applications. Biotechnol. Adv. 2021, 47, 107696 DOI: 10.1016/j.biotechadv.2021.107696Google ScholarThere is no corresponding record for this reference.
- 95Guo, H.-B.; Perminov, A.; Bekele, S. AlphaFold2 models indicate that protein sequence determines both structure and dynamics. Sci. Rep. 2022, 12, 10696 DOI: 10.1038/s41598-022-14382-9Google ScholarThere is no corresponding record for this reference.
- 96Agarwal, V.; McShan, A. C. The power and pitfalls of AlphaFold2 for structure prediction beyond rigid globular proteins. Nat. Chem. Biol. 2024, 20, 950– 959, DOI: 10.1038/s41589-024-01638-wGoogle ScholarThere is no corresponding record for this reference.
- 97Abramson, J.; Adler, J.; Dunger, J. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493– 500, DOI: 10.1038/s41586-024-07487-wGoogle ScholarThere is no corresponding record for this reference.
- 98Friedland, G. D.; Linares, A. J.; Smith, C. A.; Kortemme, T. A Simple Model of Backbone Flexibility Improves Modeling of Side-chain Conformational Variability. J. Mol. Biol. 2008, 380, 757– 774, DOI: 10.1016/j.jmb.2008.05.006Google ScholarThere is no corresponding record for this reference.
- 99Kellogg, E. H.; Leaver-Fay, A.; Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins:Struct., Funct., Bioinf. 2011, 79, 830– 838, DOI: 10.1002/prot.22921Google ScholarThere is no corresponding record for this reference.
- 100Durham, E.; Dorr, B.; Woetzel, N.; Staritzbichler, R.; Meiler, J. Solvent accessible surface area approximations for rapid and accurate protein structure prediction. J. Mol. Model. 2009, 15, 1093– 1108, DOI: 10.1007/s00894-009-0454-9Google ScholarThere is no corresponding record for this reference.
- 101Bertalan, É.; Lešnik, S.; Bren, U.; Bondar, A.-N. Protein-water hydrogen-bond networks of G protein-coupled receptors: Graph-based analyses of static structures and molecular dynamics. J. Struct. Biol. 2020, 212, 107634 DOI: 10.1016/j.jsb.2020.107634Google ScholarThere is no corresponding record for this reference.
- 102Weiss, G. A.; Watanabe, C. K.; Zhong, A.; Goddard, A.; Sidhu, S. S. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8950– 8954, DOI: 10.1073/pnas.160252097Google ScholarThere is no corresponding record for this reference.
- 103Cunningham, B. C.; Wells, J. A. High-Resolution Epitope Mapping of hGH-Receptor Interactions by Alanine-Scanning Mutagenesis. Science 1989, 244, 1081– 1085, DOI: 10.1126/science.2471267Google ScholarThere is no corresponding record for this reference.
- 104Liu, H.; Song, L.; Meng, X. Proline-Mediated Enhancement in Evolvability of Disulfide-Rich Peptides for Discovering Protein Binders. J. Am. Chem. Soc. 2025, 147, 24870– 24883, DOI: 10.1021/jacs.5c07075Google ScholarThere is no corresponding record for this reference.
- 105Holden, J. K.; Pavlovicz, R.; Gobbi, A.; Song, Y.; Cunningham, C. N. Computational Site Saturation Mutagenesis of Canonical and Non-Canonical Amino Acids to Probe Protein-Peptide Interactions. Front. Mol. Biosci. 2022, 9, 848689 DOI: 10.3389/fmolb.2022.848689Google ScholarThere is no corresponding record for this reference.
- 106Spina, S. C.; Bailey, J.; Kimmel, B. Bind, catalyze, and quantify: a modern protein and enzyme engineering toolbox of genetically encoded non-canonical amino acids Protein Eng. Des. Sel. 2026gzag007 DOI: 10.1093/protein/gzag007 .Google ScholarThere is no corresponding record for this reference.
- 107Chen, Y.; Clay, N.; Phan, N. Molecular Matchmakers: Bioconjugation Techniques Enhance Prodrug Potency for Immunotherapy. Mol. Pharmaceutics 2025, 22, 58– 80, DOI: 10.1021/acs.molpharmaceut.4c00867Google ScholarThere is no corresponding record for this reference.
- 108Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind Database: Collection of Binding Affinities for Protein–Ligand Complexes with Known Three-Dimensional Structures. J. Med. Chem. 2004, 47, 2977– 2980, DOI: 10.1021/jm030580lGoogle ScholarThere is no corresponding record for this reference.
- 109Liu, Z.; Su, M.; Han, L. Forging the Basis for Developing Protein–Ligand Interaction Scoring Functions. Acc. Chem. Res. 2017, 50, 302– 309, DOI: 10.1021/acs.accounts.6b00491Google ScholarThere is no corresponding record for this reference.
- 110King, B. R.; Sumida, K. H.; Caruso, J. L.; Baker, D.; Zalatan, J. G. Computational Stabilization of a Non-Heme Iron Enzyme Enables Efficient Evolution of New Function. Angew. Chem., Int. Ed. 2025, 64, e202414705 DOI: 10.1002/anie.202414705Google ScholarThere is no corresponding record for this reference.
- 111Howlader, M. T. H.; Kagawa, Y.; Miyakawa, A. Alanine Scanning Analyses of the Three Major Loops in Domain II of Bacillus thuringiensis Mosquitocidal Toxin Cry4Aa. Appl. Environ. Microbiol. 2010, 76, 860– 865, DOI: 10.1128/AEM.02175-09Google ScholarThere is no corresponding record for this reference.
- 112Paul, R.; Kasahara, K.; Sasaki, J. Unveiling the affinity–stability relationship in anti-measles virus antibodies: a computational approach for hotspots prediction. Front. Mol. Biosci. 2024, 10, 1302737 DOI: 10.3389/fmolb.2023.1302737Google ScholarThere is no corresponding record for this reference.
- 113Romero-Rivera, A.; Garcia-Borràs, M.; Osuna, S. Role of Conformational Dynamics in the Evolution of Retro-Aldolase Activity. ACS Catal. 2017, 7, 8524– 8532, DOI: 10.1021/acscatal.7b02954Google ScholarThere is no corresponding record for this reference.
- 114Lemkul, J. A. Introductory Tutorials for Simulating Protein Dynamics with GROMACS. J. Phys. Chem. B 2024, 128, 9418– 9435, DOI: 10.1021/acs.jpcb.4c04901Google ScholarThere is no corresponding record for this reference.
- 115Sanbonmatsu, K. Y.; Joseph, S.; Tung, C.-S. Simulating movement of tRNA into the ribosome during decoding. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15854– 15859, DOI: 10.1073/pnas.0503456102Google ScholarThere is no corresponding record for this reference.
- 116Li, R.; Macnamara, L.; Leuchter, J.; Alexander, R.; Cho, S. MD Simulations of tRNA and Aminoacyl-tRNA Synthetases: Dynamics, Folding, Binding, and Allostery. Int. J. Mol. Sci. 2015, 16, 15872– 15902, DOI: 10.3390/ijms160715872Google ScholarThere is no corresponding record for this reference.
- 117Patel, S.; Hosur, R. V. Replica exchange molecular dynamics simulations reveal self-association sites in M-Crystallin caused by mutations provide insights of cataract. Sci. Rep. 2021, 11, 23270 DOI: 10.1038/s41598-021-02728-8Google ScholarThere is no corresponding record for this reference.
- 118Stelzl, L. S.; Hummer, G. Kinetics from Replica Exchange Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 3927– 3935, DOI: 10.1021/acs.jctc.7b00372Google ScholarThere is no corresponding record for this reference.
- 119Feig, M.; Nawrocki, G.; Yu, I.; Wang, P.; Sugita, Y. Challenges and opportunities in connecting simulations with experiments via molecular dynamics of cellular environments. J. Phys. Conf. Ser. 2018, 1036, 012010 DOI: 10.1088/1742-6596/1036/1/012010Google ScholarThere is no corresponding record for this reference.
- 120Kumari, I.; Sandhu, P.; Ahmed, M.; Akhter, Y. Molecular Dynamics Simulations, Challenges and Opportunities: A Biologist’s Prospective. Curr. Protein Pept. Sci. 2017, 18, 1163– 1179, DOI: 10.2174/1389203718666170622074741Google ScholarThere is no corresponding record for this reference.
- 121Senn, H. M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed. 2009, 48, 1198– 1229, DOI: 10.1002/anie.200802019Google ScholarThere is no corresponding record for this reference.
- 122Lopes, P. E. M.; Guvench, O.; MacKerell, A. D. Current Status of Protein Force Fields for Molecular Dynamics Simulations. In Molecular Modeling of Proteins; Kukol, A., Ed.; Springer: New York, New York, NY, 2015; Vol. 1215, pp 47– 71.Google ScholarThere is no corresponding record for this reference.
- 123McMillin, D. R. Interatomic Repulsion and the Pauli Principle. J. Chem. Educ. 2021, 98, 2912– 2918, DOI: 10.1021/acs.jchemed.1c00326Google ScholarThere is no corresponding record for this reference.
- 124Guvench, O.; MacKerell, A. D. Comparison of Protein Force Fields for Molecular Dynamics Simulations. In Molecular Modeling of Proteins; Kukol, A., Ed.; Humana Press: Totowa, NJ, 2008; Vol. 443, pp 63– 88.Google ScholarThere is no corresponding record for this reference.
- 125Warshel, A.; Sharma, P. K.; Kato, M. Electrostatic Basis for Enzyme Catalysis. Chem. Rev. 2006, 106, 3210– 3235, DOI: 10.1021/cr0503106Google ScholarThere is no corresponding record for this reference.
- 126Van Der Kamp, M. W.; Mulholland, A. J. Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Computational Enzymology. Biochemistry 2013, 52, 2708– 2728, DOI: 10.1021/bi400215wGoogle ScholarThere is no corresponding record for this reference.
- 127Magliery, T. J. Protein stability: computation, sequence statistics, and new experimental methods. Curr. Opin. Struct. Biol. 2015, 33, 161– 168, DOI: 10.1016/j.sbi.2015.09.002Google ScholarThere is no corresponding record for this reference.
- 128Singh, A.; Upadhyay, V.; Upadhyay, A. K.; Singh, S. M.; Panda, A. K. Protein recovery from inclusion bodies of Escherichia coli using mild solubilization process. Microb. Cell Factories 2015, 14, 41 DOI: 10.1186/s12934-015-0222-8Google ScholarThere is no corresponding record for this reference.
- 129Sormanni, P.; Aprile, F. A.; Vendruscolo, M. The CamSol Method of Rational Design of Protein Mutants with Enhanced Solubility. J. Mol. Biol. 2015, 427, 478– 490, DOI: 10.1016/j.jmb.2014.09.026Google ScholarThere is no corresponding record for this reference.
- 130Li, B.; Ming, D. GATSol, an enhanced predictor of protein solubility through the synergy of 3D structure graph and large language modeling. BMC Bioinf. 2024, 25, 204 DOI: 10.1186/s12859-024-05820-8Google ScholarThere is no corresponding record for this reference.
- 131Tan, Y.; Zheng, J.; Hong, L.; Zhou, B. ProtSolM: Protein Solubility Prediction with Multi-modal Features, arXiv:2406.19744. arXiv.org e-Print archive. https://arxiv.org/abs/2406.19744. 2024.Google ScholarThere is no corresponding record for this reference.
- 132Ghosh, D.; Biswas, A.; Radhakrishna, M. Advanced computational approaches to understand protein aggregation. Biophys. Rev. 2024, 5, 021302 DOI: 10.1063/5.0180691Google ScholarThere is no corresponding record for this reference.
- 133Oeller, M.; Kang, R.; Bell, R. Sequence-based prediction of pH-dependent protein solubility using CamSol. Brief. Bioinform. 2023, 24, bbad004 DOI: 10.1093/bib/bbad004Google ScholarThere is no corresponding record for this reference.
- 134Kimmel, B. R.; Mrksich, M. Development of an Enzyme-Inhibitor Reaction Using Cellular Retinoic Acid Binding Protein II for One-Pot Megamolecule Assembly. Chem. - Eur. J. 2021, 27, 17843– 17848, DOI: 10.1002/chem.202103059Google ScholarThere is no corresponding record for this reference.
- 135Kimmel, B. R.; Modica, J. A.; Parker, K.; Dravid, V.; Mrksich, M. Solid-Phase Synthesis of Megamolecules. J. Am. Chem. Soc. 2020, 142, 4534– 4538, DOI: 10.1021/jacs.9b12003Google ScholarThere is no corresponding record for this reference.
- 136Adomanis, R.; Phan, N.; Walter, G.; Kimmel, B. R. Modular Nanobody Conjugates with Controlled Topology Using Genetically Encoded Non-canonical Amino Acids. Preprint at https://doi.org/10.1101/2025.11.27.691038. 2025.Google ScholarThere is no corresponding record for this reference.
- 137Rosace, A.; Bennett, A.; Oeller, M. Automated optimization of solubility and conformational stability of antibodies and proteins. Nat. Commun. 2023, 14, 1937 DOI: 10.1038/s41467-023-37668-6Google ScholarThere is no corresponding record for this reference.
- 138Kuriata, A.; Iglesias, V.; Pujols, J. Aggrescan3D (A3D) 2.0: prediction and engineering of protein solubility. Nucleic Acids Res. 2019, 47, W300– W307, DOI: 10.1093/nar/gkz321Google ScholarThere is no corresponding record for this reference.
- 139Hirsch, M.; Desai, R. R.; Annaswamy, S.; Keatinge-Clay, A. T. Mutagenesis Supports AlphaFold Prediction of How Modular Polyketide Synthase Acyl Carrier Proteins Dock With Downstream Ketosynthases. Proteins:Struct., Funct., Bioinf. 2024, 92, 1375– 1384, DOI: 10.1002/prot.26733Google ScholarThere is no corresponding record for this reference.
- 140Araki, M.; Ekimoto, T.; Takemura, K. Molecular Dynamics Unveils Multiple-Site Binding of Inhibitors with Reduced Activity on the Surface of Dihydrofolate Reductase. J. Am. Chem. Soc. 2024, 146, 28685– 28695, DOI: 10.1021/jacs.4c04648Google ScholarThere is no corresponding record for this reference.
- 141Pimtawong, T.; Ren, J.; Lee, J.; Lee, H.-M.; Na, D. A review on computational models for predicting protein solubility. J. Microbiol. 2025, 63, 2408001 DOI: 10.71150/jm.2408001Google ScholarThere is no corresponding record for this reference.
- 142Navarro, S.; Ventura, S. Computational methods to predict protein aggregation. Curr. Opin. Struct. Biol. 2022, 73, 102343 DOI: 10.1016/j.sbi.2022.102343Google ScholarThere is no corresponding record for this reference.
- 143Prediction and Evaluation of Protein Aggregation with Computational Methods. In Methods in Molecular Biology; Springer US: New York, NY, 2025; pp 299– 314 DOI: 10.1007/978-1-0716-4196-5_17 .Google ScholarThere is no corresponding record for this reference.
- 144Santos, J.; Pujols, J.; Pallarès, I.; Iglesias, V.; Ventura, S. Computational prediction of protein aggregation: Advances in proteomics, conformation-specific algorithms and biotechnological applications. Comput. Struct. Biotechnol. J. 2020, 18, 1403– 1413, DOI: 10.1016/j.csbj.2020.05.026Google ScholarThere is no corresponding record for this reference.
- 145Arnold, F. H. Design by Directed Evolution. Acc. Chem. Res. 1998, 31, 125– 131, DOI: 10.1021/ar960017fGoogle ScholarThere is no corresponding record for this reference.
- 146Arnold, F. H. Directed evolution: Creating biocatalysts for the future. Chem. Eng. Sci. 1996, 51, 5091– 5102, DOI: 10.1016/S0009-2509(96)00288-6Google ScholarThere is no corresponding record for this reference.
- 147Cobb, R. E.; Chao, R.; Zhao, H. Directed evolution: Past, present, and future. AIChE J. 2013, 59, 1432– 1440, DOI: 10.1002/aic.13995Google ScholarThere is no corresponding record for this reference.
- 148Yang, J.; Lal, R. G.; Bowden, J. C. Active learning-assisted directed evolution. Nat. Commun. 2025, 16, 714 DOI: 10.1038/s41467-025-55987-8Google ScholarThere is no corresponding record for this reference.
- 149Terashi, G.; Wang, X.; Maddhuri Venkata Subramaniya, S. R.; Tesmer, J. J. G.; Kihara, D. Residue-wise local quality estimation for protein models from cryo-EM maps. Nat. Methods 2022, 19, 1116– 1125, DOI: 10.1038/s41592-022-01574-4Google ScholarThere is no corresponding record for this reference.
- 150Graille, M.; Sacquin-Mora, S.; Taly, A. Best Practices of Using AI-Based Models in Crystallography and Their Impact in Structural Biology. J. Chem. Inf. Model. 2023, 63, 3637– 3646, DOI: 10.1021/acs.jcim.3c00381Google ScholarThere is no corresponding record for this reference.
- 151Wang, X.; Zhu, H.; Terashi, G.; Taluja, M.; Kihara, D. DiffModeler: large macromolecular structure modeling for cryo-EM maps using a diffusion model. Nat. Methods 2024, 21, 2307– 2317, DOI: 10.1038/s41592-024-02479-0Google ScholarThere is no corresponding record for this reference.
- 152Serapian, S. A.; Crosby, J.; Crump, M. P.; Van Der Kamp, M. W. Path to Actinorhodin: Regio- and Stereoselective Ketone Reduction by a Type II Polyketide Ketoreductase Revealed in Atomistic Detail. JACS Au 2022, 2, 972– 984, DOI: 10.1021/jacsau.2c00086Google ScholarThere is no corresponding record for this reference.
- 153Shukla, V. K.; Karunanithy, G.; Vallurupalli, P.; Hansen, D. F. A combined NMR and deep neural network approach for enhancing the spectral resolution of aromatic side chains in proteins. Sci. Adv. 2024, 10, eadr2155 DOI: 10.1126/sciadv.adr2155Google ScholarThere is no corresponding record for this reference.
- 154Drake, Z. C.; Fowler, A. G.; Blum, A. A.; Lindert, S. Enhanced Protein Complex Prediction via Rosetta, AlphaFold, and Nondifferential Covalent Labeling Mass Spectrometry. J. Phys. Chem. B 2025, 129, 6489– 6497, DOI: 10.1021/acs.jpcb.5c02872Google ScholarThere is no corresponding record for this reference.
- 155Lee, C. Y.; Hubrich, D.; Varga, J. K. Systematic discovery of protein interaction interfaces using AlphaFold and experimental validation. Mol. Syst. Biol. 2024, 20, 75– 97, DOI: 10.1038/s44320-023-00005-6Google ScholarThere is no corresponding record for this reference.
- 156Koehler Leman, J.; Künze, G. Recent Advances in NMR Protein Structure Prediction with ROSETTA. Int. J. Mol. Sci. 2023, 24, 7835, DOI: 10.3390/ijms24097835Google ScholarThere is no corresponding record for this reference.
- 157Alshammari, M.; Wriggers, W.; Sun, J.; He, J. Refinement of AlphaFold2 models against experimental and hybrid cryo-EM density maps. QRB Discovery 2022, 3, e16 DOI: 10.1017/qrd.2022.13Google ScholarThere is no corresponding record for this reference.
- 158Humphreys, I. R.; Pei, J.; Baek, M. Computed structures of core eukaryotic protein complexes. Science 2021, 374, eabm4805 DOI: 10.1126/science.abm4805Google ScholarThere is no corresponding record for this reference.
- 159Bordin, N.; Sillitoe, I.; Nallapareddy, V. AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms. Commun. Biol. 2023, 6, 160 DOI: 10.1038/s42003-023-04488-9Google ScholarThere is no corresponding record for this reference.
- 160Wang, H.; Wang, J. How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci. 2017, 26, 32– 39, DOI: 10.1002/pro.3022Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACS Engineering Au
© 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
Figure 1

Figure 1. From sequence to protein structure and conformational behavior. (A) Biological information transfer follows a deterministic pathway from DNA to RNA to protein, linking the encoded sequence information to the emergent molecular function and dynamics. (B) Input amino acid sequences serve as the basis for predictive modeling frameworks. (C) Sequence-informed A.I./ML frameworks trained on sequence and structural ensemble data learn the mapping between linear sequences and conformation space. (D) The resulting structural ensemble offers a data-driven view of protein flexibility and structural diversity derived directly from the amino acid sequence. Reprinted or adapted with permission under a CC-BY 3.0 License from Ille et al. (14) Copyright 2025 AIP Publishing.
Figure 2

Figure 2. Conceptual view of the protein functional universe. The diagram maps the relationships among sequence, structure, and function spaces. Each circle represents an individual protein defined by its amino acid sequence, 3D folds, and biological activity. The blue circles correspond to proteins accessible through natural evolution or traditional protein engineering, primarily clustered within well-explored regions (yellow). Gray circles indicate proteins that remain uncharacterized and lie within the unexplored sequence–structure–function space. The red circles represent proteins accessible through ML-driven de novo design, which extends exploration beyond natural boundaries into previously inaccessible regions. In this framework, sequence space (top layer) is linked to structure space (middle layer) and ultimately to function space (bottom layer), with A.I. methods systematically probing across all three layers. Reprinted or adapted with permission under a CC-BY 4.0 License from Zhang et al. (22) Copyright 2025 MDPI.
Figure 3
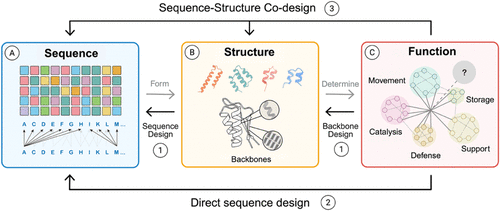
Figure 3. Overview of the current protein design dogma. Traditional protein science is often described as a one-way flow in which (A) amino acid sequences give rise to (B) folded structures, which in turn underpin (C) biological function. Modern de novo design inverts this logic: researchers now begin with the desired function and work backward to identify compatible folds and sequences. Current computational frameworks align with three broad strategies: (1) two-stage design, in which structural generators such as ROSETTA, RoseTTAFold, or PyRosetta first propose candidate protein backbones that are then optimized by sequence design engines; (2) sequence-driven methods, exemplified by AlphaFold2 and ColabFold, which predict protein structures directly from amino acid sequence information and are widely used to validate or filter design candidates; and (3) coguided approaches, including multitrack RoseTTAFold variants (RF, RFNA, RFAA) and diffusion-based models (RFDiffusion), which integrate amino acid sequence and protein structure generation simultaneously. These complementary strategies extend the protein design beyond natural sequence–structure relationships, enabling a function-first exploration of protein space. Reprinted or adapted with permission under a CC-BY 4.0 License from Zhang et al. (22) Copyright 2025 MDPI.
Figure 4

Figure 4. Overview of the A.I.-driven protein design toolbox. According to their functional roles in A.I.-driven generative protein design, the protein design toolbox can be divided into five categories: (A) structure prediction frameworks (e.g., AlphaFold2, RoseTTAFold) that validate fold accuracy; (B) de novo backbone generators (RFDiffusion, RFDiffusionAA) that embed motifs or active sites into novel folds; (C) fixed-backbone sequence designers (LigandMPNN) that optimize sequences against a defined structural context; (D) sequence generation models (ProteinMPNN), which not only perform fixed-backbone optimization but also function as a generative sampler of amino acid sequences; and (E) sequence–structure cogeneration and refinement frameworks (PLACER), which jointly optimize side chains, ligands, and active-site geometry. Reprinted or adapted with permission under a CC-BY 4.0 License from Zhang et al. (22) Copyright 2025 MDPI.
Figure 5
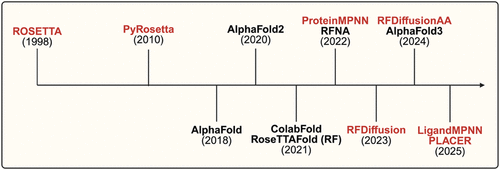
Figure 5. Timeline of major developments in protein structure prediction (black) and design methodologies (red). Following early innovations such as ROSETTA (1998) and PyRosetta (2010), the field saw nearly two decades of incremental progress before the emergence of transformative A.I.-based models such as AlphaFold2 (2020). Since then, breakthroughs in generative frameworks, including ProteinMPNN, RFDiffusion, and LigandMPNN, have rapidly expanded, marking a shift toward integrated prediction-design pipelines.
Figure 6

Figure 6. Computational strategies for evaluating amino acid sequence perturbations. (A) Structural stability analysis introduces mutations into a sequence and applies ab initio folding to predict conformational shifts, highlighting favorable and unfavorable perturbations. (B) Binding affinity analysis docks protein constituents, incorporates mutations, and estimates changes in binding free energy (ΔΔG) to evaluate the interaction stability. (C) Interface hotspot probing systematically mutates residues at binding interfaces to pinpoint the positions that are most critical for binding energetics.
Figure 7

Figure 7. Computational evaluation of the biological and functional properties of proteins. (A) Molecular dynamics and catalysis simulate mutated proteins in solvated environments to capture conformational flexibility and catalytic changes through trajectory analyses. Hybrid pipelines that integrate molecular dynamics (MD) with ROSETTA and directed evolution have yielded efficient de novo and redesigned biocatalysts, such as HG3.17 and BH32.14, whose catalytic power emerges from MD-guided active-site reorganization and solvent shielding. (B) Solubility analysis predicts the effects of amino acid sequence variation on protein solubility by comparing mutant distributions to wild-type benchmarks. CamSol-based workflows enable the rational optimization of both solubility and conformational stability, as demonstrated for six antibodies (including two approved therapeutics), enhancing developability without compromising binding. (137) (C) Aggregation propensity assesses structural and sequence features to identify residues or motifs that drive aggregation, distinguishing soluble variants from aggregation-prone variants. Using Aggrescan3D, researchers computationally minimized aggregation hotspots to engineer green fluorescent protein (GFP) mutants with significantly improved solubility and reduced aggregation, resulting in a fast-folding, aggregation-resistant variant. (138) Together, these approaches extend computational evaluation to capture dynamic solubility and aggregation behaviors that critically influence protein performance in physiological and industrial contexts.
Figure 8

Figure 8. Conceptual framework contrasting traditional and A.I.-assisted directed evolution (DE) workflows. The diagram is divided into two pathways: the upper route represents conventional DE, where (A) natural sequence diversity is explored, (B) mutational libraries are generated, (C) variants are expressed, and (E) high-throughput screening identifies improved candidates through iterative experimental cycles. The lower route introduces A.I./ML-assisted or hybrid methodologies, in which (D) supervised models with uncertainty quantification learn the sequence-fitness landscape and use acquisition functions to propose new variants, balancing exploration (high uncertainty) and exploitation (high predicted fitness). These feedback-driven optimization strategies accelerate variant discovery with a reduced screening effort. Combined approaches, such as active learning-assisted directed evolution (ALDE), have yielded (F) optimized protoglobin-based biocatalysts for nonnative cyclopropanation reactions, enhancing their activity, selectivity, and stability while minimizing experimental costs. (148)
Figure 9
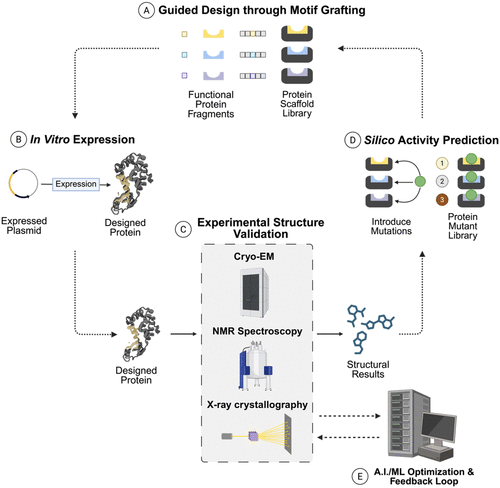
Figure 9. Experimental–computational pipeline for protein engineering. (A) Protein mutant libraries are generated by introducing sequence variations across the regions of interest. AlphaFold-guided domain-motif design (e.g., FBXO23-STX1B) has revealed novel regulatory interfaces relevant to therapeutic target discovery. (155) (B) Mutants are expressed in Escherichia coli, yeast, or mammalian systems to generate protein ensembles for screening; such expression-labeling pipelines support enzyme and biocatalyst development used in pharmaceuticals and green chemistry. (154) (C) Structural characterization via cryo-EM, NMR, and X-ray crystallography (and hybrid methods) refined with predictive models such as AlphaFold2 or ROSETTA resolves folding and conformational dynamics, as shown in ribosomal complex refinements in NMR-ROSETTA modeling of ubiquitin. (156,157) (D) Functional screening evaluates the activity and binding properties of mutant sets, such as hydroxyl radical footprinting, to identify active-site or interface residues that control activity and stability, as applied to Hsp90-co-chaperone systems and engineered oxidoreductase. (154) (E) Finally, A.I./ML integration combines experimental data with modeling to predict next-generation variants, accelerating industrial enzyme design, antibody optimization, and biosensor development.
References
This article references 160 other publications.
- 1Yu, Y.; Hu, C.; Xia, L.; Wang, J. Artificial Metalloenzyme Design with Unnatural Amino Acids and Non-Native Cofactors. ACS Catal. 2018, 8, 1851– 1863, DOI: 10.1021/acscatal.7b03754There is no corresponding record for this reference.
- 2Mirts, E. N.; Bhagi-Damodaran, A.; Lu, Y. Understanding and Modulating Metalloenzymes with Unnatural Amino Acids, Non-Native Metal Ions, and Non-Native Metallocofactors. Acc. Chem. Res. 2019, 52, 935– 944, DOI: 10.1021/acs.accounts.9b00011There is no corresponding record for this reference.
- 3Mann, S. I.; Nayak, A.; Gassner, G. T.; Therien, M. J.; DeGrado, W. F. De Novo Design, Solution Characterization, and Crystallographic Structure of an Abiological Mn–Porphyrin-Binding Protein Capable of Stabilizing a Mn(V) Species. J. Am. Chem. Soc. 2021, 143, 252– 259, DOI: 10.1021/jacs.0c10136There is no corresponding record for this reference.
- 4Bergman, M. T.; Xiao, X.; Hall, C. K. In Silico Design and Analysis of Plastic-Binding Peptides. J. Phys. Chem. B 2023, 127, 8370– 8381, DOI: 10.1021/acs.jpcb.3c04319There is no corresponding record for this reference.
- 5García-Moreno, P. J. Recent advances in the production of emulsifying peptides with the aid of proteomics and bioinformatics. Curr. Opin. Food Sci. 2023, 51, 101039 DOI: 10.1016/j.cofs.2023.101039There is no corresponding record for this reference.
- 6Ndochinwa, G. O.; Wang, Q. Y.; Okoro, N. O. New advances in protein engineering for industrial applications: Key takeaways. Open Life Sci. 2024, 19, 20220856 DOI: 10.1515/biol-2022-0856There is no corresponding record for this reference.
- 7Marcos, E.; Silva, D. Essentials of de novo protein design: Methods and applications. WIREs Comput. Mol. Sci. 2018, 8 (6), e1374 DOI: 10.1002/wcms.1374There is no corresponding record for this reference.
- 8Huang, P.-S.; Boyken, S. E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320– 327, DOI: 10.1038/nature19946There is no corresponding record for this reference.
- 9Woolfson, D. N. A Brief History of De Novo Protein Design: Minimal, Rational, and Computational. J. Mol. Biol. 2021, 433, 167160 DOI: 10.1016/j.jmb.2021.167160There is no corresponding record for this reference.
- 10Dill, K. A.; Ozkan, S. B.; Shell, M. S.; Weikl, T. R. Protein Folding Problem. Annu. Rev. Biophys. 2008, 37, 289– 316, DOI: 10.1146/annurev.biophys.37.092707.153558There is no corresponding record for this reference.
- 11Kocher, C. D.; Dill, K. A. Origins of life: The Protein Folding Problem all over again?. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2315000121 DOI: 10.1073/pnas.2315000121There is no corresponding record for this reference.
- 12Chen, S.-J.; Hassan, M.; Jernigan, R. L. Protein folds vs. protein folding: Differing questions, different challenges. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2214423119 DOI: 10.1073/pnas.2214423119There is no corresponding record for this reference.
- 13Kiss, G.; Çelebi-Ölçüm, N.; Moretti, R.; Baker, D.; Houk, K. N. Computational Enzyme Design. Angew. Chem., Int. Ed. 2013, 52, 5700– 5725, DOI: 10.1002/anie.201204077There is no corresponding record for this reference.
- 14Ille, A. M.; Anas, E.; Mathews, M. B.; Burley, S. K. From sequence to protein structure and conformational dynamics with artificial intelligence/machine learning. Struct. Dyn. 2025, 12, 030902 DOI: 10.1063/4.0000765There is no corresponding record for this reference.
- 15Anfinsen, C. B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223– 230, DOI: 10.1126/science.181.4096.223There is no corresponding record for this reference.
- 16Voigt, C. A.; Mayo, S. L.; Arnold, F. H.; Wang, Z.-G. Computational method to reduce the search space for directed protein evolution. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 3778– 3783, DOI: 10.1073/pnas.051614498There is no corresponding record for this reference.
- 17Kuhlman, B.; Baker, D. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10383– 10388, DOI: 10.1073/pnas.97.19.10383There is no corresponding record for this reference.
- 18Sleator, R. D. Solving the protein folding problem···. FEBS Lett. 2024, 598, 2831– 2835, DOI: 10.1002/1873-3468.15043There is no corresponding record for this reference.
- 19Watson, J. L.; Juergens, D.; Bennett, N. R. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089– 1100, DOI: 10.1038/s41586-023-06415-8There is no corresponding record for this reference.
- 20Leveson-Gower, R. B. Designing Enzymatic Reactivity with an Expanded Palette. ChemBioChem 2025, 26, e202500076 DOI: 10.1002/cbic.202500076There is no corresponding record for this reference.
- 21Hartman, M. C. T. Non-canonical Amino Acid Substrates of Escherichia coli Aminoacyl-tRNA Synthetases. ChemBioChem 2022, 23, e202100299 DOI: 10.1002/cbic.202100299There is no corresponding record for this reference.
- 22Zhang, G.; Liu, C.; Lu, J.; Zhang, S.; Zhu, L. The Role of AI-Driven De Novo Protein Design in the Exploration of the Protein Functional Universe. Biology 2025, 14, 1268 DOI: 10.3390/biology14091268There is no corresponding record for this reference.
- 23Rohl, C. A.; Strauss, C. E. M.; Misura, K. M. S.; Baker, D. Protein Structure Prediction Using Rosetta. Methods Enzymol. 2004, 383, 66– 93, DOI: 10.1016/S0076-6879(04)83004-0There is no corresponding record for this reference.
- 24Simons, K. T.; Kooperberg, C.; Huang, E.; Baker, D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and bayesian scoring functions. J. Mol. Biol. 1997, 268, 209– 225, DOI: 10.1006/jmbi.1997.0959There is no corresponding record for this reference.
- 25Leman, J. K.; Weitzner, B. D.; Lewis, S. M. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 2020, 17, 665– 680, DOI: 10.1038/s41592-020-0848-2There is no corresponding record for this reference.
- 26Das, R.; Baker, D. Macromolecular Modeling with Rosetta. Annu. Rev. Biochem. 2008, 77, 363– 382, DOI: 10.1146/annurev.biochem.77.062906.171838There is no corresponding record for this reference.
- 27Kaufmann, K. W.; Meiler, J. Using RosettaLigand for Small Molecule Docking into Comparative Models. PLoS One 2012, 7, e50769 DOI: 10.1371/journal.pone.0050769There is no corresponding record for this reference.
- 28Lemmon, G.; Kaufmann, K.; Meiler, J. Prediction of HIV-1 Protease/Inhibitor Affinity using RosettaLigand. Chem. Biol. Drug Des. 2012, 79, 888– 896, DOI: 10.1111/j.1747-0285.2012.01356.xThere is no corresponding record for this reference.
- 29Chaudhury, S.; Lyskov, S.; Gray, J. J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 2010, 26, 689– 691, DOI: 10.1093/bioinformatics/btq007There is no corresponding record for this reference.
- 30Le, K. H.; Adolf-Bryfogle, J.; Klima, J. C. PyRosetta Jupyter Notebooks Teach Biomolecular Structure Prediction and Design. Biophysicist 2021, 2, 108– 122, DOI: 10.35459/tbp.2019.000147There is no corresponding record for this reference.
- 31Ford, A. S.; Weitzner, B. D.; Bahl, C. D. Integration of the Rosetta suite with the python software stack via reproducible packaging and core programming interfaces for distributed simulation. Protein Sci. 2020, 29, 43– 51, DOI: 10.1002/pro.3721There is no corresponding record for this reference.
- 32Van Stappen, C.; Deng, Y.; Liu, Y. Designing Artificial Metalloenzymes by Tuning of the Environment beyond the Primary Coordination Sphere. Chem. Rev. 2022, 122, 11974– 12045, DOI: 10.1021/acs.chemrev.2c00106There is no corresponding record for this reference.
- 33Tivon, B.; Wiese, J.; Müller, M. P. Computational Design of Lysine Targeting Covalent Binders Using Rosetta. J. Chem. Inf. Model. 2025, 65, 5612– 5622, DOI: 10.1021/acs.jcim.5c00212There is no corresponding record for this reference.
- 34Jumper, J.; Evans, R.; Pritzel, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583– 589, DOI: 10.1038/s41586-021-03819-2There is no corresponding record for this reference.
- 35Tunyasuvunakool, K.; Adler, J.; Wu, Z. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590– 596, DOI: 10.1038/s41586-021-03828-1There is no corresponding record for this reference.
- 36Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)─Round XIV. Proteins:Struct., Funct., Bioinf. 2021, 89, 1607– 1617, DOI: 10.1002/prot.26237There is no corresponding record for this reference.
- 37Kovalevskiy, O.; Mateos-Garcia, J.; Tunyasuvunakool, K. AlphaFold two years on: Validation and impact. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2315002121 DOI: 10.1073/pnas.2315002121There is no corresponding record for this reference.
- 38Schneider, B.; Sweeney, B. A.; Bateman, A. When will RNA get its AlphaFold moment?. Nucleic Acids Res. 2023, 51, 9522– 9532, DOI: 10.1093/nar/gkad726There is no corresponding record for this reference.
- 39Terwilliger, T. C.; Liebschner, D.; Croll, T. I. AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nat. Methods 2024, 21, 110– 116, DOI: 10.1038/s41592-023-02087-4There is no corresponding record for this reference.
- 40Mirdita, M.; Schütze, K.; Moriwaki, Y. ColabFold: making protein folding accessible to all. Nat. Methods 2022, 19, 679– 682, DOI: 10.1038/s41592-022-01488-1There is no corresponding record for this reference.
- 41Kim, G.; Lee, S.; Levy Karin, E. Easy and accurate protein structure prediction using ColabFold. Nat. Protoc. 2025, 20, 620– 642, DOI: 10.1038/s41596-024-01060-5There is no corresponding record for this reference.
- 42Kalogeropoulos, K.; Bohn, M. F.; Jenkins, D. E. A comparative study of protein structure prediction tools for challenging targets: Snake venom toxins. Toxicon 2024, 238, 107559 DOI: 10.1016/j.toxicon.2023.107559There is no corresponding record for this reference.
- 43Baek, M.; DiMaio, F.; Anishchenko, I. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871– 876, DOI: 10.1126/science.abj8754There is no corresponding record for this reference.
- 44Baek, M.; McHugh, R.; Anishchenko, I. Accurate prediction of protein–nucleic acid complexes using RoseTTAFoldNA. Nat. Methods 2024, 21, 117– 121, DOI: 10.1038/s41592-023-02086-5There is no corresponding record for this reference.
- 45Krishna, R.; Wang, J.; Ahern, W. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science 2024, 384, eadl2528 DOI: 10.1126/science.adl2528There is no corresponding record for this reference.
- 46Liu, S.; Wu, K.; Chen, C. Obtaining protein foldability information from computational models of AlphaFold2 and RoseTTAFold. Comput. Struct. Biotechnol. J. 2022, 20, 4481– 4489, DOI: 10.1016/j.csbj.2022.08.034There is no corresponding record for this reference.
- 47Wayment-Steele, H. K.; Ojoawo, A.; Otten, R. Predicting multiple conformations via sequence clustering and AlphaFold2. Nature 2024, 625, 832– 839, DOI: 10.1038/s41586-023-06832-9There is no corresponding record for this reference.
- 48Casadevall, G.; Duran, C.; Osuna, S. AlphaFold2 and Deep Learning for Elucidating Enzyme Conformational Flexibility and Its Application for Design. JACS Au 2023, 3, 1554– 1562, DOI: 10.1021/jacsau.3c00188There is no corresponding record for this reference.
- 49Vallejo, W.; Díaz-Uribe, C.; Fajardo, C. Google Colab and Virtual Simulations: Practical e-Learning Tools to Support the Teaching of Thermodynamics and to Introduce Coding to Students. ACS Omega 2022, 7, 7421– 7429, DOI: 10.1021/acsomega.2c00362There is no corresponding record for this reference.
- 50Adiyaman, R.; Edmunds, N. S.; Genc, A. G.; Alharbi, S. M. A.; McGuffin, L. J. Improvement of protein tertiary and quaternary structure predictions using the ReFOLD refinement method and the AlphaFold2 recycling process. Bioinforma. Adv. 2023, 3 (1), vbad078 DOI: 10.1093/bioadv/vbad078There is no corresponding record for this reference.
- 51Ahern, W.; Yim, J.; Tischer, D. Atom level enzyme active site scaffolding using RFdiffusion2. Nat. Methods 2026, 23, 96– 105, DOI: 10.1038/s41592-025-02975-xThere is no corresponding record for this reference.
- 52Wang, W.; Feng, C.; Han, R. trRosettaRNA: automated prediction of RNA 3D structure with transformer network. Nat. Commun. 2023, 14, 7266 DOI: 10.1038/s41467-023-42528-4There is no corresponding record for this reference.
- 53Corso, G.; Stärk, H.; Jing, B.; Barzilay, R.; Jaakkola, T. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking, arXiv:2210.01776. arXiv.org e-Print archive. https://arxiv.org/abs/2210.01776. 2023.There is no corresponding record for this reference.
- 54Alamdari, S. Protein generation with evolutionary diffusion: sequence is all you need. Preprint at https://doi.org/10.1101/2023.09.11.556673. 2023.There is no corresponding record for this reference.
- 55Chu, A. E.; Kim, J.; Cheng, L. An all-atom protein generative model. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2311500121 DOI: 10.1073/pnas.2311500121There is no corresponding record for this reference.
- 56Dauparas, J. Robust deep learning based protein sequence design using ProteinMPNN.There is no corresponding record for this reference.
- 57Sumida, K. H.; Núñez-Franco, R.; Kalvet, I. Improving Protein Expression, Stability, and Function with ProteinMPNN. J. Am. Chem. Soc. 2024, 146, 2054– 2061, DOI: 10.1021/jacs.3c10941There is no corresponding record for this reference.
- 58De Haas, R. J.; Brunette, N.; Goodson, A. Rapid and automated design of two-component protein nanomaterials using ProteinMPNN. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2314646121 DOI: 10.1073/pnas.2314646121There is no corresponding record for this reference.
- 59Dauparas, J.; Lee, G. R.; Pecoraro, R. Atomic context-conditioned protein sequence design using LigandMPNN. Nat. Methods 2025, 22, 717– 723, DOI: 10.1038/s41592-025-02626-1There is no corresponding record for this reference.
- 60Clark-Elsayed, A. Comparing LigandMPNN and Directed Evolution for Altering the Effector-Binding Site in the RamR Transcription Factor.There is no corresponding record for this reference.
- 61An, L.; Said, M.; Tran, L. Binding and sensing diverse small molecules using shape-complementary pseudocycles. Science 2024, 385, 276– 282, DOI: 10.1126/science.adn3780There is no corresponding record for this reference.
- 62Agu, P. C.; Afiukwa, C. A.; Orji, O. U. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 2023, 13, 13398 DOI: 10.1038/s41598-023-40160-2There is no corresponding record for this reference.
- 63Anishchenko, I. Modeling protein-small molecule conformational ensembles with ChemNet. Preprint at https://doi.org/10.1101/2024.09.25.614868. 2024.There is no corresponding record for this reference.
- 64Lauko, A.; Pellock, S. J.; Sumida, K. H. Computational design of serine hydrolases. Science 2025, 388, eadu2454 DOI: 10.1126/science.adu2454There is no corresponding record for this reference.
- 65Park, H.; Zhou, G.; Baek, M.; Baker, D.; DiMaio, F. Force Field Optimization Guided by Small Molecule Crystal Lattice Data Enables Consistent Sub-Angstrom Protein–Ligand Docking. J. Chem. Theory Comput. 2021, 17, 2000– 2010, DOI: 10.1021/acs.jctc.0c01184There is no corresponding record for this reference.
- 66Garcia, M.; Dixit, S. M.; Rocklin, G. J. Evaluating zero-shot prediction of protein design success by AlphaFold, ESMFold, and ProteinMPNN.There is no corresponding record for this reference.
- 67Kong, Z. ProtFlow: Flow Matching-based Protein Sequence Design with Comprehensive Protein Semantic Distribution Learning and High-quality Generation.There is no corresponding record for this reference.
- 68Elnaggar, A.; Heinzinger, M.; Dallago, C. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112– 7127, DOI: 10.1109/TPAMI.2021.3095381There is no corresponding record for this reference.
- 69Madani, A. ProGen: Language Modeling for Protein Generation, arXiv:2004.03497. arXiv.org e-Print archive. https://arxiv.org/abs/2004.03497. 2020.There is no corresponding record for this reference.
- 70Nijkamp, E.; Ruffolo, J. A.; Weinstein, E. N.; Naik, N.; Madani, A. ProGen2: Exploring the boundaries of protein language models. Cell Syst. 2023, 14, 968– 978.e3, DOI: 10.1016/j.cels.2023.10.002There is no corresponding record for this reference.
- 71Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 2022, 13, 4348 DOI: 10.1038/s41467-022-32007-7There is no corresponding record for this reference.
- 72Nguyen, E.; Poli, M.; Durrant, M. G. Sequence modeling and design from molecular to genome scale with Evo. Science 2024, 386, eado9336 DOI: 10.1126/science.ado9336There is no corresponding record for this reference.
- 73Dalla-Torre, H.; Gonzalez, L.; Mendoza-Revilla, J. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nat. Methods 2025, 22, 287– 297, DOI: 10.1038/s41592-024-02523-zThere is no corresponding record for this reference.
- 74Avsec, Ž.; Latysheva, N.; Cheng, J. Advancing regulatory variant effect prediction with AlphaGenome. Nature 2026, 649, 1206– 1218, DOI: 10.1038/s41586-025-10014-0There is no corresponding record for this reference.
- 75Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction.There is no corresponding record for this reference.
- 76Chai Discovery. Chai-1: Decoding the molecular interactions of life. Preprint at https://doi.org/10.1101/2024.10.10.615955. 2024.There is no corresponding record for this reference.
- 77Ingraham, J. B.; Baranov, M.; Costello, Z. Illuminating protein space with a programmable generative model. Nature 2023, 623, 1070– 1078, DOI: 10.1038/s41586-023-06728-8There is no corresponding record for this reference.
- 78Mille-Fragoso, L. S. Efficient generation of epitope-targeted de novo antibodies with Germinal.There is no corresponding record for this reference.
- 79Pacesa, M.; Nickel, L.; Schellhaas, C. One-shot design of functional protein binders with BindCraft. Nature 2025, 646, 483– 492, DOI: 10.1038/s41586-025-09429-6There is no corresponding record for this reference.
- 80BoltzGen: Toward Universal Binder Design.There is no corresponding record for this reference.
- 81Zhang, O. ODesign: A World Model for Biomolecular Interaction Design, arXiv:2510.22304. arXiv.org e-Print archive. https://arxiv.org/abs/2510.22304. 2025.There is no corresponding record for this reference.
- 82Parks, M. Blind Virtual Screening at Scale: A Scalable End-to-End Pipeline for Blind Docking and Affinity Prediction.There is no corresponding record for this reference.
- 83John, P. S. BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery, arXiv:2411.10548. arXiv.org e-Print archive. https://arxiv.org/abs/2411.10548. 2025.There is no corresponding record for this reference.
- 84Silke, D.; Iskander, J.; Pan, J. ProteinDJ : A high-performance and modular protein design pipeline. Protein Sci. 2026, 35, e70464 DOI: 10.1002/pro.70464There is no corresponding record for this reference.
- 85González-Rodríguez, N.; Chacón-Sánchez, C.; Llorca, O.; Fernández-Leiro, R. Automated and modular protein binder design with BinderFlow. PLOS Comput. Biol. 2025, 21, e1013747 DOI: 10.1371/journal.pcbi.1013747There is no corresponding record for this reference.
- 86Danny, B. Ovo, an Open-Source Ecosystem for De Novo Protein Design.There is no corresponding record for this reference.
- 87Baker, D. An exciting but challenging road ahead for computational enzyme design. Protein Sci. 2010, 19, 1817– 1819, DOI: 10.1002/pro.481There is no corresponding record for this reference.
- 88Beadle, B. M.; Shoichet, B. K. Structural Bases of Stability–function Tradeoffs in Enzymes. J. Mol. Biol. 2002, 321, 285– 296, DOI: 10.1016/S0022-2836(02)00599-5There is no corresponding record for this reference.
- 89Barlow, K. A.; Conchúir, S. Ó.; Thompson, S. Flex ddG: Rosetta Ensemble-Based Estimation of Changes in Protein–Protein Binding Affinity upon Mutation. J. Phys. Chem. B 2018, 122, 5389– 5399, DOI: 10.1021/acs.jpcb.7b11367There is no corresponding record for this reference.
- 90Shringari, S. R.; Giannakoulias, S.; Ferrie, J. J.; Petersson, E. J. Rosetta custom score functions accurately predict ΔΔG of mutations at protein–protein interfaces using machine learning. Chem. Commun. 2020, 56, 6774– 6777, DOI: 10.1039/D0CC01959CThere is no corresponding record for this reference.
- 91Smith, S. T.; Meiler, J. Assessing multiple score functions in Rosetta for drug discovery. PLoS One 2020, 15, e0240450 DOI: 10.1371/journal.pone.0240450There is no corresponding record for this reference.
- 92Alford, R. F.; Leaver-Fay, A.; Jeliazkov, J. R. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031– 3048, DOI: 10.1021/acs.jctc.7b00125There is no corresponding record for this reference.
- 93Tyka, M. D.; Keedy, D. A.; André, I. Alternate States of Proteins Revealed by Detailed Energy Landscape Mapping. J. Mol. Biol. 2011, 405, 607– 618, DOI: 10.1016/j.jmb.2010.11.008There is no corresponding record for this reference.
- 94Planas-Iglesias, J.; Marques, S. M.; Pinto, G. P. Computational design of enzymes for biotechnological applications. Biotechnol. Adv. 2021, 47, 107696 DOI: 10.1016/j.biotechadv.2021.107696There is no corresponding record for this reference.
- 95Guo, H.-B.; Perminov, A.; Bekele, S. AlphaFold2 models indicate that protein sequence determines both structure and dynamics. Sci. Rep. 2022, 12, 10696 DOI: 10.1038/s41598-022-14382-9There is no corresponding record for this reference.
- 96Agarwal, V.; McShan, A. C. The power and pitfalls of AlphaFold2 for structure prediction beyond rigid globular proteins. Nat. Chem. Biol. 2024, 20, 950– 959, DOI: 10.1038/s41589-024-01638-wThere is no corresponding record for this reference.
- 97Abramson, J.; Adler, J.; Dunger, J. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493– 500, DOI: 10.1038/s41586-024-07487-wThere is no corresponding record for this reference.
- 98Friedland, G. D.; Linares, A. J.; Smith, C. A.; Kortemme, T. A Simple Model of Backbone Flexibility Improves Modeling of Side-chain Conformational Variability. J. Mol. Biol. 2008, 380, 757– 774, DOI: 10.1016/j.jmb.2008.05.006There is no corresponding record for this reference.
- 99Kellogg, E. H.; Leaver-Fay, A.; Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins:Struct., Funct., Bioinf. 2011, 79, 830– 838, DOI: 10.1002/prot.22921There is no corresponding record for this reference.
- 100Durham, E.; Dorr, B.; Woetzel, N.; Staritzbichler, R.; Meiler, J. Solvent accessible surface area approximations for rapid and accurate protein structure prediction. J. Mol. Model. 2009, 15, 1093– 1108, DOI: 10.1007/s00894-009-0454-9There is no corresponding record for this reference.
- 101Bertalan, É.; Lešnik, S.; Bren, U.; Bondar, A.-N. Protein-water hydrogen-bond networks of G protein-coupled receptors: Graph-based analyses of static structures and molecular dynamics. J. Struct. Biol. 2020, 212, 107634 DOI: 10.1016/j.jsb.2020.107634There is no corresponding record for this reference.
- 102Weiss, G. A.; Watanabe, C. K.; Zhong, A.; Goddard, A.; Sidhu, S. S. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8950– 8954, DOI: 10.1073/pnas.160252097There is no corresponding record for this reference.
- 103Cunningham, B. C.; Wells, J. A. High-Resolution Epitope Mapping of hGH-Receptor Interactions by Alanine-Scanning Mutagenesis. Science 1989, 244, 1081– 1085, DOI: 10.1126/science.2471267There is no corresponding record for this reference.
- 104Liu, H.; Song, L.; Meng, X. Proline-Mediated Enhancement in Evolvability of Disulfide-Rich Peptides for Discovering Protein Binders. J. Am. Chem. Soc. 2025, 147, 24870– 24883, DOI: 10.1021/jacs.5c07075There is no corresponding record for this reference.
- 105Holden, J. K.; Pavlovicz, R.; Gobbi, A.; Song, Y.; Cunningham, C. N. Computational Site Saturation Mutagenesis of Canonical and Non-Canonical Amino Acids to Probe Protein-Peptide Interactions. Front. Mol. Biosci. 2022, 9, 848689 DOI: 10.3389/fmolb.2022.848689There is no corresponding record for this reference.
- 106Spina, S. C.; Bailey, J.; Kimmel, B. Bind, catalyze, and quantify: a modern protein and enzyme engineering toolbox of genetically encoded non-canonical amino acids Protein Eng. Des. Sel. 2026gzag007 DOI: 10.1093/protein/gzag007 .There is no corresponding record for this reference.
- 107Chen, Y.; Clay, N.; Phan, N. Molecular Matchmakers: Bioconjugation Techniques Enhance Prodrug Potency for Immunotherapy. Mol. Pharmaceutics 2025, 22, 58– 80, DOI: 10.1021/acs.molpharmaceut.4c00867There is no corresponding record for this reference.
- 108Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind Database: Collection of Binding Affinities for Protein–Ligand Complexes with Known Three-Dimensional Structures. J. Med. Chem. 2004, 47, 2977– 2980, DOI: 10.1021/jm030580lThere is no corresponding record for this reference.
- 109Liu, Z.; Su, M.; Han, L. Forging the Basis for Developing Protein–Ligand Interaction Scoring Functions. Acc. Chem. Res. 2017, 50, 302– 309, DOI: 10.1021/acs.accounts.6b00491There is no corresponding record for this reference.
- 110King, B. R.; Sumida, K. H.; Caruso, J. L.; Baker, D.; Zalatan, J. G. Computational Stabilization of a Non-Heme Iron Enzyme Enables Efficient Evolution of New Function. Angew. Chem., Int. Ed. 2025, 64, e202414705 DOI: 10.1002/anie.202414705There is no corresponding record for this reference.
- 111Howlader, M. T. H.; Kagawa, Y.; Miyakawa, A. Alanine Scanning Analyses of the Three Major Loops in Domain II of Bacillus thuringiensis Mosquitocidal Toxin Cry4Aa. Appl. Environ. Microbiol. 2010, 76, 860– 865, DOI: 10.1128/AEM.02175-09There is no corresponding record for this reference.
- 112Paul, R.; Kasahara, K.; Sasaki, J. Unveiling the affinity–stability relationship in anti-measles virus antibodies: a computational approach for hotspots prediction. Front. Mol. Biosci. 2024, 10, 1302737 DOI: 10.3389/fmolb.2023.1302737There is no corresponding record for this reference.
- 113Romero-Rivera, A.; Garcia-Borràs, M.; Osuna, S. Role of Conformational Dynamics in the Evolution of Retro-Aldolase Activity. ACS Catal. 2017, 7, 8524– 8532, DOI: 10.1021/acscatal.7b02954There is no corresponding record for this reference.
- 114Lemkul, J. A. Introductory Tutorials for Simulating Protein Dynamics with GROMACS. J. Phys. Chem. B 2024, 128, 9418– 9435, DOI: 10.1021/acs.jpcb.4c04901There is no corresponding record for this reference.
- 115Sanbonmatsu, K. Y.; Joseph, S.; Tung, C.-S. Simulating movement of tRNA into the ribosome during decoding. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15854– 15859, DOI: 10.1073/pnas.0503456102There is no corresponding record for this reference.
- 116Li, R.; Macnamara, L.; Leuchter, J.; Alexander, R.; Cho, S. MD Simulations of tRNA and Aminoacyl-tRNA Synthetases: Dynamics, Folding, Binding, and Allostery. Int. J. Mol. Sci. 2015, 16, 15872– 15902, DOI: 10.3390/ijms160715872There is no corresponding record for this reference.
- 117Patel, S.; Hosur, R. V. Replica exchange molecular dynamics simulations reveal self-association sites in M-Crystallin caused by mutations provide insights of cataract. Sci. Rep. 2021, 11, 23270 DOI: 10.1038/s41598-021-02728-8There is no corresponding record for this reference.
- 118Stelzl, L. S.; Hummer, G. Kinetics from Replica Exchange Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 3927– 3935, DOI: 10.1021/acs.jctc.7b00372There is no corresponding record for this reference.
- 119Feig, M.; Nawrocki, G.; Yu, I.; Wang, P.; Sugita, Y. Challenges and opportunities in connecting simulations with experiments via molecular dynamics of cellular environments. J. Phys. Conf. Ser. 2018, 1036, 012010 DOI: 10.1088/1742-6596/1036/1/012010There is no corresponding record for this reference.
- 120Kumari, I.; Sandhu, P.; Ahmed, M.; Akhter, Y. Molecular Dynamics Simulations, Challenges and Opportunities: A Biologist’s Prospective. Curr. Protein Pept. Sci. 2017, 18, 1163– 1179, DOI: 10.2174/1389203718666170622074741There is no corresponding record for this reference.
- 121Senn, H. M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed. 2009, 48, 1198– 1229, DOI: 10.1002/anie.200802019There is no corresponding record for this reference.
- 122Lopes, P. E. M.; Guvench, O.; MacKerell, A. D. Current Status of Protein Force Fields for Molecular Dynamics Simulations. In Molecular Modeling of Proteins; Kukol, A., Ed.; Springer: New York, New York, NY, 2015; Vol. 1215, pp 47– 71.There is no corresponding record for this reference.
- 123McMillin, D. R. Interatomic Repulsion and the Pauli Principle. J. Chem. Educ. 2021, 98, 2912– 2918, DOI: 10.1021/acs.jchemed.1c00326There is no corresponding record for this reference.
- 124Guvench, O.; MacKerell, A. D. Comparison of Protein Force Fields for Molecular Dynamics Simulations. In Molecular Modeling of Proteins; Kukol, A., Ed.; Humana Press: Totowa, NJ, 2008; Vol. 443, pp 63– 88.There is no corresponding record for this reference.
- 125Warshel, A.; Sharma, P. K.; Kato, M. Electrostatic Basis for Enzyme Catalysis. Chem. Rev. 2006, 106, 3210– 3235, DOI: 10.1021/cr0503106There is no corresponding record for this reference.
- 126Van Der Kamp, M. W.; Mulholland, A. J. Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Computational Enzymology. Biochemistry 2013, 52, 2708– 2728, DOI: 10.1021/bi400215wThere is no corresponding record for this reference.
- 127Magliery, T. J. Protein stability: computation, sequence statistics, and new experimental methods. Curr. Opin. Struct. Biol. 2015, 33, 161– 168, DOI: 10.1016/j.sbi.2015.09.002There is no corresponding record for this reference.
- 128Singh, A.; Upadhyay, V.; Upadhyay, A. K.; Singh, S. M.; Panda, A. K. Protein recovery from inclusion bodies of Escherichia coli using mild solubilization process. Microb. Cell Factories 2015, 14, 41 DOI: 10.1186/s12934-015-0222-8There is no corresponding record for this reference.
- 129Sormanni, P.; Aprile, F. A.; Vendruscolo, M. The CamSol Method of Rational Design of Protein Mutants with Enhanced Solubility. J. Mol. Biol. 2015, 427, 478– 490, DOI: 10.1016/j.jmb.2014.09.026There is no corresponding record for this reference.
- 130Li, B.; Ming, D. GATSol, an enhanced predictor of protein solubility through the synergy of 3D structure graph and large language modeling. BMC Bioinf. 2024, 25, 204 DOI: 10.1186/s12859-024-05820-8There is no corresponding record for this reference.
- 131Tan, Y.; Zheng, J.; Hong, L.; Zhou, B. ProtSolM: Protein Solubility Prediction with Multi-modal Features, arXiv:2406.19744. arXiv.org e-Print archive. https://arxiv.org/abs/2406.19744. 2024.There is no corresponding record for this reference.
- 132Ghosh, D.; Biswas, A.; Radhakrishna, M. Advanced computational approaches to understand protein aggregation. Biophys. Rev. 2024, 5, 021302 DOI: 10.1063/5.0180691There is no corresponding record for this reference.
- 133Oeller, M.; Kang, R.; Bell, R. Sequence-based prediction of pH-dependent protein solubility using CamSol. Brief. Bioinform. 2023, 24, bbad004 DOI: 10.1093/bib/bbad004There is no corresponding record for this reference.
- 134Kimmel, B. R.; Mrksich, M. Development of an Enzyme-Inhibitor Reaction Using Cellular Retinoic Acid Binding Protein II for One-Pot Megamolecule Assembly. Chem. - Eur. J. 2021, 27, 17843– 17848, DOI: 10.1002/chem.202103059There is no corresponding record for this reference.
- 135Kimmel, B. R.; Modica, J. A.; Parker, K.; Dravid, V.; Mrksich, M. Solid-Phase Synthesis of Megamolecules. J. Am. Chem. Soc. 2020, 142, 4534– 4538, DOI: 10.1021/jacs.9b12003There is no corresponding record for this reference.
- 136Adomanis, R.; Phan, N.; Walter, G.; Kimmel, B. R. Modular Nanobody Conjugates with Controlled Topology Using Genetically Encoded Non-canonical Amino Acids. Preprint at https://doi.org/10.1101/2025.11.27.691038. 2025.There is no corresponding record for this reference.
- 137Rosace, A.; Bennett, A.; Oeller, M. Automated optimization of solubility and conformational stability of antibodies and proteins. Nat. Commun. 2023, 14, 1937 DOI: 10.1038/s41467-023-37668-6There is no corresponding record for this reference.
- 138Kuriata, A.; Iglesias, V.; Pujols, J. Aggrescan3D (A3D) 2.0: prediction and engineering of protein solubility. Nucleic Acids Res. 2019, 47, W300– W307, DOI: 10.1093/nar/gkz321There is no corresponding record for this reference.
- 139Hirsch, M.; Desai, R. R.; Annaswamy, S.; Keatinge-Clay, A. T. Mutagenesis Supports AlphaFold Prediction of How Modular Polyketide Synthase Acyl Carrier Proteins Dock With Downstream Ketosynthases. Proteins:Struct., Funct., Bioinf. 2024, 92, 1375– 1384, DOI: 10.1002/prot.26733There is no corresponding record for this reference.
- 140Araki, M.; Ekimoto, T.; Takemura, K. Molecular Dynamics Unveils Multiple-Site Binding of Inhibitors with Reduced Activity on the Surface of Dihydrofolate Reductase. J. Am. Chem. Soc. 2024, 146, 28685– 28695, DOI: 10.1021/jacs.4c04648There is no corresponding record for this reference.
- 141Pimtawong, T.; Ren, J.; Lee, J.; Lee, H.-M.; Na, D. A review on computational models for predicting protein solubility. J. Microbiol. 2025, 63, 2408001 DOI: 10.71150/jm.2408001There is no corresponding record for this reference.
- 142Navarro, S.; Ventura, S. Computational methods to predict protein aggregation. Curr. Opin. Struct. Biol. 2022, 73, 102343 DOI: 10.1016/j.sbi.2022.102343There is no corresponding record for this reference.
- 143Prediction and Evaluation of Protein Aggregation with Computational Methods. In Methods in Molecular Biology; Springer US: New York, NY, 2025; pp 299– 314 DOI: 10.1007/978-1-0716-4196-5_17 .There is no corresponding record for this reference.
- 144Santos, J.; Pujols, J.; Pallarès, I.; Iglesias, V.; Ventura, S. Computational prediction of protein aggregation: Advances in proteomics, conformation-specific algorithms and biotechnological applications. Comput. Struct. Biotechnol. J. 2020, 18, 1403– 1413, DOI: 10.1016/j.csbj.2020.05.026There is no corresponding record for this reference.
- 145Arnold, F. H. Design by Directed Evolution. Acc. Chem. Res. 1998, 31, 125– 131, DOI: 10.1021/ar960017fThere is no corresponding record for this reference.
- 146Arnold, F. H. Directed evolution: Creating biocatalysts for the future. Chem. Eng. Sci. 1996, 51, 5091– 5102, DOI: 10.1016/S0009-2509(96)00288-6There is no corresponding record for this reference.
- 147Cobb, R. E.; Chao, R.; Zhao, H. Directed evolution: Past, present, and future. AIChE J. 2013, 59, 1432– 1440, DOI: 10.1002/aic.13995There is no corresponding record for this reference.
- 148Yang, J.; Lal, R. G.; Bowden, J. C. Active learning-assisted directed evolution. Nat. Commun. 2025, 16, 714 DOI: 10.1038/s41467-025-55987-8There is no corresponding record for this reference.
- 149Terashi, G.; Wang, X.; Maddhuri Venkata Subramaniya, S. R.; Tesmer, J. J. G.; Kihara, D. Residue-wise local quality estimation for protein models from cryo-EM maps. Nat. Methods 2022, 19, 1116– 1125, DOI: 10.1038/s41592-022-01574-4There is no corresponding record for this reference.
- 150Graille, M.; Sacquin-Mora, S.; Taly, A. Best Practices of Using AI-Based Models in Crystallography and Their Impact in Structural Biology. J. Chem. Inf. Model. 2023, 63, 3637– 3646, DOI: 10.1021/acs.jcim.3c00381There is no corresponding record for this reference.
- 151Wang, X.; Zhu, H.; Terashi, G.; Taluja, M.; Kihara, D. DiffModeler: large macromolecular structure modeling for cryo-EM maps using a diffusion model. Nat. Methods 2024, 21, 2307– 2317, DOI: 10.1038/s41592-024-02479-0There is no corresponding record for this reference.
- 152Serapian, S. A.; Crosby, J.; Crump, M. P.; Van Der Kamp, M. W. Path to Actinorhodin: Regio- and Stereoselective Ketone Reduction by a Type II Polyketide Ketoreductase Revealed in Atomistic Detail. JACS Au 2022, 2, 972– 984, DOI: 10.1021/jacsau.2c00086There is no corresponding record for this reference.
- 153Shukla, V. K.; Karunanithy, G.; Vallurupalli, P.; Hansen, D. F. A combined NMR and deep neural network approach for enhancing the spectral resolution of aromatic side chains in proteins. Sci. Adv. 2024, 10, eadr2155 DOI: 10.1126/sciadv.adr2155There is no corresponding record for this reference.
- 154Drake, Z. C.; Fowler, A. G.; Blum, A. A.; Lindert, S. Enhanced Protein Complex Prediction via Rosetta, AlphaFold, and Nondifferential Covalent Labeling Mass Spectrometry. J. Phys. Chem. B 2025, 129, 6489– 6497, DOI: 10.1021/acs.jpcb.5c02872There is no corresponding record for this reference.
- 155Lee, C. Y.; Hubrich, D.; Varga, J. K. Systematic discovery of protein interaction interfaces using AlphaFold and experimental validation. Mol. Syst. Biol. 2024, 20, 75– 97, DOI: 10.1038/s44320-023-00005-6There is no corresponding record for this reference.
- 156Koehler Leman, J.; Künze, G. Recent Advances in NMR Protein Structure Prediction with ROSETTA. Int. J. Mol. Sci. 2023, 24, 7835, DOI: 10.3390/ijms24097835There is no corresponding record for this reference.
- 157Alshammari, M.; Wriggers, W.; Sun, J.; He, J. Refinement of AlphaFold2 models against experimental and hybrid cryo-EM density maps. QRB Discovery 2022, 3, e16 DOI: 10.1017/qrd.2022.13There is no corresponding record for this reference.
- 158Humphreys, I. R.; Pei, J.; Baek, M. Computed structures of core eukaryotic protein complexes. Science 2021, 374, eabm4805 DOI: 10.1126/science.abm4805There is no corresponding record for this reference.
- 159Bordin, N.; Sillitoe, I.; Nallapareddy, V. AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms. Commun. Biol. 2023, 6, 160 DOI: 10.1038/s42003-023-04488-9There is no corresponding record for this reference.
- 160Wang, H.; Wang, J. How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci. 2017, 26, 32– 39, DOI: 10.1002/pro.3022There is no corresponding record for this reference.