



This publication is Open Access under the license indicated. Learn More
Latent HIV Reservoirs in the Central Nervous System: Mechanisms, Barriers, and Therapeutic ApproachesClick to copy article linkArticle link copied!
- Yohannes MatthewYohannes MatthewDepartment of Biochemistry and Molecular Biology, Drexel University College of Medicine, Philadelphia, Pennsylvania 19102, United StatesDepartment of Pharmacology and Physiology, Drexel University College of Medicine, Philadelphia, Pennsylvania 19102, United StatesMore by Yohannes Matthew
- Nicholas FoleyNicholas FoleyDepartment of Biological Chemistry and Molecular Pharmacology, Blavatnik Institute, Harvard Medical School, Boston, Massachusetts 02115, United StatesMore by Nicholas Foley
- Daniel T. ClaiborneDaniel T. ClaiborneHIV Cure & Viral Diseases Center, The Wistar Institute, 3601 Spruce St, Philadelphia, Pennsylvania 19104, United StatesMore by Daniel T. Claiborne
- Zachary KlaseZachary KlaseDepartment of Pharmacology and Physiology, Drexel University College of Medicine, Philadelphia, Pennsylvania 19102, United StatesMore by Zachary Klase
- Alexej Dick*Alexej Dick*Email: [email protected]. Phone: 215-762-7234.Department of Biochemistry and Molecular Biology, Drexel University College of Medicine, Philadelphia, Pennsylvania 19102, United StatesMore by Alexej Dick
ACS Infectious Diseases
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
Despite advancements in antiretroviral therapy (ART), HIV-1 remains incurable due to latent viral reservoirs. These reservoirs are located in distinct areas, such as the central nervous system (CNS). The CNS reservoirs flourish inside unique cell types, including myeloid cells such as microglia, perivascular macrophages, and astrocytes. These reservoirs are established early in infection, evade immune detection, and pose a significant challenge to the delivery of therapeutic agents. Although current ARTs can suppress viral transcription, the latently infected CNS cells can produce low-level persistent neuroinflammation and contribute to HIV-associated neurocognitive disorders (HAND). Multiple molecular mechanisms underlie the establishment and maintenance of CNS HIV reservoirs, including epigenetic modifications, transcriptional repression, and limited penetration of antiretroviral drugs across the blood–brain barrier (BBB). Specifically, latency involves transcriptional silencing through histone deacetylation and histone methylation, as well as the recruitment of repressive transcriptional complexes. Therapeutically targeting these mechanisms is critical for latency reversal and reservoir eradication. Two strategies, “shock and kill” and “block and lock”, take advantage of these mechanisms. The “shock and kill” method utilizes latency-reversing agents (LRAs) to stimulate transcriptional reactivation, exposing infected cells for immune clearance. Notably, several LRAs, including Vorinostat, JQ1, and Bryostatin-1, have been shown to penetrate the BBB and exhibit promising latency-reversal activity. However, their clinical efficacy is limited by incomplete reservoir reactivation and potential neurotoxicity. Emerging therapeutic targets, such as the transcription factor RUNX1, show significant promise for both potent HIV reactivation and lack of neurotoxicity. To enhance CNS reservoir targeting, novel strategies leveraging viral vectors or lipid nanoparticles are being explored. Overall, a comprehensive understanding of HIV-1 latency mechanisms in the CNS, coupled with the strategic development of BBB-penetrant, non-neurotoxic LRAs and adjunct immune therapies, is critical. Future therapeutic regimens will likely require a multifaceted approach to eradicate HIV-1 reservoirs safely and effectively within the CNS, ultimately progressing toward a functional cure.
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Overview of HIV-1 Infection and Latent Reservoirs
Figure 1
Figure 1. HIV-1 Entry and Transcriptional Regulation in Target Cells. (A) Overview of the HIV-1 replication cycle. Key steps in the HIV-1 replication include viral entry (fusion), integration into the host genome, transcription, translation into viral proteins, virion budding, and maturation. The illustration provides insights into the transcriptional activation mechanisms in actively infected CD4+ T-cells. (B) Histone acetyltransferases (HATs) incorporate acetyl groups to lysine residues on histones positioned at nucleosomes Nuc-0 and Nuc-1, promoting euchromatin formation and enhancing transcriptional accessibility. RNA polymerase II (RNA Pol II) assembles at the 5′ long terminal repeat (LTR) region of the HIV-1 genome, where associated factors contribute to the formation of the preinitiation complex (PIC). During transcription elongation, the HIV Tat protein binds to the trans-activation response (TAR) element of the nascent viral mRNA. This interaction recruits the positive transcription elongation factor b (P-TEFb) complex, which phosphorylates the DRB sensitivity-inducing factor (DSIF) and the negative elongation factor (NELF), thereby preventing transcriptional pausing. Additionally, the C-terminal domain (CTD) of RNA polymerase II undergoes phosphorylation, facilitating efficient transcription elongation. (C) Mechanisms of transcriptional repression in latent infection. In contrast to active transcription, histone methyltransferases (HMTs) add methyl groups to histone residues, thereby driving the formation of heterochromatin that limits transcriptional accessibility. Consequently, despite RNA polymerase II and the preinitiation complex (PIC) assembling at the HIV-1 LTR, transcription is effectively suppressed, maintaining viral latency. Therapeutic LRAs target these repressive mechanisms; for example, Vorinostat inhibits histone deacetylases (HDACs) to promote acetylation, JQ1 inhibits BRD4 to release P-TEFb, and other upcoming therapies inhibit transcriptional repression at the 5′ LTR. P: Phosphate, Ac: Acetyl group, Me: Methyl, HAT: Histone Acetyl Transferases, HMT: Histone Methyl Transferases. Created with BioRender.com.
Entry and Establishment of Latent Infection in the CNS
Figure 2
Figure 2. (A) Cross-sectional representation of the Neurovascular Unit (NVU), the Structural Features of the BBB, and HIV-1 CNS infiltration. This figure illustrates the neurovascular unit and highlights its key structural components, including brain microvascular endothelial cells (BMECs), pericytes, and astrocytic endfeet. The figure emphasizes the limited ability of antiretroviral therapy molecules to cross the BBB due to efflux by transporters such as P-glycoproteins (P-gp), multidrug resistance protein (MRP), and breast cancer resistance protein (BCRP) located on the luminal side of the BMECs. A clear concentration gradient is evident, with ART levels highest in the bloodstream and substantially lower in endothelial cells and the CNS. ART: antiretroviral therapeutic, P-Gp: p-glycoprotein, BCRP: breast cancer resistance protein, MRP2: multidrug resistance protein. (B) Overview of the organization and functional characteristics of the BBB, emphasizing its role in HIV-1 infection within the CNS. HIV-1-infected CD4+ T-cells cross the BBB via tight junctions between brain microvascular endothelial cells. Subsequently, CD4+ T lymphocytes infect astrocytes through a CD4-independent mechanism. Endothelial cells are closely associated with astrocytic end feet and pericytes, collectively maintaining the BBB’s selective permeability. The figure illustrates the limitations of antiretroviral therapy penetration into the CNS, highlighting how ART molecules are unable to cross the BBB, primarily due to the presence of tight junctions and active efflux transporters, ABC: ATP Binding Cassette. Created with BioRender.com.
Genetic Diversity of HIV in the CNS
The CNS as a Critical Barrier to HIV Eradication
Mechanisms of HIV Transcriptional Control
Therapeutic Challenges in the CNS
Heterogeneous Reservoirs and the Need for LRAs
The Unmet Need for BBB-Penetrant LRAs
| ART | plasma concentration | cerebrospinal fluid (CSF) concentration | refs |
|---|---|---|---|
| maraviroc (CCR5 inhibitor) | 21.4–478.0 ng/mL | 1.83–12.2 ng/mL | (67) |
| enfurvirtide (entry inhibitor) | 3.7 μmol/mL | not determined | (12,67,68) |
| nevirapine (non-nucleoside reverse transcriptase inhibitor - NNRTI) | 7.5–16.9 μmol/mL | 1.3–10.9 μmol/mL | (12,67,68) |
| raltegravir (integrase inhibitor) | 37.0–5180.0 ng/mL | 2.0–126 ng/mL | (67) |
| abacavir (nucleoside reverse transcriptase inhibitor - NRTI) | 5.2–11.0 μmol/mL | 0.5–1.8 μmol/mL | (12,67,68) |
| indinavir (protease inhibitor) | 12.2–13.0 μmol/mL | 0.03–0.66 μmol/mL | (67) |
| LRA | plasma concentration | cerebrospinal fluid concentration | refs |
|---|---|---|---|
| disulfiram (disulfide) depletes PTEN levels. this prevents increased AKT phosphorylation and activation of a signaling pathway that leads to latent HIV-1 expression. | rapidly reduced in blood to diethyldithiocarbamate (DDC) within minutes (in vitro) | preclinical distribution: brain shows the lowest/least detectable levels in early distribution studies. | (69−71) |
| alprazolam (benzodiazepine) inhibitor of the RUNX1 transcription factor that negatively regulates HIV-1 transcription. Potentiates STAT5 recruitment to the viral promoter. | blood (median) 0.024 mg/kg | brain (median): 0.059 mg/kg (brain:blood ratio (median): 2.21) | (72−75) |
| decitabine (methyltransferase inhibitor) Blocks the addition of methyl groups, which modulates the expression of HIV after the addition of lysine residues on histone. | ∼1.3–1.6 μM (standard i.v. dose in humans) | lower than plasma, typically 27–58% of plasma levels in animal models. | (76−79) |
| vorinostat (SAHA) (HDAC inhibitor) Can inhibit Histone Deacetylase Activating Complex (HDAC), which allows for the binding of RNAPII (RNA Polymerase II) and subsequent transcriptional activation. | plasma Cmax (oral 400 mg) ∼1.2 μM | in the ventricular-CSF sampling cohort: mean CSF ∼ 75.4 nM | (69,80−83) |
| JQ1 (BRD4 inhibitor) BRD4 agonist acts as an inhibitor of the BET family of proteins. Specifically, JQ1 prevents BRD4 from binding to the HIV promoter, thereby allowing Tat to recruit and stimulate HIV expression. | plasma Cmax 34 μg/mL at 15 min after 50 mg/kg i.p. in mouse PK. | AUCbrain/AUCplasma = 0.98 (98%) after 50 mg/kg i.p. | (84−86) |
| Bryostatin-1 (PKC (protein kinase C) agonist) activates protein kinase C (PKC) alpha and delta. Stimulated the transcription of the LTR by activating the transcription factor NF-kB. | plasma Cmax (mouse 15 μg/m2 i.v. tail vein) ∼ 2.5 nM | peak brain concentration ∼ 0.2 nM at ∼1 h postdose; peak brain concentrations >8% of peak blood plasma. | (87−89) |
Examples of HIV BBB-Permeable LRAs
Vorinostat (Suberoylanilide Hydroxamic Acid) (SAHA)
Bryostatin-1
JQ1 (a BET Inhibitor)
Disulfiram
Current HIV-1 Cure Strategies and Their Limitations in the CNS
“Shock and Kill” Approaches
Figure 3
Figure 3. Contrasting Paradigms of HIV Latency Management: LRAs (″Shock and Kill″) versus LPAs (″Block and Lock″). Illustration of two distinct therapeutic strategies for managing HIV latency. (A) (″Shock and Kill″) depicts a latently infected CD4+ T-cell undergoing viral reactivation upon receiving an LRA signal. Following reactivation, an immune effector cell (specifically, a CD8+ T-cell) recognizes and targets the infected cell, inducing apoptosis and clearance. (B) (″Block and Lock″) illustrates an alternative strategy, where a latently infected CD4+ T-cell receives a latency-promoting agent (LPA) signal, reinforcing transcriptional silencing. This mechanism highlights the inhibitory action of the compound didehydro-cortistatin A (dCA) on critical HIV transcription elongation factors, including Tat, CDK9, and Cyclin T1, thereby maintaining durable latency without inducing viral production. LRA: latency reversal agent, LPA: latency promoting agent, BRD4: Bromodomain 4, MHC: Major histocompatibility class I, TCR: T cell receptor, PKC: Protein Kinase C, HDAC: Histone Deacetylase Inhibitor, dCA: didehydro-cortistatin A, CDK9: Cyclin-dependent kinase 9, Ac: Acetyl group. Created with BioRender.com.
Adequate clearance of HIV in the CNS must be established. Reactivation of viral transcription in the CNS without proper viral clearance may come with detrimental side effects on neuronal health and could lead to HIV-associated neurocognitive disorders. (14) Identification of LRAs that can cross the BBB and effectively clear the virus from the CNS is critical to advancing “shock and kill”.
Combinations and interactions between different LRA classes should be investigated. Targeting multiple pathways involved in latency may be necessary to fully reactivate the virus in diverse cell types. Establishing more diverse in vitro models could benefit this. Further research on combinations should investigate the efficacy of latency reversal across multiple cell models, potential drug interactions, toxicity, and the impact of these therapies on the host’s immune system.
Identification of robust in vivo biomarkers of latency reversal may be critical in evaluating efficacy. At present, HIV RNA measurements are commonly used to assess LRA activity. However, RNA induction may not reliably reflect full productive transcription or virion release. His limitation was highlighted in clinical studies of vorinostat, where increases in HIV RNA did not translate into measurable changes in free virus, potentially limiting immune recognition and clearance.
“Block and lock” Approaches
Novel Targets for Latency Reversal in the CNS
Immune-Based Strategies Targeting CNS Reservoirs: Broadly Neutralizing Antibodies (bNAbs) and Viral Rebound Control
Figure 4
Figure 4. Broadly Neutralizing Antibodies (bNAbs) and Control of HIV Viral Rebound. (A) Schematic representation of a bNAb, highlighting key structural components including the variable region, fragment antigen-binding (Fab) region, and fragment crystallizable (Fc) region. (B) Illustration of the HIV envelope glycoproteins gp120 and gp41, with specific epitopes targeted by individual bNAbs. These epitopes include regions within the variable loops (V2, V3), glycan-dependent sites, and the membrane-proximal external region (MPER). (C) Immune effector cells involved in antibody-mediated clearance mechanisms, specifically antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), demonstrate how bNAbs facilitate immune recognition and subsequent elimination of HIV-infected cells. (D) Schematic showing bNAb’s binding to HIV-1 virions following transcriptional reactivation of integrated, previously latent provirus in CD4+ T-cells. LRA: Latency reversal agent, LPA: Latency promoting agent, bNAb: Broadly neutralizing antibody, ADCC: Antibody-dependent cellular cytotoxicity, ADCP: Antibody-dependent cellular phagocytosis, VH: Variable Heavy, VL: Variable Light, CH: Constant Heavy, CL: Constant Light, Fab: Fragment antigen binding domain, FcyR: Fc-γ receptor, GP120: Glycoprotein 120. Created with BioRender.com.
bNAbs and Post-Latency Viral Rebound
Epigenetic and Chromatin Remodeling Factors
Figure 5
Figure 5. Overview of HIV Latency Reversal Agents. (A) Cytokine reactivation of HIV transcription is generally mediated through increased NF-kB and NFAT binding of the LTR, recruiting RNAPII. (B) Disulfiram can block PTEN/AKT signaling by inhibiting phosphatase, removing phosphate from PIP3. This increases P-TEFb, which mediates Tat-dependent HIV transcription. (C) LRAs can activate host-mediated transcription of HIV. PKC agonists increase NF-kB activity by enhancing its binding to the LTR. Benzodiazepines disrupt CBFβ binding to RUNX1, increasing transcription. (D) BRD4 inhibitor competitively block the binding of P-TEFb to BRD4, allowing P-TEFb to bind with Tat and complete Tat-dependent transcription. Methyltransferase inhibitors block the addition of methyl groups, leading to a more transcriptionally active epigenetic environment. HDAC inhibitors block histone deacetylation, leading to a more transcriptionally active epigenetic environment. ECM: Extracellular matrix, DMNT1: DNA methyltransferase 1, HDAC: Histone Deacetylase, RNAPII: RNA Polymerase II, NFAT: Nuclear Factor of Activated T-cells, NF-kB: Nuclear factor-kappa B, IKK: I kappa B kinase, P-TEFb: Positive Transcription Elongation Factor b, BRD4: Bromodomain-containing protein 4. Created with BioRender.com.
The Wnt/β-Catenin Pathway in HIV Latency
Highlighting RUNX1, an Emerging Target
Stem Cell Transplants and Gene Editing Strategies
Toll-like Receptors in HIV-1 Latency Reversal
Figure 6
Figure 6. TLR-mediated HIV Latency Reversal. A schematic illustration of the Toll-like receptors (TLRs) involved in HIV latency reversal in latently infected CD4+ T-cells. TLRs 2, 4, 5, and 6 are depicted on the cellular surface, whereas TLRs 3, 7, and 9 are localized within endosomal compartments. Pathogen-associated molecular patterns (PAMPs), including microbial and viral motifs, interact with these TLRs, initiating intracellular signaling cascades. Activation of these pathways leads to nuclear translocation of transcription factors such as AP-1, NF-κB, and IRF3, facilitating their binding to the HIV 5′ LTR region and resulting in transcriptional reactivation of latent HIV. TLR: Toll-Like Receptor, NF-kB: NF-Kappa β, LPS: Lipopolysaccharide. Created with BioRender.com.
Evidence Linking TLRs to HIV Reactivation
| receptor | agonist | refs |
|---|---|---|
| TLR 1 | lipoproteins | (135,136,140) |
| PAM3CSK4 | ||
| TLR 2 | SMU-Z1 | (135,141) |
| Aβ | ||
| biglycan | ||
| endoplasmin (HSP90B1) | ||
| HeatShockProteins (HSP60, HSP70) | ||
| HMGB1 | ||
| hyaluronan | ||
| monosodium urate crystals | ||
| α-synuclein | ||
| surfactant protein A | ||
| fibronectin | ||
| versican | ||
| TLR 3 | polyinosinic: polycytidylic acid Poly (I:C). | (135,139) |
| bacterial rRNA. | ||
| TLR4 | lipopolysaccharide (LPS). | (135,139) |
| Aβ | ||
| αA-crystallin, αB-crystallin | ||
| endoplasmin (Hsp90b1) | ||
| fibronectin | ||
| heparan sulfate | ||
| HSP60 | ||
| HSP70 | ||
| HSP72 | ||
| hyaluronan | ||
| lysozyme | ||
| monosodium urate crystals | ||
| peroxiredoxin 1 | ||
| resistin | ||
| S100 protein | ||
| surfactant protein A | ||
| tenascin C. | ||
| TLR5 | flagellin. | (139) |
| TLR6 | fibroblast-stimulating lipopeptide (FSL-1). | (139) |
| TLR7 | imiquimod | (135,138,139) |
| gardiquim | ||
| resiquimod | ||
| GS-962 | ||
| miRNA: (Let-7B, miR-146a-5p, miR-340–3p, miR-132–5p). | ||
| TLR8 | imiquimod | (135,139) |
| gardiquimod | ||
| resiquimod | ||
| ssRNA40 | ||
| miRNA: (miR-27, miR-21, miR-340–3p and miR-132–5p). | ||
| TLR9 | ODN2006. | (135,139) |
| DNA. | ||
| mtDNA. | ||
| chromatin-IgG complex. |
Implications for HIV Cure Strategies
Pharmacokinetics and BBB Penetration
Safety, Neurotoxicity, and Off-Target Effects
| 1. | Neurotoxicity and BBB Considerations: Although BBB penetration for LRAs is desirable, off-target effects in the CNS can compromise neuronal health. Because neurons exhibit limited regenerative capacity, even temporary neurotoxicity can lead to long-term functional deficits. Thus, safer, more specific molecules are needed. | ||||
| 2. | Known Toxicity Profiles: Multiple LRAs have demonstrated toxicity across various cell types, including benzodiazepines, Bryostatin-1, JQ1, and disulfiram. These compounds exhibit duality: while they might protect against HIV-associated neurocognitive impairment, they can also induce small molecule-mediated neurotoxicity if not carefully dosed. (85) | ||||
Specific Examples
JQ1
Vorinostat
Benzodiazepines
Disulfiram
Emerging Approaches and Future Directions
Author Information
- Alexej Dick - Department of Biochemistry and Molecular Biology, Drexel University College of Medicine, Philadelphia, Pennsylvania 19102, United States;
 https://orcid.org/0000-0003-0580-1897;
https://orcid.org/0000-0003-0580-1897;
Y.M. and N.F. contributed equally to this work.
This work was supported by the National Institutes of Health and the Wallace H. Coulter Foundation. Daniel Claiborne is funded by R01-AI186810. Zachary Klase is funded by R01-DA057337, R61-DA061825, and R21-DA063133. Alexj Dick, Nicholas Foley, and Yohannes Matthew are funded by RO1-900048 and by the Wallace H. Coulter Foundation (284247).
References
This article references 149 other publications.
- 1Payagala, S.; Pozniak, A. The global burden of HIV. Clin. Dermatol. 2024, 42 (2), 119– 127, DOI: 10.1016/j.clindermatol.2024.02.001Google ScholarThere is no corresponding record for this reference.
- 2Alum, E. U.; Uti, D. E.; Ugwu, O. P. C.; Alum, B. N. Toward a cure - Advancing HIV/AIDs treatment modalities beyond antiretroviral therapy: A Review. Medicine 2024, 103 (27), e38768 DOI: 10.1097/md.0000000000038768Google ScholarThere is no corresponding record for this reference.
- 3Nguyen, K.; Karn, J. The sounds of silencing: dynamic epigenetic control of HIV latency. Curr. Opin. HIV AIDS 2024, 19 (3), 102 DOI: 10.1097/COH.0000000000000850Google ScholarThere is no corresponding record for this reference.
- 4Gao, C.; Meng, J.; Xiao, X.; Wang, M.; Williams, A. B.; Wang, H. Antiretroviral therapy improves neurocognitive impairment in people living with HIV? A meta-analysis. Int. J. Nurs. Sci. 2020, 7 (2), 238– 247, DOI: 10.1016/j.ijnss.2020.03.007Google ScholarThere is no corresponding record for this reference.
- 5Mastrorosa, I.; Pinnetti, C.; Brita, A. C.; Mondi, A.; Lorenzini, P.; Del Duca, G.; Vergori, A.; Mazzotta, V.; Gagliardini, R.; Camici, M. Declining Prevalence of Human Immunodeficiency Virus (HIV)-Associated Neurocognitive Disorders in Recent Years and Associated Factors in a Large Cohort of Antiretroviral Therapy-Treated Individuals With HIV. Clin. Infect. Dis. 2023, 76 (3), e629– e637, DOI: 10.1093/cid/ciac658Google ScholarThere is no corresponding record for this reference.
- 6Chawla, A.; Wang, C.; Patton, C.; Murray, M.; Punekar, Y.; de Ruiter, A.; Steinhart, C. A Review of Long-Term Toxicity of Antiretroviral Treatment Regimens and Implications for an Aging Population. Infect. Dis. Ther. 2018, 7 (2), 183– 195, DOI: 10.1007/s40121-018-0201-6Google ScholarThere is no corresponding record for this reference.
- 7Apostolova, N.; Funes, H. A.; Blas-Garcia, A.; Galindo, M. J.; Alvarez, A.; Esplugues, J. V. Efavirenz and the CNS: what we already know and questions that need to be answered. J. Antimicrob. Chemother. 2015, 70 (10), 2693– 2708, DOI: 10.1093/jac/dkv183Google ScholarThere is no corresponding record for this reference.
- 8Venter, W. D. F.; Moorhouse, M.; Sokhela, S.; Fairlie, L.; Mashabane, N.; Masenya, M.; Serenata, C.; Akpomiemie, G.; Qavi, A.; Chandiwana, N. Dolutegravir plus Two Different Prodrugs of Tenofovir to Treat HIV. N. Engl. J. Med. 2019, 381 (9), 803– 815, DOI: 10.1056/NEJMoa1902824Google ScholarThere is no corresponding record for this reference.
- 9Churchill, M. J.; Cowley, D. J.; Wesselingh, S. L.; Gorry, P. R.; Gray, L. R.; Churchill, M. J.; Cowley, D. J.; Wesselingh, S. L.; Gorry, P. R.; Gray, L. R. HIV-1 transcriptional regulation in the central nervous system and implications for HIV cure research. J. NeuroVirol. 2014, 21 (3), 290– 300, DOI: 10.1007/s13365-014-0271-5Google ScholarThere is no corresponding record for this reference.
- 10Margolis, D. M.; Archin, N. M. Proviral Latency, Persistent Human Immunodeficiency Virus Infection, and the Development of Latency Reversing Agents. J. Infect. Dis. 2017, 215, S111– S118, DOI: 10.1093/infdis/jiw618Google ScholarThere is no corresponding record for this reference.
- 11Kreider, E. F.; Bar, K. J. HIV-1 Reservoir Persistence and Decay: Implications for Cure Strategies. Curr. HIV/AIDS Rep. 2022, 19 (3), 194– 206, DOI: 10.1007/s11904-022-00604-2Google ScholarThere is no corresponding record for this reference.
- 12Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The Paradox of HIV Blood–Brain Barrier Penetrance and Antiretroviral Drug Delivery Deficiencies. Trends Neurosci. 2020, 43 (9), 695, DOI: 10.1016/j.tins.2020.06.007Google ScholarThere is no corresponding record for this reference.
- 13Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.-J.; Castagna, A.; Richmond, G. J.; Sinclair, G. I.; Siripassorn, K.; Ruane, P. J.; Berhe, M.; Wang, H. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2022, 386 (19), 1793– 1803, DOI: 10.1056/NEJMoa2115542Google ScholarThere is no corresponding record for this reference.
- 14Ash, M. K.; Al-Harthi, L.; Schneider, J. R. HIV in the Brain: Identifying Viral Reservoirs and Addressing the Challenges of an HIV Cure. Vaccines 2021, 9 (8), 867, DOI: 10.3390/vaccines9080867Google ScholarThere is no corresponding record for this reference.
- 15Wang, J.; Crawford, K.; Yuan, M.; Wang, H.; Gorry, P. R.; Gabuzda, D. Regulation of CC chemokine receptor 5 and CD4 expression and human immunodeficiency virus type 1 replication in human macrophages and microglia by T helper type 2 cytokines. J. Infect. Dis. 2002, 185 (7), 885– 897, DOI: 10.1086/339522Google ScholarThere is no corresponding record for this reference.
- 16Bertrand, L.; Nair, M.; Toborek, M. Solving the Blood-Brain Barrier Challenge for the Effective Treatment of HIV Replication in the Central Nervous System. Curr. Pharm. Des. 2016, 22 (35), 5477– 5486, DOI: 10.2174/1381612822666160726113001Google ScholarThere is no corresponding record for this reference.
- 17Heaton, R. K.; Franklin, D. R.; Ellis, R. J.; McCutchan, J. A.; Letendre, S. L.; LeBlanc, S.; Corkran, S. H.; Duarte, N. A.; Clifford, D. B.; Woods, S. P. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J. NeuroVirol. 2011, 17 (1), 3– 16, DOI: 10.1007/s13365-010-0006-1Google ScholarThere is no corresponding record for this reference.
- 18Henrich, T. J.; Kuritzkes, D. R. HIV-1 entry inhibitors: recent development and clinical use. Curr. Opin. Virol. 2013, 3 (1), 51– 57, DOI: 10.1016/j.coviro.2012.12.002Google ScholarThere is no corresponding record for this reference.
- 19Sturdevant, C. B.; Joseph, S. B.; Schnell, G.; Price, R. W.; Swanstrom, R.; Spudich, S. Compartmentalized Replication of R5 T Cell-Tropic HIV-1 in the Central Nervous System Early in the Course of Infection. PLoS Pathog. 2015, 11 (3), e1004720 DOI: 10.1371/journal.ppat.1004720Google ScholarThere is no corresponding record for this reference.
- 20Valdebenito, S.; Castellano, P.; Ajasin, D.; Eugenin, E. A. Astrocytes are HIV reservoirs in the brain: A cell type with poor HIV infectivity and replication but efficient cell-to-cell viral transfer. J. Neurochem. 2021, 158 (2), 429– 443, DOI: 10.1111/jnc.15336Google ScholarThere is no corresponding record for this reference.
- 21Li, G. H.; Maric, D.; Major, E. O.; Nath, A. Productive HIV infection in astrocytes can be established via a nonclassical mechanism. Aids 2020, 34 (7), 963– 978, DOI: 10.1097/QAD.0000000000002512Google ScholarThere is no corresponding record for this reference.
- 22Kumar, A.; Abbas, W.; Herbein, G. HIV-1 Latency in Monocytes/Macrophages. Viruses 2014, 6 (4), 1837– 1860, DOI: 10.3390/v6041837Google ScholarThere is no corresponding record for this reference.
- 23Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P. W. G.; Marcello, A.; Van Lint, C.; Rohr, O.; Schwartz, C. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362 DOI: 10.3389/fcimb.2019.00362Google ScholarThere is no corresponding record for this reference.
- 24Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14 (1), 9 DOI: 10.1186/s12977-017-0335-8Google ScholarThere is no corresponding record for this reference.
- 25Sun, W.; Rassadkina, Y.; Gao, C.; Collens, S. I.; Lian, X.; Solomon, I. H.; Mukerji, S.; Yu, X. G.; Lichterfeld, M. Persistence of intact HIV-1 proviruses in the brain during antiretroviral therapy bioRxiv , Aug 14, 2023 DOI: 10.1101/2023.06.26.546135 .Google ScholarThere is no corresponding record for this reference.
- 26Dahabieh, M. S.; Battivelli, E.; Verdin, E. Understanding HIV latency: the road to an HIV cure. Annu. Rev. Med. 2015, 66, 407– 421, DOI: 10.1146/annurev-med-092112-152941Google ScholarThere is no corresponding record for this reference.
- 27Matsuda, T.; Bebenek, K.; Masutani, C.; Rogozin, I. B.; Hanaoka, F.; Kunkel, T. A. Error rate and specificity of human and murine DNA polymerase η. J. Mol. Biol. 2001, 312 (2), 335, DOI: 10.1006/jmbi.2001.4937Google ScholarThere is no corresponding record for this reference.
- 28García, M.; López-Fernández, L.; Mínguez, P.; Morón-López, S.; Restrepo, C.; Navarrete-Muñoz, M. A.; López-Bernaldo, J. C.; Benguría, A.; García, M. I.; Cabello, A. Transcriptional signature of resting-memory CD4 T cells differentiates spontaneous from treatment-induced HIV control. J. Mol. Med. 2020, 98 (8), 1093– 1105, DOI: 10.1007/s00109-020-01930-xGoogle ScholarThere is no corresponding record for this reference.
- 29Shah, S.; Alexaki, A.; Pirrone, V.; Dahiya, S.; Nonnemacher, M. R.; Wigdahl, B. Functional properties of the HIV-1 long terminal repeat containing single-nucleotide polymorphisms in Sp site III and CCAAT/enhancer binding protein site I. Virol. J. 2014, 11 (1), 92 DOI: 10.1186/1743-422X-11-92Google ScholarThere is no corresponding record for this reference.
- 30Chaillon, A.; Gianella, S.; Dellicour, S.; Rawlings, S. A.; Schlub, T. E.; Oliveira, M. F. D.; Ignacio, C.; Porrachia, M.; Vrancken, B.; Smith, D. M. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Invest. 2020, 130 (4), 1699– 1712, DOI: 10.1172/JCI134815Google ScholarThere is no corresponding record for this reference.
- 31Lamers, S. L.; Rose, R.; Maidji, E.; Agsalda-Garcia, M.; Nolan, D. J.; Fogel, G. B.; Salemi, M.; Garcia, D. L.; Bracci, P.; Yong, W. HIV DNA Is Frequently Present within Pathologic Tissues Evaluated at Autopsy from Combined Antiretroviral Therapy-Treated Patients with Undetectable Viral Loads. J. Virol. 2016, 90 (20), 8968– 8983, DOI: 10.1128/JVI.00674-16Google ScholarThere is no corresponding record for this reference.
- 32Ferretti, F.; De Zan, V.; Gerevini, S.; Turrini, F.; Boeri, E.; Gianotti, N.; Hasson, H.; Lazzarin, A.; Cinque, P. Relapse of Symptomatic Cerebrospinal Fluid HIV Escape. Curr. HIV/AIDS Rep. 2020, 17 (5), 522– 528, DOI: 10.1007/s11904-020-00526-xGoogle ScholarThere is no corresponding record for this reference.
- 33Williams, D. W.; Calderon, T. M.; Lopez, L.; Carvallo-Torres, L.; Gaskill, P. J.; Eugenin, E. A.; Morgello, S.; Berman, J. W. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS One 2013, 8 (7), e69270 DOI: 10.1371/journal.pone.0069270Google ScholarThere is no corresponding record for this reference.
- 34Joseph, S. B.; Arrildt, K. T.; Sturdevant, C. B.; Swanstrom, R. HIV-1 target cells in the CNS. J. NeuroVirol. 2015, 21 (3), 276– 289, DOI: 10.1007/s13365-014-0287-xGoogle ScholarThere is no corresponding record for this reference.
- 35Walsh, S. R.; Seaman, M. S. Broadly Neutralizing Antibodies for HIV-1 Prevention. Front. Immunol. 2021, 12, 712122 DOI: 10.3389/fimmu.2021.712122Google ScholarThere is no corresponding record for this reference.
- 36Spudich, S.; Robertson, K. R.; Bosch, R. J.; Gandhi, R. T.; Cyktor, J. C.; Mar, H.; Macatangay, B. J.; Lalama, C. M.; Rinaldo, C.; Collier, A. C. Persistent HIV-infected cells in cerebrospinal fluid are associated with poorer neurocognitive performance. J. Clin. Invest. 2019, 129 (8), 3339– 3346, DOI: 10.1172/JCI127413Google ScholarThere is no corresponding record for this reference.
- 37Dailey, A. F.; Hoots, B. E.; Hall, H. I.; Song, R.; Hayes, D.; Fulton, P., Jr.; Prejean, J.; Hernandez, A. L.; Koenig, L. J.; Valleroy, L. A. Vital Signs: Human Immunodeficiency Virus Testing and Diagnosis Delays - United States. MMWR Morb. Mortal. Wkly. Rep. 2017, 66 (47), 1300– 1306, DOI: 10.15585/mmwr.mm6647e1Google ScholarThere is no corresponding record for this reference.
- 38Le Douce, V.; Herbein, G.; Rohr, O.; Schwartz, C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 2010, 7, 32 DOI: 10.1186/1742-4690-7-32Google ScholarThere is no corresponding record for this reference.
- 39Mitchell, B. I.; Laws, E. I.; Ndhlovu, L. C. Impact of Myeloid Reservoirs in HIV Cure Trials. Curr. HIV/AIDS Rep. 2019, 16 (2), 129– 140, DOI: 10.1007/s11904-019-00438-5Google ScholarThere is no corresponding record for this reference.
- 40Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 2011, 6 (1), 4, DOI: 10.1097/COH.0b013e328340ffbbGoogle ScholarThere is no corresponding record for this reference.
- 41Karn, J.; Stoltzfus, C. M. Transcriptional and Posttranscriptional Regulation of HIV-1 Gene Expression. Cold Spring Harbor Perspect. Med. 2012, 2 (2), a006916 DOI: 10.1101/cshperspect.a006916Google ScholarThere is no corresponding record for this reference.
- 42Pearson, R.; Kim, Y. K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic Silencing of Human Immunodeficiency Virus (HIV) Transcription by Formation of Restrictive Chromatin Structures at the Viral Long Terminal Repeat Drives the Progressive Entry of HIV into Latency. J. Virol. 2008, 82 (24), 12291– 12303, DOI: 10.1128/JVI.01383-08Google ScholarThere is no corresponding record for this reference.
- 43D’Orso, I. The HIV-1 Transcriptional Program: From Initiation to Elongation Control. J. Mol. Biol. 2025, 437 (1), 168690 DOI: 10.1016/j.jmb.2024.168690Google ScholarThere is no corresponding record for this reference.
- 44Ma, Y.; Li, C.; Valente, S. Human chromatin remodelers regulating HIV-1 transcription: a target for small molecule inhibitors. Epigenet. Chromatin 2025, 18 (1), 21 DOI: 10.1186/s13072-025-00582-wGoogle ScholarThere is no corresponding record for this reference.
- 45Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4 (1), 261– 285, DOI: 10.1146/annurev-virology-101416-041646Google ScholarThere is no corresponding record for this reference.
- 46Shukla, A.; Ramirez, N. P.; D’Orso, I. HIV-1 Proviral Transcription and Latency in the New Era. Viruses 2020, 12 (5), 555 DOI: 10.3390/v12050555Google ScholarThere is no corresponding record for this reference.
- 47Valle-Casuso, J. C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29 (3), 611, DOI: 10.1016/j.cmet.2018.11.015Google ScholarThere is no corresponding record for this reference.
- 48Chevalier, M. F.; Weiss, L. The split personality of regulatory T cells in HIV infection. Blood 2013, 121 (1), 29– 37, DOI: 10.1182/blood-2012-07-409755Google ScholarThere is no corresponding record for this reference.
- 49Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A. C.; Denis, G. V.; Bradner, J.; Sebastiani, P.; Margolis, D. M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukocyte Biol. 2012, 92 (6), 1147, DOI: 10.1189/jlb.0312165Google ScholarThere is no corresponding record for this reference.
- 50Mbonye, U.; Karn, J. The cell biology of HIV-1 latency and rebound. Retrovirology 2024, 21 (1), 6 DOI: 10.1186/s12977-024-00639-wGoogle ScholarThere is no corresponding record for this reference.
- 51Tamaru, H. Confining euchromatin/heterochromatin territory: jumonji crosses the line. Genes Dev. 2010, 24 (14), 1465, DOI: 10.1101/gad.1941010Google ScholarThere is no corresponding record for this reference.
- 52Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521 (7551), 227, DOI: 10.1038/nature14226Google ScholarThere is no corresponding record for this reference.
- 53Bedwell, G. J.; Engelman, A. N. Factors that mold the nuclear landscape of HIV-1 integration. Nucleic Acids Res. 2021, 49 (2), 621, DOI: 10.1093/nar/gkaa1207Google ScholarThere is no corresponding record for this reference.
- 54Hashemi, P.; Sadowski, I. Diversity of small molecule HIV-1 latency reversing agents identified in low- and high-throughput small molecule screens. Med. Res. Rev. 2020, 40 (3), 881, DOI: 10.1002/med.21638Google ScholarThere is no corresponding record for this reference.
- 55Wierda, R. J.; Kuipers, H. F.; van Eggermond, M. C.; Benard, A.; van Leeuwen, J. C.; Carluccio, S.; Geutskens, S. B.; Jukema, J. W.; Marquez, V. E.; Quax, P. H.; van den Elsen, P. J. Epigenetic control of CCR5 transcript levels in immune cells and modulation by small molecules inhibitors. J. Cell Mol. Med. 2012, 16 (8), 1866– 1877, DOI: 10.1111/j.1582-4934.2011.01482.xGoogle ScholarThere is no corresponding record for this reference.
- 56Alamer, E.; Zhong, C.; Liu, Z.; Niu, Q.; Long, F.; Guo, L.; Gelman, B. B.; Soong, L.; Zhou, J.; Hu, H. Epigenetic Suppression of HIV in Myeloid Cells by the BRD4-Selective Small Molecule Modulator ZL0580. J. Virol. 2020, 94 (11), e01880-19 DOI: 10.1128/JVI.01880-19Google ScholarThere is no corresponding record for this reference.
- 57Le Douce, V.; Colin, L.; Redel, L.; Cherrier, T.; Herbein, G.; Aunis, D.; Rohr, O.; Van Lint, C.; Schwartz, C. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res. 2012, 40 (5), 1904– 1915, DOI: 10.1093/nar/gkr857Google ScholarThere is no corresponding record for this reference.
- 58Brown, L. S.; Foster, C. G.; Courtney, J.-M.; King, N. E.; Howells, D. W.; Sutherland, B. A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 282 DOI: 10.3389/fncel.2019.00282Google ScholarThere is no corresponding record for this reference.
- 59Marinho, M.; Novais, C.; Marques, J.; Bragança, M. Efavirenz and neuropsychiatric effects–When the treatment complicates matter further. Eur. Psychiatry 2017, 41 (S1), S237 DOI: 10.1016/j.eurpsy.2017.01.2255Google ScholarThere is no corresponding record for this reference.
- 60Wang, F.; Rademeyer, K.; Namuju, O. C.; Abdusalaamu, K.; Fisher, J.; Meya, D. B.; McRae, M.; Boulware, D. R.; Lukande, R.; Nicol, M. R. Postmortem Analysis of Dolutegravir, Tenofovir, Lamivudine, and Efavirenz Penetration in Multiple Central Nervous System Compartments. J. Infect. Dis. 2024, 230 (5), 1215– 1223, DOI: 10.1093/infdis/jiae325Google ScholarThere is no corresponding record for this reference.
- 61Banga, R.; Perreau, M. The multifaceted nature of HIV tissue reservoirs. Curr. Opin. HIV AIDS 2024, 19 (3), 116, DOI: 10.1097/COH.0000000000000851Google ScholarThere is no corresponding record for this reference.
- 62Nühn, M. M.; Gumbs, S. B. H.; Schipper, P. J.; Drosou, I.; Gharu, L.; Buchholtz, N.; Snijders, G.; Gigase, F. A. J.; Wensing, A. M. J.; Symons, J. Microglia Exhibit a Unique Intact HIV Reservoir in Human Postmortem Brain Tissue. Viruses 2025, 17 (4), 467, DOI: 10.3390/v17040467Google ScholarThere is no corresponding record for this reference.
- 63Honeycutt, J. B.; Liao, B.; Nixon, C. C.; Cleary, R. A.; Thayer, W. O.; Birath, S. L.; Swanson, M. D.; Sheridan, P.; Zakharova, O.; Prince, F. T cells establish and maintain CNS viral infection in HIV-infected humanized mice. J. Clin. Invest. 2018, 128 (7), 2862– 2876, DOI: 10.1172/JCI98968Google ScholarThere is no corresponding record for this reference.
- 64Gray, L. R.; Cowley, D.; Welsh, C.; Lu, H. K.; Brew, B. J.; Lewin, S. R.; Wesselingh, S. L.; Gorry, P. R.; Churchill, M. J. CNS-specific regulatory elements in brain-derived HIV-1 strains affect responses to latency-reversing agents with implications for cure strategies. Mol. Psychiatry 2016, 21 (4), 574, DOI: 10.1038/mp.2015.111Google ScholarThere is no corresponding record for this reference.
- 65Chan, P.; Spudich, S. Central Nervous System Effects of Early HIV Infection and Consequences of Antiretroviral Therapy Initiation during Acute HIV. Viruses 2024, 16 (7), 1082, DOI: 10.3390/v16071082Google ScholarThere is no corresponding record for this reference.
- 66Nühn, M. M.; Gumbs, S. B. H.; Buchholtz, N.; Jannink, L. M.; Gharu, L.; de Witte, L. D.; Wensing, A. M. J.; Lewin, S. R.; Nijhuis, M.; Symons, J. Shock and kill within the CNS: A promising HIV eradication approach?. J. Leukocyte Biol. 2022, 112 (5), 1297– 1315, DOI: 10.1002/JLB.5VMR0122-046RRRGoogle ScholarThere is no corresponding record for this reference.
- 67Ene, L.; Duiculescu, D.; Ruta, S. M. How much do antiretroviral drugs penetrate into the central nervous system?. J. Med. Life 2011, 4 (4), 432– 439Google ScholarThere is no corresponding record for this reference.
- 68Health, N. I. o. FDA approval of HIV medicines. 2021.Google ScholarThere is no corresponding record for this reference.
- 69McMahon, J. H.; Evans, V. A.; Lau, J. S. Y.; Symons, J.; Zerbato, J. M.; Chang, J.; Solomon, A.; Tennakoon, S.; Dantanarayana, A.; Hagenauer, M. Neurotoxicity with high-dose disulfiram and vorinostat used for HIV latency reversal. AIDS 2022, 36 (1), 75, DOI: 10.1097/QAD.0000000000003091Google ScholarThere is no corresponding record for this reference.
- 70Doyon, G.; Zerbato, J.; Mellors, J. W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. Aids 2013, 27 (2), F7– F11, DOI: 10.1097/QAD.0b013e3283570620Google ScholarThere is no corresponding record for this reference.
- 71Eneanya, D. I.; Bianchine, J. R.; Duran, D. O.; Andresen, B. D. The actions of metabolic fate of disulfiram. Annu. Rev. Pharmacol. Toxicol. 1981, 21, 575– 596, DOI: 10.1146/annurev.pa.21.040181.003043Google ScholarThere is no corresponding record for this reference.
- 72Lin, A.; Elbezanti, W. O.; Schirling, A.; Ahmed, A.; Van Duyne, R.; Cocklin, S.; Klase, Z. Alprazolam Prompts HIV-1 Transcriptional Reactivation and Enhances CTL Response Through RUNX1 Inhibition and STAT5 Activation. Front. Neurol. 2021, 12, 663793 DOI: 10.3389/fneur.2021.663793Google ScholarThere is no corresponding record for this reference.
- 73Chowdhury, Z. S.; Morshed, M. M.; Shahriar, M.; Bhuiyan, M. A.; Islam, S. M.; Bin Sayeed, M. S. The Effect of Chronic Alprazolam Intake on Memory, Attention, and Psychomotor Performance in Healthy Human Male Volunteers. Behav. Neurol. 2016, 2016, 3730940 DOI: 10.1155/2016/3730940Google ScholarThere is no corresponding record for this reference.
- 74Ait-Daoud, N.; Hamby, A. S.; Sharma, S.; Blevins, D. A Review of Alprazolam Use, Misuse, and Withdrawal. J. Addict. Med. 2018, 12 (1), 4, DOI: 10.1097/ADM.0000000000000350Google ScholarThere is no corresponding record for this reference.
- 75Skov, L.; Holm, K. M.; Linnet, K. Nitrobenzodiazepines: Postmortem brain and blood reference concentrations. Forensic Sci. Int. 2016, 268, 39– 45, DOI: 10.1016/j.forsciint.2016.09.002Google ScholarThere is no corresponding record for this reference.
- 76Jansen, Y. J. L.; Verset, G.; Schats, K.; Dam, P.-J. V.; Seremet, T.; Kockx, M.; Laethem, J.-L. B. V.; Neyns, B. Phase I clinical trial of decitabine (5-aza-2′-deoxycytidine) administered by hepatic arterial infusion in patients with unresectable liver-predominant metastases. ESMO Open 2019, 4 (2), e000464 DOI: 10.1136/esmoopen-2018-000464Google ScholarThere is no corresponding record for this reference.
- 77Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Van Driessche, B.; Corazza, F.; Gatot, J. S.; Melard, A.; Vanhulle, C.; Kabeya, K. Sequential treatment with 5-aza-2′-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8 (2), 117– 138, DOI: 10.15252/emmm.201505557Google ScholarThere is no corresponding record for this reference.
- 78Chabot, G. G.; Rivard, G. E.; Momparler, R. L. Plasma and cerebrospinal fluid pharmacokinetics of 5-Aza-2′-deoxycytidine in rabbits and dogs. Cancer Res. 1983, 43 (2), 592– 597Google ScholarThere is no corresponding record for this reference.
- 79Noël, J.-M.; Raynal, J.-P. J. I. Chapter 7 - DNA Methyltransferase Inhibitors. Drug Discovery Cancer Epigenet. 2016, 169– 190, DOI: 10.1016/B978-0-12-802208-5.00007-2Google ScholarThere is no corresponding record for this reference.
- 80Lucera, M. B.; Tilton, C. A.; Mao, H.; Dobrowolski, C.; Tabler, C. O.; Haqqani, A. A.; Karn, J.; Tilton, J. C. The Histone Deacetylase Inhibitor Vorinostat (SAHA) Increases the Susceptibility of Uninfected CD4+ T Cells to HIV by Increasing the Kinetics and Efficiency of Postentry Viral Events. J. Virol. 2014, 88 (18), 10803 DOI: 10.1128/JVI.00320-14Google ScholarThere is no corresponding record for this reference.
- 81Archin, N. M.; Kirchherr, J. L.; Sung, J. A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba AD, A. D.; Kuruc, J. D. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Invest. 2017, 127 (8), 3126 DOI: 10.1172/JCI92684Google ScholarThere is no corresponding record for this reference.
- 82Vorinostat Indications and Prescribing Information - FDA.Google ScholarThere is no corresponding record for this reference.
- 83Guntner, A. S.; Peyrl, A.; Mayr, L.; Englinger, B.; Berger, W.; Slavc, I.; Buchberger, W.; Gojo, J. Cerebrospinal fluid penetration of targeted therapeutics in pediatric brain tumor patients. Acta Neuropathol. Commun. 2020, 8 (1), 78 DOI: 10.1186/s40478-020-00953-2Google ScholarThere is no corresponding record for this reference.
- 84Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41 (1), 277 DOI: 10.1093/nar/gks976Google ScholarThere is no corresponding record for this reference.
- 85Bakshi, S.; McKee, C.; Walker, K.; Brown, C.; Chaudhry, G. R. Toxicity of JQ1 in neuronal derivatives of human umbilical cord mesenchymal stem cells. Oncotarget 2018, 9 (73), 33853 DOI: 10.18632/oncotarget.26127Google ScholarThere is no corresponding record for this reference.
- 86Matzuk, M. M.; McKeown, M. R.; Filippakopoulos, P.; Li, Q.; Ma, L.; Agno, J. E.; Lemieux, M. E.; Picaud, S.; Yu, R. N.; Qi, J. Small-Molecule Inhibition of BRDT for Male Contraception. Cell 2012, 150 (4), 673– 684, DOI: 10.1016/j.cell.2012.06.045Google ScholarThere is no corresponding record for this reference.
- 87Dental, C.; Proust, A.; Ouellet, M.; Barat, C.; Tremblay, M. J. HIV-1 Latency-Reversing Agents Prostratin and Bryostatin-1 Induce Blood-Brain Barrier Disruption/Inflammation and Modulate Leukocyte Adhesion/Transmigration. J. Immunol. 2017, 198 (3), 1229– 1241, DOI: 10.4049/jimmunol.1600742Google ScholarThere is no corresponding record for this reference.
- 88Díaz, L.; Martínez-Bonet, M.; Sánchez, J.; Fernández-Pineda, A.; Jiménez, J. L.; Muñoz, E.; Moreno, S.; Álvarez, S.; Muñoz-Fernández, M. Á. Bryostatin activates HIV-1 latent expression in human astrocytes through a PKC and NF-κB-dependent mechanism. Sci. Rep. 2015, 5, 12442 DOI: 10.1038/srep12442Google ScholarThere is no corresponding record for this reference.
- 89Nelson, T. J.; Sen, A.; Alkon, D. L.; Sun, M. K. Adduct formation in liquid chromatography-triple quadrupole mass spectrometric measurement of bryostatin 1. J. Chromatogr. B:Anal. Technol. Biomed. Life Sci. 2014, 944, 55– 62, DOI: 10.1016/j.jchromb.2013.11.020Google ScholarThere is no corresponding record for this reference.
- 90Bartholomeeusen, K.; Fujinaga, K.; Xiang, Y.; Peterlin, B. M. Histone Deacetylase Inhibitors (HDACis) That Release the Positive Transcription Elongation Factor b (P-TEFb) from Its Inhibitory Complex Also Activate HIV Transcription. J. Biol. Chem. 2013, 288 (20), 14400 DOI: 10.1074/jbc.M113.464834Google ScholarThere is no corresponding record for this reference.
- 91Palmieri, D.; Lockman, P. R.; Thomas, F. C.; Hua, E.; Herring, J.; Hargrave, E.; Johnson, M.; Flores, N.; Qian, Y.; Vega-Valle, E. Vorinostat Inhibits Brain Metastatic Colonization in a Model of Triple-Negative Breast Cancer and Induces DNA Double-Strand Breaks. Clin. Cancer Res. 2009, 15 (19), 6148, DOI: 10.1158/1078-0432.CCR-09-1039Google ScholarThere is no corresponding record for this reference.
- 92Jiang, G.; Dandekar, S. Targeting NF-κB Signaling with Protein Kinase C Agonists As an Emerging Strategy for Combating HIV Latency. AIDS Res. Hum. Retroviruses 2015, 31 (1), 4, DOI: 10.1089/aid.2014.0199Google ScholarThere is no corresponding record for this reference.
- 93Nelson, T. J.; Sun, M. K.; Lim, C.; Sen, A.; Khan, T.; Chirila, F. V.; Alkon, D. L. Bryostatin Effects on Cognitive Function and PKCε in Alzheimer’s Disease Phase IIa and Expanded Access Trials. J. Alzheimer’s Dis. 2017, 58 (2), 521– 535, DOI: 10.3233/JAD-170161Google ScholarThere is no corresponding record for this reference.
- 94Fuller, R. K.; Gordis, E. Does disulfiram have a role in alcoholism treatment today?. Addiction 2004, 99 (1), 21, DOI: 10.1111/j.1360-0443.2004.00597.xGoogle ScholarThere is no corresponding record for this reference.
- 95Abner, E.; Jordan, A. HIV ″shock and kill″ therapy: In need of revision. Antiviral Res. 2019, 166, 19– 34, DOI: 10.1016/j.antiviral.2019.03.008Google ScholarThere is no corresponding record for this reference.
- 96Mukim, A.; Smith, D. M.; Deshmukh, S.; Qazi, A. A.; Beliakova-Bethell, N. A camptothetin analog, topotecan, promotes HIV latency via interference with HIV transcription and RNA splicing. J. Virol. 2023, 97 (2), e01630-22 DOI: 10.1128/jvi.01630-22Google ScholarThere is no corresponding record for this reference.
- 97Mousseau, G.; Kessing, C. F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S. T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. mBio 2015, 6 (4), e00465 DOI: 10.1128/mBio.00465-15Google ScholarThere is no corresponding record for this reference.
- 98Li, C.; Mori, L.; Valente, S. T. The Block-and-Lock Strategy for Human Immunodeficiency Virus Cure: Lessons Learned from Didehydro-Cortistatin A. J. Infect. Dis. 2021, 223, S46– S53, DOI: 10.1093/infdis/jiaa681Google ScholarThere is no corresponding record for this reference.
- 99Peters, R. J.; Stevenson, M. Irreversible Loss of HIV-1 Proviral Competence in Myeloid Cells upon Suppression of NF-κB Activity. J. Virol. 2022, 96 (12), e00484-22 DOI: 10.1128/jvi.00484-22Google ScholarThere is no corresponding record for this reference.
- 100Debrabander, Q.; Hensley, K. S.; Psomas, C. K.; Bramer, W.; Mahmoudi, T.; Welzen, B. J. v.; Verbon, A.; Rokx, C. The efficacy and tolerability of latency-reversing agents in reactivating the HIV-1 reservoir in clinical studies: a systematic review. J. Virus Erad. 2023, 9 (3), 100342 DOI: 10.1016/j.jve.2023.100342Google ScholarThere is no corresponding record for this reference.
- 101Liu, Y.; Liu, H.; Kim, B. O.; Gattone, V. H.; Li, J. L.; Nath, A.; Blum, J.; He, J. J. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: Requirement for the human mannose receptor. J. Virol. 2004, 78 (8), 4120– 4133, DOI: 10.1128/JVI.78.8.4120-4133.2004Google ScholarThere is no corresponding record for this reference.
- 102Liu, Y. B.; Cao, W.; Sun, M.; Li, T. S. Broadly neutralizing antibodies for HIV-1: efficacies, challenges and opportunities. Emerging Microbes Infect. 2020, 9 (1), 194– 206, DOI: 10.1080/22221751.2020.1713707Google ScholarThere is no corresponding record for this reference.
- 103Wen, J.; Cheever, T.; Wang, L.; Wu, D.; Reed, J.; Mascola, J.; Chen, X. J.; Liu, C. P.; Pegu, A.; Sacha, J. B. Improved delivery of broadly neutralizing antibodies by nanocapsules suppresses SHIV infection in the CNS of infant rhesus macaques. PLoS Pathog. 2021, 17 (7), e1009738 DOI: 10.1371/journal.ppat.1009738Google ScholarThere is no corresponding record for this reference.
- 104Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33 (9), 941– 951, DOI: 10.1038/nbt.3330Google ScholarThere is no corresponding record for this reference.
- 105Halper-Stromberg, A.; Lu, C. L.; Klein, F.; Horwitz, J. A.; Bournazos, S.; Nogueira, L.; Eisenreich, T. R.; Liu, C.; Gazumyan, A.; Schaefer, U. Broadly Neutralizing Antibodies and Viral Inducers Decrease Rebound from HIV-1 Latent Reservoirs in Humanized Mice. Cell 2014, 158 (5), 989– 999, DOI: 10.1016/j.cell.2014.07.043Google ScholarThere is no corresponding record for this reference.
- 106Gunst, J. D.; Hojen, J. F.; Sogaard, O. S. Broadly neutralizing antibodies combined with latency-reversing agents or immune modulators as strategy for HIV-1 remission. Curr. Opin. HIV AIDS 2020, 15 (5), 309– 315, DOI: 10.1097/COH.0000000000000641Google ScholarThere is no corresponding record for this reference.
- 107Julg, B.; Walker-Sperling, V. E. K.; Wagh, K.; Aid, M.; Stephenson, K. E.; Zash, R.; Liu, J. Y.; Nkolola, J. P.; Hoyt, A.; Castro, M. Safety and antiviral effect of a triple combination of HIV-1 broadly neutralizing antibodies: a phase 1/2a trial. Nat. Med. 2024, 30 (12), 3534– 3543, DOI: 10.1038/s41591-024-03247-5Google ScholarThere is no corresponding record for this reference.
- 108Mbonye, U.; Karn, J. Control of HIV Latency by Epigenetic and Non-Epigenetic Mechanisms. Curr. HIV Res. 2011, 9 (8), 554, DOI: 10.2174/157016211798998736Google ScholarThere is no corresponding record for this reference.
- 109Duggan, N. N.; Dragic, T.; Chanda, S. K.; Pache, L. Breaking the Silence: Regulation of HIV Transcription and Latency on the Road to a Cure. Viruses 2023, 15 (12), 2435, DOI: 10.3390/v15122435Google ScholarThere is no corresponding record for this reference.
- 110Archin, N. M.; Bateson, R.; Tripathy, M. K.; Crooks, A. M.; Yang, K.-H.; Dahl, N. P.; Kearney, M. F.; Anderson, E. M.; Coffin, J. M.; Strain, M. C. HIV-1 Expression Within Resting CD4+ T Cells After Multiple Doses of Vorinostat. J. Infect. Dis. 2014, 210 (5), 728, DOI: 10.1093/infdis/jiu155Google ScholarThere is no corresponding record for this reference.
- 111Kinloch, N. N.; Shahid, A.; Dong, W.; Kirkby, D.; Jones, B. R.; Beelen, C. J.; MacMillan, D.; Lee, G. Q.; Mota, T. M.; Sudderuddin, H. HIV reservoirs are dominated by genetically younger and clonally enriched proviruses. mBio 2023, 14 (6), e02417-23 DOI: 10.1128/mbio.02417-23Google ScholarThere is no corresponding record for this reference.
- 112Battistini, A.; Sgarbanti, M. HIV-1 Latency: An Update of Molecular Mechanisms and Therapeutic Strategies. Viruses 2014, 6 (4), 1715, DOI: 10.3390/v6041715Google ScholarThere is no corresponding record for this reference.
- 113Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Driessche, B. V.; Corazza, F.; Gatot, J. S.; Melard, A.; Vanhulle, C.; Kabeya, K. Sequential treatment with 5-aza-2′-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2015, 8 (2), 117– 138, DOI: 10.15252/emmm.201505557Google ScholarThere is no corresponding record for this reference.
- 114Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B. M. Bromodomain and Extra-terminal (BET) Bromodomain Inhibition Activate Transcription via Transient Release of Positive Transcription Elongation Factor b (P-TEFb) from 7SK Small Nuclear Ribonucleoprotein. J. Biol. Chem. 2012, 287 (43), 36609– 36616, DOI: 10.1074/jbc.M112.410746Google ScholarThere is no corresponding record for this reference.
- 115Henderson, L. J.; Sharma, A.; Monaco, M. C.; Major, E. O.; Al-Harthi, L. Human immunodeficiency virus type 1 (HIV-1) transactivator of transcription through its intact core and cysteine-rich domains inhibits Wnt/β-catenin signaling in astrocytes: relevance to HIV neuropathogenesis. J. Neurosci. 2012, 32 (46), 16306– 16313, DOI: 10.1523/JNEUROSCI.3145-12.2012Google ScholarThere is no corresponding record for this reference.
- 116Al-Harthi, L. Interplay between Wnt/β-catenin signaling and HIV: virologic and biologic consequences in the CNS. J. Neuroimmune Pharmacol. 2012, 7 (4), 731– 739, DOI: 10.1007/s11481-012-9411-yGoogle ScholarThere is no corresponding record for this reference.
- 117Barbian, H. J.; Seaton, M. S.; Narasipura, S. D.; Wallace, J.; Rajan, R.; Sha, B. E.; Al-Harthi, L. β-catenin regulates HIV latency and modulates HIV reactivation. PLoS Pathog. 2022, 18 (3), e1010354 DOI: 10.1371/journal.ppat.1010354Google ScholarThere is no corresponding record for this reference.
- 118Wen, J.; Li, X.; Zhao, Q. X.; Yang, X. F.; Wu, M. L.; Yan, Q.; Chang, J.; Wang, H.; Jin, X.; Su, X. Pharmacological suppression of glycogen synthase kinase-3 reactivates HIV-1 from latency via activating Wnt/β-catenin/TCF1 axis in CD4(+) T cells. Emerging Microbes Infect. 2022, 11 (1), 391– 405, DOI: 10.1080/22221751.2022.2026198Google ScholarThere is no corresponding record for this reference.
- 119Mavigner, M.; Zanoni, M.; Tharp, G. K.; Habib, J.; Mattingly, C. R.; Lichterfeld, M.; Nega, M. T.; Vanderford, T. H.; Bosinger, S. E.; Chahroudi, A. Pharmacological Modulation of the Wnt/β-Catenin Pathway Inhibits Proliferation and Promotes Differentiation of Long-Lived Memory CD4(+) T Cells in Antiretroviral Therapy-Suppressed Simian Immunodeficiency Virus-Infected Macaques. J. Virol. 2019, 94 (1), e01094-19 DOI: 10.1128/JVI.01094-19Google ScholarThere is no corresponding record for this reference.
- 120Tibebe, H.; Marquez, D.; McGraw, A.; Gagliardi, S.; Sullivan, C.; Hillmer, G.; Narayan, K.; Izumi, C.; Keating, A.; Izumi, T. Targeting Latent HIV Reservoirs: Effectiveness of Combination Therapy with HDAC and PARP Inhibitors. Viruses 2025, 17 (3), 400, DOI: 10.3390/v17030400Google ScholarThere is no corresponding record for this reference.
- 121Huang, Y.; Luo, D.; Chen, X.; Zhang, D.; Huang, Z.; Xiao, S. HIV-Related Stress Experienced by Newly Diagnosed People Living with HIV in China: A 1-Year Longitudinal Study. Int. J. Environ. Res. Public Health 2020, 17 (8), 2681, DOI: 10.3390/ijerph17082681Google ScholarThere is no corresponding record for this reference.
- 122Saloner, R.; Grelotti, D. J.; Tyree, G.; Sundermann, E. E.; Ma, Q.; Letendre, S.; Heaton, R. K.; Cherner, M. Benzodiazepine Use is Associated with an Increased Risk of Neurocognitive Impairment in People Living with HIV. J. Acquired Immune Defic. Syndr. 2019, 82 (5), 475, DOI: 10.1097/QAI.0000000000002183Google ScholarThere is no corresponding record for this reference.
- 123Olfson, M.; King, M.; Schoenbaum, M. Benzodiazepine use in the United States. JAMA Psychiatry 2015, 72 (2), 136– 142, DOI: 10.1001/jamapsychiatry.2014.1763Google ScholarThere is no corresponding record for this reference.
- 124Cunningham, L.; Finckbeiner, S.; Hyde, R. K.; Southall, N.; Marugan, J.; Yedavalli, V. R. K.; Dehdashti, S. J.; Reinhold, W. C.; Alemu, L.; Zhao, L. Identification of benzodiazepine Ro5–3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1–CBFβ interaction. Proc. Natl. Acad. Sci. U.S.A. 2012, 109 (36), 14592 DOI: 10.1073/pnas.1200037109Google ScholarThere is no corresponding record for this reference.
- 125Klase, Z.; Yedavalli, V.; Houzet, L.; Perkins, M.; Maldarelli, F.; Brenchley, J.; Strebel, K.; Liu, P.; Jeang, K. T. Activation of HIV-1 from Latent Infection via Synergy of RUNX1 Inhibitor Ro5–3335 and SAHA. PLoS Pathog. 2014, 10 (3), e1003997 DOI: 10.1371/journal.ppat.1003997Google ScholarThere is no corresponding record for this reference.
- 126Kierdorf, K.; Prinz, M. Factors regulating microglia activation. Front. Cell Neurosci. 2013, 7, 44, DOI: 10.3389/fncel.2013.00044Google ScholarThere is no corresponding record for this reference.
- 127Capriotti, Z.; Klase, Z. Innate immune memory in chronic HIV and HIV-associated neurocognitive disorders (HAND): potential mechanisms and clinical implications. J. NeuroVirol. 2024, 30, 451, DOI: 10.1007/s13365-024-01239-2Google ScholarThere is no corresponding record for this reference.
- 128Elbezanti, W.; Lin, A. G.; Schirling, A.; Jackson, A.; Marshall, M.; Van Duyne, R.; Maldarelli, F.; Sardo, L.; Klase, Z. Benzodiazepines Drive Alteration of Chromatin at the Integrated HIV-1 LTR. Viruses 2020, 12 (2), 191 DOI: 10.3390/v12020191Google ScholarThere is no corresponding record for this reference.
- 129Castillo, A. A.; McElrath, C.; Marshall, G.; Stevenson, M.; Castillo, A. A.; McElrath, C.; Marshall, G.; Stevenson, M. Myeloid Cell Reservoirs: Role in HIV-Host Interplay and Strategies for Myeloid Reservoir Elimination. Curr. Clin. Microbiol. Rep. 2024, 11 (4), 209– 219, DOI: 10.1007/s40588-024-00234-9Google ScholarThere is no corresponding record for this reference.
- 130Didigu, C. A.; Wilen, C. B.; Wang, J.; Duong, J.; Secreto, A. J.; Danet-Desnoyers, G. A.; Riley, J. L.; Gregory, P. D.; June, C. H.; Holmes, M. C.; Doms, R. W. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood 2014, 123 (1), 61– 69, DOI: 10.1182/blood-2013-08-521229Google ScholarThere is no corresponding record for this reference.
- 131Tebas, P.; Stein, D.; Tang, W. W.; Frank, I.; Wang, S. Q.; Lee, G.; Spratt, S. K.; Surosky, R. T.; Giedlin, M. A.; Nichol, G. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370 (10), 901– 910, DOI: 10.1056/NEJMoa1300662Google ScholarThere is no corresponding record for this reference.
- 132Tebas, P.; Jadlowsky, J. K.; Shaw, P. A.; Tian, L.; Esparza, E.; Brennan, A. L.; Kim, S.; Naing, S. Y.; Richardson, M. W.; Vogel, A. N. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J. Clin. Invest. 2021, 131 (7), e144486 DOI: 10.1172/JCI144486Google ScholarThere is no corresponding record for this reference.
- 133Letchumanan, P.; Theva Das, K. The role of genetic diversity, epigenetic regulation, and sex-based differences in HIV cure research: a comprehensive review. Epigenet. Chromatin 2025, 18 (1), 1 DOI: 10.1186/s13072-024-00564-4Google ScholarThere is no corresponding record for this reference.
- 134Macedo, A. B.; Novis, C. L.; Bosque, A. Targeting Cellular and Tissue HIV Reservoirs With Toll-Like Receptor Agonists. Front. Immunol. 2019, 10, 2450 DOI: 10.3389/fimmu.2019.02450Google ScholarThere is no corresponding record for this reference.
- 135Acioglu, C.; Heary, R. F.; Elkabes, S. Roles of neuronal toll-like receptors in neuropathic pain and central nervous system injuries and diseases. Brain, Behav., Immun. 2022, 102, 163– 178, DOI: 10.1016/j.bbi.2022.02.016Google ScholarThere is no corresponding record for this reference.
- 136Kielian, T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J. Neurosci. Res. 2006, 83 (5), 711– 730, DOI: 10.1002/jnr.20767Google ScholarThere is no corresponding record for this reference.
- 137Thibault, S.; Imbeault, M.; Tardif, M. R.; Tremblay, M. J. TLR5 stimulation is sufficient to trigger reactivation of latent HIV-1 provirus in T lymphoid cells and activate virus gene expression in central memory CD4 < SUP>+</SUP> T cells. Virology 2009, 389 (1–2), 20– 25, DOI: 10.1016/j.virol.2009.04.019Google ScholarThere is no corresponding record for this reference.
- 138Tsai, A.; Irrinki, A.; Kaur, J.; Cihlar, T.; Kukolj, G.; Sloan, D. D.; Murry, J. P. Toll-Like Receptor 7 Agonist GS-9620 Induces HIV Expression and HIV-Specific Immunity in Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J. Virol. 2017, 91 (8), e02166-16 DOI: 10.1128/JVI.02166-16Google ScholarThere is no corresponding record for this reference.
- 139Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14, 1 DOI: 10.1186/s12977-017-0335-8Google ScholarThere is no corresponding record for this reference.
- 140Omueti, K. O.; Beyer, J. M.; Johnson, C. M.; Lyle, E. A.; Tapping, R. I. Domain exchange between human Toll-like receptors 1 and 6 reveals a region required for lipopeptide discrimination. J. Biol. Chem. 2005, 280 (44), 36616– 36625, DOI: 10.1074/jbc.M504320200Google ScholarThere is no corresponding record for this reference.
- 141Duan, S. Q.; Liu, S. W. Targeting NK Cells for HIV-1 Treatment and Reservoir Clearance. Front. Immunol. 2022, 13, 842746 DOI: 10.3389/fimmu.2022.842746Google ScholarThere is no corresponding record for this reference.
- 142Lu, C. L.; Murakowski, D. K.; Bournazos, S.; Schoofs, T.; Sarkar, D.; Halper-Stromberg, A.; Horwitz, J. A.; Nogueira, L.; Golijanin, J.; Gazumyan, A. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science 2016, 352 (6288), 1001– 1004, DOI: 10.1126/science.aaf1279Google ScholarThere is no corresponding record for this reference.
- 143Merck & Co. Product Information. Zolinza (vorinostat). 2006.Google ScholarThere is no corresponding record for this reference.
- 144Thorlund, K.; Horwitz, M. S.; Fife, B. T.; Lester, R.; Cameron, D. W. Landscape review of current HIV ’kick and kill’ cure research - some kicking, not enough killing. BMC Infect. Dis. 2017, 17, 595 DOI: 10.1186/s12879-017-2683-3Google ScholarThere is no corresponding record for this reference.
- 145Jones, R. B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G. L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M. A.; Trocha, A. Histone Deacetylase Inhibitors Impair the Elimination of HIV-Infected Cells by Cytotoxic T-Lymphocytes. PLoS Pathog. 2014, 10 (8), e1004287 DOI: 10.1371/journal.ppat.1004287Google ScholarThere is no corresponding record for this reference.
- 146Li, Y. Y.; Hong, J. X.; Zhang, L. Q. The Rational Combination Strategy of Immunomodulatory Latency Reversing Agents and Novel Immunotherapy to Achieve HIV-1 Cure. Infect. Dis. Immun. 2022, 2 (4), 263– 273, DOI: 10.1097/ID9.0000000000000045Google ScholarThere is no corresponding record for this reference.
- 147Mastrangelo, A.; Gama, L.; Cinque, P. Strategies to target the central nervous system HIV reservoir. Curr. Opin. HIV AIDS 2024, 19 (3), 133– 140, DOI: 10.1097/COH.0000000000000847Google ScholarThere is no corresponding record for this reference.
- 148Morris, E. C.; Neelapu, S. S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22 (2), 85– 96, DOI: 10.1038/s41577-021-00547-6Google ScholarThere is no corresponding record for this reference.
- 149Kapoor, A.; Tan, C. S. Immunotherapeutics to Treat HIV in the Central Nervous System. Curr. Hiv/Aids Rep. 2020, 17 (5), 499– 506, DOI: 10.1007/s11904-020-00519-wGoogle ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACS Infectious Diseases
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
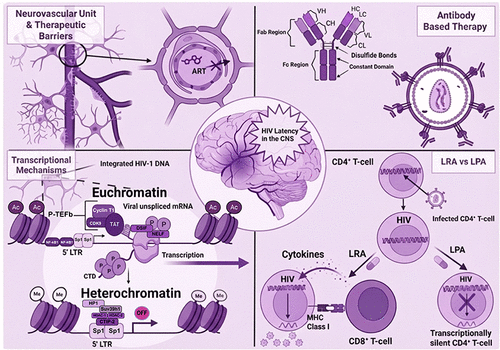
Figure 1

Figure 1. HIV-1 Entry and Transcriptional Regulation in Target Cells. (A) Overview of the HIV-1 replication cycle. Key steps in the HIV-1 replication include viral entry (fusion), integration into the host genome, transcription, translation into viral proteins, virion budding, and maturation. The illustration provides insights into the transcriptional activation mechanisms in actively infected CD4+ T-cells. (B) Histone acetyltransferases (HATs) incorporate acetyl groups to lysine residues on histones positioned at nucleosomes Nuc-0 and Nuc-1, promoting euchromatin formation and enhancing transcriptional accessibility. RNA polymerase II (RNA Pol II) assembles at the 5′ long terminal repeat (LTR) region of the HIV-1 genome, where associated factors contribute to the formation of the preinitiation complex (PIC). During transcription elongation, the HIV Tat protein binds to the trans-activation response (TAR) element of the nascent viral mRNA. This interaction recruits the positive transcription elongation factor b (P-TEFb) complex, which phosphorylates the DRB sensitivity-inducing factor (DSIF) and the negative elongation factor (NELF), thereby preventing transcriptional pausing. Additionally, the C-terminal domain (CTD) of RNA polymerase II undergoes phosphorylation, facilitating efficient transcription elongation. (C) Mechanisms of transcriptional repression in latent infection. In contrast to active transcription, histone methyltransferases (HMTs) add methyl groups to histone residues, thereby driving the formation of heterochromatin that limits transcriptional accessibility. Consequently, despite RNA polymerase II and the preinitiation complex (PIC) assembling at the HIV-1 LTR, transcription is effectively suppressed, maintaining viral latency. Therapeutic LRAs target these repressive mechanisms; for example, Vorinostat inhibits histone deacetylases (HDACs) to promote acetylation, JQ1 inhibits BRD4 to release P-TEFb, and other upcoming therapies inhibit transcriptional repression at the 5′ LTR. P: Phosphate, Ac: Acetyl group, Me: Methyl, HAT: Histone Acetyl Transferases, HMT: Histone Methyl Transferases. Created with BioRender.com.
Figure 2

Figure 2. (A) Cross-sectional representation of the Neurovascular Unit (NVU), the Structural Features of the BBB, and HIV-1 CNS infiltration. This figure illustrates the neurovascular unit and highlights its key structural components, including brain microvascular endothelial cells (BMECs), pericytes, and astrocytic endfeet. The figure emphasizes the limited ability of antiretroviral therapy molecules to cross the BBB due to efflux by transporters such as P-glycoproteins (P-gp), multidrug resistance protein (MRP), and breast cancer resistance protein (BCRP) located on the luminal side of the BMECs. A clear concentration gradient is evident, with ART levels highest in the bloodstream and substantially lower in endothelial cells and the CNS. ART: antiretroviral therapeutic, P-Gp: p-glycoprotein, BCRP: breast cancer resistance protein, MRP2: multidrug resistance protein. (B) Overview of the organization and functional characteristics of the BBB, emphasizing its role in HIV-1 infection within the CNS. HIV-1-infected CD4+ T-cells cross the BBB via tight junctions between brain microvascular endothelial cells. Subsequently, CD4+ T lymphocytes infect astrocytes through a CD4-independent mechanism. Endothelial cells are closely associated with astrocytic end feet and pericytes, collectively maintaining the BBB’s selective permeability. The figure illustrates the limitations of antiretroviral therapy penetration into the CNS, highlighting how ART molecules are unable to cross the BBB, primarily due to the presence of tight junctions and active efflux transporters, ABC: ATP Binding Cassette. Created with BioRender.com.
Figure 3
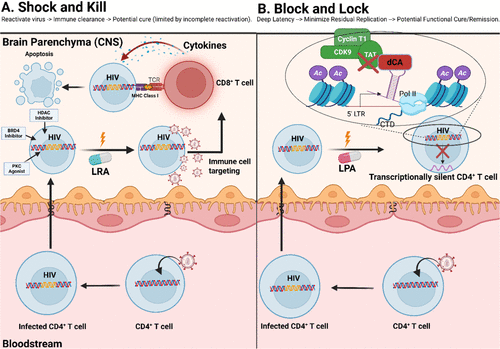
Figure 3. Contrasting Paradigms of HIV Latency Management: LRAs (″Shock and Kill″) versus LPAs (″Block and Lock″). Illustration of two distinct therapeutic strategies for managing HIV latency. (A) (″Shock and Kill″) depicts a latently infected CD4+ T-cell undergoing viral reactivation upon receiving an LRA signal. Following reactivation, an immune effector cell (specifically, a CD8+ T-cell) recognizes and targets the infected cell, inducing apoptosis and clearance. (B) (″Block and Lock″) illustrates an alternative strategy, where a latently infected CD4+ T-cell receives a latency-promoting agent (LPA) signal, reinforcing transcriptional silencing. This mechanism highlights the inhibitory action of the compound didehydro-cortistatin A (dCA) on critical HIV transcription elongation factors, including Tat, CDK9, and Cyclin T1, thereby maintaining durable latency without inducing viral production. LRA: latency reversal agent, LPA: latency promoting agent, BRD4: Bromodomain 4, MHC: Major histocompatibility class I, TCR: T cell receptor, PKC: Protein Kinase C, HDAC: Histone Deacetylase Inhibitor, dCA: didehydro-cortistatin A, CDK9: Cyclin-dependent kinase 9, Ac: Acetyl group. Created with BioRender.com.
Figure 4
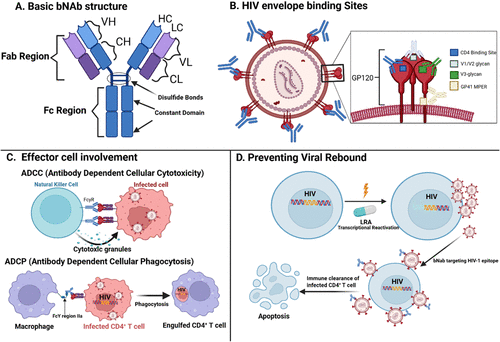
Figure 4. Broadly Neutralizing Antibodies (bNAbs) and Control of HIV Viral Rebound. (A) Schematic representation of a bNAb, highlighting key structural components including the variable region, fragment antigen-binding (Fab) region, and fragment crystallizable (Fc) region. (B) Illustration of the HIV envelope glycoproteins gp120 and gp41, with specific epitopes targeted by individual bNAbs. These epitopes include regions within the variable loops (V2, V3), glycan-dependent sites, and the membrane-proximal external region (MPER). (C) Immune effector cells involved in antibody-mediated clearance mechanisms, specifically antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), demonstrate how bNAbs facilitate immune recognition and subsequent elimination of HIV-infected cells. (D) Schematic showing bNAb’s binding to HIV-1 virions following transcriptional reactivation of integrated, previously latent provirus in CD4+ T-cells. LRA: Latency reversal agent, LPA: Latency promoting agent, bNAb: Broadly neutralizing antibody, ADCC: Antibody-dependent cellular cytotoxicity, ADCP: Antibody-dependent cellular phagocytosis, VH: Variable Heavy, VL: Variable Light, CH: Constant Heavy, CL: Constant Light, Fab: Fragment antigen binding domain, FcyR: Fc-γ receptor, GP120: Glycoprotein 120. Created with BioRender.com.
Figure 5
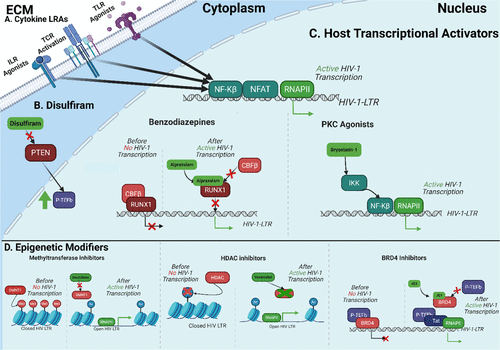
Figure 5. Overview of HIV Latency Reversal Agents. (A) Cytokine reactivation of HIV transcription is generally mediated through increased NF-kB and NFAT binding of the LTR, recruiting RNAPII. (B) Disulfiram can block PTEN/AKT signaling by inhibiting phosphatase, removing phosphate from PIP3. This increases P-TEFb, which mediates Tat-dependent HIV transcription. (C) LRAs can activate host-mediated transcription of HIV. PKC agonists increase NF-kB activity by enhancing its binding to the LTR. Benzodiazepines disrupt CBFβ binding to RUNX1, increasing transcription. (D) BRD4 inhibitor competitively block the binding of P-TEFb to BRD4, allowing P-TEFb to bind with Tat and complete Tat-dependent transcription. Methyltransferase inhibitors block the addition of methyl groups, leading to a more transcriptionally active epigenetic environment. HDAC inhibitors block histone deacetylation, leading to a more transcriptionally active epigenetic environment. ECM: Extracellular matrix, DMNT1: DNA methyltransferase 1, HDAC: Histone Deacetylase, RNAPII: RNA Polymerase II, NFAT: Nuclear Factor of Activated T-cells, NF-kB: Nuclear factor-kappa B, IKK: I kappa B kinase, P-TEFb: Positive Transcription Elongation Factor b, BRD4: Bromodomain-containing protein 4. Created with BioRender.com.
Figure 6
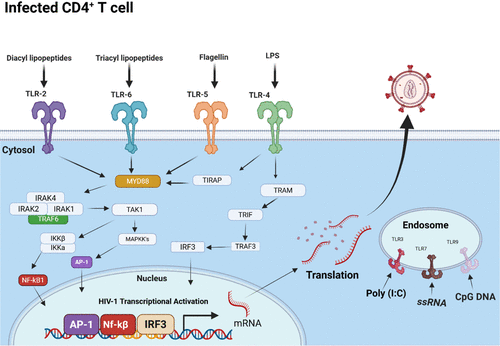
Figure 6. TLR-mediated HIV Latency Reversal. A schematic illustration of the Toll-like receptors (TLRs) involved in HIV latency reversal in latently infected CD4+ T-cells. TLRs 2, 4, 5, and 6 are depicted on the cellular surface, whereas TLRs 3, 7, and 9 are localized within endosomal compartments. Pathogen-associated molecular patterns (PAMPs), including microbial and viral motifs, interact with these TLRs, initiating intracellular signaling cascades. Activation of these pathways leads to nuclear translocation of transcription factors such as AP-1, NF-κB, and IRF3, facilitating their binding to the HIV 5′ LTR region and resulting in transcriptional reactivation of latent HIV. TLR: Toll-Like Receptor, NF-kB: NF-Kappa β, LPS: Lipopolysaccharide. Created with BioRender.com.
References
This article references 149 other publications.
- 1Payagala, S.; Pozniak, A. The global burden of HIV. Clin. Dermatol. 2024, 42 (2), 119– 127, DOI: 10.1016/j.clindermatol.2024.02.001There is no corresponding record for this reference.
- 2Alum, E. U.; Uti, D. E.; Ugwu, O. P. C.; Alum, B. N. Toward a cure - Advancing HIV/AIDs treatment modalities beyond antiretroviral therapy: A Review. Medicine 2024, 103 (27), e38768 DOI: 10.1097/md.0000000000038768There is no corresponding record for this reference.
- 3Nguyen, K.; Karn, J. The sounds of silencing: dynamic epigenetic control of HIV latency. Curr. Opin. HIV AIDS 2024, 19 (3), 102 DOI: 10.1097/COH.0000000000000850There is no corresponding record for this reference.
- 4Gao, C.; Meng, J.; Xiao, X.; Wang, M.; Williams, A. B.; Wang, H. Antiretroviral therapy improves neurocognitive impairment in people living with HIV? A meta-analysis. Int. J. Nurs. Sci. 2020, 7 (2), 238– 247, DOI: 10.1016/j.ijnss.2020.03.007There is no corresponding record for this reference.
- 5Mastrorosa, I.; Pinnetti, C.; Brita, A. C.; Mondi, A.; Lorenzini, P.; Del Duca, G.; Vergori, A.; Mazzotta, V.; Gagliardini, R.; Camici, M. Declining Prevalence of Human Immunodeficiency Virus (HIV)-Associated Neurocognitive Disorders in Recent Years and Associated Factors in a Large Cohort of Antiretroviral Therapy-Treated Individuals With HIV. Clin. Infect. Dis. 2023, 76 (3), e629– e637, DOI: 10.1093/cid/ciac658There is no corresponding record for this reference.
- 6Chawla, A.; Wang, C.; Patton, C.; Murray, M.; Punekar, Y.; de Ruiter, A.; Steinhart, C. A Review of Long-Term Toxicity of Antiretroviral Treatment Regimens and Implications for an Aging Population. Infect. Dis. Ther. 2018, 7 (2), 183– 195, DOI: 10.1007/s40121-018-0201-6There is no corresponding record for this reference.
- 7Apostolova, N.; Funes, H. A.; Blas-Garcia, A.; Galindo, M. J.; Alvarez, A.; Esplugues, J. V. Efavirenz and the CNS: what we already know and questions that need to be answered. J. Antimicrob. Chemother. 2015, 70 (10), 2693– 2708, DOI: 10.1093/jac/dkv183There is no corresponding record for this reference.
- 8Venter, W. D. F.; Moorhouse, M.; Sokhela, S.; Fairlie, L.; Mashabane, N.; Masenya, M.; Serenata, C.; Akpomiemie, G.; Qavi, A.; Chandiwana, N. Dolutegravir plus Two Different Prodrugs of Tenofovir to Treat HIV. N. Engl. J. Med. 2019, 381 (9), 803– 815, DOI: 10.1056/NEJMoa1902824There is no corresponding record for this reference.
- 9Churchill, M. J.; Cowley, D. J.; Wesselingh, S. L.; Gorry, P. R.; Gray, L. R.; Churchill, M. J.; Cowley, D. J.; Wesselingh, S. L.; Gorry, P. R.; Gray, L. R. HIV-1 transcriptional regulation in the central nervous system and implications for HIV cure research. J. NeuroVirol. 2014, 21 (3), 290– 300, DOI: 10.1007/s13365-014-0271-5There is no corresponding record for this reference.
- 10Margolis, D. M.; Archin, N. M. Proviral Latency, Persistent Human Immunodeficiency Virus Infection, and the Development of Latency Reversing Agents. J. Infect. Dis. 2017, 215, S111– S118, DOI: 10.1093/infdis/jiw618There is no corresponding record for this reference.
- 11Kreider, E. F.; Bar, K. J. HIV-1 Reservoir Persistence and Decay: Implications for Cure Strategies. Curr. HIV/AIDS Rep. 2022, 19 (3), 194– 206, DOI: 10.1007/s11904-022-00604-2There is no corresponding record for this reference.
- 12Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The Paradox of HIV Blood–Brain Barrier Penetrance and Antiretroviral Drug Delivery Deficiencies. Trends Neurosci. 2020, 43 (9), 695, DOI: 10.1016/j.tins.2020.06.007There is no corresponding record for this reference.
- 13Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.-J.; Castagna, A.; Richmond, G. J.; Sinclair, G. I.; Siripassorn, K.; Ruane, P. J.; Berhe, M.; Wang, H. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2022, 386 (19), 1793– 1803, DOI: 10.1056/NEJMoa2115542There is no corresponding record for this reference.
- 14Ash, M. K.; Al-Harthi, L.; Schneider, J. R. HIV in the Brain: Identifying Viral Reservoirs and Addressing the Challenges of an HIV Cure. Vaccines 2021, 9 (8), 867, DOI: 10.3390/vaccines9080867There is no corresponding record for this reference.
- 15Wang, J.; Crawford, K.; Yuan, M.; Wang, H.; Gorry, P. R.; Gabuzda, D. Regulation of CC chemokine receptor 5 and CD4 expression and human immunodeficiency virus type 1 replication in human macrophages and microglia by T helper type 2 cytokines. J. Infect. Dis. 2002, 185 (7), 885– 897, DOI: 10.1086/339522There is no corresponding record for this reference.
- 16Bertrand, L.; Nair, M.; Toborek, M. Solving the Blood-Brain Barrier Challenge for the Effective Treatment of HIV Replication in the Central Nervous System. Curr. Pharm. Des. 2016, 22 (35), 5477– 5486, DOI: 10.2174/1381612822666160726113001There is no corresponding record for this reference.
- 17Heaton, R. K.; Franklin, D. R.; Ellis, R. J.; McCutchan, J. A.; Letendre, S. L.; LeBlanc, S.; Corkran, S. H.; Duarte, N. A.; Clifford, D. B.; Woods, S. P. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J. NeuroVirol. 2011, 17 (1), 3– 16, DOI: 10.1007/s13365-010-0006-1There is no corresponding record for this reference.
- 18Henrich, T. J.; Kuritzkes, D. R. HIV-1 entry inhibitors: recent development and clinical use. Curr. Opin. Virol. 2013, 3 (1), 51– 57, DOI: 10.1016/j.coviro.2012.12.002There is no corresponding record for this reference.
- 19Sturdevant, C. B.; Joseph, S. B.; Schnell, G.; Price, R. W.; Swanstrom, R.; Spudich, S. Compartmentalized Replication of R5 T Cell-Tropic HIV-1 in the Central Nervous System Early in the Course of Infection. PLoS Pathog. 2015, 11 (3), e1004720 DOI: 10.1371/journal.ppat.1004720There is no corresponding record for this reference.
- 20Valdebenito, S.; Castellano, P.; Ajasin, D.; Eugenin, E. A. Astrocytes are HIV reservoirs in the brain: A cell type with poor HIV infectivity and replication but efficient cell-to-cell viral transfer. J. Neurochem. 2021, 158 (2), 429– 443, DOI: 10.1111/jnc.15336There is no corresponding record for this reference.
- 21Li, G. H.; Maric, D.; Major, E. O.; Nath, A. Productive HIV infection in astrocytes can be established via a nonclassical mechanism. Aids 2020, 34 (7), 963– 978, DOI: 10.1097/QAD.0000000000002512There is no corresponding record for this reference.
- 22Kumar, A.; Abbas, W.; Herbein, G. HIV-1 Latency in Monocytes/Macrophages. Viruses 2014, 6 (4), 1837– 1860, DOI: 10.3390/v6041837There is no corresponding record for this reference.
- 23Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P. W. G.; Marcello, A.; Van Lint, C.; Rohr, O.; Schwartz, C. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362 DOI: 10.3389/fcimb.2019.00362There is no corresponding record for this reference.
- 24Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14 (1), 9 DOI: 10.1186/s12977-017-0335-8There is no corresponding record for this reference.
- 25Sun, W.; Rassadkina, Y.; Gao, C.; Collens, S. I.; Lian, X.; Solomon, I. H.; Mukerji, S.; Yu, X. G.; Lichterfeld, M. Persistence of intact HIV-1 proviruses in the brain during antiretroviral therapy bioRxiv , Aug 14, 2023 DOI: 10.1101/2023.06.26.546135 .There is no corresponding record for this reference.
- 26Dahabieh, M. S.; Battivelli, E.; Verdin, E. Understanding HIV latency: the road to an HIV cure. Annu. Rev. Med. 2015, 66, 407– 421, DOI: 10.1146/annurev-med-092112-152941There is no corresponding record for this reference.
- 27Matsuda, T.; Bebenek, K.; Masutani, C.; Rogozin, I. B.; Hanaoka, F.; Kunkel, T. A. Error rate and specificity of human and murine DNA polymerase η. J. Mol. Biol. 2001, 312 (2), 335, DOI: 10.1006/jmbi.2001.4937There is no corresponding record for this reference.
- 28García, M.; López-Fernández, L.; Mínguez, P.; Morón-López, S.; Restrepo, C.; Navarrete-Muñoz, M. A.; López-Bernaldo, J. C.; Benguría, A.; García, M. I.; Cabello, A. Transcriptional signature of resting-memory CD4 T cells differentiates spontaneous from treatment-induced HIV control. J. Mol. Med. 2020, 98 (8), 1093– 1105, DOI: 10.1007/s00109-020-01930-xThere is no corresponding record for this reference.
- 29Shah, S.; Alexaki, A.; Pirrone, V.; Dahiya, S.; Nonnemacher, M. R.; Wigdahl, B. Functional properties of the HIV-1 long terminal repeat containing single-nucleotide polymorphisms in Sp site III and CCAAT/enhancer binding protein site I. Virol. J. 2014, 11 (1), 92 DOI: 10.1186/1743-422X-11-92There is no corresponding record for this reference.
- 30Chaillon, A.; Gianella, S.; Dellicour, S.; Rawlings, S. A.; Schlub, T. E.; Oliveira, M. F. D.; Ignacio, C.; Porrachia, M.; Vrancken, B.; Smith, D. M. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Invest. 2020, 130 (4), 1699– 1712, DOI: 10.1172/JCI134815There is no corresponding record for this reference.
- 31Lamers, S. L.; Rose, R.; Maidji, E.; Agsalda-Garcia, M.; Nolan, D. J.; Fogel, G. B.; Salemi, M.; Garcia, D. L.; Bracci, P.; Yong, W. HIV DNA Is Frequently Present within Pathologic Tissues Evaluated at Autopsy from Combined Antiretroviral Therapy-Treated Patients with Undetectable Viral Loads. J. Virol. 2016, 90 (20), 8968– 8983, DOI: 10.1128/JVI.00674-16There is no corresponding record for this reference.
- 32Ferretti, F.; De Zan, V.; Gerevini, S.; Turrini, F.; Boeri, E.; Gianotti, N.; Hasson, H.; Lazzarin, A.; Cinque, P. Relapse of Symptomatic Cerebrospinal Fluid HIV Escape. Curr. HIV/AIDS Rep. 2020, 17 (5), 522– 528, DOI: 10.1007/s11904-020-00526-xThere is no corresponding record for this reference.
- 33Williams, D. W.; Calderon, T. M.; Lopez, L.; Carvallo-Torres, L.; Gaskill, P. J.; Eugenin, E. A.; Morgello, S.; Berman, J. W. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS One 2013, 8 (7), e69270 DOI: 10.1371/journal.pone.0069270There is no corresponding record for this reference.
- 34Joseph, S. B.; Arrildt, K. T.; Sturdevant, C. B.; Swanstrom, R. HIV-1 target cells in the CNS. J. NeuroVirol. 2015, 21 (3), 276– 289, DOI: 10.1007/s13365-014-0287-xThere is no corresponding record for this reference.
- 35Walsh, S. R.; Seaman, M. S. Broadly Neutralizing Antibodies for HIV-1 Prevention. Front. Immunol. 2021, 12, 712122 DOI: 10.3389/fimmu.2021.712122There is no corresponding record for this reference.
- 36Spudich, S.; Robertson, K. R.; Bosch, R. J.; Gandhi, R. T.; Cyktor, J. C.; Mar, H.; Macatangay, B. J.; Lalama, C. M.; Rinaldo, C.; Collier, A. C. Persistent HIV-infected cells in cerebrospinal fluid are associated with poorer neurocognitive performance. J. Clin. Invest. 2019, 129 (8), 3339– 3346, DOI: 10.1172/JCI127413There is no corresponding record for this reference.
- 37Dailey, A. F.; Hoots, B. E.; Hall, H. I.; Song, R.; Hayes, D.; Fulton, P., Jr.; Prejean, J.; Hernandez, A. L.; Koenig, L. J.; Valleroy, L. A. Vital Signs: Human Immunodeficiency Virus Testing and Diagnosis Delays - United States. MMWR Morb. Mortal. Wkly. Rep. 2017, 66 (47), 1300– 1306, DOI: 10.15585/mmwr.mm6647e1There is no corresponding record for this reference.
- 38Le Douce, V.; Herbein, G.; Rohr, O.; Schwartz, C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 2010, 7, 32 DOI: 10.1186/1742-4690-7-32There is no corresponding record for this reference.
- 39Mitchell, B. I.; Laws, E. I.; Ndhlovu, L. C. Impact of Myeloid Reservoirs in HIV Cure Trials. Curr. HIV/AIDS Rep. 2019, 16 (2), 129– 140, DOI: 10.1007/s11904-019-00438-5There is no corresponding record for this reference.
- 40Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 2011, 6 (1), 4, DOI: 10.1097/COH.0b013e328340ffbbThere is no corresponding record for this reference.
- 41Karn, J.; Stoltzfus, C. M. Transcriptional and Posttranscriptional Regulation of HIV-1 Gene Expression. Cold Spring Harbor Perspect. Med. 2012, 2 (2), a006916 DOI: 10.1101/cshperspect.a006916There is no corresponding record for this reference.
- 42Pearson, R.; Kim, Y. K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic Silencing of Human Immunodeficiency Virus (HIV) Transcription by Formation of Restrictive Chromatin Structures at the Viral Long Terminal Repeat Drives the Progressive Entry of HIV into Latency. J. Virol. 2008, 82 (24), 12291– 12303, DOI: 10.1128/JVI.01383-08There is no corresponding record for this reference.
- 43D’Orso, I. The HIV-1 Transcriptional Program: From Initiation to Elongation Control. J. Mol. Biol. 2025, 437 (1), 168690 DOI: 10.1016/j.jmb.2024.168690There is no corresponding record for this reference.
- 44Ma, Y.; Li, C.; Valente, S. Human chromatin remodelers regulating HIV-1 transcription: a target for small molecule inhibitors. Epigenet. Chromatin 2025, 18 (1), 21 DOI: 10.1186/s13072-025-00582-wThere is no corresponding record for this reference.
- 45Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4 (1), 261– 285, DOI: 10.1146/annurev-virology-101416-041646There is no corresponding record for this reference.
- 46Shukla, A.; Ramirez, N. P.; D’Orso, I. HIV-1 Proviral Transcription and Latency in the New Era. Viruses 2020, 12 (5), 555 DOI: 10.3390/v12050555There is no corresponding record for this reference.
- 47Valle-Casuso, J. C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29 (3), 611, DOI: 10.1016/j.cmet.2018.11.015There is no corresponding record for this reference.
- 48Chevalier, M. F.; Weiss, L. The split personality of regulatory T cells in HIV infection. Blood 2013, 121 (1), 29– 37, DOI: 10.1182/blood-2012-07-409755There is no corresponding record for this reference.
- 49Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A. C.; Denis, G. V.; Bradner, J.; Sebastiani, P.; Margolis, D. M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukocyte Biol. 2012, 92 (6), 1147, DOI: 10.1189/jlb.0312165There is no corresponding record for this reference.
- 50Mbonye, U.; Karn, J. The cell biology of HIV-1 latency and rebound. Retrovirology 2024, 21 (1), 6 DOI: 10.1186/s12977-024-00639-wThere is no corresponding record for this reference.
- 51Tamaru, H. Confining euchromatin/heterochromatin territory: jumonji crosses the line. Genes Dev. 2010, 24 (14), 1465, DOI: 10.1101/gad.1941010There is no corresponding record for this reference.
- 52Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521 (7551), 227, DOI: 10.1038/nature14226There is no corresponding record for this reference.
- 53Bedwell, G. J.; Engelman, A. N. Factors that mold the nuclear landscape of HIV-1 integration. Nucleic Acids Res. 2021, 49 (2), 621, DOI: 10.1093/nar/gkaa1207There is no corresponding record for this reference.
- 54Hashemi, P.; Sadowski, I. Diversity of small molecule HIV-1 latency reversing agents identified in low- and high-throughput small molecule screens. Med. Res. Rev. 2020, 40 (3), 881, DOI: 10.1002/med.21638There is no corresponding record for this reference.
- 55Wierda, R. J.; Kuipers, H. F.; van Eggermond, M. C.; Benard, A.; van Leeuwen, J. C.; Carluccio, S.; Geutskens, S. B.; Jukema, J. W.; Marquez, V. E.; Quax, P. H.; van den Elsen, P. J. Epigenetic control of CCR5 transcript levels in immune cells and modulation by small molecules inhibitors. J. Cell Mol. Med. 2012, 16 (8), 1866– 1877, DOI: 10.1111/j.1582-4934.2011.01482.xThere is no corresponding record for this reference.
- 56Alamer, E.; Zhong, C.; Liu, Z.; Niu, Q.; Long, F.; Guo, L.; Gelman, B. B.; Soong, L.; Zhou, J.; Hu, H. Epigenetic Suppression of HIV in Myeloid Cells by the BRD4-Selective Small Molecule Modulator ZL0580. J. Virol. 2020, 94 (11), e01880-19 DOI: 10.1128/JVI.01880-19There is no corresponding record for this reference.
- 57Le Douce, V.; Colin, L.; Redel, L.; Cherrier, T.; Herbein, G.; Aunis, D.; Rohr, O.; Van Lint, C.; Schwartz, C. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res. 2012, 40 (5), 1904– 1915, DOI: 10.1093/nar/gkr857There is no corresponding record for this reference.
- 58Brown, L. S.; Foster, C. G.; Courtney, J.-M.; King, N. E.; Howells, D. W.; Sutherland, B. A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 282 DOI: 10.3389/fncel.2019.00282There is no corresponding record for this reference.
- 59Marinho, M.; Novais, C.; Marques, J.; Bragança, M. Efavirenz and neuropsychiatric effects–When the treatment complicates matter further. Eur. Psychiatry 2017, 41 (S1), S237 DOI: 10.1016/j.eurpsy.2017.01.2255There is no corresponding record for this reference.
- 60Wang, F.; Rademeyer, K.; Namuju, O. C.; Abdusalaamu, K.; Fisher, J.; Meya, D. B.; McRae, M.; Boulware, D. R.; Lukande, R.; Nicol, M. R. Postmortem Analysis of Dolutegravir, Tenofovir, Lamivudine, and Efavirenz Penetration in Multiple Central Nervous System Compartments. J. Infect. Dis. 2024, 230 (5), 1215– 1223, DOI: 10.1093/infdis/jiae325There is no corresponding record for this reference.
- 61Banga, R.; Perreau, M. The multifaceted nature of HIV tissue reservoirs. Curr. Opin. HIV AIDS 2024, 19 (3), 116, DOI: 10.1097/COH.0000000000000851There is no corresponding record for this reference.
- 62Nühn, M. M.; Gumbs, S. B. H.; Schipper, P. J.; Drosou, I.; Gharu, L.; Buchholtz, N.; Snijders, G.; Gigase, F. A. J.; Wensing, A. M. J.; Symons, J. Microglia Exhibit a Unique Intact HIV Reservoir in Human Postmortem Brain Tissue. Viruses 2025, 17 (4), 467, DOI: 10.3390/v17040467There is no corresponding record for this reference.
- 63Honeycutt, J. B.; Liao, B.; Nixon, C. C.; Cleary, R. A.; Thayer, W. O.; Birath, S. L.; Swanson, M. D.; Sheridan, P.; Zakharova, O.; Prince, F. T cells establish and maintain CNS viral infection in HIV-infected humanized mice. J. Clin. Invest. 2018, 128 (7), 2862– 2876, DOI: 10.1172/JCI98968There is no corresponding record for this reference.
- 64Gray, L. R.; Cowley, D.; Welsh, C.; Lu, H. K.; Brew, B. J.; Lewin, S. R.; Wesselingh, S. L.; Gorry, P. R.; Churchill, M. J. CNS-specific regulatory elements in brain-derived HIV-1 strains affect responses to latency-reversing agents with implications for cure strategies. Mol. Psychiatry 2016, 21 (4), 574, DOI: 10.1038/mp.2015.111There is no corresponding record for this reference.
- 65Chan, P.; Spudich, S. Central Nervous System Effects of Early HIV Infection and Consequences of Antiretroviral Therapy Initiation during Acute HIV. Viruses 2024, 16 (7), 1082, DOI: 10.3390/v16071082There is no corresponding record for this reference.
- 66Nühn, M. M.; Gumbs, S. B. H.; Buchholtz, N.; Jannink, L. M.; Gharu, L.; de Witte, L. D.; Wensing, A. M. J.; Lewin, S. R.; Nijhuis, M.; Symons, J. Shock and kill within the CNS: A promising HIV eradication approach?. J. Leukocyte Biol. 2022, 112 (5), 1297– 1315, DOI: 10.1002/JLB.5VMR0122-046RRRThere is no corresponding record for this reference.
- 67Ene, L.; Duiculescu, D.; Ruta, S. M. How much do antiretroviral drugs penetrate into the central nervous system?. J. Med. Life 2011, 4 (4), 432– 439There is no corresponding record for this reference.
- 68Health, N. I. o. FDA approval of HIV medicines. 2021.There is no corresponding record for this reference.
- 69McMahon, J. H.; Evans, V. A.; Lau, J. S. Y.; Symons, J.; Zerbato, J. M.; Chang, J.; Solomon, A.; Tennakoon, S.; Dantanarayana, A.; Hagenauer, M. Neurotoxicity with high-dose disulfiram and vorinostat used for HIV latency reversal. AIDS 2022, 36 (1), 75, DOI: 10.1097/QAD.0000000000003091There is no corresponding record for this reference.
- 70Doyon, G.; Zerbato, J.; Mellors, J. W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. Aids 2013, 27 (2), F7– F11, DOI: 10.1097/QAD.0b013e3283570620There is no corresponding record for this reference.
- 71Eneanya, D. I.; Bianchine, J. R.; Duran, D. O.; Andresen, B. D. The actions of metabolic fate of disulfiram. Annu. Rev. Pharmacol. Toxicol. 1981, 21, 575– 596, DOI: 10.1146/annurev.pa.21.040181.003043There is no corresponding record for this reference.
- 72Lin, A.; Elbezanti, W. O.; Schirling, A.; Ahmed, A.; Van Duyne, R.; Cocklin, S.; Klase, Z. Alprazolam Prompts HIV-1 Transcriptional Reactivation and Enhances CTL Response Through RUNX1 Inhibition and STAT5 Activation. Front. Neurol. 2021, 12, 663793 DOI: 10.3389/fneur.2021.663793There is no corresponding record for this reference.
- 73Chowdhury, Z. S.; Morshed, M. M.; Shahriar, M.; Bhuiyan, M. A.; Islam, S. M.; Bin Sayeed, M. S. The Effect of Chronic Alprazolam Intake on Memory, Attention, and Psychomotor Performance in Healthy Human Male Volunteers. Behav. Neurol. 2016, 2016, 3730940 DOI: 10.1155/2016/3730940There is no corresponding record for this reference.
- 74Ait-Daoud, N.; Hamby, A. S.; Sharma, S.; Blevins, D. A Review of Alprazolam Use, Misuse, and Withdrawal. J. Addict. Med. 2018, 12 (1), 4, DOI: 10.1097/ADM.0000000000000350There is no corresponding record for this reference.
- 75Skov, L.; Holm, K. M.; Linnet, K. Nitrobenzodiazepines: Postmortem brain and blood reference concentrations. Forensic Sci. Int. 2016, 268, 39– 45, DOI: 10.1016/j.forsciint.2016.09.002There is no corresponding record for this reference.
- 76Jansen, Y. J. L.; Verset, G.; Schats, K.; Dam, P.-J. V.; Seremet, T.; Kockx, M.; Laethem, J.-L. B. V.; Neyns, B. Phase I clinical trial of decitabine (5-aza-2′-deoxycytidine) administered by hepatic arterial infusion in patients with unresectable liver-predominant metastases. ESMO Open 2019, 4 (2), e000464 DOI: 10.1136/esmoopen-2018-000464There is no corresponding record for this reference.
- 77Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Van Driessche, B.; Corazza, F.; Gatot, J. S.; Melard, A.; Vanhulle, C.; Kabeya, K. Sequential treatment with 5-aza-2′-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8 (2), 117– 138, DOI: 10.15252/emmm.201505557There is no corresponding record for this reference.
- 78Chabot, G. G.; Rivard, G. E.; Momparler, R. L. Plasma and cerebrospinal fluid pharmacokinetics of 5-Aza-2′-deoxycytidine in rabbits and dogs. Cancer Res. 1983, 43 (2), 592– 597There is no corresponding record for this reference.
- 79Noël, J.-M.; Raynal, J.-P. J. I. Chapter 7 - DNA Methyltransferase Inhibitors. Drug Discovery Cancer Epigenet. 2016, 169– 190, DOI: 10.1016/B978-0-12-802208-5.00007-2There is no corresponding record for this reference.
- 80Lucera, M. B.; Tilton, C. A.; Mao, H.; Dobrowolski, C.; Tabler, C. O.; Haqqani, A. A.; Karn, J.; Tilton, J. C. The Histone Deacetylase Inhibitor Vorinostat (SAHA) Increases the Susceptibility of Uninfected CD4+ T Cells to HIV by Increasing the Kinetics and Efficiency of Postentry Viral Events. J. Virol. 2014, 88 (18), 10803 DOI: 10.1128/JVI.00320-14There is no corresponding record for this reference.
- 81Archin, N. M.; Kirchherr, J. L.; Sung, J. A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba AD, A. D.; Kuruc, J. D. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Invest. 2017, 127 (8), 3126 DOI: 10.1172/JCI92684There is no corresponding record for this reference.
- 82Vorinostat Indications and Prescribing Information - FDA.There is no corresponding record for this reference.
- 83Guntner, A. S.; Peyrl, A.; Mayr, L.; Englinger, B.; Berger, W.; Slavc, I.; Buchberger, W.; Gojo, J. Cerebrospinal fluid penetration of targeted therapeutics in pediatric brain tumor patients. Acta Neuropathol. Commun. 2020, 8 (1), 78 DOI: 10.1186/s40478-020-00953-2There is no corresponding record for this reference.
- 84Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41 (1), 277 DOI: 10.1093/nar/gks976There is no corresponding record for this reference.
- 85Bakshi, S.; McKee, C.; Walker, K.; Brown, C.; Chaudhry, G. R. Toxicity of JQ1 in neuronal derivatives of human umbilical cord mesenchymal stem cells. Oncotarget 2018, 9 (73), 33853 DOI: 10.18632/oncotarget.26127There is no corresponding record for this reference.
- 86Matzuk, M. M.; McKeown, M. R.; Filippakopoulos, P.; Li, Q.; Ma, L.; Agno, J. E.; Lemieux, M. E.; Picaud, S.; Yu, R. N.; Qi, J. Small-Molecule Inhibition of BRDT for Male Contraception. Cell 2012, 150 (4), 673– 684, DOI: 10.1016/j.cell.2012.06.045There is no corresponding record for this reference.
- 87Dental, C.; Proust, A.; Ouellet, M.; Barat, C.; Tremblay, M. J. HIV-1 Latency-Reversing Agents Prostratin and Bryostatin-1 Induce Blood-Brain Barrier Disruption/Inflammation and Modulate Leukocyte Adhesion/Transmigration. J. Immunol. 2017, 198 (3), 1229– 1241, DOI: 10.4049/jimmunol.1600742There is no corresponding record for this reference.
- 88Díaz, L.; Martínez-Bonet, M.; Sánchez, J.; Fernández-Pineda, A.; Jiménez, J. L.; Muñoz, E.; Moreno, S.; Álvarez, S.; Muñoz-Fernández, M. Á. Bryostatin activates HIV-1 latent expression in human astrocytes through a PKC and NF-κB-dependent mechanism. Sci. Rep. 2015, 5, 12442 DOI: 10.1038/srep12442There is no corresponding record for this reference.
- 89Nelson, T. J.; Sen, A.; Alkon, D. L.; Sun, M. K. Adduct formation in liquid chromatography-triple quadrupole mass spectrometric measurement of bryostatin 1. J. Chromatogr. B:Anal. Technol. Biomed. Life Sci. 2014, 944, 55– 62, DOI: 10.1016/j.jchromb.2013.11.020There is no corresponding record for this reference.
- 90Bartholomeeusen, K.; Fujinaga, K.; Xiang, Y.; Peterlin, B. M. Histone Deacetylase Inhibitors (HDACis) That Release the Positive Transcription Elongation Factor b (P-TEFb) from Its Inhibitory Complex Also Activate HIV Transcription. J. Biol. Chem. 2013, 288 (20), 14400 DOI: 10.1074/jbc.M113.464834There is no corresponding record for this reference.
- 91Palmieri, D.; Lockman, P. R.; Thomas, F. C.; Hua, E.; Herring, J.; Hargrave, E.; Johnson, M.; Flores, N.; Qian, Y.; Vega-Valle, E. Vorinostat Inhibits Brain Metastatic Colonization in a Model of Triple-Negative Breast Cancer and Induces DNA Double-Strand Breaks. Clin. Cancer Res. 2009, 15 (19), 6148, DOI: 10.1158/1078-0432.CCR-09-1039There is no corresponding record for this reference.
- 92Jiang, G.; Dandekar, S. Targeting NF-κB Signaling with Protein Kinase C Agonists As an Emerging Strategy for Combating HIV Latency. AIDS Res. Hum. Retroviruses 2015, 31 (1), 4, DOI: 10.1089/aid.2014.0199There is no corresponding record for this reference.
- 93Nelson, T. J.; Sun, M. K.; Lim, C.; Sen, A.; Khan, T.; Chirila, F. V.; Alkon, D. L. Bryostatin Effects on Cognitive Function and PKCε in Alzheimer’s Disease Phase IIa and Expanded Access Trials. J. Alzheimer’s Dis. 2017, 58 (2), 521– 535, DOI: 10.3233/JAD-170161There is no corresponding record for this reference.
- 94Fuller, R. K.; Gordis, E. Does disulfiram have a role in alcoholism treatment today?. Addiction 2004, 99 (1), 21, DOI: 10.1111/j.1360-0443.2004.00597.xThere is no corresponding record for this reference.
- 95Abner, E.; Jordan, A. HIV ″shock and kill″ therapy: In need of revision. Antiviral Res. 2019, 166, 19– 34, DOI: 10.1016/j.antiviral.2019.03.008There is no corresponding record for this reference.
- 96Mukim, A.; Smith, D. M.; Deshmukh, S.; Qazi, A. A.; Beliakova-Bethell, N. A camptothetin analog, topotecan, promotes HIV latency via interference with HIV transcription and RNA splicing. J. Virol. 2023, 97 (2), e01630-22 DOI: 10.1128/jvi.01630-22There is no corresponding record for this reference.
- 97Mousseau, G.; Kessing, C. F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S. T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. mBio 2015, 6 (4), e00465 DOI: 10.1128/mBio.00465-15There is no corresponding record for this reference.
- 98Li, C.; Mori, L.; Valente, S. T. The Block-and-Lock Strategy for Human Immunodeficiency Virus Cure: Lessons Learned from Didehydro-Cortistatin A. J. Infect. Dis. 2021, 223, S46– S53, DOI: 10.1093/infdis/jiaa681There is no corresponding record for this reference.
- 99Peters, R. J.; Stevenson, M. Irreversible Loss of HIV-1 Proviral Competence in Myeloid Cells upon Suppression of NF-κB Activity. J. Virol. 2022, 96 (12), e00484-22 DOI: 10.1128/jvi.00484-22There is no corresponding record for this reference.
- 100Debrabander, Q.; Hensley, K. S.; Psomas, C. K.; Bramer, W.; Mahmoudi, T.; Welzen, B. J. v.; Verbon, A.; Rokx, C. The efficacy and tolerability of latency-reversing agents in reactivating the HIV-1 reservoir in clinical studies: a systematic review. J. Virus Erad. 2023, 9 (3), 100342 DOI: 10.1016/j.jve.2023.100342There is no corresponding record for this reference.
- 101Liu, Y.; Liu, H.; Kim, B. O.; Gattone, V. H.; Li, J. L.; Nath, A.; Blum, J.; He, J. J. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: Requirement for the human mannose receptor. J. Virol. 2004, 78 (8), 4120– 4133, DOI: 10.1128/JVI.78.8.4120-4133.2004There is no corresponding record for this reference.
- 102Liu, Y. B.; Cao, W.; Sun, M.; Li, T. S. Broadly neutralizing antibodies for HIV-1: efficacies, challenges and opportunities. Emerging Microbes Infect. 2020, 9 (1), 194– 206, DOI: 10.1080/22221751.2020.1713707There is no corresponding record for this reference.
- 103Wen, J.; Cheever, T.; Wang, L.; Wu, D.; Reed, J.; Mascola, J.; Chen, X. J.; Liu, C. P.; Pegu, A.; Sacha, J. B. Improved delivery of broadly neutralizing antibodies by nanocapsules suppresses SHIV infection in the CNS of infant rhesus macaques. PLoS Pathog. 2021, 17 (7), e1009738 DOI: 10.1371/journal.ppat.1009738There is no corresponding record for this reference.
- 104Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33 (9), 941– 951, DOI: 10.1038/nbt.3330There is no corresponding record for this reference.
- 105Halper-Stromberg, A.; Lu, C. L.; Klein, F.; Horwitz, J. A.; Bournazos, S.; Nogueira, L.; Eisenreich, T. R.; Liu, C.; Gazumyan, A.; Schaefer, U. Broadly Neutralizing Antibodies and Viral Inducers Decrease Rebound from HIV-1 Latent Reservoirs in Humanized Mice. Cell 2014, 158 (5), 989– 999, DOI: 10.1016/j.cell.2014.07.043There is no corresponding record for this reference.
- 106Gunst, J. D.; Hojen, J. F.; Sogaard, O. S. Broadly neutralizing antibodies combined with latency-reversing agents or immune modulators as strategy for HIV-1 remission. Curr. Opin. HIV AIDS 2020, 15 (5), 309– 315, DOI: 10.1097/COH.0000000000000641There is no corresponding record for this reference.
- 107Julg, B.; Walker-Sperling, V. E. K.; Wagh, K.; Aid, M.; Stephenson, K. E.; Zash, R.; Liu, J. Y.; Nkolola, J. P.; Hoyt, A.; Castro, M. Safety and antiviral effect of a triple combination of HIV-1 broadly neutralizing antibodies: a phase 1/2a trial. Nat. Med. 2024, 30 (12), 3534– 3543, DOI: 10.1038/s41591-024-03247-5There is no corresponding record for this reference.
- 108Mbonye, U.; Karn, J. Control of HIV Latency by Epigenetic and Non-Epigenetic Mechanisms. Curr. HIV Res. 2011, 9 (8), 554, DOI: 10.2174/157016211798998736There is no corresponding record for this reference.
- 109Duggan, N. N.; Dragic, T.; Chanda, S. K.; Pache, L. Breaking the Silence: Regulation of HIV Transcription and Latency on the Road to a Cure. Viruses 2023, 15 (12), 2435, DOI: 10.3390/v15122435There is no corresponding record for this reference.
- 110Archin, N. M.; Bateson, R.; Tripathy, M. K.; Crooks, A. M.; Yang, K.-H.; Dahl, N. P.; Kearney, M. F.; Anderson, E. M.; Coffin, J. M.; Strain, M. C. HIV-1 Expression Within Resting CD4+ T Cells After Multiple Doses of Vorinostat. J. Infect. Dis. 2014, 210 (5), 728, DOI: 10.1093/infdis/jiu155There is no corresponding record for this reference.
- 111Kinloch, N. N.; Shahid, A.; Dong, W.; Kirkby, D.; Jones, B. R.; Beelen, C. J.; MacMillan, D.; Lee, G. Q.; Mota, T. M.; Sudderuddin, H. HIV reservoirs are dominated by genetically younger and clonally enriched proviruses. mBio 2023, 14 (6), e02417-23 DOI: 10.1128/mbio.02417-23There is no corresponding record for this reference.
- 112Battistini, A.; Sgarbanti, M. HIV-1 Latency: An Update of Molecular Mechanisms and Therapeutic Strategies. Viruses 2014, 6 (4), 1715, DOI: 10.3390/v6041715There is no corresponding record for this reference.
- 113Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Driessche, B. V.; Corazza, F.; Gatot, J. S.; Melard, A.; Vanhulle, C.; Kabeya, K. Sequential treatment with 5-aza-2′-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2015, 8 (2), 117– 138, DOI: 10.15252/emmm.201505557There is no corresponding record for this reference.
- 114Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B. M. Bromodomain and Extra-terminal (BET) Bromodomain Inhibition Activate Transcription via Transient Release of Positive Transcription Elongation Factor b (P-TEFb) from 7SK Small Nuclear Ribonucleoprotein. J. Biol. Chem. 2012, 287 (43), 36609– 36616, DOI: 10.1074/jbc.M112.410746There is no corresponding record for this reference.
- 115Henderson, L. J.; Sharma, A.; Monaco, M. C.; Major, E. O.; Al-Harthi, L. Human immunodeficiency virus type 1 (HIV-1) transactivator of transcription through its intact core and cysteine-rich domains inhibits Wnt/β-catenin signaling in astrocytes: relevance to HIV neuropathogenesis. J. Neurosci. 2012, 32 (46), 16306– 16313, DOI: 10.1523/JNEUROSCI.3145-12.2012There is no corresponding record for this reference.
- 116Al-Harthi, L. Interplay between Wnt/β-catenin signaling and HIV: virologic and biologic consequences in the CNS. J. Neuroimmune Pharmacol. 2012, 7 (4), 731– 739, DOI: 10.1007/s11481-012-9411-yThere is no corresponding record for this reference.
- 117Barbian, H. J.; Seaton, M. S.; Narasipura, S. D.; Wallace, J.; Rajan, R.; Sha, B. E.; Al-Harthi, L. β-catenin regulates HIV latency and modulates HIV reactivation. PLoS Pathog. 2022, 18 (3), e1010354 DOI: 10.1371/journal.ppat.1010354There is no corresponding record for this reference.
- 118Wen, J.; Li, X.; Zhao, Q. X.; Yang, X. F.; Wu, M. L.; Yan, Q.; Chang, J.; Wang, H.; Jin, X.; Su, X. Pharmacological suppression of glycogen synthase kinase-3 reactivates HIV-1 from latency via activating Wnt/β-catenin/TCF1 axis in CD4(+) T cells. Emerging Microbes Infect. 2022, 11 (1), 391– 405, DOI: 10.1080/22221751.2022.2026198There is no corresponding record for this reference.
- 119Mavigner, M.; Zanoni, M.; Tharp, G. K.; Habib, J.; Mattingly, C. R.; Lichterfeld, M.; Nega, M. T.; Vanderford, T. H.; Bosinger, S. E.; Chahroudi, A. Pharmacological Modulation of the Wnt/β-Catenin Pathway Inhibits Proliferation and Promotes Differentiation of Long-Lived Memory CD4(+) T Cells in Antiretroviral Therapy-Suppressed Simian Immunodeficiency Virus-Infected Macaques. J. Virol. 2019, 94 (1), e01094-19 DOI: 10.1128/JVI.01094-19There is no corresponding record for this reference.
- 120Tibebe, H.; Marquez, D.; McGraw, A.; Gagliardi, S.; Sullivan, C.; Hillmer, G.; Narayan, K.; Izumi, C.; Keating, A.; Izumi, T. Targeting Latent HIV Reservoirs: Effectiveness of Combination Therapy with HDAC and PARP Inhibitors. Viruses 2025, 17 (3), 400, DOI: 10.3390/v17030400There is no corresponding record for this reference.
- 121Huang, Y.; Luo, D.; Chen, X.; Zhang, D.; Huang, Z.; Xiao, S. HIV-Related Stress Experienced by Newly Diagnosed People Living with HIV in China: A 1-Year Longitudinal Study. Int. J. Environ. Res. Public Health 2020, 17 (8), 2681, DOI: 10.3390/ijerph17082681There is no corresponding record for this reference.
- 122Saloner, R.; Grelotti, D. J.; Tyree, G.; Sundermann, E. E.; Ma, Q.; Letendre, S.; Heaton, R. K.; Cherner, M. Benzodiazepine Use is Associated with an Increased Risk of Neurocognitive Impairment in People Living with HIV. J. Acquired Immune Defic. Syndr. 2019, 82 (5), 475, DOI: 10.1097/QAI.0000000000002183There is no corresponding record for this reference.
- 123Olfson, M.; King, M.; Schoenbaum, M. Benzodiazepine use in the United States. JAMA Psychiatry 2015, 72 (2), 136– 142, DOI: 10.1001/jamapsychiatry.2014.1763There is no corresponding record for this reference.
- 124Cunningham, L.; Finckbeiner, S.; Hyde, R. K.; Southall, N.; Marugan, J.; Yedavalli, V. R. K.; Dehdashti, S. J.; Reinhold, W. C.; Alemu, L.; Zhao, L. Identification of benzodiazepine Ro5–3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1–CBFβ interaction. Proc. Natl. Acad. Sci. U.S.A. 2012, 109 (36), 14592 DOI: 10.1073/pnas.1200037109There is no corresponding record for this reference.
- 125Klase, Z.; Yedavalli, V.; Houzet, L.; Perkins, M.; Maldarelli, F.; Brenchley, J.; Strebel, K.; Liu, P.; Jeang, K. T. Activation of HIV-1 from Latent Infection via Synergy of RUNX1 Inhibitor Ro5–3335 and SAHA. PLoS Pathog. 2014, 10 (3), e1003997 DOI: 10.1371/journal.ppat.1003997There is no corresponding record for this reference.
- 126Kierdorf, K.; Prinz, M. Factors regulating microglia activation. Front. Cell Neurosci. 2013, 7, 44, DOI: 10.3389/fncel.2013.00044There is no corresponding record for this reference.
- 127Capriotti, Z.; Klase, Z. Innate immune memory in chronic HIV and HIV-associated neurocognitive disorders (HAND): potential mechanisms and clinical implications. J. NeuroVirol. 2024, 30, 451, DOI: 10.1007/s13365-024-01239-2There is no corresponding record for this reference.
- 128Elbezanti, W.; Lin, A. G.; Schirling, A.; Jackson, A.; Marshall, M.; Van Duyne, R.; Maldarelli, F.; Sardo, L.; Klase, Z. Benzodiazepines Drive Alteration of Chromatin at the Integrated HIV-1 LTR. Viruses 2020, 12 (2), 191 DOI: 10.3390/v12020191There is no corresponding record for this reference.
- 129Castillo, A. A.; McElrath, C.; Marshall, G.; Stevenson, M.; Castillo, A. A.; McElrath, C.; Marshall, G.; Stevenson, M. Myeloid Cell Reservoirs: Role in HIV-Host Interplay and Strategies for Myeloid Reservoir Elimination. Curr. Clin. Microbiol. Rep. 2024, 11 (4), 209– 219, DOI: 10.1007/s40588-024-00234-9There is no corresponding record for this reference.
- 130Didigu, C. A.; Wilen, C. B.; Wang, J.; Duong, J.; Secreto, A. J.; Danet-Desnoyers, G. A.; Riley, J. L.; Gregory, P. D.; June, C. H.; Holmes, M. C.; Doms, R. W. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood 2014, 123 (1), 61– 69, DOI: 10.1182/blood-2013-08-521229There is no corresponding record for this reference.
- 131Tebas, P.; Stein, D.; Tang, W. W.; Frank, I.; Wang, S. Q.; Lee, G.; Spratt, S. K.; Surosky, R. T.; Giedlin, M. A.; Nichol, G. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370 (10), 901– 910, DOI: 10.1056/NEJMoa1300662There is no corresponding record for this reference.
- 132Tebas, P.; Jadlowsky, J. K.; Shaw, P. A.; Tian, L.; Esparza, E.; Brennan, A. L.; Kim, S.; Naing, S. Y.; Richardson, M. W.; Vogel, A. N. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J. Clin. Invest. 2021, 131 (7), e144486 DOI: 10.1172/JCI144486There is no corresponding record for this reference.
- 133Letchumanan, P.; Theva Das, K. The role of genetic diversity, epigenetic regulation, and sex-based differences in HIV cure research: a comprehensive review. Epigenet. Chromatin 2025, 18 (1), 1 DOI: 10.1186/s13072-024-00564-4There is no corresponding record for this reference.
- 134Macedo, A. B.; Novis, C. L.; Bosque, A. Targeting Cellular and Tissue HIV Reservoirs With Toll-Like Receptor Agonists. Front. Immunol. 2019, 10, 2450 DOI: 10.3389/fimmu.2019.02450There is no corresponding record for this reference.
- 135Acioglu, C.; Heary, R. F.; Elkabes, S. Roles of neuronal toll-like receptors in neuropathic pain and central nervous system injuries and diseases. Brain, Behav., Immun. 2022, 102, 163– 178, DOI: 10.1016/j.bbi.2022.02.016There is no corresponding record for this reference.
- 136Kielian, T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J. Neurosci. Res. 2006, 83 (5), 711– 730, DOI: 10.1002/jnr.20767There is no corresponding record for this reference.
- 137Thibault, S.; Imbeault, M.; Tardif, M. R.; Tremblay, M. J. TLR5 stimulation is sufficient to trigger reactivation of latent HIV-1 provirus in T lymphoid cells and activate virus gene expression in central memory CD4 < SUP>+</SUP> T cells. Virology 2009, 389 (1–2), 20– 25, DOI: 10.1016/j.virol.2009.04.019There is no corresponding record for this reference.
- 138Tsai, A.; Irrinki, A.; Kaur, J.; Cihlar, T.; Kukolj, G.; Sloan, D. D.; Murry, J. P. Toll-Like Receptor 7 Agonist GS-9620 Induces HIV Expression and HIV-Specific Immunity in Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J. Virol. 2017, 91 (8), e02166-16 DOI: 10.1128/JVI.02166-16There is no corresponding record for this reference.
- 139Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14, 1 DOI: 10.1186/s12977-017-0335-8There is no corresponding record for this reference.
- 140Omueti, K. O.; Beyer, J. M.; Johnson, C. M.; Lyle, E. A.; Tapping, R. I. Domain exchange between human Toll-like receptors 1 and 6 reveals a region required for lipopeptide discrimination. J. Biol. Chem. 2005, 280 (44), 36616– 36625, DOI: 10.1074/jbc.M504320200There is no corresponding record for this reference.
- 141Duan, S. Q.; Liu, S. W. Targeting NK Cells for HIV-1 Treatment and Reservoir Clearance. Front. Immunol. 2022, 13, 842746 DOI: 10.3389/fimmu.2022.842746There is no corresponding record for this reference.
- 142Lu, C. L.; Murakowski, D. K.; Bournazos, S.; Schoofs, T.; Sarkar, D.; Halper-Stromberg, A.; Horwitz, J. A.; Nogueira, L.; Golijanin, J.; Gazumyan, A. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science 2016, 352 (6288), 1001– 1004, DOI: 10.1126/science.aaf1279There is no corresponding record for this reference.
- 143Merck & Co. Product Information. Zolinza (vorinostat). 2006.There is no corresponding record for this reference.
- 144Thorlund, K.; Horwitz, M. S.; Fife, B. T.; Lester, R.; Cameron, D. W. Landscape review of current HIV ’kick and kill’ cure research - some kicking, not enough killing. BMC Infect. Dis. 2017, 17, 595 DOI: 10.1186/s12879-017-2683-3There is no corresponding record for this reference.
- 145Jones, R. B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G. L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M. A.; Trocha, A. Histone Deacetylase Inhibitors Impair the Elimination of HIV-Infected Cells by Cytotoxic T-Lymphocytes. PLoS Pathog. 2014, 10 (8), e1004287 DOI: 10.1371/journal.ppat.1004287There is no corresponding record for this reference.
- 146Li, Y. Y.; Hong, J. X.; Zhang, L. Q. The Rational Combination Strategy of Immunomodulatory Latency Reversing Agents and Novel Immunotherapy to Achieve HIV-1 Cure. Infect. Dis. Immun. 2022, 2 (4), 263– 273, DOI: 10.1097/ID9.0000000000000045There is no corresponding record for this reference.
- 147Mastrangelo, A.; Gama, L.; Cinque, P. Strategies to target the central nervous system HIV reservoir. Curr. Opin. HIV AIDS 2024, 19 (3), 133– 140, DOI: 10.1097/COH.0000000000000847There is no corresponding record for this reference.
- 148Morris, E. C.; Neelapu, S. S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22 (2), 85– 96, DOI: 10.1038/s41577-021-00547-6There is no corresponding record for this reference.
- 149Kapoor, A.; Tan, C. S. Immunotherapeutics to Treat HIV in the Central Nervous System. Curr. Hiv/Aids Rep. 2020, 17 (5), 499– 506, DOI: 10.1007/s11904-020-00519-wThere is no corresponding record for this reference.