



This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Electrochemical N-Propargylation of N-Heterocycles via Decarboxylation of Allenoic AcidsClick to copy article linkArticle link copied!
- Sarah AbdounSarah AbdounBiomolécules: Conception, Isolement, Synthèse (BioCIS), CNRS UMR 8076, Université Paris-Saclay, 17 avenue des Sciences, 91400 Orsay, FranceMore by Sarah Abdoun
- Christophe BourChristophe BourInstitut de Chimie Moléculaire et des Matériaux d’Orsay (ICMMO), CNRS UMR 8182, Université Paris-Saclay, 17 avenue des Sciences, 91400 Orsay, FranceMore by Christophe Bour
- Robert J. Mayer*Robert J. Mayer*Email: [email protected]Technical University of Munich, School of Natural Sciences, Department Chemie, 85748 Garching, GermanyMore by Robert J. Mayer
- Marie Vayer*Marie Vayer*Email: [email protected]Biomolécules: Conception, Isolement, Synthèse (BioCIS), CNRS UMR 8076, Université Paris-Saclay, 17 avenue des Sciences, 91400 Orsay, FranceMore by Marie Vayer
ACS Organic & Inorganic Au
© 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.

Non-Commercial (NC): Only non-commercial uses of the work are permitted.

No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
Broadly applicable and selective C–N bond formation remains a cornerstone challenge in organic synthesis. Although electrochemical methods have recently emerged as an efficient way for achieving decarboxylative C–N coupling, transforming allenoic acids into N-functionalized products remains elusive. Herein, we report the development of an electrochemical strategy for the decarboxylative N-propargylation of N-heterocycles using readily available 2,3-allenoic acids. By correlating the oxidation potentials of allenoic acids and nucleophilic coupling partners, we derived predictive criteria for anticipating reaction efficiency across a broad substrate scope. Mechanistic studies (cyclic voltammetry, in situ infrared (IR) kinetics, and density functional theory (DFT) calculations) support a two-step sequence involving oxidative decarboxylation to an allenyl radical rapidly oxidized to a highly electrophilic allenyl/propargyl cation and regioselective heterocycle attack yielding N-propargylated products. This work showcases a straightforward method for accessing reactive allenyl cation intermediates and expands the toolkit for sustainable electrosynthetic C–N bond formation.
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Introduction
Scheme 1
Results and Discussion
Verification of the Hypothesis and Optimization
Figure 1
Figure 1. Identification of reaction conditions based on cyclic voltammetry. [a]Cyclic voltammogram of allenoic acid 1a (purple curve) and pyrazole 2a (blue curve) in MeCN with nBu4NPF6 (0.1 M) as the electrolyte using a platinum disk (d = 2 mm) as the working electrode, a platinum wire as the counter electrode, and a nonaqueous Ag/AgNO3 electrode (3.0 M in MeCN) as the reference. All cyclic voltammetry experiments scan from 0.0 V vs the Ag/Ag+ reference electrode in the anodic direction at a scan rate of 50 mV/s. The cyclic voltammetry traces were presented with the IUPAC convention. [b]Initial conditions: 1a (0.2 mmol), pyrazole 2a (1.5 equiv), 2,4,6-collidine (1.5 equiv), nBu4NPF6 (0.05 M), DCM (3.0 mL), undivided cell, C-SK-50 anode, Ni cathode, CCE = 5 mA, 2.2 F/mol. [c]Isolated yield. [d]Surface degradation of the C anode was observed. [e]Faradaic efficiency.
Scope and Limitations
Scheme 2
[a] Two major oxidation peaks were observed on the CV of 1i (see the SI for CV graphs). [b] A broad, ill-defined oxidation wave was observed, which precluded the determination of an oxidation potential for 1j.
Scheme 3
[a]22 was observed in the crude reaction mixture with a similar conversion as 21, but it could not be isolated in the pure form, presumably due to decomposition. [b] 3 equiv of the nucleophile was used. [c] The reaction was performed in a 9:1 (v/v) mixture of DCM/alcohol.
Scheme 4
Mechanistic Studies
Figure 2
Figure 2. Overview of the mechanistic experiments. (A) Divided-cell experiments were carried out in IKA Pro-Divide equipment that includes two PTFE cells separated from each other by a glass frit (pore size 10–16 μm) equipped with an O-ring. A graphite SK-50 electrode (8 mm wide, 50 mm length, 1 mm thickness) was used as the anode, and a nickel electrode (8 mm wide, 50 mm length, 1 mm thickness) was used as the cathode. (B) Constant-potential electrolysis experiments were performed using IKA ElectraSyn equipment equipped with graphite as the working electrode, nickel as the counter electrode, and Ag/AgCl wire in 3.0 M KCl as the reference electrode. Constant-potential electrolysis was conducted at various potentials and terminated once a total charge of 1.0 F/mol had passed through the circuit. The reaction yield was determined by NMR using mesitylene as an internal standard. (C) Chirality transfer experiments using standard conditions. (D) IR kinetics were followed using a Mettler-Toledo ReactIR 15 system and a 6.3 mm AgX Fiber probe with 78 scans per spectrum. Data was acquired and processed with Mettler-Toledo iC IR software with solvent subtraction of DCM and electrolyte subtraction of nBu4NPF6. The absorbance at 1689 cm–1 was recorded.
Figure 3
Figure 3. Computational exploration of the reaction mechanism at the SMD(CH2Cl2)/ωB97XD/def2-TZVPP//SMD(CH2Cl2)/ωB97XD/def2-SVP level of theory. (A) Competing pathways for electrochemical oxidation; all Gibbs energies are reported in kJ mol–1. Electrochemical steps are highlighted with blue arrows, and the oxidation potentials are calculated vs the saturated calomel electrode (SCE). (B, C) Evaluation of addition of 2a to radical intermediates D and E. (D) Gibbs energy profile for the reaction of cation F as allenyl vs propargyl cations with nucleophile 2a.
Conclusion
Experimental Methods
General Information
General Procedure for the N-Alkylation of Heterocycles with Allenoic Acids (Cond. A)
General Procedure for the N-Alkylation of Heterocycles with Allenoic Acids (Cond. B)
General Procedure for the Propargylation of Other Nucleophiles from Allenoic Acid 1a
Data Availability
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.5c00117.
Experimental procedures and characterization data, additional experimental details, and 1H, 13C, and 19F NMR spectra (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Robert J. Mayer - Technical University of Munich, School of Natural Sciences, Department Chemie, 85748 Garching, Germany;
 https://orcid.org/0000-0002-9864-7042;
https://orcid.org/0000-0002-9864-7042;
- Marie Vayer - Biomolécules: Conception, Isolement, Synthèse (BioCIS), CNRS UMR 8076, Université Paris-Saclay, 17 avenue des Sciences, 91400 Orsay, France;https://orcid.org/0000-0002-3870-5378;
- Christophe Bour - Institut de Chimie Moléculaire et des Matériaux d’Orsay (ICMMO), CNRS UMR 8182, Université Paris-Saclay, 17 avenue des Sciences, 91400 Orsay, France;https://orcid.org/0000-0001-6733-5284
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Acknowledgments
Financial support for this work was provided by the French National Research Agency (ANR-24-CE07-3221), the ANR PIA funding (ANR-20-IDEES-0002), and as part of the France 2030 program ANR11-IDEX-0003, awarded by the Graduate School Chemistry of the Université Paris-Saclay. M.V. thanks the CNRS. R.J.M. thanks the Fonds der Chemischen Industrie and the Emmy-Noether Program of the DFG (553844165) for financial support. Computations were performed on resources provided by the Leibniz Supercomputing Centre (LRZ). C.B. thanks the French National Research Agency (ANR-21-CE07-0027). Dr. Laurence Grimaud is acknowledged for helpful discussions.
References
This article references 41 other publications.
- 1(a) Bansal, Y.; Silakari, O. The Therapeutic Journey of Benzimidazoles: A Review. Bioorg. J. Med. Chem. 2012, 20, 6208– 6236, DOI: 10.1016/j.bmc.2012.09.013Google ScholarThere is no corresponding record for this reference.(b) Küçükgüzel, Ş.; Şenkardeş, S. Recent Advances in Bioactive Pyrazoles. Eur. J. Med. Chem. 2015, 97, 786– 815, DOI: 10.1016/j.ejmech.2014.11.059Google ScholarThere is no corresponding record for this reference.(c) Ríos, M.-C.; Portilla, J. Recent Advances in Synthesis and Properties of Pyrazoles. Chemistry 2022, 4, 940– 968, DOI: 10.3390/chemistry4030065Google ScholarThere is no corresponding record for this reference.(d) Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K. K.; Jonnalagadda, S. B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909– 1951, DOI: 10.3390/molecules25081909Google ScholarThere is no corresponding record for this reference.
- 2(a) Ruiz-Castillo, P.; Buchwald, S. L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564– 12649, DOI: 10.1021/acs.chemrev.6b00512Google ScholarThere is no corresponding record for this reference.(b) Dorel, R.; Grugel, C. P.; Haydl, A. M. The Buchwald–Hartwig Amination After 25 Years. Angew. Chem., Int. Ed. 2019, 58, 17118– 17129, DOI: 10.1002/anie.201904795Google ScholarThere is no corresponding record for this reference.
- 3(a) Monnier, F.; Taillefer, M. Catalytic C–C, C–N, and C–O Ullmann-Type Coupling Reactions. Angew. Chem., Int. Ed. 2009, 48, 6954– 6971, DOI: 10.1002/anie.200804497Google ScholarThere is no corresponding record for this reference.(b) Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Copper Catalysed Ullmann Type Chemistry: from Mechanistic Aspects to Modern Development. Chem. Soc. Rev. 2014, 43, 3525– 3550, DOI: 10.1039/C3CS60289CGoogle ScholarThere is no corresponding record for this reference.(c) Yang, Q.; Zhao, Y.; Ma, D. Cu-Mediated Ullmann-Type Cross-Coupling and Industrial Applications in Route Design, Process Development, and Scale-up of Pharmaceutical and Agrochemical Processes. Org. Process Res. Dev. 2022, 26, 1690– 1750, DOI: 10.1021/acs.oprd.2c00050Google ScholarThere is no corresponding record for this reference.
- 4(a) West, M. J.; Fyfe, J. W. B.; Vantourout, J. C.; Watson, A. J. B. Mechanistic Development and Recent Applications of the Chan-Lam Amination. Chem. Rev. 2019, 119, 12491– 12523, DOI: 10.1021/acs.chemrev.9b00491Google ScholarThere is no corresponding record for this reference.(b) Chen, J.; Li, J.; Dong, Z. A Review on the Latest Progress of Chan-Lam Coupling Reaction. Adv. Synth. Catal. 2020, 362, 3311– 3331, DOI: 10.1002/adsc.202000495Google ScholarThere is no corresponding record for this reference.(c) Devi, P. S.; Saranya, S.; Anilkumar, G. Recent Advances in Chan–Lam Coupling Reaction. Catal. Sci. Technol. 2024, 14, 2320– 2351, DOI: 10.1039/D4CY00034JGoogle ScholarThere is no corresponding record for this reference.
- 5Zhang, R.; Song, C.; Sui, Z.; Yuan, Y.; Gu, Y.; Chen, C. Recent Advances in Carbon-Nitrogen/Carbon-Oxygen Bond Formation Under Transition-Metal-Free Conditions. Chem. Rec. 2023, 23, e202300020 DOI: 10.1002/tcr.202300020Google ScholarThere is no corresponding record for this reference.
- 6(a) Gomez, S.; Peters, J. A.; Maschmeyer, T. The Reductive Amination of Aldehydes and Ketones and the Hydrogenation of Nitriles: Mechanistic Aspects and Selectivity Control. Adv. Synth. Catal. 2002, 344, 1037– 1057, DOI: 10.1002/1615-4169(200212)344:10<1037::AID-ADSC1037>3.0.CO;2-3Google ScholarThere is no corresponding record for this reference.(b) Afanasyev, O. I.; Kuchuk, E.; Usanov, D. L.; Chusov, D. Reductive Amination in the Synthesis of Pharmaceuticals. Chem. Rev. 2019, 119, 11857– 11911, DOI: 10.1021/acs.chemrev.9b00383Google ScholarThere is no corresponding record for this reference.
- 7Ghosh, A. K.; Sarkar, A.; Brindisi, M. The Curtius Rearrangement: Mechanistic Insight and Recent Applications in Natural Product Syntheses. Org. Biomol. Chem. 2018, 16, 2006– 2027, DOI: 10.1039/C8OB00138CGoogle ScholarThere is no corresponding record for this reference.
- 8(a) Swamy, K. C. K.; Kumar, N. N. B.; Balaraman, E.; Kumar, K. V. P. P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551– 2651, DOI: 10.1021/cr800278zGoogle ScholarThere is no corresponding record for this reference.(b) Fletcher, S. The Mitsunobu Reaction in the 21st Century. Org. Chem. Front. 2015, 2, 739– 752, DOI: 10.1039/C5QO00016EGoogle ScholarThere is no corresponding record for this reference.
- 9(a) Yan, M.; Kawamata, Y.; Baran, P. S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230– 13319, DOI: 10.1021/acs.chemrev.7b00397Google ScholarThere is no corresponding record for this reference.(b) Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Electrocatalysis as an Enabling Technology for Organic Synthesis. Chem. Soc. Rev. 2021, 50, 7941– 8002, DOI: 10.1039/D1CS00223FGoogle ScholarThere is no corresponding record for this reference.
- 10(a) Kim, J. E.; Choi, S.; Balamurugan, M.; Jang, J. H.; Nam, K. T. Electrochemical C–N Bond Formation for Sustainable Amine Synthesis. Trends Chem. 2020, 2, 1004– 1019, DOI: 10.1016/j.trechm.2020.09.003Google ScholarThere is no corresponding record for this reference.(b) Liu, C.; Liu, J.; Li, W.; Lu, H.; Zhang, Y. Recent Advances in Electrochemical C–H Bond Amination. Org. Chem. Front. 2023, 10, 5309– 5330, DOI: 10.1039/D3QO01159CGoogle ScholarThere is no corresponding record for this reference.(c) Li, X.; Yuan, X.; Hu, J.; Li, Y.; Bao, H. Radical Decarboxylative Carbon–Nitrogen Bond Formation. Molecules 2023, 28, 4249– 4285, DOI: 10.3390/molecules28104249Google ScholarThere is no corresponding record for this reference.(d) Sitter, J. D.; Lemus-Rivera, E. E.; Vannucci, A. K. Insights into Reactivity Trends for Electrochemical C–N Bond Formations. Org. Biomol. Chem. 2023, 21, 4290– 4296, DOI: 10.1039/D3OB00236EGoogle ScholarThere is no corresponding record for this reference.
- 11(a) Leech, M. C.; Lam, K. Electrosynthesis Using Carboxylic Acid Derivatives: New Tricks for Old Reactions. Acc. Chem. Res. 2020, 53, 121– 134, DOI: 10.1021/acs.accounts.9b00586Google ScholarThere is no corresponding record for this reference.(b) Ramadoss, V.; Zheng, Y.; Shao, X.; Tian, L.; Wang, Y. Advances in Electrochemical Decarboxylative Transformation Reactions. Chem.─Eur. J. 2021, 27, 3213– 3228, DOI: 10.1002/chem.202001764Google ScholarThere is no corresponding record for this reference.(c) Chen, N.; Ye, Z.; Zhang, F. Recent Progress on Electrochemical Synthesis Involving Carboxylic Acids. Org. Biomol. Chem. 2021, 19, 5501– 5520, DOI: 10.1039/D1OB00420DGoogle ScholarThere is no corresponding record for this reference.
- 12Kolbe, H. Untersuchungen über die Elektrolyse organischer Verbindungen. Liebigs Ann. Chem. 1849, 69, 257– 294, DOI: 10.1002/jlac.18490690302Google ScholarThere is no corresponding record for this reference.
- 13Shao, X.; Zheng, Y.; Tian, L.; Martín-Torres, I.; Echavarren, A. M.; Wang, Y. Decarboxylative Csp3-N Bond Formation by Electrochemical Oxidation of Amino Acids. Org. Lett. 2019, 21, 9262– 9267, DOI: 10.1021/acs.orglett.9b03696Google ScholarThere is no corresponding record for this reference.
- 14Sheng, T.; Zhang, H.-J.; Shang, M.; He, C.; Vantourout, J. C.; Baran, P. S. Electrochemical Decarboxylative N-Alkylation of Heterocycles. Org. Lett. 2020, 22, 7594– 7598, DOI: 10.1021/acs.orglett.0c02799Google ScholarThere is no corresponding record for this reference.
- 15Chen, R.; Yuan, H.; Wang, Y.; Chen, H.; Zhang, Y. Aerobic Electrochemical Csp3–N Coupling between Aliphatic Carboxylic Acids and N-Heterocycles. Organometallics 2023, 42, 1– 5, DOI: 10.1021/acs.organomet.2c00455Google ScholarThere is no corresponding record for this reference.
- 16(a) Borah, A. J.; Yan, G. Decarboxylative Functionalization of Cinnamic Acids. Org. Biomol. Chem. 2015, 13, 8094– 8115, DOI: 10.1039/C5OB00727EGoogle ScholarThere is no corresponding record for this reference.(b) Chen, L.; Zhang, L.; Yan, G.; Huang, D. Recent Advances of Cinnamic Acids in Organic Synthesis. Asian J. Org. Chem. 2020, 9, 842– 862, DOI: 10.1002/ajoc.202000217Google ScholarThere is no corresponding record for this reference.
- 17(a) Qian, P.; Bi, M.-X.; Su, J.-H.; Zha, Z.-G.; Wang, Z.-Y. Electrosynthesis of (E)-Vinyl Sulfones Directly from Cinnamic Acids and Sodium Sulfinates via Decarboxylative Sulfono Functionalization. J. Org. Chem. 2016, 81, 4876– 4882, DOI: 10.1021/acs.joc.6b00661Google ScholarThere is no corresponding record for this reference.(b) Zhao, Y.; Lai, Y.-L.; Du, K.-S.; Lin, D.-Z.; Huang, J.-M. Electrochemical Decarboxylative Sulfonylation of Cinnamic Acids with Aromatic Sulfonylhydrazides to Vinyl Sulfones. J. Org. Chem. 2017, 82, 9655– 9661, DOI: 10.1021/acs.joc.7b01741Google ScholarThere is no corresponding record for this reference.
- 18Yang, S.-M.; He, T.-J.; Lin, D.-Z.; Huang, J.-M. Electrosynthesis of (E)-Vinyl Thiocyanates from Cinnamic Acids via Decarboxylative Coupling Reaction. Org. Lett. 2019, 21, 1958– 1962, DOI: 10.1021/acs.orglett.8b04136Google ScholarThere is no corresponding record for this reference.
- 19Chen, X.; Huang, Y.-G.; Zhong, W.-Q.; Huang, J.-M. Electrochemical Decarboxylative Silylation of α,β-Unsaturated Carboxylic Acids. Org. Lett. 2023, 25, 4562– 4566, DOI: 10.1021/acs.orglett.3c01592Google ScholarThere is no corresponding record for this reference.
- 20Zhang, Z.; Shen, Y.; Zhao, Y.; Wu, J. Electrochemical Phosphorylation of α,β-Unsaturated Carboxylic Acids via Decarboxylative Cross-Coupling Reaction. Chem. Commun. 2025, 61, 12034– 12037, DOI: 10.1039/D5CC03160EGoogle ScholarThere is no corresponding record for this reference.
- 21(a) Li, F.-Y.; Lin, D.-Z.; He, T.-J.; Zhong, W.-Q.; Huang, J.-M. Electrochemical Decarboxylative Trifluoromethylation of α, β-Unsaturated Carboxylic Acids with CF3SO2Na. ChemCatChem 2019, 11, 2350– 2354, DOI: 10.1002/cctc.201900438Google ScholarThere is no corresponding record for this reference.(b) Yamamoto, Y.; Goto, N.; Uchida, H.; Yasui, T. Electrochemical Decarboxylative Trifluoromethylation of Cinnamic Acids Revisited: A Combined Experimental and Computational Study. Chem. - Asian J. 2025, 20, e202400967 DOI: 10.1002/asia.202400967Google ScholarThere is no corresponding record for this reference.
- 22(a) da Silva, G. Mystery of 1-Vinylpropargyl Formation from Acetylene Addition to the Propargyl Radical: An Open-and-Shut Case. J. Phys. Chem. A 2017, 121, 2086– 2095, DOI: 10.1021/acs.jpca.6b12996Google ScholarThere is no corresponding record for this reference.(b) Muresan, M.; Subramanian, H.; Sibi, M. P.; Green, J. R. Propargyl Radicals in Organic Synthesis. Eur. J. Org. Chem. 2021, 2021, 3359– 3375, DOI: 10.1002/ejoc.202100367Google ScholarThere is no corresponding record for this reference.
- 23
For examples selected of Pd-catalyzed cyclization from 2,3-allenoic acids see:
(a) Ma, S.; Yu, Z. Pd(II)-Catalyzed Coupling Cyclization of 2,3-Allenoic Acids with Allylic Halides. An Efficient Methodology for the Synthesis of β-Allylic Butenolides. J. Org. Chem. 2003, 68, 6149– 6152, DOI: 10.1021/jo034511qGoogle ScholarThere is no corresponding record for this reference.(b) Ma, S.; Yu, Z. Efficient Synthesis of 4-(3′-Furanyl)butenolide Derivatives via PdII-Catalyzed Oxidative Heterodimeric Cyclization Reaction of 2,3-Allenoic Acids and 1,2-Allenyl Ketones. Chem.─Eur. J. 2004, 10, 2078– 2087, DOI: 10.1002/chem.200305341Google ScholarThere is no corresponding record for this reference.(c) Ma, S.; Gu, Z. PdCl2-Catalyzed Two-Component Cross-Coupling Cyclization of 2,3-Allenoic Acids with 2,3-Allenols. An Efficient Synthesis of 4-(1‘,3‘-Dien-2‘-yl)-2(5H)-furanone Derivatives. J. Am. Chem. Soc. 2005, 127, 6182– 6183, DOI: 10.1021/ja0500815Google ScholarThere is no corresponding record for this reference.(d) Wang, B.; Ren, M.; Iqbal, N.; Mu, X.; Bäckvall, J.-E.; Yang, B. Palladium-Catalyzed Dehydrogenative Carbonylative Esterification of Allenoic Acids for the Synthesis of γ-Butyrolactone Derivatives. Org. Lett. 2024, 26, 2430– 2434, DOI: 10.1021/acs.orglett.4c00572Google ScholarThere is no corresponding record for this reference. - 24Fan, J.; Fu, C.; Wu, X.; Ma, S. Rh-Catalyzed Cyclization of 2,3-Allenoic Acids in the Presence of 2,3-Allenols. Chem. Commun. 2021, 57, 10411– 10414, DOI: 10.1039/D1CC04367FGoogle ScholarThere is no corresponding record for this reference.
- 25
For selected examples of metal catalyzed radical cyclization from 2,3-allenoic acids see:
(a) Yu, Q.; Ma, S. Copper-Catalyzed Cyclic Oxytrifluoromethylation of 2,3-Allenoic Acids to Trifluoromethylated Butenolides. Chem.─Eur. J. 2013, 19, 13304– 13308, DOI: 10.1002/chem.201302169Google ScholarThere is no corresponding record for this reference.(b) Pan, S.; Huang, Y.; Xu, X.-H.; Qing, F.-L. Copper-Assisted Oxidative Trifluoromethylthiolation of 2,3-Allenoic Acids with AgSCF3. Org. Lett. 2017, 19, 4624– 4627, DOI: 10.1021/acs.orglett.7b02249Google ScholarThere is no corresponding record for this reference.(c) Zhong, T.; Zheng, X.; Yin, C.; Shen, Q.; Yu, C. Copper-Catalyzed Phosphorylation of 2,3-Allenoic Acids and Phosphine Oxide: Access to Phosphorylated Butenolides. J. Org. Chem. 2021, 86, 9699– 9710, DOI: 10.1021/acs.joc.1c00998Google ScholarThere is no corresponding record for this reference.(d) Shi, Y.; Fu, C.; Zheng, J.; Ma, S. Photocatalytic Chemoselective Cyclic Oxysulfonylation of 2,3-Allenoic Acids. Org. Lett. 2024, 26, 5182– 5186, DOI: 10.1021/acs.orglett.4c01730Google ScholarThere is no corresponding record for this reference. - 26(a) Vitaku, E.; Smith, D. T.; Njardarson, J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257– 10274, DOI: 10.1021/jm501100bGoogle ScholarThere is no corresponding record for this reference.(b) Marshall, C. M.; Federice, J. G.; Bell, C. N.; Cox, P. B.; Njardarson, J. T. An Update on the Nitrogen Heterocycle Compositions and Properties of U.S. FDA-Approved Pharmaceuticals (2013–2023). J. Med. Chem. 2024, 67, 11622– 11655, DOI: 10.1021/acs.jmedchem.4c01122Google ScholarThere is no corresponding record for this reference.
- 27Yan, W.; Wang, Q.; Chen, Y.; Petersen, J. L.; Shi, X. Iron-Catalyzed C–O Bond Activation for the Synthesis of Propargyl-1,2,3-triazoles and 1,1-Bis-triazoles. Org. Lett. 2010, 12, 3308– 3311, DOI: 10.1021/ol101082vGoogle ScholarThere is no corresponding record for this reference.
- 28González-Pelayo, S.; López, L. A. Microwave-Assisted Generation and Capture by Azoles of ortho-Quinone Methide Intermediates under Aqueous Conditions. Eur. J. Org. Chem. 2017, 2017, 6003– 6007, DOI: 10.1002/ejoc.201701183Google ScholarThere is no corresponding record for this reference.
- 29Wang, Y.; Wang, S.; Shan, W.; Shao, Z. Direct asymmetric N-propargylation of indoles and carbazoles catalyzed by lithium SPINOL phosphate. Nat. Commun. 2020, 11, 226 DOI: 10.1038/s41467-019-13886-9Google ScholarThere is no corresponding record for this reference.
- 30Kotsinaris, A.; Kyriacou, G.; Lambrou, C. Electrochemical Reduction of Dichloromethane to Higher Hydrocarbons. J. Appl. Electrochem. 1998, 28, 613– 616, DOI: 10.1023/A:1003202203067Google ScholarThere is no corresponding record for this reference.
- 31Hu, T.; Fagué, V.; Bouyssi, D.; Monteiro, N.; Amgoune, A. Hydride-Free Reduction of Propargyl Electrophiles: a Nickel-Catalyzed Photoredox Strategy for Allene Synthesis. Green Chem. 2024, 26, 6124– 6130, DOI: 10.1039/D4GC00984CGoogle ScholarThere is no corresponding record for this reference.
- 32
The oxidation potential of BHT into the corresponding radical cation was determined by DFT as Eox = 1.15 V (vs Ag/AgCl).
Gavilán-Arriazu, E. M.; Alaniz, R. D.; Charoen-amornkitt, P.; Fernández, J. M.; Pierini, G. D.; Rodriguez, S. A. Study of the BHT Oxidation Mechanism Coupling Theory and Experiment. ACS Org. Inorg. Au 2024, 4, 692– 704, DOI: 10.1021/acsorginorgau.4c00067Google ScholarThere is no corresponding record for this reference. - 33
The difference of reactivity between 4-bromopyrazole and 3-bromopyrazole could be explained by the difference of basicity and nucleophilicity of the nitrogen atoms due to the electronic effect of the bromine in the 3-position. The basicity of various pyrazoles was studied and discussed in this paper
Marín-Luna, M.; Alkorta, I.; Elguero, J. A theoretical study of the gas phase (proton affinity) and aqueous (pKa) basicity of a series of 150 pyrazoles. New J. Chem. 2015, 39, 2861– 2871, DOI: 10.1039/C4NJ02201GGoogle ScholarThere is no corresponding record for this reference. - 34Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378– 6396, DOI: 10.1021/jp810292nGoogle ScholarThere is no corresponding record for this reference.
- 35Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615– 6620, DOI: 10.1039/b810189bGoogle ScholarThere is no corresponding record for this reference.
- 36Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297– 3305, DOI: 10.1039/b508541aGoogle ScholarThere is no corresponding record for this reference.
- 37Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford CT, 2016.Google ScholarThere is no corresponding record for this reference.
- 38
A similar situation has been discussed computationally in:
Wei, Q.; Lee, Y.; Liang, W.; Chen, X.; Mu, B.-S.; Cui, X.-Y.; Wu, W.; Bai, S.; Liu, Z. Photocatalytic Direct Borylation of Carboxylic Acids. Nat. Commun. 2022, 13, 7112 DOI: 10.1038/s41467-022-34833-1Google ScholarThere is no corresponding record for this reference. - 39(a) Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P. G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735– 6748, DOI: 10.1002/ejoc.201900956Google ScholarThere is no corresponding record for this reference.(b) Kütt, A.; Tshepelevitsh, S.; Saame, J.; Lõkov, M.; Kaljurand, I.; Selberg, S.; Leito, I. Strengths of Acids in Acetonitrile. Eur. J. Org. Chem. 2021, 2021, 1407– 1419, DOI: 10.1002/ejoc.202001649Google ScholarThere is no corresponding record for this reference.
- 40
For a discussion of competing kinetic vs thermodynamic product formation of allenyl/propargyl cations, see:
Mayr, H.; Schneider, R. Ab-initio-MO-Studie Methyl- und Phenyl-substituierter Allenyl-Kationen. Chem. Ber. 1982, 115, 3470– 3478, DOI: 10.1002/cber.19821151103Google ScholarThere is no corresponding record for this reference. - 41
For a discussion of the loss of selectivity upon reaching diffusion control, see:
Mayr, H.; Ofial, A. R. The Reactivity–Selectivity Principle: An Imperishable Myth in Organic Chemistry. Angew. Chem., Int. Ed. 2006, 45, 1844– 1854, DOI: 10.1002/anie.200503273Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACS Organic & Inorganic Au
© 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
Non-Commercial (NC): Only non-commercial uses of the work are permitted.
No Derivatives (ND): Derivative works may be created for non-commercial purposes, but sharing is prohibited.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
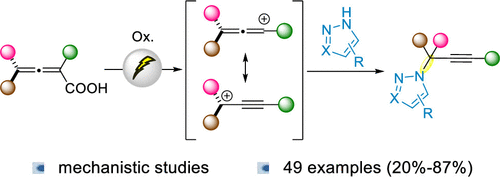
Scheme 1
 Scheme 1. Synthesis of Propargylic N-Heterocycles and Reactivity of Carboxylic Acids
Scheme 1. Synthesis of Propargylic N-Heterocycles and Reactivity of Carboxylic AcidsFigure 1
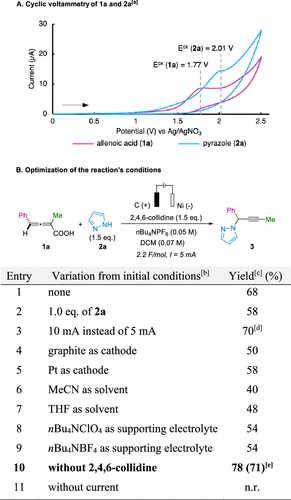
Figure 1. Identification of reaction conditions based on cyclic voltammetry. [a]Cyclic voltammogram of allenoic acid 1a (purple curve) and pyrazole 2a (blue curve) in MeCN with nBu4NPF6 (0.1 M) as the electrolyte using a platinum disk (d = 2 mm) as the working electrode, a platinum wire as the counter electrode, and a nonaqueous Ag/AgNO3 electrode (3.0 M in MeCN) as the reference. All cyclic voltammetry experiments scan from 0.0 V vs the Ag/Ag+ reference electrode in the anodic direction at a scan rate of 50 mV/s. The cyclic voltammetry traces were presented with the IUPAC convention. [b]Initial conditions: 1a (0.2 mmol), pyrazole 2a (1.5 equiv), 2,4,6-collidine (1.5 equiv), nBu4NPF6 (0.05 M), DCM (3.0 mL), undivided cell, C-SK-50 anode, Ni cathode, CCE = 5 mA, 2.2 F/mol. [c]Isolated yield. [d]Surface degradation of the C anode was observed. [e]Faradaic efficiency.
Scheme 2
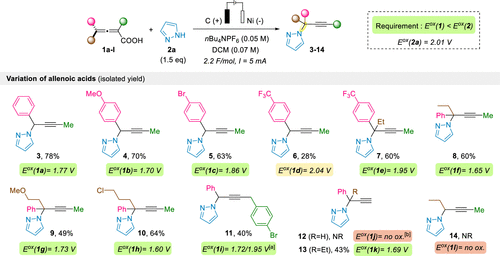 Scheme 2. Electrochemical Propargylation of Pyrazole with Various Allenoic Acids
Scheme 2. Electrochemical Propargylation of Pyrazole with Various Allenoic Acids[a] Two major oxidation peaks were observed on the CV of 1i (see the SI for CV graphs). [b] A broad, ill-defined oxidation wave was observed, which precluded the determination of an oxidation potential for 1j.
Scheme 3
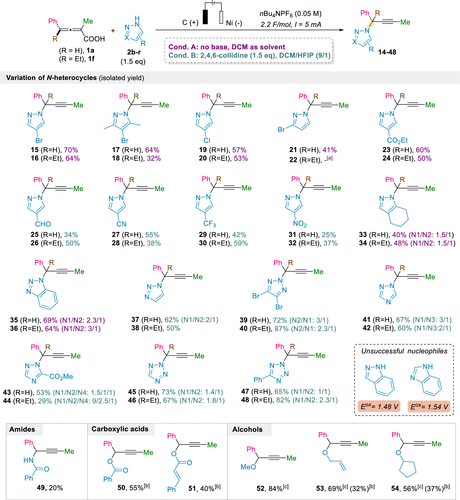 Scheme 3. Scope of N-Heterocycles for the Electrochemical Propargylation of Allenoic Acids and Extension to Other Nucleophiles
Scheme 3. Scope of N-Heterocycles for the Electrochemical Propargylation of Allenoic Acids and Extension to Other Nucleophiles[a]22 was observed in the crude reaction mixture with a similar conversion as 21, but it could not be isolated in the pure form, presumably due to decomposition. [b] 3 equiv of the nucleophile was used. [c] The reaction was performed in a 9:1 (v/v) mixture of DCM/alcohol.
Scheme 4
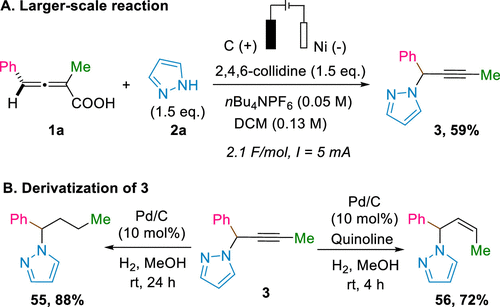 Scheme 4. Synthetic Utility of the Electrochemical Propargylation of N-Heterocycles
Scheme 4. Synthetic Utility of the Electrochemical Propargylation of N-HeterocyclesFigure 2
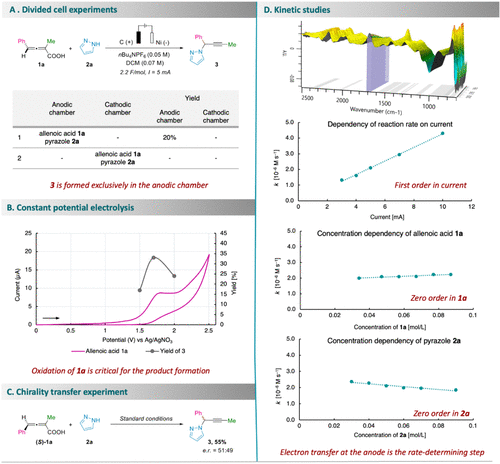
Figure 2. Overview of the mechanistic experiments. (A) Divided-cell experiments were carried out in IKA Pro-Divide equipment that includes two PTFE cells separated from each other by a glass frit (pore size 10–16 μm) equipped with an O-ring. A graphite SK-50 electrode (8 mm wide, 50 mm length, 1 mm thickness) was used as the anode, and a nickel electrode (8 mm wide, 50 mm length, 1 mm thickness) was used as the cathode. (B) Constant-potential electrolysis experiments were performed using IKA ElectraSyn equipment equipped with graphite as the working electrode, nickel as the counter electrode, and Ag/AgCl wire in 3.0 M KCl as the reference electrode. Constant-potential electrolysis was conducted at various potentials and terminated once a total charge of 1.0 F/mol had passed through the circuit. The reaction yield was determined by NMR using mesitylene as an internal standard. (C) Chirality transfer experiments using standard conditions. (D) IR kinetics were followed using a Mettler-Toledo ReactIR 15 system and a 6.3 mm AgX Fiber probe with 78 scans per spectrum. Data was acquired and processed with Mettler-Toledo iC IR software with solvent subtraction of DCM and electrolyte subtraction of nBu4NPF6. The absorbance at 1689 cm–1 was recorded.
Figure 3
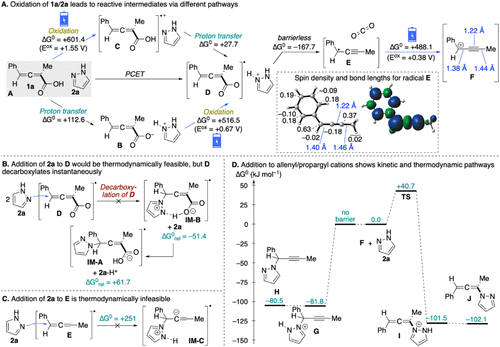
Figure 3. Computational exploration of the reaction mechanism at the SMD(CH2Cl2)/ωB97XD/def2-TZVPP//SMD(CH2Cl2)/ωB97XD/def2-SVP level of theory. (A) Competing pathways for electrochemical oxidation; all Gibbs energies are reported in kJ mol–1. Electrochemical steps are highlighted with blue arrows, and the oxidation potentials are calculated vs the saturated calomel electrode (SCE). (B, C) Evaluation of addition of 2a to radical intermediates D and E. (D) Gibbs energy profile for the reaction of cation F as allenyl vs propargyl cations with nucleophile 2a.
References
This article references 41 other publications.
- 1(a) Bansal, Y.; Silakari, O. The Therapeutic Journey of Benzimidazoles: A Review. Bioorg. J. Med. Chem. 2012, 20, 6208– 6236, DOI: 10.1016/j.bmc.2012.09.013There is no corresponding record for this reference.(b) Küçükgüzel, Ş.; Şenkardeş, S. Recent Advances in Bioactive Pyrazoles. Eur. J. Med. Chem. 2015, 97, 786– 815, DOI: 10.1016/j.ejmech.2014.11.059There is no corresponding record for this reference.(c) Ríos, M.-C.; Portilla, J. Recent Advances in Synthesis and Properties of Pyrazoles. Chemistry 2022, 4, 940– 968, DOI: 10.3390/chemistry4030065There is no corresponding record for this reference.(d) Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K. K.; Jonnalagadda, S. B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909– 1951, DOI: 10.3390/molecules25081909There is no corresponding record for this reference.
- 2(a) Ruiz-Castillo, P.; Buchwald, S. L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564– 12649, DOI: 10.1021/acs.chemrev.6b00512There is no corresponding record for this reference.(b) Dorel, R.; Grugel, C. P.; Haydl, A. M. The Buchwald–Hartwig Amination After 25 Years. Angew. Chem., Int. Ed. 2019, 58, 17118– 17129, DOI: 10.1002/anie.201904795There is no corresponding record for this reference.
- 3(a) Monnier, F.; Taillefer, M. Catalytic C–C, C–N, and C–O Ullmann-Type Coupling Reactions. Angew. Chem., Int. Ed. 2009, 48, 6954– 6971, DOI: 10.1002/anie.200804497There is no corresponding record for this reference.(b) Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Copper Catalysed Ullmann Type Chemistry: from Mechanistic Aspects to Modern Development. Chem. Soc. Rev. 2014, 43, 3525– 3550, DOI: 10.1039/C3CS60289CThere is no corresponding record for this reference.(c) Yang, Q.; Zhao, Y.; Ma, D. Cu-Mediated Ullmann-Type Cross-Coupling and Industrial Applications in Route Design, Process Development, and Scale-up of Pharmaceutical and Agrochemical Processes. Org. Process Res. Dev. 2022, 26, 1690– 1750, DOI: 10.1021/acs.oprd.2c00050There is no corresponding record for this reference.
- 4(a) West, M. J.; Fyfe, J. W. B.; Vantourout, J. C.; Watson, A. J. B. Mechanistic Development and Recent Applications of the Chan-Lam Amination. Chem. Rev. 2019, 119, 12491– 12523, DOI: 10.1021/acs.chemrev.9b00491There is no corresponding record for this reference.(b) Chen, J.; Li, J.; Dong, Z. A Review on the Latest Progress of Chan-Lam Coupling Reaction. Adv. Synth. Catal. 2020, 362, 3311– 3331, DOI: 10.1002/adsc.202000495There is no corresponding record for this reference.(c) Devi, P. S.; Saranya, S.; Anilkumar, G. Recent Advances in Chan–Lam Coupling Reaction. Catal. Sci. Technol. 2024, 14, 2320– 2351, DOI: 10.1039/D4CY00034JThere is no corresponding record for this reference.
- 5Zhang, R.; Song, C.; Sui, Z.; Yuan, Y.; Gu, Y.; Chen, C. Recent Advances in Carbon-Nitrogen/Carbon-Oxygen Bond Formation Under Transition-Metal-Free Conditions. Chem. Rec. 2023, 23, e202300020 DOI: 10.1002/tcr.202300020There is no corresponding record for this reference.
- 6(a) Gomez, S.; Peters, J. A.; Maschmeyer, T. The Reductive Amination of Aldehydes and Ketones and the Hydrogenation of Nitriles: Mechanistic Aspects and Selectivity Control. Adv. Synth. Catal. 2002, 344, 1037– 1057, DOI: 10.1002/1615-4169(200212)344:10<1037::AID-ADSC1037>3.0.CO;2-3There is no corresponding record for this reference.(b) Afanasyev, O. I.; Kuchuk, E.; Usanov, D. L.; Chusov, D. Reductive Amination in the Synthesis of Pharmaceuticals. Chem. Rev. 2019, 119, 11857– 11911, DOI: 10.1021/acs.chemrev.9b00383There is no corresponding record for this reference.
- 7Ghosh, A. K.; Sarkar, A.; Brindisi, M. The Curtius Rearrangement: Mechanistic Insight and Recent Applications in Natural Product Syntheses. Org. Biomol. Chem. 2018, 16, 2006– 2027, DOI: 10.1039/C8OB00138CThere is no corresponding record for this reference.
- 8(a) Swamy, K. C. K.; Kumar, N. N. B.; Balaraman, E.; Kumar, K. V. P. P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551– 2651, DOI: 10.1021/cr800278zThere is no corresponding record for this reference.(b) Fletcher, S. The Mitsunobu Reaction in the 21st Century. Org. Chem. Front. 2015, 2, 739– 752, DOI: 10.1039/C5QO00016EThere is no corresponding record for this reference.
- 9(a) Yan, M.; Kawamata, Y.; Baran, P. S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230– 13319, DOI: 10.1021/acs.chemrev.7b00397There is no corresponding record for this reference.(b) Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Electrocatalysis as an Enabling Technology for Organic Synthesis. Chem. Soc. Rev. 2021, 50, 7941– 8002, DOI: 10.1039/D1CS00223FThere is no corresponding record for this reference.
- 10(a) Kim, J. E.; Choi, S.; Balamurugan, M.; Jang, J. H.; Nam, K. T. Electrochemical C–N Bond Formation for Sustainable Amine Synthesis. Trends Chem. 2020, 2, 1004– 1019, DOI: 10.1016/j.trechm.2020.09.003There is no corresponding record for this reference.(b) Liu, C.; Liu, J.; Li, W.; Lu, H.; Zhang, Y. Recent Advances in Electrochemical C–H Bond Amination. Org. Chem. Front. 2023, 10, 5309– 5330, DOI: 10.1039/D3QO01159CThere is no corresponding record for this reference.(c) Li, X.; Yuan, X.; Hu, J.; Li, Y.; Bao, H. Radical Decarboxylative Carbon–Nitrogen Bond Formation. Molecules 2023, 28, 4249– 4285, DOI: 10.3390/molecules28104249There is no corresponding record for this reference.(d) Sitter, J. D.; Lemus-Rivera, E. E.; Vannucci, A. K. Insights into Reactivity Trends for Electrochemical C–N Bond Formations. Org. Biomol. Chem. 2023, 21, 4290– 4296, DOI: 10.1039/D3OB00236EThere is no corresponding record for this reference.
- 11(a) Leech, M. C.; Lam, K. Electrosynthesis Using Carboxylic Acid Derivatives: New Tricks for Old Reactions. Acc. Chem. Res. 2020, 53, 121– 134, DOI: 10.1021/acs.accounts.9b00586There is no corresponding record for this reference.(b) Ramadoss, V.; Zheng, Y.; Shao, X.; Tian, L.; Wang, Y. Advances in Electrochemical Decarboxylative Transformation Reactions. Chem.─Eur. J. 2021, 27, 3213– 3228, DOI: 10.1002/chem.202001764There is no corresponding record for this reference.(c) Chen, N.; Ye, Z.; Zhang, F. Recent Progress on Electrochemical Synthesis Involving Carboxylic Acids. Org. Biomol. Chem. 2021, 19, 5501– 5520, DOI: 10.1039/D1OB00420DThere is no corresponding record for this reference.
- 12Kolbe, H. Untersuchungen über die Elektrolyse organischer Verbindungen. Liebigs Ann. Chem. 1849, 69, 257– 294, DOI: 10.1002/jlac.18490690302There is no corresponding record for this reference.
- 13Shao, X.; Zheng, Y.; Tian, L.; Martín-Torres, I.; Echavarren, A. M.; Wang, Y. Decarboxylative Csp3-N Bond Formation by Electrochemical Oxidation of Amino Acids. Org. Lett. 2019, 21, 9262– 9267, DOI: 10.1021/acs.orglett.9b03696There is no corresponding record for this reference.
- 14Sheng, T.; Zhang, H.-J.; Shang, M.; He, C.; Vantourout, J. C.; Baran, P. S. Electrochemical Decarboxylative N-Alkylation of Heterocycles. Org. Lett. 2020, 22, 7594– 7598, DOI: 10.1021/acs.orglett.0c02799There is no corresponding record for this reference.
- 15Chen, R.; Yuan, H.; Wang, Y.; Chen, H.; Zhang, Y. Aerobic Electrochemical Csp3–N Coupling between Aliphatic Carboxylic Acids and N-Heterocycles. Organometallics 2023, 42, 1– 5, DOI: 10.1021/acs.organomet.2c00455There is no corresponding record for this reference.
- 16(a) Borah, A. J.; Yan, G. Decarboxylative Functionalization of Cinnamic Acids. Org. Biomol. Chem. 2015, 13, 8094– 8115, DOI: 10.1039/C5OB00727EThere is no corresponding record for this reference.(b) Chen, L.; Zhang, L.; Yan, G.; Huang, D. Recent Advances of Cinnamic Acids in Organic Synthesis. Asian J. Org. Chem. 2020, 9, 842– 862, DOI: 10.1002/ajoc.202000217There is no corresponding record for this reference.
- 17(a) Qian, P.; Bi, M.-X.; Su, J.-H.; Zha, Z.-G.; Wang, Z.-Y. Electrosynthesis of (E)-Vinyl Sulfones Directly from Cinnamic Acids and Sodium Sulfinates via Decarboxylative Sulfono Functionalization. J. Org. Chem. 2016, 81, 4876– 4882, DOI: 10.1021/acs.joc.6b00661There is no corresponding record for this reference.(b) Zhao, Y.; Lai, Y.-L.; Du, K.-S.; Lin, D.-Z.; Huang, J.-M. Electrochemical Decarboxylative Sulfonylation of Cinnamic Acids with Aromatic Sulfonylhydrazides to Vinyl Sulfones. J. Org. Chem. 2017, 82, 9655– 9661, DOI: 10.1021/acs.joc.7b01741There is no corresponding record for this reference.
- 18Yang, S.-M.; He, T.-J.; Lin, D.-Z.; Huang, J.-M. Electrosynthesis of (E)-Vinyl Thiocyanates from Cinnamic Acids via Decarboxylative Coupling Reaction. Org. Lett. 2019, 21, 1958– 1962, DOI: 10.1021/acs.orglett.8b04136There is no corresponding record for this reference.
- 19Chen, X.; Huang, Y.-G.; Zhong, W.-Q.; Huang, J.-M. Electrochemical Decarboxylative Silylation of α,β-Unsaturated Carboxylic Acids. Org. Lett. 2023, 25, 4562– 4566, DOI: 10.1021/acs.orglett.3c01592There is no corresponding record for this reference.
- 20Zhang, Z.; Shen, Y.; Zhao, Y.; Wu, J. Electrochemical Phosphorylation of α,β-Unsaturated Carboxylic Acids via Decarboxylative Cross-Coupling Reaction. Chem. Commun. 2025, 61, 12034– 12037, DOI: 10.1039/D5CC03160EThere is no corresponding record for this reference.
- 21(a) Li, F.-Y.; Lin, D.-Z.; He, T.-J.; Zhong, W.-Q.; Huang, J.-M. Electrochemical Decarboxylative Trifluoromethylation of α, β-Unsaturated Carboxylic Acids with CF3SO2Na. ChemCatChem 2019, 11, 2350– 2354, DOI: 10.1002/cctc.201900438There is no corresponding record for this reference.(b) Yamamoto, Y.; Goto, N.; Uchida, H.; Yasui, T. Electrochemical Decarboxylative Trifluoromethylation of Cinnamic Acids Revisited: A Combined Experimental and Computational Study. Chem. - Asian J. 2025, 20, e202400967 DOI: 10.1002/asia.202400967There is no corresponding record for this reference.
- 22(a) da Silva, G. Mystery of 1-Vinylpropargyl Formation from Acetylene Addition to the Propargyl Radical: An Open-and-Shut Case. J. Phys. Chem. A 2017, 121, 2086– 2095, DOI: 10.1021/acs.jpca.6b12996There is no corresponding record for this reference.(b) Muresan, M.; Subramanian, H.; Sibi, M. P.; Green, J. R. Propargyl Radicals in Organic Synthesis. Eur. J. Org. Chem. 2021, 2021, 3359– 3375, DOI: 10.1002/ejoc.202100367There is no corresponding record for this reference.
- 23
For examples selected of Pd-catalyzed cyclization from 2,3-allenoic acids see:
(a) Ma, S.; Yu, Z. Pd(II)-Catalyzed Coupling Cyclization of 2,3-Allenoic Acids with Allylic Halides. An Efficient Methodology for the Synthesis of β-Allylic Butenolides. J. Org. Chem. 2003, 68, 6149– 6152, DOI: 10.1021/jo034511qThere is no corresponding record for this reference.(b) Ma, S.; Yu, Z. Efficient Synthesis of 4-(3′-Furanyl)butenolide Derivatives via PdII-Catalyzed Oxidative Heterodimeric Cyclization Reaction of 2,3-Allenoic Acids and 1,2-Allenyl Ketones. Chem.─Eur. J. 2004, 10, 2078– 2087, DOI: 10.1002/chem.200305341There is no corresponding record for this reference.(c) Ma, S.; Gu, Z. PdCl2-Catalyzed Two-Component Cross-Coupling Cyclization of 2,3-Allenoic Acids with 2,3-Allenols. An Efficient Synthesis of 4-(1‘,3‘-Dien-2‘-yl)-2(5H)-furanone Derivatives. J. Am. Chem. Soc. 2005, 127, 6182– 6183, DOI: 10.1021/ja0500815There is no corresponding record for this reference.(d) Wang, B.; Ren, M.; Iqbal, N.; Mu, X.; Bäckvall, J.-E.; Yang, B. Palladium-Catalyzed Dehydrogenative Carbonylative Esterification of Allenoic Acids for the Synthesis of γ-Butyrolactone Derivatives. Org. Lett. 2024, 26, 2430– 2434, DOI: 10.1021/acs.orglett.4c00572There is no corresponding record for this reference. - 24Fan, J.; Fu, C.; Wu, X.; Ma, S. Rh-Catalyzed Cyclization of 2,3-Allenoic Acids in the Presence of 2,3-Allenols. Chem. Commun. 2021, 57, 10411– 10414, DOI: 10.1039/D1CC04367FThere is no corresponding record for this reference.
- 25
For selected examples of metal catalyzed radical cyclization from 2,3-allenoic acids see:
(a) Yu, Q.; Ma, S. Copper-Catalyzed Cyclic Oxytrifluoromethylation of 2,3-Allenoic Acids to Trifluoromethylated Butenolides. Chem.─Eur. J. 2013, 19, 13304– 13308, DOI: 10.1002/chem.201302169There is no corresponding record for this reference.(b) Pan, S.; Huang, Y.; Xu, X.-H.; Qing, F.-L. Copper-Assisted Oxidative Trifluoromethylthiolation of 2,3-Allenoic Acids with AgSCF3. Org. Lett. 2017, 19, 4624– 4627, DOI: 10.1021/acs.orglett.7b02249There is no corresponding record for this reference.(c) Zhong, T.; Zheng, X.; Yin, C.; Shen, Q.; Yu, C. Copper-Catalyzed Phosphorylation of 2,3-Allenoic Acids and Phosphine Oxide: Access to Phosphorylated Butenolides. J. Org. Chem. 2021, 86, 9699– 9710, DOI: 10.1021/acs.joc.1c00998There is no corresponding record for this reference.(d) Shi, Y.; Fu, C.; Zheng, J.; Ma, S. Photocatalytic Chemoselective Cyclic Oxysulfonylation of 2,3-Allenoic Acids. Org. Lett. 2024, 26, 5182– 5186, DOI: 10.1021/acs.orglett.4c01730There is no corresponding record for this reference. - 26(a) Vitaku, E.; Smith, D. T.; Njardarson, J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257– 10274, DOI: 10.1021/jm501100bThere is no corresponding record for this reference.(b) Marshall, C. M.; Federice, J. G.; Bell, C. N.; Cox, P. B.; Njardarson, J. T. An Update on the Nitrogen Heterocycle Compositions and Properties of U.S. FDA-Approved Pharmaceuticals (2013–2023). J. Med. Chem. 2024, 67, 11622– 11655, DOI: 10.1021/acs.jmedchem.4c01122There is no corresponding record for this reference.
- 27Yan, W.; Wang, Q.; Chen, Y.; Petersen, J. L.; Shi, X. Iron-Catalyzed C–O Bond Activation for the Synthesis of Propargyl-1,2,3-triazoles and 1,1-Bis-triazoles. Org. Lett. 2010, 12, 3308– 3311, DOI: 10.1021/ol101082vThere is no corresponding record for this reference.
- 28González-Pelayo, S.; López, L. A. Microwave-Assisted Generation and Capture by Azoles of ortho-Quinone Methide Intermediates under Aqueous Conditions. Eur. J. Org. Chem. 2017, 2017, 6003– 6007, DOI: 10.1002/ejoc.201701183There is no corresponding record for this reference.
- 29Wang, Y.; Wang, S.; Shan, W.; Shao, Z. Direct asymmetric N-propargylation of indoles and carbazoles catalyzed by lithium SPINOL phosphate. Nat. Commun. 2020, 11, 226 DOI: 10.1038/s41467-019-13886-9There is no corresponding record for this reference.
- 30Kotsinaris, A.; Kyriacou, G.; Lambrou, C. Electrochemical Reduction of Dichloromethane to Higher Hydrocarbons. J. Appl. Electrochem. 1998, 28, 613– 616, DOI: 10.1023/A:1003202203067There is no corresponding record for this reference.
- 31Hu, T.; Fagué, V.; Bouyssi, D.; Monteiro, N.; Amgoune, A. Hydride-Free Reduction of Propargyl Electrophiles: a Nickel-Catalyzed Photoredox Strategy for Allene Synthesis. Green Chem. 2024, 26, 6124– 6130, DOI: 10.1039/D4GC00984CThere is no corresponding record for this reference.
- 32
The oxidation potential of BHT into the corresponding radical cation was determined by DFT as Eox = 1.15 V (vs Ag/AgCl).
Gavilán-Arriazu, E. M.; Alaniz, R. D.; Charoen-amornkitt, P.; Fernández, J. M.; Pierini, G. D.; Rodriguez, S. A. Study of the BHT Oxidation Mechanism Coupling Theory and Experiment. ACS Org. Inorg. Au 2024, 4, 692– 704, DOI: 10.1021/acsorginorgau.4c00067There is no corresponding record for this reference. - 33
The difference of reactivity between 4-bromopyrazole and 3-bromopyrazole could be explained by the difference of basicity and nucleophilicity of the nitrogen atoms due to the electronic effect of the bromine in the 3-position. The basicity of various pyrazoles was studied and discussed in this paper
Marín-Luna, M.; Alkorta, I.; Elguero, J. A theoretical study of the gas phase (proton affinity) and aqueous (pKa) basicity of a series of 150 pyrazoles. New J. Chem. 2015, 39, 2861– 2871, DOI: 10.1039/C4NJ02201GThere is no corresponding record for this reference. - 34Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378– 6396, DOI: 10.1021/jp810292nThere is no corresponding record for this reference.
- 35Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615– 6620, DOI: 10.1039/b810189bThere is no corresponding record for this reference.
- 36Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297– 3305, DOI: 10.1039/b508541aThere is no corresponding record for this reference.
- 37Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford CT, 2016.There is no corresponding record for this reference.
- 38
A similar situation has been discussed computationally in:
Wei, Q.; Lee, Y.; Liang, W.; Chen, X.; Mu, B.-S.; Cui, X.-Y.; Wu, W.; Bai, S.; Liu, Z. Photocatalytic Direct Borylation of Carboxylic Acids. Nat. Commun. 2022, 13, 7112 DOI: 10.1038/s41467-022-34833-1There is no corresponding record for this reference. - 39(a) Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P. G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735– 6748, DOI: 10.1002/ejoc.201900956There is no corresponding record for this reference.(b) Kütt, A.; Tshepelevitsh, S.; Saame, J.; Lõkov, M.; Kaljurand, I.; Selberg, S.; Leito, I. Strengths of Acids in Acetonitrile. Eur. J. Org. Chem. 2021, 2021, 1407– 1419, DOI: 10.1002/ejoc.202001649There is no corresponding record for this reference.
- 40
For a discussion of competing kinetic vs thermodynamic product formation of allenyl/propargyl cations, see:
Mayr, H.; Schneider, R. Ab-initio-MO-Studie Methyl- und Phenyl-substituierter Allenyl-Kationen. Chem. Ber. 1982, 115, 3470– 3478, DOI: 10.1002/cber.19821151103There is no corresponding record for this reference. - 41
For a discussion of the loss of selectivity upon reaching diffusion control, see:
Mayr, H.; Ofial, A. R. The Reactivity–Selectivity Principle: An Imperishable Myth in Organic Chemistry. Angew. Chem., Int. Ed. 2006, 45, 1844– 1854, DOI: 10.1002/anie.200503273There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.5c00117.
Experimental procedures and characterization data, additional experimental details, and 1H, 13C, and 19F NMR spectra (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.