


Atomic-Scale Representation and Statistical Learning of Tensorial Properties
- Andrea GrisafiAndrea GrisafiLaboratory of Computational Science and Modeling, IMX, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, SwitzerlandMore by Andrea Grisafi
- ,
- David M. WilkinsDavid M. WilkinsLaboratory of Computational Science and Modeling, IMX, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, SwitzerlandMore by David M. Wilkins
- ,
- Michael J. WillattMichael J. WillattLaboratory of Computational Science and Modeling, IMX, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, SwitzerlandMore by Michael J. Willatt
- , and
- Michele Ceriotti *Michele Ceriotti*E-mail: [email protected]Laboratory of Computational Science and Modeling, IMX, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, SwitzerlandMore by Michele Ceriotti

This publication is free to access through this site. Learn More

Abstract
This chapter discusses the importance of incorporating three-dimensional symmetries in the context of statistical learning models geared towards the interpolation of the tensorial properties of atomic-scale structures. We focus on Gaussian process regression, and in particular on the construction of structural representations, and the associated kernel functions, that are endowed with the geometric covariance properties compatible with those of the learning targets. We summarize the general formulation of such a symmetry-adapted Gaussian process regression model, and how it can be implemented based on a scheme that generalizes the popular smooth overlap of atomic positions representation. We give examples of the performance of this framework when learning the polarizability, the hyperpolarizability, and the ground-state electron density of a molecule.
This publication is licensed for personal use by The American Chemical Society.
Introduction
Figure 1
{kind=link}
Linear Regression


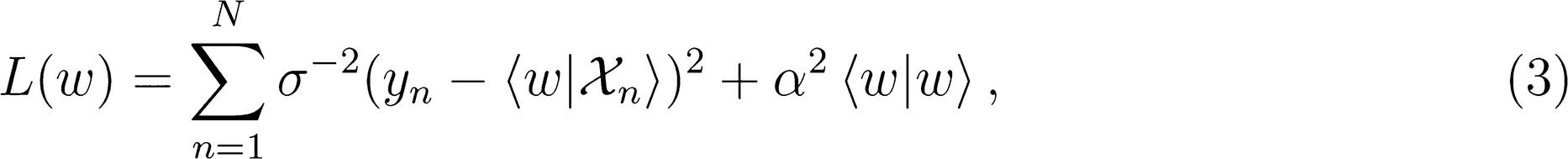



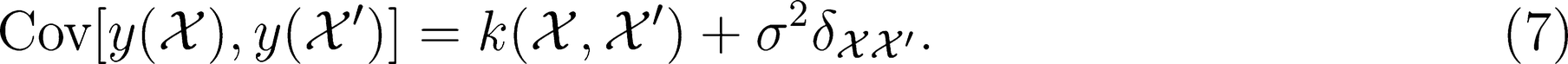
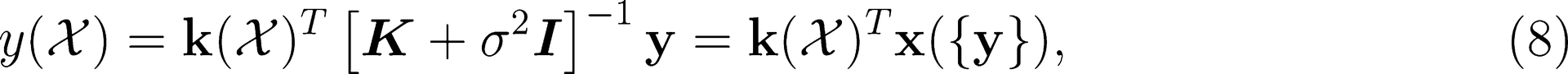
Tensors, Symmetries and Correlations
Translations

Rotations
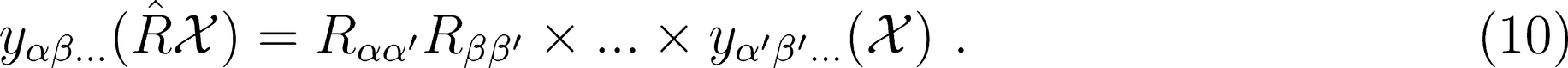
Reflections
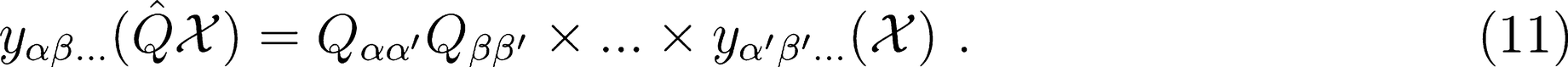
Covariant Descriptors
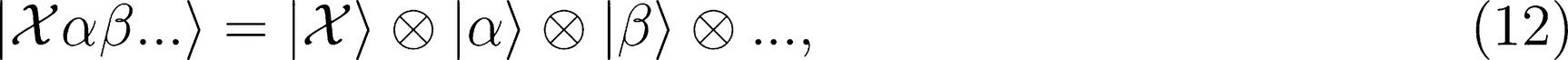


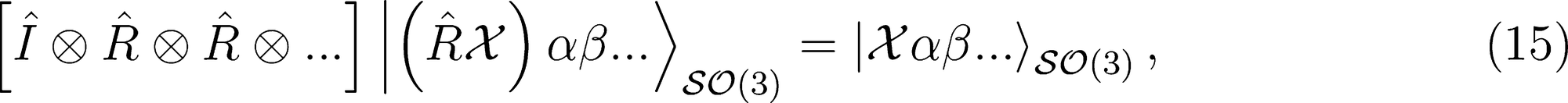
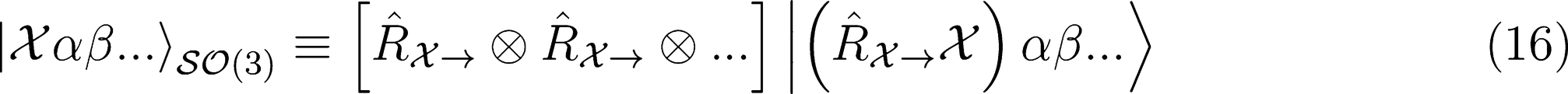
Figure 2
{kind=link}
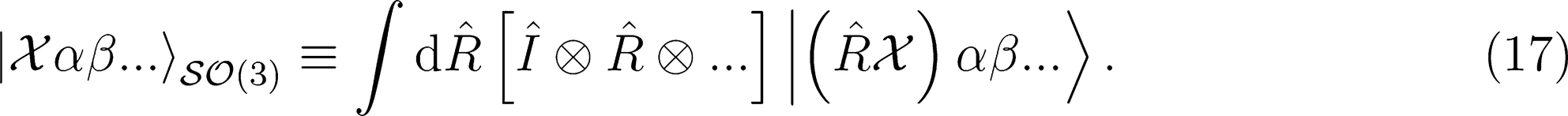
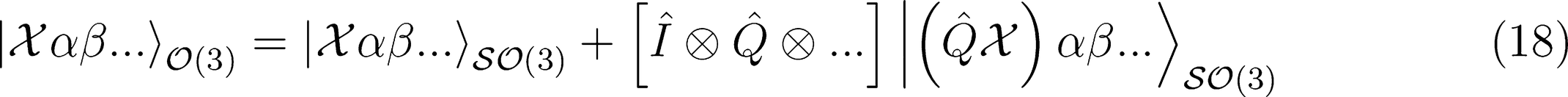
Covariant Regression
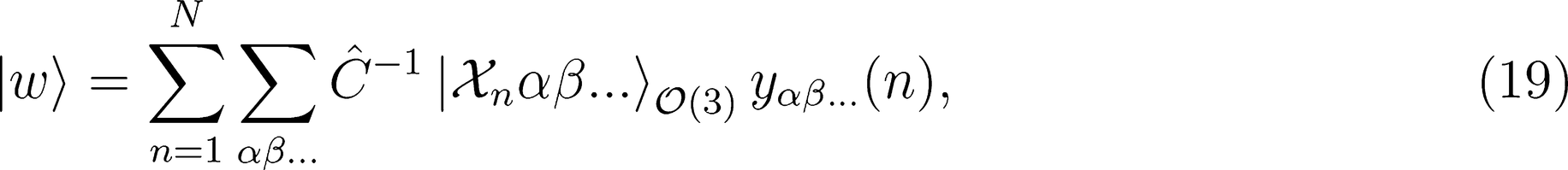
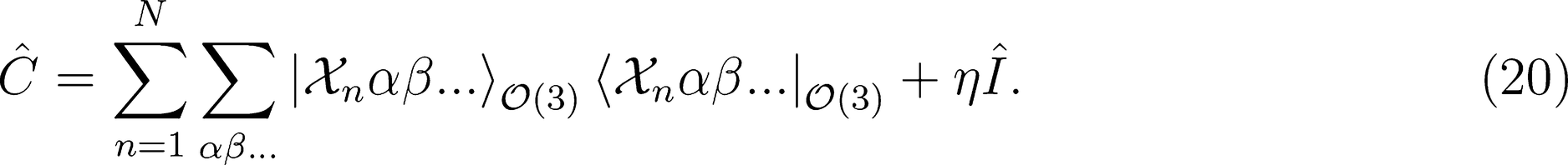
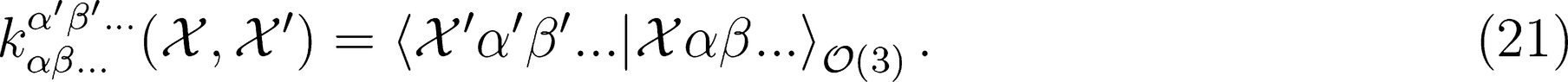
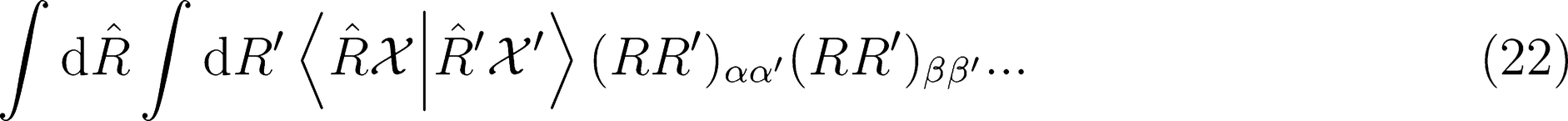

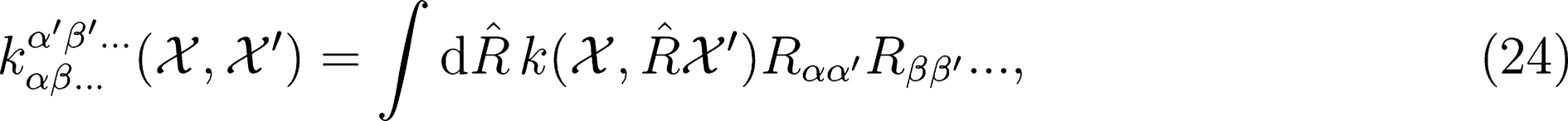
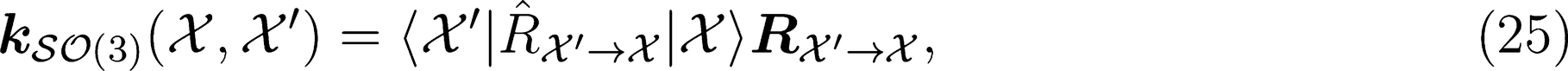
 . This strategy has been successfully used in the learning of electronic multipoles of organic molecules as well as for predicting optical response functions of water molecules in their liquid environments (
. This strategy has been successfully used in the learning of electronic multipoles of organic molecules as well as for predicting optical response functions of water molecules in their liquid environments (Figure 3
{kind=link}
Spherical Representation
 . Spherical harmonics form a complete basis set of the SO(3) group. In particular, each λ-component of the tensor spans an orthogonal subspace of dimension 2λ + 1. For instance, the 9 components of a rank-2 tensor separate out into a term (proportional to the trace) that transforms like a scalar, three terms that transform like
. Spherical harmonics form a complete basis set of the SO(3) group. In particular, each λ-component of the tensor spans an orthogonal subspace of dimension 2λ + 1. For instance, the 9 components of a rank-2 tensor separate out into a term (proportional to the trace) that transforms like a scalar, three terms that transform like  , and five terms that transform like
, and five terms that transform like  . When using a spherical representation, the kernel matrix is block diagonal, which greatly reduces the number of non-zero entries, and makes it possible to learn separately the different components. An additional advantage is that the possible symmetry of the tensor can be naturally incorporated by retaining only the spherical components λ that have the same parity as the tensor rank r. For instance, the λ = 1 component of a symmetric rank-2 tensor vanishes identically, meaning that only the 6 surviving elements of the tensor need to be considered when doing the regression. Especially for high rank tensors, this property means that the number of components can be cut down significantly.
. When using a spherical representation, the kernel matrix is block diagonal, which greatly reduces the number of non-zero entries, and makes it possible to learn separately the different components. An additional advantage is that the possible symmetry of the tensor can be naturally incorporated by retaining only the spherical components λ that have the same parity as the tensor rank r. For instance, the λ = 1 component of a symmetric rank-2 tensor vanishes identically, meaning that only the 6 surviving elements of the tensor need to be considered when doing the regression. Especially for high rank tensors, this property means that the number of components can be cut down significantly.
 . Its symmetry-adapted counterpart, which is covariant in SO(3), is
. Its symmetry-adapted counterpart, which is covariant in SO(3), is 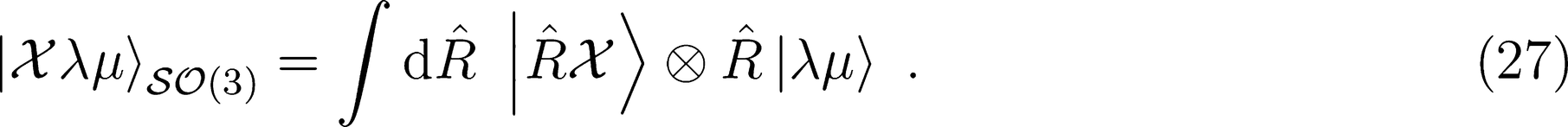
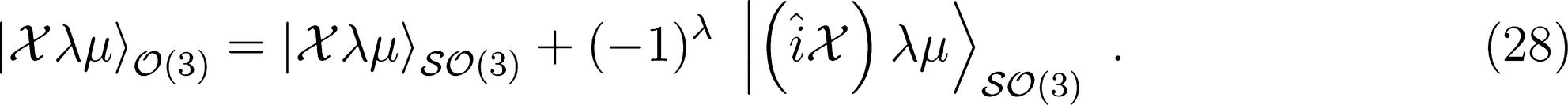
 :
: 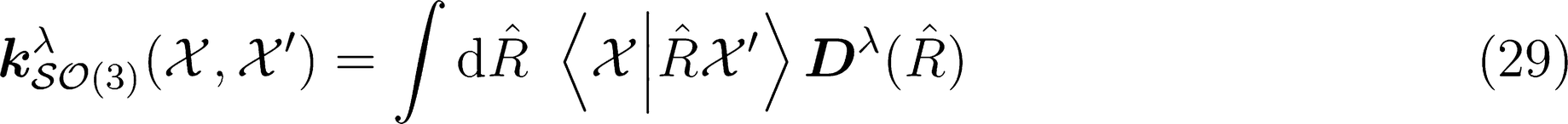
SOAP Representation
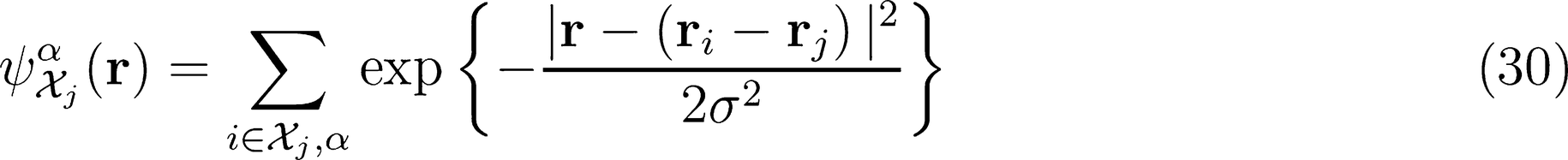

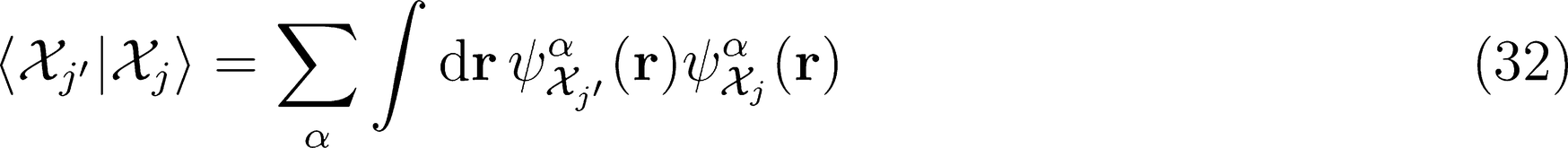
λ-SOAP(1) Representation
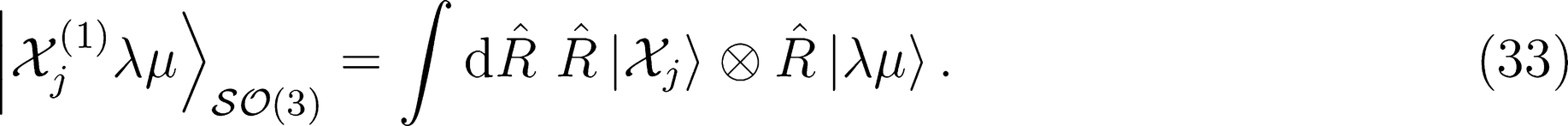
 can be understood as a rotational average of the environmental density which is rigidly attached to a spherical harmonic of order λ,
can be understood as a rotational average of the environmental density which is rigidly attached to a spherical harmonic of order λ, 
 on a basis of spherical harmonics, in which the integral over rotations can be performed analytically,
on a basis of spherical harmonics, in which the integral over rotations can be performed analytically, 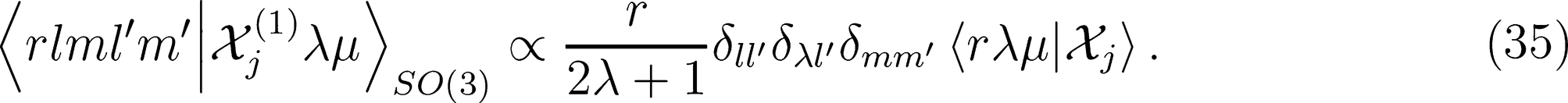
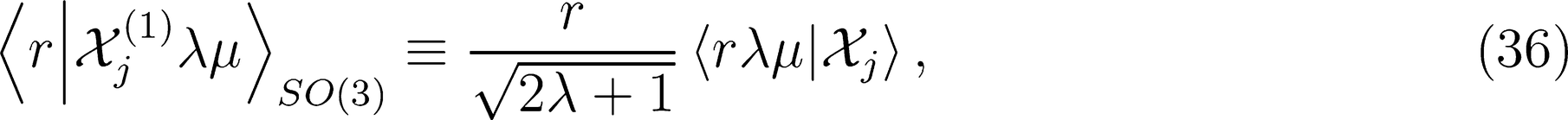
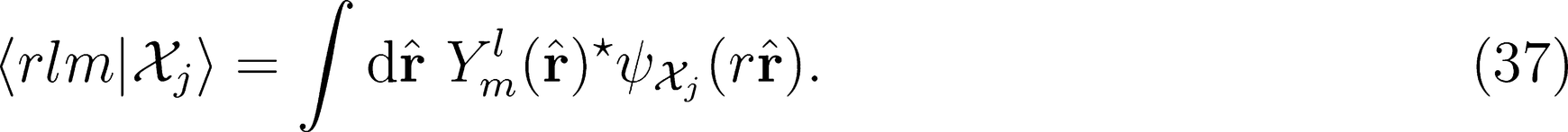
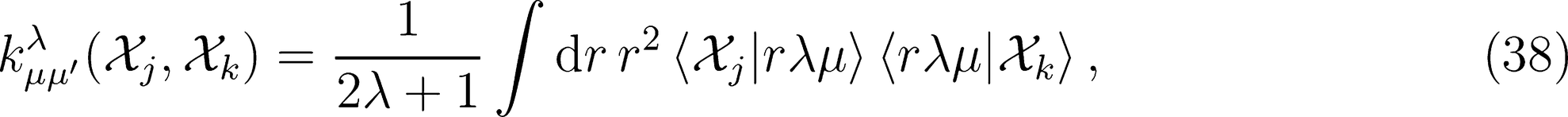
 :
: 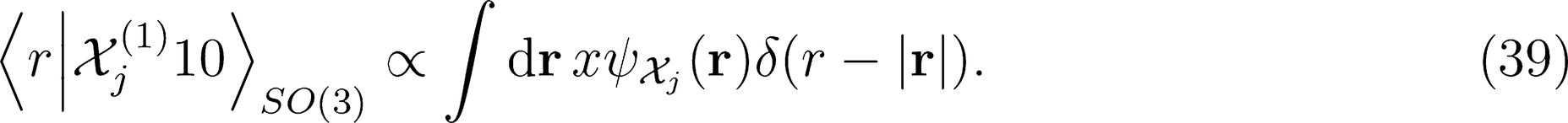
λ-SOAP(2) Representation
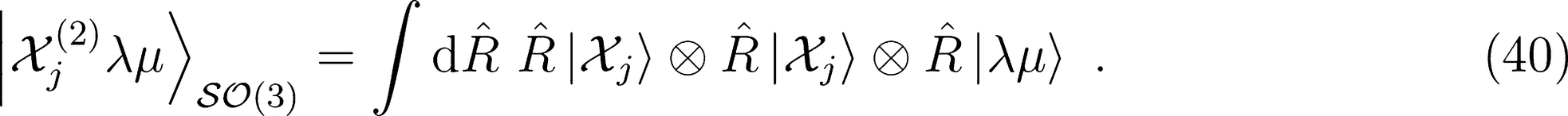
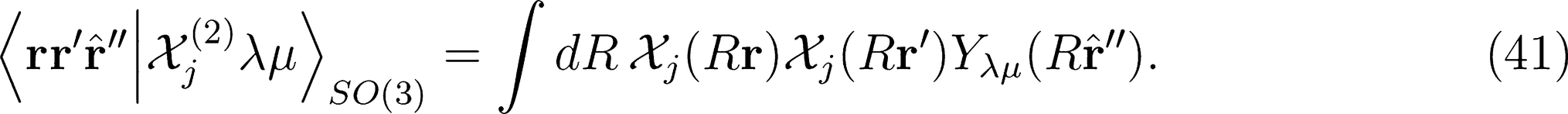
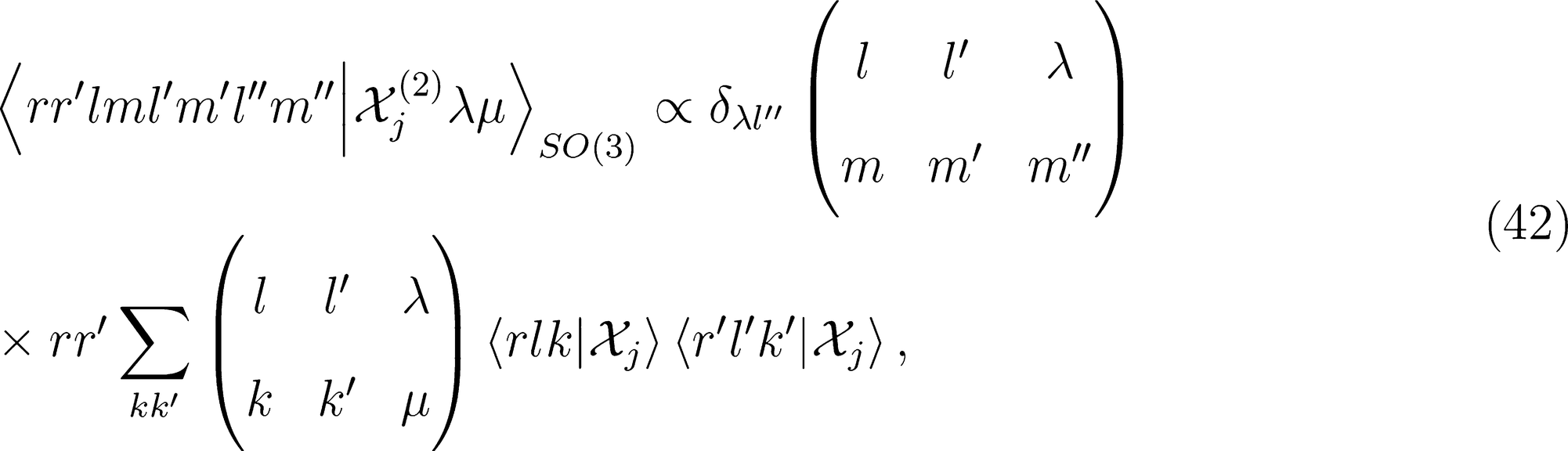
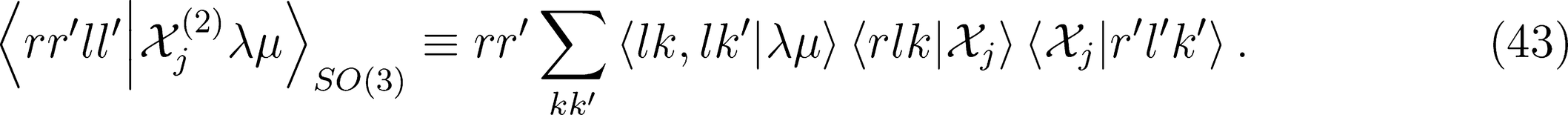
 that is needed for λ-SOAP(2) linear regression. Note that 〈lk, lk'|λµ〉 is zero unless k + k′ = µ, that the indices l, l' and λ must satisfy the inequality |l − l'| ≤ λ ≤ l + l' and that the representation is invariant under transposition of r and r'.
that is needed for λ-SOAP(2) linear regression. Note that 〈lk, lk'|λµ〉 is zero unless k + k′ = µ, that the indices l, l' and λ must satisfy the inequality |l − l'| ≤ λ ≤ l + l' and that the representation is invariant under transposition of r and r'.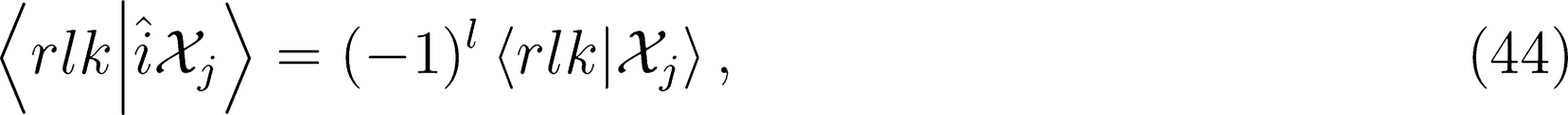
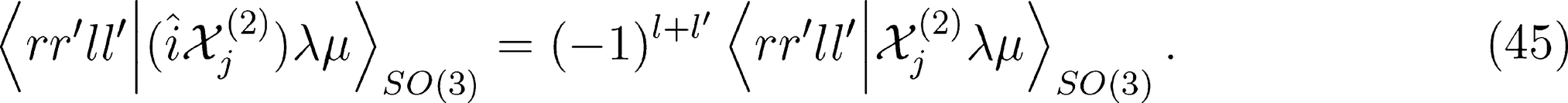
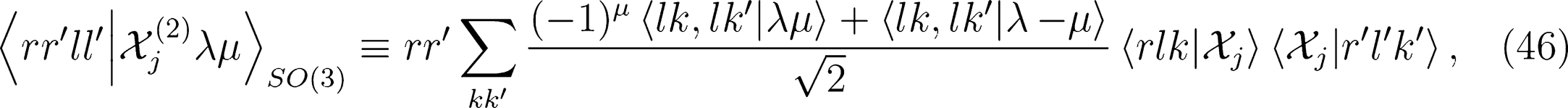
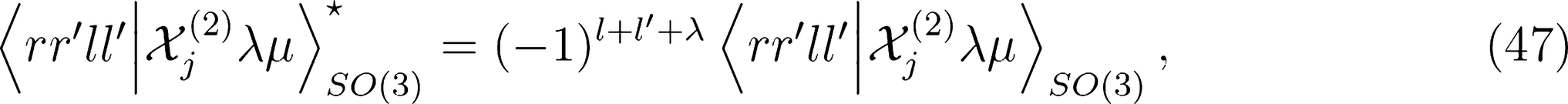
 for ν < 0 and |λ0〉 for ν = 0). One can therefore discard all imaginary components of the representation to enforce inversion invariance.
for ν < 0 and |λ0〉 for ν = 0). One can therefore discard all imaginary components of the representation to enforce inversion invariance.Non-linearity
 kets would require re-projecting the product onto the irreducible representations of the group, which would be as cumbersome as increasing the body order exponent ν. One obvious solution to this problem is to multiply the spherical kernel of order λ by its scalar and rotationally invariant counterpart, which can then be raised to an integer power ζ without breaking the tensorial nature of the kernel. For any generic order ν and ν′ in structural correlations, this procedure consists in considering the tensor product
kets would require re-projecting the product onto the irreducible representations of the group, which would be as cumbersome as increasing the body order exponent ν. One obvious solution to this problem is to multiply the spherical kernel of order λ by its scalar and rotationally invariant counterpart, which can then be raised to an integer power ζ without breaking the tensorial nature of the kernel. For any generic order ν and ν′ in structural correlations, this procedure consists in considering the tensor product 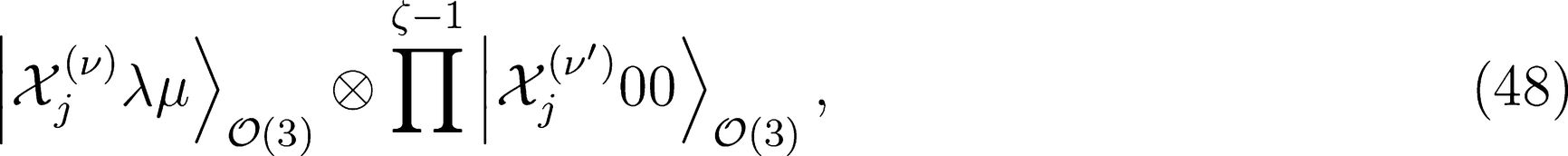
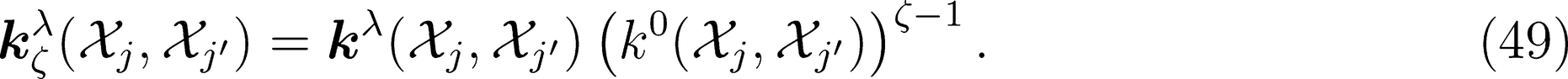
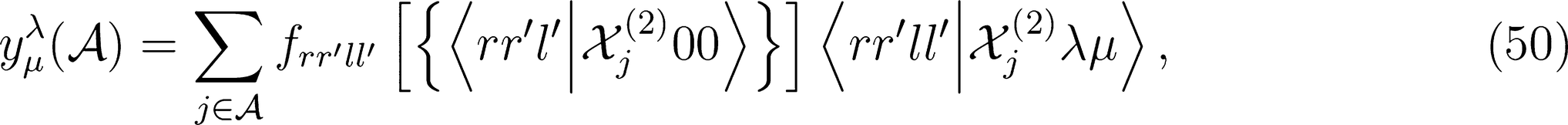
Implementation
 . In turn, computing this quantity requires the evaluation of the density expansion coefficients 〈rlm|Xj〉. In practice, the continuous variable r can be replaced by an expansion over a discrete set of orthogonal radial functions Rn(r) that are defined within the spherical cutoff rcut. For this reason, we will refer, from now on, to the density expansion coefficients as 〈nlm|Xj〉.
. In turn, computing this quantity requires the evaluation of the density expansion coefficients 〈rlm|Xj〉. In practice, the continuous variable r can be replaced by an expansion over a discrete set of orthogonal radial functions Rn(r) that are defined within the spherical cutoff rcut. For this reason, we will refer, from now on, to the density expansion coefficients as 〈nlm|Xj〉.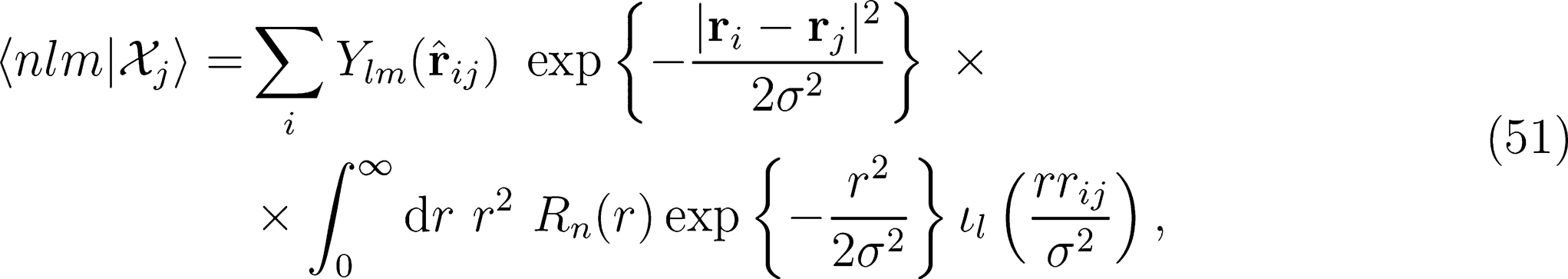
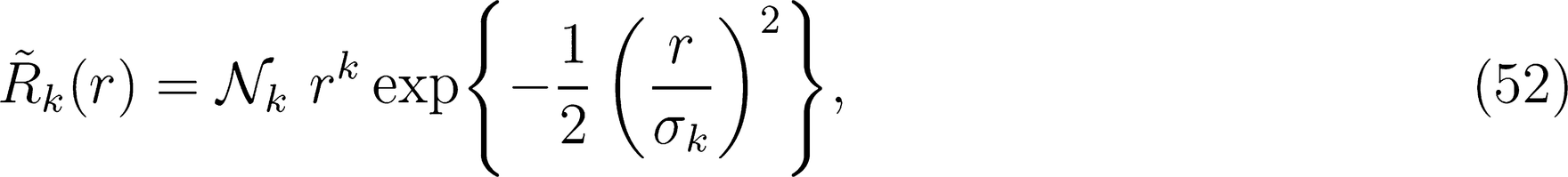
 . The set of Gaussian widths {σk} can be chosen to effectively span the radial interval involved in the environment definition. For instance, one can take
. The set of Gaussian widths {σk} can be chosen to effectively span the radial interval involved in the environment definition. For instance, one can take  , obtaining functions that have equally-spaced peaks between 0 and rcut. The explicit functional form of the primitive radial integrals is
, obtaining functions that have equally-spaced peaks between 0 and rcut. The explicit functional form of the primitive radial integrals is 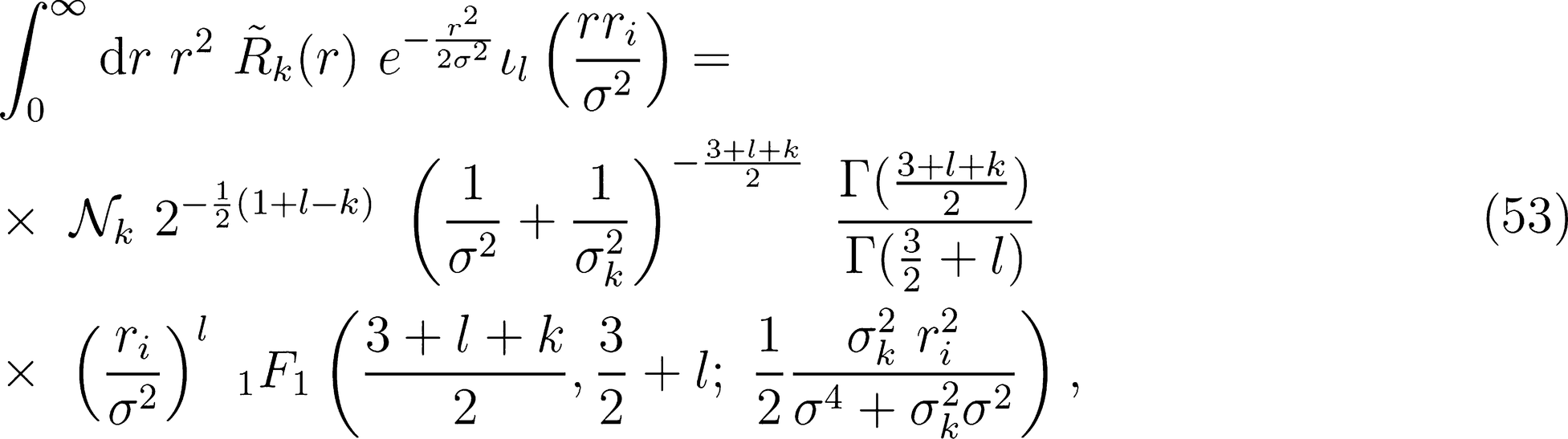
Examples
Dielectric Response Series
 . By construction this tensor is symmetric, meaning that it can be decomposed into two spherical components, 3 of λ = 1 symmetry and 7 of λ = 3 symmetry. The total number of components to be learned is thus 10, consistently with the number of non-equivalent components of the Cartesian tensor. The dataset is made of 1000 configurations, of which 800 are randomly selected to train the regression model, while the remaining 200 are using to test the prediction performances. λ-SOAP(2) kernels that are adapted to SO(3) and O(3) group symmetry were constructed using a Gaussian smearing of σ = 0.3 Å and an environment cutoff of rcut = 4.0 Å.
. By construction this tensor is symmetric, meaning that it can be decomposed into two spherical components, 3 of λ = 1 symmetry and 7 of λ = 3 symmetry. The total number of components to be learned is thus 10, consistently with the number of non-equivalent components of the Cartesian tensor. The dataset is made of 1000 configurations, of which 800 are randomly selected to train the regression model, while the remaining 200 are using to test the prediction performances. λ-SOAP(2) kernels that are adapted to SO(3) and O(3) group symmetry were constructed using a Gaussian smearing of σ = 0.3 Å and an environment cutoff of rcut = 4.0 Å.Figure 4
{kind=link}
Electronic Charge Densities

Figure 5
{kind=link}
Conclusions
Acknowledgments
The Authors acknowledge support by the European Research Council under the European Union’s Horizon 2020 research and innovation programme (Grant Agreement No. 677013-HBMAP).
References
This chapter references 36 other publications.
- 1Williams, C. K. I.; Rasmussen, C. E. Gaussian Processes for Machine Learning; MIT Press, 2006.Google ScholarThere is no corresponding record for this reference.
- 2Bartók A. P. Payne M. C. Kondor R. Csányi G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons Phys. Rev. Lett. 2010 104 136403Google Scholar2Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the ElectronsBartok, Albert P.; Payne, Mike C.; Kondor, Risi; Csanyi, GaborPhysical Review Letters (2010), 104 (13), 136403/1-136403/4CODEN: PRLTAO; ISSN:0031-9007. (American Physical Society)We introduce a class of interat. potential models that can be automatically generated from data consisting of the energies and forces experienced by atoms, as derived from quantum mech. calcns. The models do not have a fixed functional form and hence are capable of modeling complex potential energy landscapes. They are systematically improvable with more data. We apply the method to bulk crystals, and test it by calcg. properties at high temps. Using the interat. potential to generate the long mol. dynamics trajectories required for such calcns. saves orders of magnitude in computational cost.
- 3Jain A. Ong S. P. Hautier G. Chen W. Richards W. D. Dacek S. Cholia S. Gunter D. Skinner D. Ceder G. Persson K. A. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation APL Mater. 2013 1 011002Google Scholar3Commentary: The Materials Project: A materials genome approach to accelerating materials innovationJain, Anubhav; Ong, Shyue Ping; Hautier, Geoffroy; Chen, Wei; Richards, William Davidson; Dacek, Stephen; Cholia, Shreyas; Gunter, Dan; Skinner, David; Ceder, Gerbrand; Persson, Kristin A.APL Materials (2013), 1 (1), 011002/1-011002/11CODEN: AMPADS; ISSN:2166-532X. (American Institute of Physics)Accelerating the discovery of advanced materials is essential for human welfare and sustainable, clean energy. In this paper, we introduce the Materials Project (www.materialsproject.org), a core program of the Materials Genome Initiative that uses high-throughput computing to uncover the properties of all known inorg. materials. This open dataset can be accessed through multiple channels for both interactive exploration and data mining. The Materials Project also seeks to create open-source platforms for developing robust, sophisticated materials analyses. Future efforts will enable users to perform rapid-prototyping'' of new materials in silico, and provide researchers with new avenues for cost-effective, data-driven materials design. (c) 2013 American Institute of Physics.
- 4Calderon C. E. Plata J. J. Toher C. Oses C. Levy O. Fornari M. Natan A. Mehl M. J. Hart G. Nardelli M. B. Curtarolo S. The AFLOW standard for high-throughput materials science calculations Comput. Mater. Sci. 2015 108 233 238Google Scholar4The AFLOW standard for high-throughput materials science calculationsCalderon, Camilo E.; Plata, Jose J.; Toher, Cormac; Oses, Corey; Levy, Ohad; Fornari, Marco; Natan, Amir; Mehl, Michael J.; Hart, Gus; Buongiorno Nardelli, Marco; Curtarolo, StefanoComputational Materials Science (2015), 108 (Part_A), 233-238CODEN: CMMSEM; ISSN:0927-0256. (Elsevier B.V.)The Automatic-Flow (AFLOW) std. for the high-throughput construction of materials science electronic structure databases is described. Electronic structure calcns. of solid state materials depend on a large no. of parameters which must be understood by researchers, and must be reported by originators to ensure reproducibility and enable collaborative database expansion. We therefore describe std. parameter values for k-point grid d., basis set plane wave kinetic energy cut-off, exchange-correlation functionals, pseudopotentials, DFT+U parameters, and convergence criteria used in AFLOW calcns.
- 5Ward L. Wolverton C. Atomistic calculations and materials informatics: A review Curr. Opin. Solid State Mater. Sci. 2017 21 167 176Google Scholar5Atomistic calculations and materials informatics: A reviewWard, Logan; Wolverton, ChrisCurrent Opinion in Solid State & Materials Science (2017), 21 (3), 167-176CODEN: COSSFX; ISSN:1359-0286. (Elsevier Ltd.)A review. In recent years, there has been a large effort in the materials science community to employ materials informatics to accelerate materials discovery or to develop new understanding of materials behavior. Materials informatics methods utilize machine learning techniques to ext. new knowledge or predictive models out of existing materials data. In this review, we discuss major advances in the intersection between data science and atom-scale calcns. with a particular focus on studies of solid-state, inorg. materials. The examples discussed in this review cover methods for accelerating the calcn. of computationally-expensive properties, identifying promising regions for materials discovery based on existing data, and extg. chem. intuition automatically from datasets. We also identify key issues in this field, such as limited distribution of software necessary to utilize these techniques, and opportunities for areas of research that would help lead to the wider adoption of materials informatics in the atomistic calcns. community.
- 6Li Z. Kermode J. R. De Vita A. Molecular Dynamics with On-the-Fly Machine Learning of Quantum-Mechanical Forces Phys. Rev. Lett. 2015 114 096405Google Scholar6Molecular dynamics with on-the-fly machine learning of quantum-mechanical forcesLi, Zhenwei; Kermode, James R.; De Vita, AlessandroPhysical Review Letters (2015), 114 (9), 096405/1-096405/5CODEN: PRLTAO; ISSN:0031-9007. (American Physical Society)We present a mol. dynamics scheme which combines first-principles and machine-learning (ML) techniques in a single information-efficient approach. Forces on atoms are either predicted by Bayesian inference or, if necessary, computed by on-the-fly quantum-mech. (QM) calcns. and added to a growing ML database, whose completeness is, thus, never required. As a result, the scheme is accurate and general, while progressively fewer QM calls are needed when a new chem. process is encountered for the second and subsequent times, as demonstrated by tests on cryst. and molten silicon.
- 7Glielmo A. Sollich P. De Vita A. Accurate interatomic force fields via machine learning with covariant kernels Phys. Rev. B 2017 95 214302Google Scholar7Accurate interatomic force fields via machine learning with covariant kernelsGlielmo, Aldo; Sollich, Peter; De Vita, AlessandroPhysical Review B (2017), 95 (21), 214302/1-214302/10CODEN: PRBHB7; ISSN:2469-9969. (American Physical Society)A review. We present a novel scheme to accurately predict at. forces as vector quantities, rather than sets of scalar components, by Gaussian process (GP) regression. This is based on matrix-valued kernel functions, on which we impose the requirements that the predicted force rotates with the target configuration and is independent of any rotations applied to the configuration database entries. We show that such covariant GP kernels can be obtained by integration over the elements of the rotation group SO(d) for the relevant dimensionality d. Remarkably, in specific cases the integration can be carried out anal. and yields a conservative force field that can be recast into a pair interaction form. Finally, we show that restricting the integration to a summation over the elements of a finite point group relevant to the target system is sufficient to recover an accurate GP. The accuracy of our kernels in predicting quantum-mech. forces in real materials is investigated by tests on pure and defective Ni, Fe, and Si cryst. systems.
- 8Glielmo A. Zeni C. De Vita A. Efficient nonparametric n-body force fields from machine learning Phys. Rev. B 2018 97 184307Google Scholar8Efficient nonparametric n-body force fields from machine learningGlielmo, Aldo; Zeni, Claudio; De Vita, AlessandroPhysical Review B (2018), 97 (18), 184307CODEN: PRBHB7; ISSN:2469-9969. (American Physical Society)A review. We provide a definition and explicit expressions for n-body Gaussian process (GP) kernels, which can learn any interat. interaction occurring in a phys. system, up to n-body contributions, for any value of n. The series is complete, as it can be shown that the "universal approximator" squared exponential kernel can be written as a sum of n-body kernels. These recipes enable the choice of optimally efficient force models for each target system, as confirmed by extensive testing on various materials. We furthermore describe how the n-body kernels can be "mapped" on equiv. representations that provide database-size-independent predictions and are thus crucially more efficient. We explicitly carry out this mapping procedure for the first nontrivial (three-body) kernel of the series, and we show that this reproduces the GP-predicted forces with meV/Å accuracy while being orders of magnitude faster. These results pave the way to using novel force models (here named "M-FFs") that are computationally as fast as their corresponding std. parametrized n-body force fields, while retaining the nonparametric character, the ease of training and validation, and the accuracy of the best recently proposed machine-learning potentials.
- 9Yuan Y. Mills M. J. Popelier P. L. Multipolar electrostatics based on the Kriging machine learning method: an application to serine J. Mol. Model. 2014 20 2172Google Scholar9Multipolar electrostatics based on the Kriging machine learning method: an application to serineYuan Yongna; Mills Matthew J L; Popelier Paul L AJournal of molecular modeling (2014), 20 (4), 2172 ISSN:.A multipolar, polarizable electrostatic method for future use in a novel force field is described. Quantum Chemical Topology (QCT) is used to partition the electron density of a chemical system into atoms, then the machine learning method Kriging is used to build models that relate the multipole moments of the atoms to the positions of their surrounding nuclei. The pilot system serine is used to study both the influence of the level of theory and the set of data generator methods used. The latter consists of: (i) sampling of protein structures deposited in the Protein Data Bank (PDB), or (ii) normal mode distortion along either (a) Cartesian coordinates, or (b) redundant internal coordinates. Wavefunctions for the sampled geometries were obtained at the HF/6-31G(d,p), B3LYP/apc-1, and MP2/cc-pVDZ levels of theory, prior to calculation of the atomic multipole moments by volume integration. The average absolute error (over an independent test set of conformations) in the total atom-atom electrostatic interaction energy of serine, using Kriging models built with the three data generator methods is 11.3 kJ mol-1 (PDB), 8.2 kJ mol-1 (Cartesian distortion), and 10.1 kJ mol-1 (redundant internal distortion) at the HF/6-31G(d,p) level. At the B3LYP/apc-1 level, the respective errors are 7.7 kJ mol-1, 6.7 kJ mol-1, and 4.9 kJ mol-1, while at the MP2/cc-pVDZ level they are 6.5 kJ mol-1, 5.3 kJ mol-1, and 4.0 kJ mol-1. The ranges of geometries generated by the redundant internal coordinate distortion and by extraction from the PDB are much wider than the range generated by Cartesian distortion. The atomic multipole moment and electrostatic interaction energy predictions for the B3LYP/apc-1 and MP2/cc-pVDZ levels are similar, and both are better than the corresponding predictions at the HF/6-31G(d,p) level.
- 10Bereau T. Andrienko D. von Lilienfeld O. A. Transferable Atomic Multipole Machine Learning Models for Small Organic Molecules J. Chem. Theory Comput. 2015 11 3225 3233Google Scholar10Transferable Atomic Multipole Machine Learning Models for Small Organic MoleculesBereau, Tristan; Andrienko, Denis; von Lilienfeld, O. AnatoleJournal of Chemical Theory and Computation (2015), 11 (7), 3225-3233CODEN: JCTCCE; ISSN:1549-9618. (American Chemical Society)Accurate representation of the mol. electrostatic potential, which is often expanded in distributed multipole moments, is crucial for an efficient evaluation of intermol. interactions. Here we introduce a machine learning model for multipole coeffs. of atom types H, C, O, N, S, F, and Cl in any mol. conformation. The model is trained on quantum-chem. results for atoms in varying chem. environments drawn from thousands of org. mols. Multipoles in systems with neutral, cationic, and anionic mol. charge states are treated with individual models. The models' predictive accuracy and applicability are illustrated by evaluating intermol. interaction energies of nearly 1,000 dimers and the cohesive energy of the benzene crystal.
- 11Bereau T. DiStasio R. A. Tkatchenko A. von Lilienfeld O. A. Non-covalent interactions across organic and biological subsets of chemical space: Physics-based potentials parametrized from machine learning J. Chem. Phys. 2018 148 241706Google Scholar11Non-covalent interactions across organic and biological subsets of chemical space: Physics-based potentials parametrized from machine learningBereau, Tristan; Di Stasio, Robert A.; Tkatchenko, Alexandre; von Lilienfeld, O. AnatoleJournal of Chemical Physics (2018), 148 (24), 241706/1-241706/14CODEN: JCPSA6; ISSN:0021-9606. (American Institute of Physics)Classical intermol. potentials typically require an extensive parametrization procedure for any new compd. considered. To do away with prior parametrization, we propose a combination of physics-based potentials with machine learning (ML), coined IPML, which is transferable across small neutral org. and biol. relevant mols. ML models provide on-the-fly predictions for environment-dependent local at. properties: electrostatic multipole coeffs. (significant error redn. compared to previously reported), the population and decay rate of valence at. densities, and polarizabilities across conformations and chem. compns. of H, C, N, and O atoms. These parameters enable accurate calcns. of intermol. contributions-electrostatics, charge penetration, repulsion, induction/polarization, and many-body dispersion. Unlike other potentials, this model is transferable in its ability to handle new mols. and conformations without explicit prior parametrization: All local at. properties are predicted from ML, leaving only eight global parameters-optimized once and for all across compds. We validate IPML on various gas-phase dimers at and away from equil. sepn., where we obtain mean abs. errors between 0.4 and 0.7 kcal/mol for several chem. and conformationally diverse datasets representative of non-covalent interactions in biol. relevant mols. We further focus on hydrogen-bonded complexes - essential but challenging due to their directional nature - where datasets of DNA base pairs and amino acids yield an extremely encouraging 1.4 kcal/mol error. Finally, and as a first look, we consider IPML for denser systems: water clusters, supramol. host-guest complexes, and the benzene crystal. (c) 2018 American Institute of Physics.
- 12Liang C. Tocci G. Wilkins D. M. Grisafi A. Roke S. Ceriotti M. Solvent fluctuations and nuclear quantum effects modulate the molecular hyperpolarizability of water Phys. Rev. B 2017 96 041407Google Scholar12Solvent fluctuations and nuclear quantum effects modulate the molecular hyperpolarizability of waterLiang, Chungwen; Tocci, Gabriele; Wilkins, David M.; Grisafi, Andrea; Roke, Sylvie; Ceriotti, MichelePhysical Review B (2017), 96 (4), 041407/1-041407/6CODEN: PRBHB7; ISSN:2469-9969. (American Physical Society)Second-harmonic scattering (SHS) expts. provide a unique approach to probe noncentrosym. environments in aq. media, from bulk solns. to interfaces, living cells, and tissue. A central assumption made in analyzing SHS expts. is that each mol. scatters light according to a const. mol. hyperpolarizability tensor β(2). Here, we investigate the dependence of the mol. hyperpolarizability of water on its environment and internal geometric distortions, in order to test the hypothesis of const. β(2). We use quantum chem. calcns. of the hyperpolarizability of a mol. embedded in point-charge environments obtained from simulations of bulk water. We demonstrate that both the heterogeneity of the solvent configurations and the quantum mech. fluctuations of the mol. geometry introduce large variations in the nonlinear optical response of water. This finding has the potential to change the way SHS expts. are interpreted: In particular, isotopic differences between H2O and D2O could explain recent SHS observations. Finally, we show that a machine-learning framework can predict accurately the fluctuations of the mol. hyperpolarizability. This model accounts for the microscopic inhomogeneity of the solvent and represents a step towards quant. modeling of SHS expts.
- 13Grisafi A. Wilkins D. M. Csányi G. Ceriotti M. Symmetry-Adapted Machine Learning for Tensorial Properties of Atomistic Systems Phys. Rev. Lett. 2018 120 036002Google Scholar13Symmetry-Adapted Machine Learning for Tensorial Properties of Atomistic SystemsGrisafi, Andrea; Wilkins, David M.; Csanyi, Gabor; Ceriotti, MichelePhysical Review Letters (2018), 120 (3), 036002CODEN: PRLTAO; ISSN:1079-7114. (American Physical Society)A review. Statistical learning methods show great promise in providing an accurate prediction of materials and mol. properties, while minimizing the need for computationally demanding electronic structure calcns. The accuracy and transferability of these models are increased significantly by encoding into the learning procedure the fundamental symmetries of rotational and permutational invariance of scalar properties. However, the prediction of tensorial properties requires that the model respects the appropriate geometric transformations, rather than invariance, when the ref. frame is rotated. We introduce a formalism that extends existing schemes and makes it possible to perform machine learning of tensorial properties of arbitrary rank, and for general mol. geometries. To demonstrate it, we derive a tensor kernel adapted to rotational symmetry, which is the natural generalization of the smooth overlap of at. positions kernel commonly used for the prediction of scalar properties at the at. scale. The performance and generality of the approach is demonstrated by learning the instantaneous response to an external elec. field of water oligomers of increasing complexity, from the isolated mol. to the condensed phase.
- 14Wilkins D. M. Grisafi A. Yang Y. Lao K. U. DiStasio R. A. Ceriotti M. Accurate molecular polarizabilities with coupled cluster theory and machine learning Proc. Natl. Acad. Sci. 2019 116 3401 3406Google Scholar14Accurate molecular polarizabilities with coupled cluster theory and machine learningWilkins, David M.; Grisafi, Andrea; Yang, Yang; Lao, Ka Un; Di Stasio, Robert A., Jr.; Ceriotti, MicheleProceedings of the National Academy of Sciences of the United States of America (2019), 116 (9), 3401-3406CODEN: PNASA6; ISSN:0027-8424. (National Academy of Sciences)The mol. dipole polarizability describes the tendency of a mol. to change its dipole moment in response to an applied elec. field. This quantity governs key intra- and intermol. interactions, such as induction and dispersion; plays a vital role in detg. the spectroscopic signatures of mols.; and is an essential ingredient in polarizable force fields. Compared with other ground-state properties, an accurate prediction of the mol. polarizability is considerably more difficult, as this response quantity is quite sensitive to the underlying electronic structure description. In this work, we present highly accurate quantum mech. calcns. of the static dipole polarizability tensors of 7,211 small org. mols. computed using linear response coupled cluster singles and doubles theory (LR-CCSD). Using a symmetry-adapted machine-learning approach, we demonstrate that it is possible to predict the LR-CCSD mol. polarizabilities of these small mols. with an error that is an order of magnitude smaller than that of hybrid d. functional theory (DFT) at a negligible computational cost. The resultant model is robust and transferable, yielding mol. polarizabilities for a diverse set of 52 larger mols. (including challenging conjugated systems, carbohydrates, small drugs, amino acids, nucleobases, and hydrocarbon isomers) at an accuracy that exceeds that of hybrid DFT. The atom-centered decompn. implicit in our machine-learning approach offers some insight into the shortcomings of DFT in the prediction of this fundamental quantity of interest.
- 15Christensen A. S. Faber F. A. von Lilienfeld O. A. Operators in quantum machine learning: Response properties in chemical space J. Chem. Phys. 2019 150 064105Google Scholar15Operators in quantum machine learning: Response properties in chemical spaceChristensen, Anders S.; Faber, Felix A.; von Lilienfeld, O. AnatoleJournal of Chemical Physics (2019), 150 (6), 064105/1-064105/12CODEN: JCPSA6; ISSN:0021-9606. (American Institute of Physics)The role of response operators is well established in quantum mechanics. We investigate their use for universal quantum machine learning models of response properties in mols. After introducing a theor. basis, we present and discuss numerical evidence based on measuring the potential energy's response with respect to at. displacement and to elec. fields. Prediction errors for corresponding properties, at. forces, and dipole moments improve in a systematic fashion with training set size and reach high accuracy for small training sets. Prediction of normal modes and IR-spectra of some small mols. demonstrates the usefulness of this approach for chem. (c) 2019 American Institute of Physics.
- 16Brockherde F. Vogt L. Li L. Tuckerman M. E. Burke K. Mu¨ller K.-R. Bypassing the Kohn-Sham equations with machine learning Nat. Commun. 2017 8 872Google Scholar16Bypassing the Kohn-Sham equations with machine learningBrockherde Felix; Muller Klaus-Robert; Brockherde Felix; Vogt Leslie; Tuckerman Mark E; Li Li; Burke Kieron; Tuckerman Mark E; Tuckerman Mark E; Burke Kieron; Muller Klaus-Robert; Muller Klaus-RobertNature communications (2017), 8 (1), 872 ISSN:.Last year, at least 30,000 scientific papers used the Kohn-Sham scheme of density functional theory to solve electronic structure problems in a wide variety of scientific fields. Machine learning holds the promise of learning the energy functional via examples, bypassing the need to solve the Kohn-Sham equations. This should yield substantial savings in computer time, allowing larger systems and/or longer time-scales to be tackled, but attempts to machine-learn this functional have been limited by the need to find its derivative. The present work overcomes this difficulty by directly learning the density-potential and energy-density maps for test systems and various molecules. We perform the first molecular dynamics simulation with a machine-learned density functional on malonaldehyde and are able to capture the intramolecular proton transfer process. Learning density models now allows the construction of accurate density functionals for realistic molecular systems.Machine learning allows electronic structure calculations to access larger system sizes and, in dynamical simulations, longer time scales. Here, the authors perform such a simulation using a machine-learned density functional that avoids direct solution of the Kohn-Sham equations.
- 17Alred J. M. Bets K. V. Xie Y. Yakobson B. I. Machine learning electron density in sulfur crosslinked carbon nanotubes Compos. Sci. Technol. 2018 166 3 9Google Scholar17Machine learning electron density in sulfur crosslinked carbon nanotubesAlred, John M.; Bets, Ksenia V.; Xie, Yu; Yakobson, Boris I.Composites Science and Technology (2018), 166 (), 3-9CODEN: CSTCEH; ISSN:0266-3538. (Elsevier Ltd.)Mech. strengthening of composite materials that include carbon nanotubes (CNT) requires strong inter-bonding to achieve significant CNT-CNT or CNT-matrix load transfer. The same principle is applicable to the improvement of CNT bundles and calls for covalent crosslinks between individual tubes. In this work, sulfur crosslinks are studied using a combination of d. functional theory (DFT) and classical mol. dynamics (MD). Atomic chains of at least two sulfur atoms or more are shown to be stable between both zigzag and armchair CNTs. All types of crosslinked CNTs exhibit significantly improved load transfer. Moreover, sulfur crosslinks show evidence of a cooperative self-healing mechanism allowing for links to rebond once broken leading to sustained load transfer under shear loading. Addnl., a general approach for utilizing machine learning for assessing the ground state electron d. is developed and applied to these sulfur crosslinked CNTs.
- 18Grisafi A. Fabrizio A. Meyer B. Wilkins D. M. Corminboeuf C. Ceriotti M. Transferable Machine-Learning Model of the Electron Density ACS Cent. Sci. 2019 5 57 64Google Scholar18Transferable Machine-Learning Model of the Electron DensityGrisafi, Andrea; Fabrizio, Alberto; Meyer, Benjamin; Wilkins, David M.; Corminboeuf, Clemence; Ceriotti, MicheleACS Central Science (2019), 5 (1), 57-64CODEN: ACSCII; ISSN:2374-7951. (American Chemical Society)The electronic charge d. plays a central role in detg. the behavior of matter at the at. scale, but its computational evaluation requires demanding electronic-structure calcns. We introduce an atom-centered, symmetry-adapted framework to machine-learn the valence charge d. based on a small no. of ref. calcns. The model is highly transferable, meaning it can be trained on electronic-structure data of small mols. and used to predict the charge d. of larger compds. with low, linear-scaling cost. Applications are shown for various hydrocarbon mols. of increasing complexity and flexibility, and demonstrate the accuracy of the model when predicting the d. on octane and octatetraene after training exclusively on butane and butadiene. This transferable, data-driven model can be used to interpret expts., accelerate electronic structure calcns., and compute electrostatic interactions in mols. and condensed-phase systems.
- 19Braams B. J. Bowman J. M. Permutationally invariant potential energy surfaces in high dimensionality Int. Rev. Phys. Chem. 2009 28 577 606Google Scholar19Permutationally invariant potential energy surfaces in high dimensionalityBraams, Bastiaan J.; Bowman, Joel M.International Reviews in Physical Chemistry (2009), 28 (4), 577-606CODEN: IRPCDL; ISSN:0144-235X. (Taylor & Francis Ltd.)We review recent progress in developing potential energy and dipole moment surfaces for polyat. systems with up to 10 atoms. The emphasis is on global linear least squares fitting of tens of thousands of scattered ab initio energies using a special, compact fitting basis of permutationally invariant polynomials in Morse-type variables of all the internuclear distances. The computational mathematics underlying this approach is reviewed first, followed by a review of the practical approaches used to obtain the data for the fits. A straightforward symmetrization approach is also given, mainly for pedagogical purposes. The methods are illustrated for potential energy surfaces for CH+5, (H2O)2 and CH3CHO. The relationship of this approach to other approaches is also briefly reviewed.
- 20Behler J. Parrinello M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces Phys. Rev. Lett. 2007 98 146401Google Scholar20Generalized Neural-Network Representation of High-Dimensional Potential-Energy SurfacesBehler, Jorg; Parrinello, MichelePhysical Review Letters (2007), 98 (14), 146401/1-146401/4CODEN: PRLTAO; ISSN:0031-9007. (American Physical Society)The accurate description of chem. processes often requires the use of computationally demanding methods like d.-functional theory (DFT), making long simulations of large systems unfeasible. In this Letter we introduce a new kind of neural-network representation of DFT potential-energy surfaces, which provides the energy and forces as a function of all at. positions in systems of arbitrary size and is several orders of magnitude faster than DFT. The high accuracy of the method is demonstrated for bulk silicon and compared with empirical potentials and DFT. The method is general and can be applied to all types of periodic and nonperiodic systems.
- 21Bartók A. P. Kondor R. Csányi G. On representing chemical environments Phys. Rev. B 2013 87 184115Google Scholar21On representing chemical environmentsBartok, Albert P.; Kondor, Risi; Csanyi, GaborPhysical Review B: Condensed Matter and Materials Physics (2013), 87 (18), 184115/1-184115/16CODEN: PRBMDO; ISSN:1098-0121. (American Physical Society)We review some recently published methods to represent at. neighborhood environments, and analyze their relative merits in terms of their faithfulness and suitability for fitting potential energy surfaces. The crucial properties that such representations (sometimes called descriptors) must have are differentiability with respect to moving the atoms and invariance to the basic symmetries of physics: rotation, reflection, translation, and permutation of atoms of the same species. We demonstrate that certain widely used descriptors that initially look quite different are specific cases of a general approach, in which a finite set of basis functions with increasing angular wave nos. are used to expand the at. neighborhood d. function. Using the example system of small clusters, we quant. show that this expansion needs to be carried to higher and higher wave nos. as the no. of neighbors increases in order to obtain a faithful representation, and that variants of the descriptors converge at very different rates. We also propose an altogether different approach, called Smooth Overlap of Atomic Positions, that sidesteps these difficulties by directly defining the similarity between any two neighborhood environments, and show that it is still closely connected to the invariant descriptors. We test the performance of the various representations by fitting models to the potential energy surface of small silicon clusters and the bulk crystal.
- 22Shapeev A. Moment Tensor Potentials: A Class of Systematically Improvable Interatomic Potentials Multiscale Model. Sim. 2016 14 1153 1173Google ScholarThere is no corresponding record for this reference.
- 23Zhang L. Han J. Wang H. Car R. E W. Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics Phys. Rev. Lett. 2018 120 143001Google Scholar23Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum MechanicsZhang, Linfeng; Han, Jiequn; Wang, Han; Car, Roberto; E, WeinanPhysical Review Letters (2018), 120 (14), 143001CODEN: PRLTAO; ISSN:1079-7114. (American Physical Society)We introduce a scheme for mol. simulations, the deep potential mol. dynamics (DPMD) method, based on a many-body potential and interat. forces generated by a carefully crafted deep neural network trained with ab initio data. The neural network model preserves all the natural symmetries in the problem. It is first-principles based in the sense that there are no ad hoc components aside from the network model. We show that the proposed scheme provides an efficient and accurate protocol in a variety of systems, including bulk materials and mols. In all these cases, DPMD gives results that are essentially indistinguishable from the original data, at a cost that scales linearly with system size.
- 24Weinert U. Spherical tensor representation Arch. Ration. Mech. Anal. 1980 74 165 196Google ScholarThere is no corresponding record for this reference.
- 25Stone A. J. Transformation between cartesian and spherical tensors Mol. Phys. 1975 29 1461 1471Google Scholar25Transformation between cartesian and spherical tensorsStone, A. J.Molecular Physics (1975), 29 (5), 1461-71CODEN: MOPHAM; ISSN:0026-8976.A std. unitary transformation is detd. for interconversion between cartesian and spherical tensors and between equations including such tensors. The effects of symmetry with respect to permutation of cartesian tensor suffixes were examd. The angle dependence of the circular intensity differential of Rayleigh scattering from a dimer was derived.
- 26De S. Bartók A. P. Csányi G. Ceriotti M. Comparing molecules and solids across structural and alchemical space Phys. Chem. Chem. Phys. 2016 18 13754 13769Google Scholar26Comparing molecules and solids across structural and alchemical spaceDe, Sandip; Bartok, Albert P.; Csanyi, Gabor; Ceriotti, MichelePhysical Chemistry Chemical Physics (2016), 18 (20), 13754-13769CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)Evaluating the (dis)similarity of cryst., disordered and mol. compds. is a crit. step in the development of algorithms to navigate automatically the configuration space of complex materials. For instance, a structural similarity metric is crucial for classifying structures, searching chem. space for better compds. and materials, and driving the next generation of machine-learning techniques for predicting the stability and properties of mols. and materials. In the last few years several strategies have been designed to compare at. coordination environments. In particular, the smooth overlap of at. positions (SOAPs) has emerged as an elegant framework to obtain translation, rotation and permutation-invariant descriptors of groups of atoms, underlying the development of various classes of machine-learned inter-at. potentials. Here we discuss how one can combine such local descriptors using a regularized entropy match (REMatch) approach to describe the similarity of both whole mol. and bulk periodic structures, introducing powerful metrics that enable the navigation of alchem. and structural complexities within a unified framework. Furthermore, using this kernel and a ridge regression method we can predict atomization energies for a database of small org. mols. with a mean abs. error below 1 kcal mol-1, reaching an important milestone in the application of machine-learning techniques for the evaluation of mol. properties.
- 27Musil F. De S. Yang J. Campbell J. E. J. Day G. G. M. Ceriotti M. Machine learning for the structure-energy-property landscapes of molecular crystals Chem. Sci. 2018 9 1289 1300Google Scholar27Machine learning for the structure-energy-property landscapes of molecular crystalsMusil, Felix; De, Sandip; Yang, Jack; Campbell, Joshua E.; Day, Graeme M.; Ceriotti, MicheleChemical Science (2018), 9 (5), 1289-1300CODEN: CSHCCN; ISSN:2041-6520. (Royal Society of Chemistry)Mol. crystals play an important role in several fields of science and technol. They frequently crystallize in different polymorphs with substantially different phys. properties. To help guide the synthesis of candidate materials, at.-scale modeling can be used to enumerate the stable polymorphs and to predict their properties, as well as to propose heuristic rules to rationalize the correlations between crystal structure and materials properties. Here we show how a recently-developed machine-learning (ML) framework can be used to achieve inexpensive and accurate predictions of the stability and properties of polymorphs, and a data-driven classification that is less biased and more flexible than typical heuristic rules. We discuss, as examples, the lattice energy and property landscapes of pentacene and two azapentacene isomers that are of interest as org. semiconductor materials. We show that we can est. force field or DFT lattice energies with sub-kJ mol-1 accuracy, using only a few hundred ref. configurations, and reduce by a factor of ten the computational effort needed to predict charge mobility in the crystal structures. The automatic structural classification of the polymorphs reveals a more detailed picture of mol. packing than that provided by conventional heuristics, and helps disentangle the role of hydrogen bonded and π-stacking interactions in detg. mol. self-assembly. This observation demonstrates that ML is not just a black-box scheme to interpolate between ref. calcns., but can also be used as a tool to gain intuitive insights into structure-property relations in mol. crystal engineering.
- 28Bartók A. P. De S. Poelking C. Bernstein N. Kermode J. R. Csányi G. Ceriotti M. Machine learning unifies the modeling of materials and molecules Sci. Adv. 2017 3Google ScholarThere is no corresponding record for this reference.
- 29Willatt M. J. Musil F. Ceriotti M. Atom-density representations for machine learning J. Chem. Phys. 2019 150 154110Google Scholar29Atom-density representations for machine learningWillatt, Michael J.; Musil, Felix; Ceriotti, MicheleJournal of Chemical Physics (2019), 150 (15), 154110/1-154110/12CODEN: JCPSA6; ISSN:0021-9606. (American Institute of Physics)The applications of machine learning techniques to chem. and materials science become more numerous by the day. The main challenge is to devise representations of at. systems that are at the same time complete and concise, so as to reduce the no. of ref. calcns. that are needed to predict the properties of different types of materials reliably. This has led to a proliferation of alternative ways to convert an at. structure into an input for a machine-learning model. We introduce an abstr. definition of chem. environments that is based on a smoothed at. d., using a bra-ket notation to emphasize basis set independence and to highlight the connections with some popular choices of representations for describing at. systems. The correlations between the spatial distribution of atoms and their chem. identities are computed as inner products between these feature kets, which can be given an explicit representation in terms of the expansion of the atom d. on orthogonal basis functions, that is equiv. to the smooth overlap of at. positions power spectrum, but also in real space, corresponding to n-body correlations of the atom d. This formalism lays the foundations for a more systematic tuning of the behavior of the representations, by introducing operators that represent the correlations between structure, compn., and the target properties. It provides a unifying picture of recent developments in the field and indicates a way forward toward more effective and computationally affordable machine-learning schemes for mols. and materials. (c) 2019 American Institute of Physics.
- 30Willatt M. J. Musil F. Ceriotti M. Feature Optimization for Atomistic Machine Learning Yields a Data-Driven Construction of the Periodic Table of the Elements Phys. Chem. Chem. Phys. 2018 20 29661 29668Google Scholar30Feature optimization for atomistic machine learning yields a data-driven construction of the periodic table of the elementsWillatt, Michael J.; Musil, Felix; Ceriotti, MichelePhysical Chemistry Chemical Physics (2018), 20 (47), 29661-29668CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)Machine-learning of at.-scale properties amts. to extg. correlations between structure, compn. and the quantity that one wants to predict. Representing the input structure in a way that best reflects such correlations makes it possible to improve the accuracy of the model for a given amt. of ref. data. When using a description of the structures that is transparent and well-principled, optimizing the representation might reveal insights into the chem. of the data set. Here, we show how one can generalize the SOAP kernel to introduce a distance-dependent wt. that accounts for the multi-scale nature of the interactions, and a description of correlations between chem. species. We show that this improves substantially the performance of ML models of mol. and materials stability, while making it easier to work with complex, multi-component systems and to extend SOAP to coarse-grained intermol. potentials. The element correlations that give the best performing model show striking similarities with the conventional periodic table of the elements, providing an inspiring example of how machine learning can rediscover, and generalize, intuitive concepts that constitute the foundations of chem.
- 31Kondor R. Zhen L. Trivedi S. Clebsch-Gordan Nets: a Fully Fourier Space Spherical Convolutional Neural Network arXiv:1806.09231 2018Google ScholarThere is no corresponding record for this reference.
- 32Kaufmann K. Baumeister W. Single-centre expansion of Gaussian basis functions and the angular decomposition of their overlap integrals J. Phys. B: At. Mol. Opt. 1989 22 1Google Scholar32Single-center expansion of Gaussian basis functions and the angular decomposition of their overlap integralsKaufmann, Karl; Baumeister, WernerJournal of Physics B: Atomic, Molecular and Optical Physics (1989), 22 (1), 1-12CODEN: JPAPEH; ISSN:0953-4075.Single-center partial-wave expansions are derived for several Gaussian-type functions: simple, solid harmonic, and spheric Gaussians. Single-center expansions for the most commonly used Cartesian Gaussians are obtained by expanding these functions in spherical Gaussians. Transformation matrixes for expanding Cartsian in spherical Gaussians are given for s-, p-, d-, and f-type functions. The single-center expansions are used to calc. the partial-wave decompn. of overlap integrals for all Gaussian-type functions specified. The formulas given are suitable for fast numerical computation, and were tested with programs developed for this purpose.
- 33Gradshteyn, I. S.; Ryzhik, I. M. Table of integrals, series, and products, 7th ed.; Elsevier/Academic Press, Amsterdam, 2007; pp xlviii+1171, Translated from the Russian, Translation edited and with a preface by Alan Jeffrey and Daniel Zwillinger, With one CD-ROM (Windows, Macintosh and UNIX).Google ScholarThere is no corresponding record for this reference.
- 34Chandrasekaran A. Kamal D. Batra R. Kim C. Chen L. Ramprasad R. Solving the electronic structure problem with machine learning Npj Comput. Mater. 2019 5 22Google ScholarThere is no corresponding record for this reference.
- 35Ceriotti M. Tribello G. A. Parrinello M. Demonstrating the Transferability and the Descriptive Power of Sketch-Map J. Chem. Theory Comput. 2013 9 1521 1532Google Scholar35Demonstrating the Transferability and the Descriptive Power of Sketch-MapCeriotti, Michele; Tribello, Gareth A.; Parrinello, MicheleJournal of Chemical Theory and Computation (2013), 9 (3), 1521-1532CODEN: JCTCCE; ISSN:1549-9618. (American Chemical Society)Increasingly, it is recognized that new automated forms of anal. are required to understand the high-dimensional output obtained from atomistic simulations. Recently, we introduced a new dimensionality redn. algorithm, sketch-map, that was designed specifically to work with data from mol. dynamics trajectories. In what follows, we provide more details on how this algorithm works and on how to set its parameters. We also test it on two well-studied Lennard-Jones clusters and show that the coordinates we ext. using this algorithm are extremely robust. In particular, we demonstrate that the coordinates constructed for one particular Lennard-Jones cluster can be used to describe the configurations adopted by a second, different cluster and even to tell apart different phases of bulk Lennard-Jonesium.
- 36Hättig C. Optimization of auxiliary basis sets for RI-MP2 and RI-CC2 calculations: Corevalence and quintuple- basis sets for H to Ar and QZVPP basis sets for Li to Kr Phys. Chem. Chem. Phys. 2005 7 59 66Google ScholarThere is no corresponding record for this reference.
Figure 1
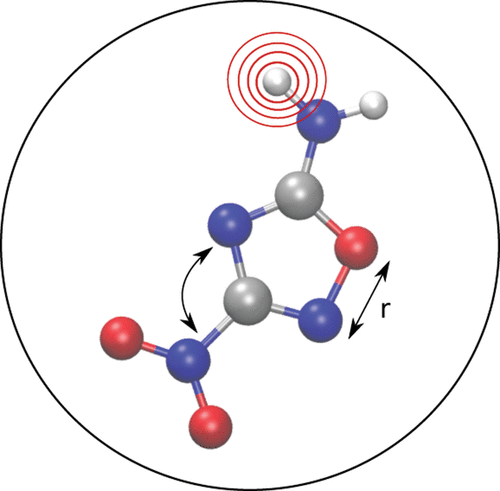 Figure 1. Structural descriptors should identify unequivocally and concisely the geometry and composition of a molecule or condensed phase.
Figure 1. Structural descriptors should identify unequivocally and concisely the geometry and composition of a molecule or condensed phase.Figure 2
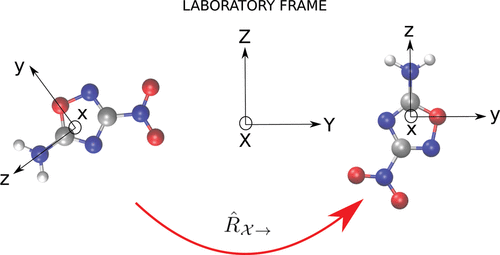 Figure 2. Provided that one can define a local reference system, it is possible to learn tensorial properties by aligning each molecule (or environment) into a fixed reference frame.
Figure 2. Provided that one can define a local reference system, it is possible to learn tensorial properties by aligning each molecule (or environment) into a fixed reference frame.Figure 3
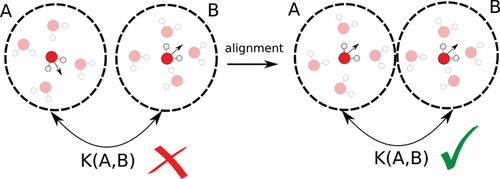 Figure 3. Representation of the reciprocal alignment between water environments.
Figure 3. Representation of the reciprocal alignment between water environments.Figure 4
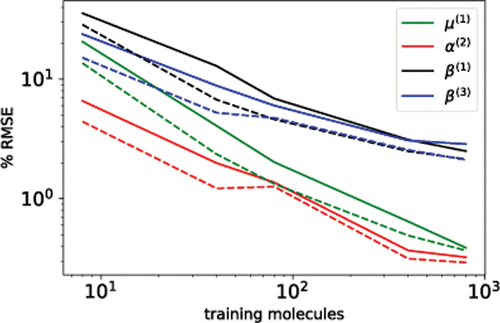 Figure 4. Learning curves of the Zundel cation dielectric response series µ,α and β as decomposed in their anisotropic (λ > 0) spherical tensor components. Full and dashed lines refer to predictions that are carried out with λ-SOAP kernel functions that are covariant in SO(3) and O(3) respectively.
Figure 4. Learning curves of the Zundel cation dielectric response series µ,α and β as decomposed in their anisotropic (λ > 0) spherical tensor components. Full and dashed lines refer to predictions that are carried out with λ-SOAP kernel functions that are covariant in SO(3) and O(3) respectively.Figure 5
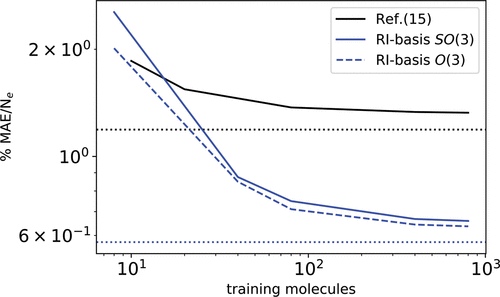 Figure 5. Learning curves of the predicted charge density of 200 randomly selected butane molecules, when considering up to 800 reference molecules to train the model. The molecular geometries and computational details are the same as in Grisafi et al. ( 18) The black full line refers to the prediction error as reported in Grisafi et al. ( 18) Blue lines refer to the result obtained with the RI-cc-pV5Z basis, both with a λ-SOAP(2) descriptor covariant in SO (3) (full) and O(3) (dashed). Dotted lines refer to the basis set error. In both cases, 100 reference atomic environments have been used to define the problem dimensionality.
Figure 5. Learning curves of the predicted charge density of 200 randomly selected butane molecules, when considering up to 800 reference molecules to train the model. The molecular geometries and computational details are the same as in Grisafi et al. ( 18) The black full line refers to the prediction error as reported in Grisafi et al. ( 18) Blue lines refer to the result obtained with the RI-cc-pV5Z basis, both with a λ-SOAP(2) descriptor covariant in SO (3) (full) and O(3) (dashed). Dotted lines refer to the basis set error. In both cases, 100 reference atomic environments have been used to define the problem dimensionality.References
CHAPTER SECTIONSThis chapter references 36 other publications.
- 1Williams, C. K. I.; Rasmussen, C. E. Gaussian Processes for Machine Learning; MIT Press, 2006.There is no corresponding record for this reference.
- 2Bartók A. P. Payne M. C. Kondor R. Csányi G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons Phys. Rev. Lett. 2010 104 1364032Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the ElectronsBartok, Albert P.; Payne, Mike C.; Kondor, Risi; Csanyi, GaborPhysical Review Letters (2010), 104 (13), 136403/1-136403/4CODEN: PRLTAO; ISSN:0031-9007. (American Physical Society)We introduce a class of interat. potential models that can be automatically generated from data consisting of the energies and forces experienced by atoms, as derived from quantum mech. calcns. The models do not have a fixed functional form and hence are capable of modeling complex potential energy landscapes. They are systematically improvable with more data. We apply the method to bulk crystals, and test it by calcg. properties at high temps. Using the interat. potential to generate the long mol. dynamics trajectories required for such calcns. saves orders of magnitude in computational cost.
- 3Jain A. Ong S. P. Hautier G. Chen W. Richards W. D. Dacek S. Cholia S. Gunter D. Skinner D. Ceder G. Persson K. A. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation APL Mater. 2013 1 0110023Commentary: The Materials Project: A materials genome approach to accelerating materials innovationJain, Anubhav; Ong, Shyue Ping; Hautier, Geoffroy; Chen, Wei; Richards, William Davidson; Dacek, Stephen; Cholia, Shreyas; Gunter, Dan; Skinner, David; Ceder, Gerbrand; Persson, Kristin A.APL Materials (2013), 1 (1), 011002/1-011002/11CODEN: AMPADS; ISSN:2166-532X. (American Institute of Physics)Accelerating the discovery of advanced materials is essential for human welfare and sustainable, clean energy. In this paper, we introduce the Materials Project (www.materialsproject.org), a core program of the Materials Genome Initiative that uses high-throughput computing to uncover the properties of all known inorg. materials. This open dataset can be accessed through multiple channels for both interactive exploration and data mining. The Materials Project also seeks to create open-source platforms for developing robust, sophisticated materials analyses. Future efforts will enable users to perform rapid-prototyping'' of new materials in silico, and provide researchers with new avenues for cost-effective, data-driven materials design. (c) 2013 American Institute of Physics.
- 4Calderon C. E. Plata J. J. Toher C. Oses C. Levy O. Fornari M. Natan A. Mehl M. J. Hart G. Nardelli M. B. Curtarolo S. The AFLOW standard for high-throughput materials science calculations Comput. Mater. Sci. 2015 108 233 2384The AFLOW standard for high-throughput materials science calculationsCalderon, Camilo E.; Plata, Jose J.; Toher, Cormac; Oses, Corey; Levy, Ohad; Fornari, Marco; Natan, Amir; Mehl, Michael J.; Hart, Gus; Buongiorno Nardelli, Marco; Curtarolo, StefanoComputational Materials Science (2015), 108 (Part_A), 233-238CODEN: CMMSEM; ISSN:0927-0256. (Elsevier B.V.)The Automatic-Flow (AFLOW) std. for the high-throughput construction of materials science electronic structure databases is described. Electronic structure calcns. of solid state materials depend on a large no. of parameters which must be understood by researchers, and must be reported by originators to ensure reproducibility and enable collaborative database expansion. We therefore describe std. parameter values for k-point grid d., basis set plane wave kinetic energy cut-off, exchange-correlation functionals, pseudopotentials, DFT+U parameters, and convergence criteria used in AFLOW calcns.
- 5Ward L. Wolverton C. Atomistic calculations and materials informatics: A review Curr. Opin. Solid State Mater. Sci. 2017 21 167 1765Atomistic calculations and materials informatics: A reviewWard, Logan; Wolverton, ChrisCurrent Opinion in Solid State & Materials Science (2017), 21 (3), 167-176CODEN: COSSFX; ISSN:1359-0286. (Elsevier Ltd.)A review. In recent years, there has been a large effort in the materials science community to employ materials informatics to accelerate materials discovery or to develop new understanding of materials behavior. Materials informatics methods utilize machine learning techniques to ext. new knowledge or predictive models out of existing materials data. In this review, we discuss major advances in the intersection between data science and atom-scale calcns. with a particular focus on studies of solid-state, inorg. materials. The examples discussed in this review cover methods for accelerating the calcn. of computationally-expensive properties, identifying promising regions for materials discovery based on existing data, and extg. chem. intuition automatically from datasets. We also identify key issues in this field, such as limited distribution of software necessary to utilize these techniques, and opportunities for areas of research that would help lead to the wider adoption of materials informatics in the atomistic calcns. community.
- 6Li Z. Kermode J. R. De Vita A. Molecular Dynamics with On-the-Fly Machine Learning of Quantum-Mechanical Forces Phys. Rev. Lett. 2015 114 0964056Molecular dynamics with on-the-fly machine learning of quantum-mechanical forcesLi, Zhenwei; Kermode, James R.; De Vita, AlessandroPhysical Review Letters (2015), 114 (9), 096405/1-096405/5CODEN: PRLTAO; ISSN:0031-9007. (American Physical Society)We present a mol. dynamics scheme which combines first-principles and machine-learning (ML) techniques in a single information-efficient approach. Forces on atoms are either predicted by Bayesian inference or, if necessary, computed by on-the-fly quantum-mech. (QM) calcns. and added to a growing ML database, whose completeness is, thus, never required. As a result, the scheme is accurate and general, while progressively fewer QM calls are needed when a new chem. process is encountered for the second and subsequent times, as demonstrated by tests on cryst. and molten silicon.
- 7Glielmo A. Sollich P. De Vita A. Accurate interatomic force fields via machine learning with covariant kernels Phys. Rev. B 2017 95 2143027Accurate interatomic force fields via machine learning with covariant kernelsGlielmo, Aldo; Sollich, Peter; De Vita, AlessandroPhysical Review B (2017), 95 (21), 214302/1-214302/10CODEN: PRBHB7; ISSN:2469-9969. (American Physical Society)A review. We present a novel scheme to accurately predict at. forces as vector quantities, rather than sets of scalar components, by Gaussian process (GP) regression. This is based on matrix-valued kernel functions, on which we impose the requirements that the predicted force rotates with the target configuration and is independent of any rotations applied to the configuration database entries. We show that such covariant GP kernels can be obtained by integration over the elements of the rotation group SO(d) for the relevant dimensionality d. Remarkably, in specific cases the integration can be carried out anal. and yields a conservative force field that can be recast into a pair interaction form. Finally, we show that restricting the integration to a summation over the elements of a finite point group relevant to the target system is sufficient to recover an accurate GP. The accuracy of our kernels in predicting quantum-mech. forces in real materials is investigated by tests on pure and defective Ni, Fe, and Si cryst. systems.
- 8Glielmo A. Zeni C. De Vita A. Efficient nonparametric n-body force fields from machine learning Phys. Rev. B 2018 97 1843078Efficient nonparametric n-body force fields from machine learningGlielmo, Aldo; Zeni, Claudio; De Vita, AlessandroPhysical Review B (2018), 97 (18), 184307CODEN: PRBHB7; ISSN:2469-9969. (American Physical Society)A review. We provide a definition and explicit expressions for n-body Gaussian process (GP) kernels, which can learn any interat. interaction occurring in a phys. system, up to n-body contributions, for any value of n. The series is complete, as it can be shown that the "universal approximator" squared exponential kernel can be written as a sum of n-body kernels. These recipes enable the choice of optimally efficient force models for each target system, as confirmed by extensive testing on various materials. We furthermore describe how the n-body kernels can be "mapped" on equiv. representations that provide database-size-independent predictions and are thus crucially more efficient. We explicitly carry out this mapping procedure for the first nontrivial (three-body) kernel of the series, and we show that this reproduces the GP-predicted forces with meV/Å accuracy while being orders of magnitude faster. These results pave the way to using novel force models (here named "M-FFs") that are computationally as fast as their corresponding std. parametrized n-body force fields, while retaining the nonparametric character, the ease of training and validation, and the accuracy of the best recently proposed machine-learning potentials.
- 9Yuan Y. Mills M. J. Popelier P. L. Multipolar electrostatics based on the Kriging machine learning method: an application to serine J. Mol. Model. 2014 20 21729Multipolar electrostatics based on the Kriging machine learning method: an application to serineYuan Yongna; Mills Matthew J L; Popelier Paul L AJournal of molecular modeling (2014), 20 (4), 2172 ISSN:.A multipolar, polarizable electrostatic method for future use in a novel force field is described. Quantum Chemical Topology (QCT) is used to partition the electron density of a chemical system into atoms, then the machine learning method Kriging is used to build models that relate the multipole moments of the atoms to the positions of their surrounding nuclei. The pilot system serine is used to study both the influence of the level of theory and the set of data generator methods used. The latter consists of: (i) sampling of protein structures deposited in the Protein Data Bank (PDB), or (ii) normal mode distortion along either (a) Cartesian coordinates, or (b) redundant internal coordinates. Wavefunctions for the sampled geometries were obtained at the HF/6-31G(d,p), B3LYP/apc-1, and MP2/cc-pVDZ levels of theory, prior to calculation of the atomic multipole moments by volume integration. The average absolute error (over an independent test set of conformations) in the total atom-atom electrostatic interaction energy of serine, using Kriging models built with the three data generator methods is 11.3 kJ mol-1 (PDB), 8.2 kJ mol-1 (Cartesian distortion), and 10.1 kJ mol-1 (redundant internal distortion) at the HF/6-31G(d,p) level. At the B3LYP/apc-1 level, the respective errors are 7.7 kJ mol-1, 6.7 kJ mol-1, and 4.9 kJ mol-1, while at the MP2/cc-pVDZ level they are 6.5 kJ mol-1, 5.3 kJ mol-1, and 4.0 kJ mol-1. The ranges of geometries generated by the redundant internal coordinate distortion and by extraction from the PDB are much wider than the range generated by Cartesian distortion. The atomic multipole moment and electrostatic interaction energy predictions for the B3LYP/apc-1 and MP2/cc-pVDZ levels are similar, and both are better than the corresponding predictions at the HF/6-31G(d,p) level.
- 10Bereau T. Andrienko D. von Lilienfeld O. A. Transferable Atomic Multipole Machine Learning Models for Small Organic Molecules J. Chem. Theory Comput. 2015 11 3225 323310Transferable Atomic Multipole Machine Learning Models for Small Organic MoleculesBereau, Tristan; Andrienko, Denis; von Lilienfeld, O. AnatoleJournal of Chemical Theory and Computation (2015), 11 (7), 3225-3233CODEN: JCTCCE; ISSN:1549-9618. (American Chemical Society)Accurate representation of the mol. electrostatic potential, which is often expanded in distributed multipole moments, is crucial for an efficient evaluation of intermol. interactions. Here we introduce a machine learning model for multipole coeffs. of atom types H, C, O, N, S, F, and Cl in any mol. conformation. The model is trained on quantum-chem. results for atoms in varying chem. environments drawn from thousands of org. mols. Multipoles in systems with neutral, cationic, and anionic mol. charge states are treated with individual models. The models' predictive accuracy and applicability are illustrated by evaluating intermol. interaction energies of nearly 1,000 dimers and the cohesive energy of the benzene crystal.
- 11Bereau T. DiStasio R. A. Tkatchenko A. von Lilienfeld O. A. Non-covalent interactions across organic and biological subsets of chemical space: Physics-based potentials parametrized from machine learning J. Chem. Phys. 2018 148 24170611Non-covalent interactions across organic and biological subsets of chemical space: Physics-based potentials parametrized from machine learningBereau, Tristan; Di Stasio, Robert A.; Tkatchenko, Alexandre; von Lilienfeld, O. AnatoleJournal of Chemical Physics (2018), 148 (24), 241706/1-241706/14CODEN: JCPSA6; ISSN:0021-9606. (American Institute of Physics)Classical intermol. potentials typically require an extensive parametrization procedure for any new compd. considered. To do away with prior parametrization, we propose a combination of physics-based potentials with machine learning (ML), coined IPML, which is transferable across small neutral org. and biol. relevant mols. ML models provide on-the-fly predictions for environment-dependent local at. properties: electrostatic multipole coeffs. (significant error redn. compared to previously reported), the population and decay rate of valence at. densities, and polarizabilities across conformations and chem. compns. of H, C, N, and O atoms. These parameters enable accurate calcns. of intermol. contributions-electrostatics, charge penetration, repulsion, induction/polarization, and many-body dispersion. Unlike other potentials, this model is transferable in its ability to handle new mols. and conformations without explicit prior parametrization: All local at. properties are predicted from ML, leaving only eight global parameters-optimized once and for all across compds. We validate IPML on various gas-phase dimers at and away from equil. sepn., where we obtain mean abs. errors between 0.4 and 0.7 kcal/mol for several chem. and conformationally diverse datasets representative of non-covalent interactions in biol. relevant mols. We further focus on hydrogen-bonded complexes - essential but challenging due to their directional nature - where datasets of DNA base pairs and amino acids yield an extremely encouraging 1.4 kcal/mol error. Finally, and as a first look, we consider IPML for denser systems: water clusters, supramol. host-guest complexes, and the benzene crystal. (c) 2018 American Institute of Physics.
- 12Liang C. Tocci G. Wilkins D. M. Grisafi A. Roke S. Ceriotti M. Solvent fluctuations and nuclear quantum effects modulate the molecular hyperpolarizability of water Phys. Rev. B 2017 96 04140712Solvent fluctuations and nuclear quantum effects modulate the molecular hyperpolarizability of waterLiang, Chungwen; Tocci, Gabriele; Wilkins, David M.; Grisafi, Andrea; Roke, Sylvie; Ceriotti, MichelePhysical Review B (2017), 96 (4), 041407/1-041407/6CODEN: PRBHB7; ISSN:2469-9969. (American Physical Society)Second-harmonic scattering (SHS) expts. provide a unique approach to probe noncentrosym. environments in aq. media, from bulk solns. to interfaces, living cells, and tissue. A central assumption made in analyzing SHS expts. is that each mol. scatters light according to a const. mol. hyperpolarizability tensor β(2). Here, we investigate the dependence of the mol. hyperpolarizability of water on its environment and internal geometric distortions, in order to test the hypothesis of const. β(2). We use quantum chem. calcns. of the hyperpolarizability of a mol. embedded in point-charge environments obtained from simulations of bulk water. We demonstrate that both the heterogeneity of the solvent configurations and the quantum mech. fluctuations of the mol. geometry introduce large variations in the nonlinear optical response of water. This finding has the potential to change the way SHS expts. are interpreted: In particular, isotopic differences between H2O and D2O could explain recent SHS observations. Finally, we show that a machine-learning framework can predict accurately the fluctuations of the mol. hyperpolarizability. This model accounts for the microscopic inhomogeneity of the solvent and represents a step towards quant. modeling of SHS expts.
- 13Grisafi A. Wilkins D. M. Csányi G. Ceriotti M. Symmetry-Adapted Machine Learning for Tensorial Properties of Atomistic Systems Phys. Rev. Lett. 2018 120 03600213Symmetry-Adapted Machine Learning for Tensorial Properties of Atomistic SystemsGrisafi, Andrea; Wilkins, David M.; Csanyi, Gabor; Ceriotti, MichelePhysical Review Letters (2018), 120 (3), 036002CODEN: PRLTAO; ISSN:1079-7114. (American Physical Society)A review. Statistical learning methods show great promise in providing an accurate prediction of materials and mol. properties, while minimizing the need for computationally demanding electronic structure calcns. The accuracy and transferability of these models are increased significantly by encoding into the learning procedure the fundamental symmetries of rotational and permutational invariance of scalar properties. However, the prediction of tensorial properties requires that the model respects the appropriate geometric transformations, rather than invariance, when the ref. frame is rotated. We introduce a formalism that extends existing schemes and makes it possible to perform machine learning of tensorial properties of arbitrary rank, and for general mol. geometries. To demonstrate it, we derive a tensor kernel adapted to rotational symmetry, which is the natural generalization of the smooth overlap of at. positions kernel commonly used for the prediction of scalar properties at the at. scale. The performance and generality of the approach is demonstrated by learning the instantaneous response to an external elec. field of water oligomers of increasing complexity, from the isolated mol. to the condensed phase.
- 14Wilkins D. M. Grisafi A. Yang Y. Lao K. U. DiStasio R. A. Ceriotti M. Accurate molecular polarizabilities with coupled cluster theory and machine learning Proc. Natl. Acad. Sci. 2019 116 3401 340614Accurate molecular polarizabilities with coupled cluster theory and machine learningWilkins, David M.; Grisafi, Andrea; Yang, Yang; Lao, Ka Un; Di Stasio, Robert A., Jr.; Ceriotti, MicheleProceedings of the National Academy of Sciences of the United States of America (2019), 116 (9), 3401-3406CODEN: PNASA6; ISSN:0027-8424. (National Academy of Sciences)The mol. dipole polarizability describes the tendency of a mol. to change its dipole moment in response to an applied elec. field. This quantity governs key intra- and intermol. interactions, such as induction and dispersion; plays a vital role in detg. the spectroscopic signatures of mols.; and is an essential ingredient in polarizable force fields. Compared with other ground-state properties, an accurate prediction of the mol. polarizability is considerably more difficult, as this response quantity is quite sensitive to the underlying electronic structure description. In this work, we present highly accurate quantum mech. calcns. of the static dipole polarizability tensors of 7,211 small org. mols. computed using linear response coupled cluster singles and doubles theory (LR-CCSD). Using a symmetry-adapted machine-learning approach, we demonstrate that it is possible to predict the LR-CCSD mol. polarizabilities of these small mols. with an error that is an order of magnitude smaller than that of hybrid d. functional theory (DFT) at a negligible computational cost. The resultant model is robust and transferable, yielding mol. polarizabilities for a diverse set of 52 larger mols. (including challenging conjugated systems, carbohydrates, small drugs, amino acids, nucleobases, and hydrocarbon isomers) at an accuracy that exceeds that of hybrid DFT. The atom-centered decompn. implicit in our machine-learning approach offers some insight into the shortcomings of DFT in the prediction of this fundamental quantity of interest.
- 15Christensen A. S. Faber F. A. von Lilienfeld O. A. Operators in quantum machine learning: Response properties in chemical space J. Chem. Phys. 2019 150 06410515Operators in quantum machine learning: Response properties in chemical spaceChristensen, Anders S.; Faber, Felix A.; von Lilienfeld, O. AnatoleJournal of Chemical Physics (2019), 150 (6), 064105/1-064105/12CODEN: JCPSA6; ISSN:0021-9606. (American Institute of Physics)The role of response operators is well established in quantum mechanics. We investigate their use for universal quantum machine learning models of response properties in mols. After introducing a theor. basis, we present and discuss numerical evidence based on measuring the potential energy's response with respect to at. displacement and to elec. fields. Prediction errors for corresponding properties, at. forces, and dipole moments improve in a systematic fashion with training set size and reach high accuracy for small training sets. Prediction of normal modes and IR-spectra of some small mols. demonstrates the usefulness of this approach for chem. (c) 2019 American Institute of Physics.
- 16Brockherde F. Vogt L. Li L. Tuckerman M. E. Burke K. Mu¨ller K.-R. Bypassing the Kohn-Sham equations with machine learning Nat. Commun. 2017 8 87216Bypassing the Kohn-Sham equations with machine learningBrockherde Felix; Muller Klaus-Robert; Brockherde Felix; Vogt Leslie; Tuckerman Mark E; Li Li; Burke Kieron; Tuckerman Mark E; Tuckerman Mark E; Burke Kieron; Muller Klaus-Robert; Muller Klaus-RobertNature communications (2017), 8 (1), 872 ISSN:.Last year, at least 30,000 scientific papers used the Kohn-Sham scheme of density functional theory to solve electronic structure problems in a wide variety of scientific fields. Machine learning holds the promise of learning the energy functional via examples, bypassing the need to solve the Kohn-Sham equations. This should yield substantial savings in computer time, allowing larger systems and/or longer time-scales to be tackled, but attempts to machine-learn this functional have been limited by the need to find its derivative. The present work overcomes this difficulty by directly learning the density-potential and energy-density maps for test systems and various molecules. We perform the first molecular dynamics simulation with a machine-learned density functional on malonaldehyde and are able to capture the intramolecular proton transfer process. Learning density models now allows the construction of accurate density functionals for realistic molecular systems.Machine learning allows electronic structure calculations to access larger system sizes and, in dynamical simulations, longer time scales. Here, the authors perform such a simulation using a machine-learned density functional that avoids direct solution of the Kohn-Sham equations.
- 17Alred J. M. Bets K. V. Xie Y. Yakobson B. I. Machine learning electron density in sulfur crosslinked carbon nanotubes Compos. Sci. Technol. 2018 166 3 917Machine learning electron density in sulfur crosslinked carbon nanotubesAlred, John M.; Bets, Ksenia V.; Xie, Yu; Yakobson, Boris I.Composites Science and Technology (2018), 166 (), 3-9CODEN: CSTCEH; ISSN:0266-3538. (Elsevier Ltd.)Mech. strengthening of composite materials that include carbon nanotubes (CNT) requires strong inter-bonding to achieve significant CNT-CNT or CNT-matrix load transfer. The same principle is applicable to the improvement of CNT bundles and calls for covalent crosslinks between individual tubes. In this work, sulfur crosslinks are studied using a combination of d. functional theory (DFT) and classical mol. dynamics (MD). Atomic chains of at least two sulfur atoms or more are shown to be stable between both zigzag and armchair CNTs. All types of crosslinked CNTs exhibit significantly improved load transfer. Moreover, sulfur crosslinks show evidence of a cooperative self-healing mechanism allowing for links to rebond once broken leading to sustained load transfer under shear loading. Addnl., a general approach for utilizing machine learning for assessing the ground state electron d. is developed and applied to these sulfur crosslinked CNTs.
- 18Grisafi A. Fabrizio A. Meyer B. Wilkins D. M. Corminboeuf C. Ceriotti M. Transferable Machine-Learning Model of the Electron Density ACS Cent. Sci. 2019 5 57 6418Transferable Machine-Learning Model of the Electron DensityGrisafi, Andrea; Fabrizio, Alberto; Meyer, Benjamin; Wilkins, David M.; Corminboeuf, Clemence; Ceriotti, MicheleACS Central Science (2019), 5 (1), 57-64CODEN: ACSCII; ISSN:2374-7951. (American Chemical Society)The electronic charge d. plays a central role in detg. the behavior of matter at the at. scale, but its computational evaluation requires demanding electronic-structure calcns. We introduce an atom-centered, symmetry-adapted framework to machine-learn the valence charge d. based on a small no. of ref. calcns. The model is highly transferable, meaning it can be trained on electronic-structure data of small mols. and used to predict the charge d. of larger compds. with low, linear-scaling cost. Applications are shown for various hydrocarbon mols. of increasing complexity and flexibility, and demonstrate the accuracy of the model when predicting the d. on octane and octatetraene after training exclusively on butane and butadiene. This transferable, data-driven model can be used to interpret expts., accelerate electronic structure calcns., and compute electrostatic interactions in mols. and condensed-phase systems.
- 19Braams B. J. Bowman J. M. Permutationally invariant potential energy surfaces in high dimensionality Int. Rev. Phys. Chem. 2009 28 577 60619Permutationally invariant potential energy surfaces in high dimensionalityBraams, Bastiaan J.; Bowman, Joel M.International Reviews in Physical Chemistry (2009), 28 (4), 577-606CODEN: IRPCDL; ISSN:0144-235X. (Taylor & Francis Ltd.)We review recent progress in developing potential energy and dipole moment surfaces for polyat. systems with up to 10 atoms. The emphasis is on global linear least squares fitting of tens of thousands of scattered ab initio energies using a special, compact fitting basis of permutationally invariant polynomials in Morse-type variables of all the internuclear distances. The computational mathematics underlying this approach is reviewed first, followed by a review of the practical approaches used to obtain the data for the fits. A straightforward symmetrization approach is also given, mainly for pedagogical purposes. The methods are illustrated for potential energy surfaces for CH+5, (H2O)2 and CH3CHO. The relationship of this approach to other approaches is also briefly reviewed.
- 20Behler J. Parrinello M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces Phys. Rev. Lett. 2007 98 14640120Generalized Neural-Network Representation of High-Dimensional Potential-Energy SurfacesBehler, Jorg; Parrinello, MichelePhysical Review Letters (2007), 98 (14), 146401/1-146401/4CODEN: PRLTAO; ISSN:0031-9007. (American Physical Society)The accurate description of chem. processes often requires the use of computationally demanding methods like d.-functional theory (DFT), making long simulations of large systems unfeasible. In this Letter we introduce a new kind of neural-network representation of DFT potential-energy surfaces, which provides the energy and forces as a function of all at. positions in systems of arbitrary size and is several orders of magnitude faster than DFT. The high accuracy of the method is demonstrated for bulk silicon and compared with empirical potentials and DFT. The method is general and can be applied to all types of periodic and nonperiodic systems.
- 21Bartók A. P. Kondor R. Csányi G. On representing chemical environments Phys. Rev. B 2013 87 18411521On representing chemical environmentsBartok, Albert P.; Kondor, Risi; Csanyi, GaborPhysical Review B: Condensed Matter and Materials Physics (2013), 87 (18), 184115/1-184115/16CODEN: PRBMDO; ISSN:1098-0121. (American Physical Society)We review some recently published methods to represent at. neighborhood environments, and analyze their relative merits in terms of their faithfulness and suitability for fitting potential energy surfaces. The crucial properties that such representations (sometimes called descriptors) must have are differentiability with respect to moving the atoms and invariance to the basic symmetries of physics: rotation, reflection, translation, and permutation of atoms of the same species. We demonstrate that certain widely used descriptors that initially look quite different are specific cases of a general approach, in which a finite set of basis functions with increasing angular wave nos. are used to expand the at. neighborhood d. function. Using the example system of small clusters, we quant. show that this expansion needs to be carried to higher and higher wave nos. as the no. of neighbors increases in order to obtain a faithful representation, and that variants of the descriptors converge at very different rates. We also propose an altogether different approach, called Smooth Overlap of Atomic Positions, that sidesteps these difficulties by directly defining the similarity between any two neighborhood environments, and show that it is still closely connected to the invariant descriptors. We test the performance of the various representations by fitting models to the potential energy surface of small silicon clusters and the bulk crystal.
- 22Shapeev A. Moment Tensor Potentials: A Class of Systematically Improvable Interatomic Potentials Multiscale Model. Sim. 2016 14 1153 1173There is no corresponding record for this reference.
- 23Zhang L. Han J. Wang H. Car R. E W. Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics Phys. Rev. Lett. 2018 120 14300123Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum MechanicsZhang, Linfeng; Han, Jiequn; Wang, Han; Car, Roberto; E, WeinanPhysical Review Letters (2018), 120 (14), 143001CODEN: PRLTAO; ISSN:1079-7114. (American Physical Society)We introduce a scheme for mol. simulations, the deep potential mol. dynamics (DPMD) method, based on a many-body potential and interat. forces generated by a carefully crafted deep neural network trained with ab initio data. The neural network model preserves all the natural symmetries in the problem. It is first-principles based in the sense that there are no ad hoc components aside from the network model. We show that the proposed scheme provides an efficient and accurate protocol in a variety of systems, including bulk materials and mols. In all these cases, DPMD gives results that are essentially indistinguishable from the original data, at a cost that scales linearly with system size.
- 24Weinert U. Spherical tensor representation Arch. Ration. Mech. Anal. 1980 74 165 196There is no corresponding record for this reference.
- 25Stone A. J. Transformation between cartesian and spherical tensors Mol. Phys. 1975 29 1461 147125Transformation between cartesian and spherical tensorsStone, A. J.Molecular Physics (1975), 29 (5), 1461-71CODEN: MOPHAM; ISSN:0026-8976.A std. unitary transformation is detd. for interconversion between cartesian and spherical tensors and between equations including such tensors. The effects of symmetry with respect to permutation of cartesian tensor suffixes were examd. The angle dependence of the circular intensity differential of Rayleigh scattering from a dimer was derived.
- 26De S. Bartók A. P. Csányi G. Ceriotti M. Comparing molecules and solids across structural and alchemical space Phys. Chem. Chem. Phys. 2016 18 13754 1376926Comparing molecules and solids across structural and alchemical spaceDe, Sandip; Bartok, Albert P.; Csanyi, Gabor; Ceriotti, MichelePhysical Chemistry Chemical Physics (2016), 18 (20), 13754-13769CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)Evaluating the (dis)similarity of cryst., disordered and mol. compds. is a crit. step in the development of algorithms to navigate automatically the configuration space of complex materials. For instance, a structural similarity metric is crucial for classifying structures, searching chem. space for better compds. and materials, and driving the next generation of machine-learning techniques for predicting the stability and properties of mols. and materials. In the last few years several strategies have been designed to compare at. coordination environments. In particular, the smooth overlap of at. positions (SOAPs) has emerged as an elegant framework to obtain translation, rotation and permutation-invariant descriptors of groups of atoms, underlying the development of various classes of machine-learned inter-at. potentials. Here we discuss how one can combine such local descriptors using a regularized entropy match (REMatch) approach to describe the similarity of both whole mol. and bulk periodic structures, introducing powerful metrics that enable the navigation of alchem. and structural complexities within a unified framework. Furthermore, using this kernel and a ridge regression method we can predict atomization energies for a database of small org. mols. with a mean abs. error below 1 kcal mol-1, reaching an important milestone in the application of machine-learning techniques for the evaluation of mol. properties.
- 27Musil F. De S. Yang J. Campbell J. E. J. Day G. G. M. Ceriotti M. Machine learning for the structure-energy-property landscapes of molecular crystals Chem. Sci. 2018 9 1289 130027Machine learning for the structure-energy-property landscapes of molecular crystalsMusil, Felix; De, Sandip; Yang, Jack; Campbell, Joshua E.; Day, Graeme M.; Ceriotti, MicheleChemical Science (2018), 9 (5), 1289-1300CODEN: CSHCCN; ISSN:2041-6520. (Royal Society of Chemistry)Mol. crystals play an important role in several fields of science and technol. They frequently crystallize in different polymorphs with substantially different phys. properties. To help guide the synthesis of candidate materials, at.-scale modeling can be used to enumerate the stable polymorphs and to predict their properties, as well as to propose heuristic rules to rationalize the correlations between crystal structure and materials properties. Here we show how a recently-developed machine-learning (ML) framework can be used to achieve inexpensive and accurate predictions of the stability and properties of polymorphs, and a data-driven classification that is less biased and more flexible than typical heuristic rules. We discuss, as examples, the lattice energy and property landscapes of pentacene and two azapentacene isomers that are of interest as org. semiconductor materials. We show that we can est. force field or DFT lattice energies with sub-kJ mol-1 accuracy, using only a few hundred ref. configurations, and reduce by a factor of ten the computational effort needed to predict charge mobility in the crystal structures. The automatic structural classification of the polymorphs reveals a more detailed picture of mol. packing than that provided by conventional heuristics, and helps disentangle the role of hydrogen bonded and π-stacking interactions in detg. mol. self-assembly. This observation demonstrates that ML is not just a black-box scheme to interpolate between ref. calcns., but can also be used as a tool to gain intuitive insights into structure-property relations in mol. crystal engineering.
- 28Bartók A. P. De S. Poelking C. Bernstein N. Kermode J. R. Csányi G. Ceriotti M. Machine learning unifies the modeling of materials and molecules Sci. Adv. 2017 3There is no corresponding record for this reference.
- 29Willatt M. J. Musil F. Ceriotti M. Atom-density representations for machine learning J. Chem. Phys. 2019 150 15411029Atom-density representations for machine learningWillatt, Michael J.; Musil, Felix; Ceriotti, MicheleJournal of Chemical Physics (2019), 150 (15), 154110/1-154110/12CODEN: JCPSA6; ISSN:0021-9606. (American Institute of Physics)The applications of machine learning techniques to chem. and materials science become more numerous by the day. The main challenge is to devise representations of at. systems that are at the same time complete and concise, so as to reduce the no. of ref. calcns. that are needed to predict the properties of different types of materials reliably. This has led to a proliferation of alternative ways to convert an at. structure into an input for a machine-learning model. We introduce an abstr. definition of chem. environments that is based on a smoothed at. d., using a bra-ket notation to emphasize basis set independence and to highlight the connections with some popular choices of representations for describing at. systems. The correlations between the spatial distribution of atoms and their chem. identities are computed as inner products between these feature kets, which can be given an explicit representation in terms of the expansion of the atom d. on orthogonal basis functions, that is equiv. to the smooth overlap of at. positions power spectrum, but also in real space, corresponding to n-body correlations of the atom d. This formalism lays the foundations for a more systematic tuning of the behavior of the representations, by introducing operators that represent the correlations between structure, compn., and the target properties. It provides a unifying picture of recent developments in the field and indicates a way forward toward more effective and computationally affordable machine-learning schemes for mols. and materials. (c) 2019 American Institute of Physics.
- 30Willatt M. J. Musil F. Ceriotti M. Feature Optimization for Atomistic Machine Learning Yields a Data-Driven Construction of the Periodic Table of the Elements Phys. Chem. Chem. Phys. 2018 20 29661 2966830Feature optimization for atomistic machine learning yields a data-driven construction of the periodic table of the elementsWillatt, Michael J.; Musil, Felix; Ceriotti, MichelePhysical Chemistry Chemical Physics (2018), 20 (47), 29661-29668CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)Machine-learning of at.-scale properties amts. to extg. correlations between structure, compn. and the quantity that one wants to predict. Representing the input structure in a way that best reflects such correlations makes it possible to improve the accuracy of the model for a given amt. of ref. data. When using a description of the structures that is transparent and well-principled, optimizing the representation might reveal insights into the chem. of the data set. Here, we show how one can generalize the SOAP kernel to introduce a distance-dependent wt. that accounts for the multi-scale nature of the interactions, and a description of correlations between chem. species. We show that this improves substantially the performance of ML models of mol. and materials stability, while making it easier to work with complex, multi-component systems and to extend SOAP to coarse-grained intermol. potentials. The element correlations that give the best performing model show striking similarities with the conventional periodic table of the elements, providing an inspiring example of how machine learning can rediscover, and generalize, intuitive concepts that constitute the foundations of chem.
- 31Kondor R. Zhen L. Trivedi S. Clebsch-Gordan Nets: a Fully Fourier Space Spherical Convolutional Neural Network arXiv:1806.09231 2018There is no corresponding record for this reference.
- 32Kaufmann K. Baumeister W. Single-centre expansion of Gaussian basis functions and the angular decomposition of their overlap integrals J. Phys. B: At. Mol. Opt. 1989 22 132Single-center expansion of Gaussian basis functions and the angular decomposition of their overlap integralsKaufmann, Karl; Baumeister, WernerJournal of Physics B: Atomic, Molecular and Optical Physics (1989), 22 (1), 1-12CODEN: JPAPEH; ISSN:0953-4075.Single-center partial-wave expansions are derived for several Gaussian-type functions: simple, solid harmonic, and spheric Gaussians. Single-center expansions for the most commonly used Cartesian Gaussians are obtained by expanding these functions in spherical Gaussians. Transformation matrixes for expanding Cartsian in spherical Gaussians are given for s-, p-, d-, and f-type functions. The single-center expansions are used to calc. the partial-wave decompn. of overlap integrals for all Gaussian-type functions specified. The formulas given are suitable for fast numerical computation, and were tested with programs developed for this purpose.
- 33Gradshteyn, I. S.; Ryzhik, I. M. Table of integrals, series, and products, 7th ed.; Elsevier/Academic Press, Amsterdam, 2007; pp xlviii+1171, Translated from the Russian, Translation edited and with a preface by Alan Jeffrey and Daniel Zwillinger, With one CD-ROM (Windows, Macintosh and UNIX).There is no corresponding record for this reference.
- 34Chandrasekaran A. Kamal D. Batra R. Kim C. Chen L. Ramprasad R. Solving the electronic structure problem with machine learning Npj Comput. Mater. 2019 5 22There is no corresponding record for this reference.
- 35Ceriotti M. Tribello G. A. Parrinello M. Demonstrating the Transferability and the Descriptive Power of Sketch-Map J. Chem. Theory Comput. 2013 9 1521 153235Demonstrating the Transferability and the Descriptive Power of Sketch-MapCeriotti, Michele; Tribello, Gareth A.; Parrinello, MicheleJournal of Chemical Theory and Computation (2013), 9 (3), 1521-1532CODEN: JCTCCE; ISSN:1549-9618. (American Chemical Society)Increasingly, it is recognized that new automated forms of anal. are required to understand the high-dimensional output obtained from atomistic simulations. Recently, we introduced a new dimensionality redn. algorithm, sketch-map, that was designed specifically to work with data from mol. dynamics trajectories. In what follows, we provide more details on how this algorithm works and on how to set its parameters. We also test it on two well-studied Lennard-Jones clusters and show that the coordinates we ext. using this algorithm are extremely robust. In particular, we demonstrate that the coordinates constructed for one particular Lennard-Jones cluster can be used to describe the configurations adopted by a second, different cluster and even to tell apart different phases of bulk Lennard-Jonesium.
- 36Hättig C. Optimization of auxiliary basis sets for RI-MP2 and RI-CC2 calculations: Corevalence and quintuple- basis sets for H to Ar and QZVPP basis sets for Li to Kr Phys. Chem. Chem. Phys. 2005 7 59 66There is no corresponding record for this reference.