



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Top-Down Characterization of Protein Anions Using Ultraviolet Photodissociation Mass SpectrometryClick to copy article linkArticle link copied!
- Hanlin RenHanlin RenDepartment of Chemistry, University of Texas at Austin, Austin, Texas 78712, United StatesMore by Hanlin Ren
- Jennifer S. Brodbelt*Jennifer S. Brodbelt*Email: [email protected]Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United StatesMore by Jennifer S. Brodbelt
Abstract
Top-down proteomics is primarily performed using electrospray ionization-tandem mass spectrometry (ESI-MS/MS) in the positive mode. Development of methods in the negative mode can potentially facilitate analysis of acidic proteome, but has been hampered by the low ionization efficiency and the lack of effective fragmentation methods for protein anions. Here, we investigate the performance of ultraviolet photodissociation (UVPD) for top-down analysis of protein anions. We employed organic bases as additives in solution to yield highly charged, nonadducted protein anions of high abundance. We compared UVPD with higher energy collisional dissociation (HCD) and activated electron photodetachment (a-EPD) for fragmentation of proteins ranging from 8.6 to 47 kDa. UVPD yielded abundant charge-reduced precursor radicals, in addition to numerous a/x, b/y and c/z fragment ions. UVPD offered 70–95% sequence coverage for proteins below 20 kDa regardless of the presence of disulfide bonds, and 30% coverage for the largest protein studied (47 kDa enolase), higher coverage than HCD and a-EPD. UVPD of deprotonated proteins exhibited several features similar to those of protonated proteins, such as minimal sensitivity to the charge state, production of abundant a/x fragment ions, and fairly uniform backbone cleavages adjacent to each residue (i.e., no prominent preferential cleavage sites).
This publication is licensed for personal use by The American Chemical Society.
Special Issue
Published as part of Journal of the American Society for Mass Spectrometry special issue “Fenn: Photoactivation and Ion Activation”.
Introduction
Experimental Section
Sample Preparation
Top-Down Mass Spectrometry
Data Analysis
Results and Discussion
Impact of Organic Bases on Ionization of Proteins in the Negative Mode
Figure 1
Figure 1. MS1 spectra of solutions containing 10 μM myoglobin and (a) 1% formic acid in positive mode, (b) with 1% NH3·H2O in negative mode, and (c) with 0.05% DBN in negative mode. Selected charge states of the protein and the heme are annotated.
MS/MS Analysis of Myoglobin
Figure 2
Figure 2. MS/MS spectra of myoglobin (20–, m/z 846, indicated by an asterisk in the UVPD spectrum). (a) HCD using NCE of 15. (c) UVPD using 1 laser pulse of 0.5 mJ. (e) a-EPD performed using UVPD (1 laser pulse of 0.3 mJ) followed by isolation of the charge-reduced 19−• radical ions and then CID using NCE of 23 (b), (d), (f) show sequence coverage and backbone cleavage maps based on relative intensities of fragment ions originating from different backbone cleavage sites derived from the corresponding HCD, UVPD, and a-EPD mass spectra.
MS/MS Analysis of Other Proteins
Figure 3
Figure 3. Sequence coverage produced by HCD, UVPD and a-EPD, as a function of precursor charge state for (a) ubiquitin, (b) myoglobin, (c) cytochrome c, (d) β-lactoglobulin, (e) carbonic anhydrase and (f) enolase.
Figure 4
Figure 4. (a) Fraction of abundance of fragment ion types generated by HCD (HCD−), UVPD (UVPD−), and a-EPD for protein anions and by UVPD for protein cations (UVPD+) with error bars showing standard deviations across six proteins and multiple charge states. (b) Violin plot showing the count of fragment ions as a function of fragment size (expressed as a percentage of the full protein length) for four MS/MS methods.
Fragmentation Features of Protein Anions
Figure 5
Figure 5. Backbone cleavage maps displaying relative intensities of fragment ions originating from different backbone cleavage sites of β lactoglobulin based on (a) HCD (NCE 14) of 14– charge state, (b) UVPD (1 laser pulse of 1.5 mJ) of 14– charge state, (c) a-EPD of 14– charge state [UVPD (1 laser pulse of 0.5 mJ) followed by isolation of the 13–• radical ions and then CID (NCE 20)], (d) HCD (NCE 14) of the 14+ charge state, and (e) UVPD (1 laser pulse of 1.5 mJ) of 14+ charge state. The backbone cleavage sites adjacent to the C66–C160 disulfide bond that resulted in the most abundant fragment ions are marked with circles and triangles on the backbone cleavage plots for UVPD (5b) and a-EPD (5c). (f) Isotope fitting results of the two of the most abundant fragments originating from cleavage adjacent to C66–C160 disulfide bond produced by UVPD ((a65 + 1)6– and b16012–) and a-EPD (c655– and b16012–) for the 14– charge state (all with score >0.8 in Prosight Native).
Comparison of UVPD of Protein Anions and Cations
Conclusions
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.5c00380.
(1) Mass spectra for optimization of ESI-MS in negative mode, (2) sequence maps and isotopic fitting results supporting identification of fragment ions, (3) bar graphs for sequence coverage and feature of fragment ions for varied charge states of proteins using HCD, UVPD (varied laser energy) and a-EPD, (4) bar graphs for preferential cleavage analysis of HCD, UVPD and a-EPD, and (5) mass spectra and bar graphs comparing positive and negative mode UVPD of all proteins (Figure S23) (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Jennifer S. Brodbelt - Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States;
 https://orcid.org/0000-0003-3207-0217;
https://orcid.org/0000-0003-3207-0217;
Acknowledgments
We acknowledge the following funding sources: NSF (Grant CHE-2203602) and the Welch Foundation (Grant F-1155).
References
This article references 70 other publications.
- 1Shuken, S. R. An Introduction to Mass Spectrometry-Based Proteomics. J. Proteome Res. 2023, 22 (7), 2151– 2171, DOI: 10.1021/acs.jproteome.2c00838Google ScholarThere is no corresponding record for this reference.
- 2Po, A.; Eyers, C. E. Top-Down Proteomics and the Challenges of True Proteoform Characterization. J. Proteome Res. 2023, 22 (12), 3663– 3675, DOI: 10.1021/acs.jproteome.3c00416Google ScholarThere is no corresponding record for this reference.
- 3Jiang, Y.; Rex, D. A. B.; Schuster, D.; Neely, B. A.; Rosano, G. L.; Volkmar, N.; Momenzadeh, A.; Peters-Clarke, T. M.; Egbert, S. B.; Kreimer, S. Comprehensive Overview of Bottom-Up Proteomics Using Mass Spectrometry. ACS Meas Sci. Au 2024, 4 (4), 338– 417, DOI: 10.1021/acsmeasuresciau.3c00068Google ScholarThere is no corresponding record for this reference.
- 4Chen, B.; Brown, K. A.; Lin, Z.; Ge, Y. Top-Down Proteomics: Ready for Prime Time?. Anal. Chem. 2018, 90 (1), 110– 127, DOI: 10.1021/acs.analchem.7b04747Google ScholarThere is no corresponding record for this reference.
- 5Meyer, J. G.; Niemi, N. M.; Pagliarini, D. J.; Coon, J. J. Quantitative shotgun proteome analysis by direct infusion. Nat. Methods 2020, 17 (12), 1222– 1228, DOI: 10.1038/s41592-020-00999-zGoogle ScholarThere is no corresponding record for this reference.
- 6Lenčo, J.; Jadeja, S.; Naplekov, D. K.; Krokhin, O. V.; Khalikova, M. A.; Chocholous, P.; Urban, J.; Broeckhoven, K.; Novakova, L.; Svec, F. Reversed-Phase Liquid Chromatography of Peptides for Bottom-Up Proteomics: A Tutorial. J. Proteome Res. 2022, 21 (12), 2846– 2892, DOI: 10.1021/acs.jproteome.2c00407Google ScholarThere is no corresponding record for this reference.
- 7Kurotani, A.; Tokmakov, A. A.; Sato, K. I.; Stefanov, V. E.; Yamada, Y.; Sakurai, T. Localization-specific distributions of protein pI in human proteome are governed by local pH and membrane charge. BMC Mol. Cell Biol. 2019, 20 (1), 36 DOI: 10.1186/s12860-019-0221-4Google ScholarThere is no corresponding record for this reference.
- 8Penanes, P. A.; Gorshkov, V.; Ivanov, M. V.; Gorshkov, M. V.; Kjeldsen, F. Potential of Negative-Ion-Mode Proteomics: An MS1-Only Approach. J. Proteome Res. 2023, 22 (8), 2734– 2742, DOI: 10.1021/acs.jproteome.3c00307Google ScholarThere is no corresponding record for this reference.
- 9Liigand, P.; Kaupmees, K.; Haav, K.; Liigand, J.; Leito, I.; Girod, M.; Antoine, R.; Kruve, A. Think Negative: Finding the Best Electrospray Ionization/MS Mode for Your Analyte. Anal. Chem. 2017, 89 (11), 5665– 5668, DOI: 10.1021/acs.analchem.7b00096Google ScholarThere is no corresponding record for this reference.
- 10Leutert, M.; Entwisle, S. W.; Villen, J. Decoding Post-Translational Modification Crosstalk With Proteomics. Mol. Cell. Proteomics 2021, 20, 100129 DOI: 10.1016/j.mcpro.2021.100129Google ScholarThere is no corresponding record for this reference.
- 11Ruhaak, L. R.; Xu, G.; Li, Q.; Goonatilleke, E.; Lebrilla, C. B. Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses. Chem. Rev. 2018, 118 (17), 7886– 7930, DOI: 10.1021/acs.chemrev.7b00732Google ScholarThere is no corresponding record for this reference.
- 12McAlister, G. C.; Russell, J. D.; Rumachik, N. G.; Hebert, A. S.; Syka, J. E.; Geer, L. Y.; Westphall, M. S.; Pagliarini, D. J.; Coon, J. J. Analysis of the acidic proteome with negative electron-transfer dissociation mass spectrometry. Anal. Chem. 2012, 84 (6), 2875– 2882, DOI: 10.1021/ac203430uGoogle ScholarThere is no corresponding record for this reference.
- 13Robinson, M. R.; Taliaferro, J. M.; Dalby, K. N.; Brodbelt, J. S. 193 nm Ultraviolet Photodissociation Mass Spectrometry for Phosphopeptide Characterization in the Positive and Negative Ion Modes. J. Proteome Res. 2016, 15 (8), 2739– 2748, DOI: 10.1021/acs.jproteome.6b00289Google ScholarThere is no corresponding record for this reference.
- 14Ewing, N. P.; Cassady, C. J. Dissociation of multiply charged negative ions for hirudin (54–65), fibrinopeptide B, and insulin A (oxidized). J. Am. Soc. Mass Spectrom. 2001, 12 (1), 105– 116, DOI: 10.1016/S1044-0305(00)00195-1Google ScholarThere is no corresponding record for this reference.
- 15Bowie, J. H.; Brinkworth, C. S.; Dua, S. Collision-induced fragmentations of the (M-H)- parent anions of underivatized peptides: an aid to structure determination and some unusual negative ion cleavages. Mass Spectrom Rev. 2002, 21 (2), 87– 107, DOI: 10.1002/mas.10022Google ScholarThere is no corresponding record for this reference.
- 16Brinkworth, C. S.; Dua, S.; McAnoy, A. M.; Bowie, J. H. Negative ion fragmentations of deprotonated peptides: backbone cleavages directed through both Asp and Glu. Rapid Commun. Mass Spectrom. 2001, 15 (20), 1965– 1973, DOI: 10.1002/rcm.457Google ScholarThere is no corresponding record for this reference.
- 17Zuo, M.-Q.; Sun, R.-X.; Fang, R.-Q.; He, S.-M.; Dong, M.-Q. Characterization of collision-induced dissociation of deprotonated peptides of 4–16 amino acids using high-resolution mass spectrometry. Int. J. Mass Spectrom. 2019, 445, 116186 DOI: 10.1016/j.ijms.2019.116186Google ScholarThere is no corresponding record for this reference.
- 18Chrisman, P. A.; McLuckey, S. A. Dissociations of disulfide-linked gaseous polypeptide/protein anions: ion chemistry with implications for protein identification and characterization. J. Proteome Res. 2002, 1 (6), 549– 557, DOI: 10.1021/pr025561zGoogle ScholarThere is no corresponding record for this reference.
- 19Kjeldsen, F.; Horning, O. B.; Jensen, S. S.; Giessing, A. M.; Jensen, O. N. Towards liquid chromatography time-scale peptide sequencing and characterization of post-translational modifications in the negative-ion mode using electron detachment dissociation tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19 (8), 1156– 1162, DOI: 10.1016/j.jasms.2008.04.031Google ScholarThere is no corresponding record for this reference.
- 20Kalli, A.; Grigorean, G.; Hakansson, K. Electron induced dissociation of singly deprotonated peptides. J. Am. Soc. Mass Spectrom. 2011, 22 (12), 2209– 2221, DOI: 10.1007/s13361-011-0233-6Google ScholarThere is no corresponding record for this reference.
- 21Yoo, H. J.; Wang, N.; Zhuang, S.; Song, H.; Hakansson, K. Negative-ion electron capture dissociation: radical-driven fragmentation of charge-increased gaseous peptide anions. J. Am. Chem. Soc. 2011, 133 (42), 16790– 16793, DOI: 10.1021/ja207736yGoogle ScholarThere is no corresponding record for this reference.
- 22Huzarska, M.; Ugalde, I.; Kaplan, D. A.; Hartmer, R.; Easterling, M. L.; Polfer, N. C. Negative electron transfer dissociation of deprotonated phosphopeptide anions: choice of radical cation reagent and competition between electron and proton transfer. Anal. Chem. 2010, 82 (7), 2873– 2878, DOI: 10.1021/ac9028592Google ScholarThere is no corresponding record for this reference.
- 23Riley, N. M.; Rush, M. J.; Rose, C. M.; Richards, A. L.; Kwiecien, N. W.; Bailey, D. J.; Hebert, A. S.; Westphall, M. S.; Coon, J. J. The Negative Mode Proteome with Activated Ion Negative Electron Transfer Dissociation (AI-NETD). Mol. Cell. Proteomics 2015, 14 (10), 2644– 2660, DOI: 10.1074/mcp.M115.049726Google ScholarThere is no corresponding record for this reference.
- 24Shaw, J. B.; Kaplan, D. A.; Brodbelt, J. S. Activated ion negative electron transfer dissociation of multiply charged peptide anions. Anal. Chem. 2013, 85 (9), 4721– 4728, DOI: 10.1021/ac4005315Google ScholarThere is no corresponding record for this reference.
- 25Madsen, J. A.; Kaoud, T. S.; Dalby, K. N.; Brodbelt, J. S. 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics 2011, 11 (7), 1329– 1334, DOI: 10.1002/pmic.201000565Google ScholarThere is no corresponding record for this reference.
- 26Greer, S. M.; Bern, M.; Becker, C.; Brodbelt, J. S. Extending Proteome Coverage by Combining MS/MS Methods and a Modified Bioinformatics Platform Adapted for Database Searching of Positive and Negative Polarity 193 nm Ultraviolet Photodissociation Mass Spectra. J. Proteome Res. 2018, 1340– 1347, DOI: 10.1021/acs.jproteome.7b00673Google ScholarThere is no corresponding record for this reference.
- 27Shaw, J. B.; Madsen, J. A.; Xu, H.; Brodbelt, J. S. Systematic comparison of ultraviolet photodissociation and electron transfer dissociation for peptide anion characterization. J. Am. Soc. Mass Spectrom. 2012, 23 (10), 1707– 1715, DOI: 10.1007/s13361-012-0424-9Google ScholarThere is no corresponding record for this reference.
- 28Brodbelt, J. S.; Morrison, L. J.; Santos, I. Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev. 2020, 120 (7), 3328– 3380, DOI: 10.1021/acs.chemrev.9b00440Google ScholarThere is no corresponding record for this reference.
- 29LeBlanc, B. M.; Moreno, R. Y.; Escobar, E. E.; Venkat Ramani, M. K.; Brodbelt, J. S.; Zhang, Y. What’s all the phos about? Insights into the phosphorylation state of the RNA polymerase II C-terminal domain via mass spectrometry. RSC Chem. Biol. 2021, 2 (4), 1084– 1095, DOI: 10.1039/D1CB00083GGoogle ScholarThere is no corresponding record for this reference.
- 30Bashyal, A.; Brodbelt, J. S. Uncommon posttranslational modifications in proteomics: ADP-ribosylation, tyrosine nitration, and tyrosine sulfation. Mass Spectrom. Rev. 2024, 43 (2), 289– 326, DOI: 10.1002/mas.21811Google ScholarThere is no corresponding record for this reference.
- 31Madsen, J. A.; Ko, B. J.; Xu, H.; Iwashkiw, J. A.; Robotham, S. A.; Shaw, J. B.; Feldman, M. F.; Brodbelt, J. S. Concurrent automated sequencing of the glycan and peptide portions of O-linked glycopeptide anions by ultraviolet photodissociation mass spectrometry. Anal. Chem. 2013, 85 (19), 9253– 9261, DOI: 10.1021/ac4021177Google ScholarThere is no corresponding record for this reference.
- 32Robinson, M. R.; Brodbelt, J. S. Integrating Weak Anion Exchange and Ultraviolet Photodissociation Mass Spectrometry with Strategic Modulation of Peptide Basicity for the Enrichment of Sulfopeptides. Anal. Chem. 2016, 88 (22), 11037– 11045, DOI: 10.1021/acs.analchem.6b02899Google ScholarThere is no corresponding record for this reference.
- 33Antoine, R.; Joly, L.; Tabarin, T.; Broyer, M.; Dugourd, P.; Lemoine, J. Photo-induced formation of radical anion peptides. Electron photodetachment dissociation experiments. Rapid Commun. Mass Spectrom. 2007, 21 (2), 265– 268, DOI: 10.1002/rcm.2810Google ScholarThere is no corresponding record for this reference.
- 34Larraillet, V.; Antoine, R.; Dugourd, P.; Lemoine, J. Activated-electron photodetachment dissociation for the structural characterization of protein polyanions. Anal. Chem. 2009, 81 (20), 8410– 8416, DOI: 10.1021/ac901304dGoogle ScholarThere is no corresponding record for this reference.
- 35Cech, N. B.; Enke, C. G. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 2001, 20 (6), 362– 387, DOI: 10.1002/mas.10008Google ScholarThere is no corresponding record for this reference.
- 36Konermann, L.; Douglas, D. J. Unfolding of proteins monitored by electrospray ionization mass spectrometry: a comparison of positive and negative ion modes. J. Am. Soc. Mass Spectrom. 1998, 9 (12), 1248– 1254, DOI: 10.1016/S1044-0305(98)00103-2Google ScholarThere is no corresponding record for this reference.
- 37Sterling, H. J.; Daly, M. P.; Feld, G. K.; Thoren, K. L.; Kintzer, A. F.; Krantz, B. A.; Williams, E. R. Effects of supercharging reagents on noncovalent complex structure in electrospray ionization from aqueous solutions. J. Am. Soc. Mass Spectrom. 2010, 21 (10), 1762– 1774, DOI: 10.1016/j.jasms.2010.06.012Google ScholarThere is no corresponding record for this reference.
- 38Kharlamova, A.; McLuckey, S. A. Negative electrospray droplet exposure to gaseous bases for the manipulation of protein charge state distributions. Anal. Chem. 2011, 83 (1), 431– 437, DOI: 10.1021/ac1027319Google ScholarThere is no corresponding record for this reference.
- 39Nišavić, M.; Hozic, A.; Hamersak, Z.; Radic, M.; Butorac, A.; Duvnjak, M.; Cindric, M. High-Efficiency Microflow and Nanoflow Negative Electrospray Ionization of Peptides Induced by Gas-Phase Proton Transfer Reactions. Anal. Chem. 2017, 89 (9), 4847– 4854, DOI: 10.1021/acs.analchem.6b04466Google ScholarThere is no corresponding record for this reference.
- 40McClory, P. J.; Hakansson, K. Corona Discharge Suppression in Negative Ion Mode Nanoelectrospray Ionization via Trifluoroethanol Addition. Anal. Chem. 2017, 89 (19), 10188– 10193, DOI: 10.1021/acs.analchem.7b01225Google ScholarThere is no corresponding record for this reference.
- 41Stevens, A.; Abramsson, M. L.; Agasid, M. T.; Allison, T. M.; Landreh, M. Stabilization of Protein Interactions through Electrospray Additives in Negative Ion Mode Native Mass Spectrometry. Anal. Chem. 2025, 97 (20), 10738– 10744, DOI: 10.1021/acs.analchem.5c00757Google ScholarThere is no corresponding record for this reference.
- 42Ogorzalek Loo, R. R.; Lakshmanan, R.; Loo, J. A. What protein charging (and supercharging) reveal about the mechanism of electrospray ionization. J. Am. Soc. Mass Spectrom. 2014, 25 (10), 1675– 1693, DOI: 10.1007/s13361-014-0965-1Google ScholarThere is no corresponding record for this reference.
- 43Greer, S. M.; Cannon, J. R.; Brodbelt, J. S. Improvement of shotgun proteomics in the negative mode by carbamylation of peptides and ultraviolet photodissociation mass spectrometry. Anal. Chem. 2014, 86 (24), 12285– 12290, DOI: 10.1021/ac5035314Google ScholarThere is no corresponding record for this reference.
- 44Le Blanc, J. C. Y.; Guevremont, R.; Siu, K. W. M. Electrospray mass spectrometry of some proteins and the aqueous solution acid/base equilibrium model in the negative ion detection mode. Int. J. Mass Spectrom. 1993, 125 (2–3), 145– 153, DOI: 10.1016/0168-1176(93)80037-FGoogle ScholarThere is no corresponding record for this reference.
- 45Ganisl, B.; Taucher, M.; Riml, C.; Breuker, K. Charge as you like! Efficient manipulation of negative ion net charge in electrospray ionization of proteins and nucleic acids. Eur. J. Mass Spectrom 2011, 17 (4), 333– 343, DOI: 10.1255/ejms.1140Google ScholarThere is no corresponding record for this reference.
- 46Madsen, J. A.; Xu, H.; Robinson, M. R.; Horton, A. P.; Shaw, J. B.; Giles, D. K.; Kaoud, T. S.; Dalby, K. N.; Trent, M. S.; Brodbelt, J. S. High-throughput database search and large-scale negative polarity liquid chromatography-tandem mass spectrometry with ultraviolet photodissociation for complex proteomic samples. Mol. Cell. Proteomics 2013, 12 (9), 2604– 2614, DOI: 10.1074/mcp.O113.028258Google ScholarThere is no corresponding record for this reference.
- 47Donnelly, D. P.; Rawlins, C. M.; DeHart, C. J.; Fornelli, L.; Schachner, L. F.; Lin, Z.; Lippens, J. L.; Aluri, K. C.; Sarin, R.; Chen, B. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16 (7), 587– 594, DOI: 10.1038/s41592-019-0457-0Google ScholarThere is no corresponding record for this reference.
- 48Roberts, D. S.; Loo, J. A.; Tsybin, Y. O.; Liu, X.; Wu, S.; Chamot-Rooke, J.; Agar, J. N.; Pasa-Tolic, L.; Smith, L. M.; Ge, Y. Top-down proteomics. Nat. Rev. Methods Primers 2024, 4 (1), 38 DOI: 10.1038/s43586-024-00318-2Google ScholarThere is no corresponding record for this reference.
- 49Larson, E. J.; Roberts, D. S.; Melby, J. A.; Buck, K. M.; Zhu, Y.; Zhou, S.; Han, L.; Zhang, Q.; Ge, Y. High-Throughput Multi-attribute Analysis of Antibody-Drug Conjugates Enabled by Trapped Ion Mobility Spectrometry and Top-Down Mass Spectrometry. Anal. Chem. 2021, 93 (29), 10013– 10021, DOI: 10.1021/acs.analchem.1c00150Google ScholarThere is no corresponding record for this reference.
- 50Riley, N. M.; Westphall, M. S.; Coon, J. J. Activated Ion-Electron Transfer Dissociation Enables Comprehensive Top-Down Protein Fragmentation. J. Proteome Res. 2017, 16 (7), 2653– 2659, DOI: 10.1021/acs.jproteome.7b00249Google ScholarThere is no corresponding record for this reference.
- 51Wei, B.; Zenaidee, M. A.; Lantz, C.; Williams, B. J.; Totten, S.; Ogorzalek Loo, R. R.; Loo, J. A. Top-down mass spectrometry and assigning internal fragments for determining disulfide bond positions in proteins. Analyst 2022, 148 (1), 26– 37, DOI: 10.1039/D2AN01517JGoogle ScholarThere is no corresponding record for this reference.
- 52Dunham, S. D.; Brodbelt, J. S. Enhancing Top-Down Analysis of Proteins by Combining Ultraviolet Photodissociation (UVPD), Proton-Transfer Charge Reduction (PTCR), and Gas-Phase Fractionation to Alleviate the Impact of Nondissociated Precursor Ions. J. Am. Soc. Mass Spectrom. 2024, 35 (2), 255– 265, DOI: 10.1021/jasms.3c00351Google ScholarThere is no corresponding record for this reference.
- 53Walker, J. N.; Lam, R.; Brodbelt, J. S. Enhanced Characterization of Histones Using 193 nm Ultraviolet Photodissociation and Proton Transfer Charge Reduction. Anal. Chem. 2023, 95 (14), 5985– 5993, DOI: 10.1021/acs.analchem.2c05765Google ScholarThere is no corresponding record for this reference.
- 54Watts, E.; Thyer, R.; Ellington, A. D.; Brodbelt, J. S. Integrated Top-Down and Bottom-Up Mass Spectrometry for Characterization of Diselenide Bridging Patterns of Synthetic Selenoproteins. Anal. Chem. 2022, 94 (32), 11175– 11184, DOI: 10.1021/acs.analchem.2c01433Google ScholarThere is no corresponding record for this reference.
- 55Brodbelt, J. S. Deciphering combinatorial post-translational modifications by top-down mass spectrometry. Curr. Opin. Chem. Biol. 2022, 70, 102180 DOI: 10.1016/j.cbpa.2022.102180Google ScholarThere is no corresponding record for this reference.
- 56Fornelli, L.; Srzentic, K.; Toby, T. K.; Doubleday, P. F.; Huguet, R.; Mullen, C.; Melani, R. D.; Dos Santos Seckler, H.; DeHart, C. J.; Weisbrod, C. R. Thorough Performance Evaluation of 213 nm Ultraviolet Photodissociation for Top-down Proteomics. Mol. Cell. Proteomics 2020, 19 (2), 405– 420, DOI: 10.1074/mcp.TIR119.001638Google ScholarThere is no corresponding record for this reference.
- 57Oates, R. N.; Lieu, L. B.; Srzentic, K.; Damoc, E.; Fornelli, L. Characterization of a Monoclonal Antibody by Native and Denaturing Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2024, 35 (9), 2197– 2208, DOI: 10.1021/jasms.4c00224Google ScholarThere is no corresponding record for this reference.
- 58Cannon, J. R.; Cammarata, M. B.; Robotham, S. A.; Cotham, V. C.; Shaw, J. B.; Fellers, R. T.; Early, B. P.; Thomas, P. M.; Kelleher, N. L.; Brodbelt, J. S. Ultraviolet photodissociation for characterization of whole proteins on a chromatographic time scale. Anal. Chem. 2014, 86 (4), 2185– 2192, DOI: 10.1021/ac403859aGoogle ScholarThere is no corresponding record for this reference.
- 59Lanzillotti, M.; Brodbelt, J. S. Comparison of Top-Down Protein Fragmentation Induced by 213 and 193 nm UVPD. J. Am. Soc. Mass Spectrom. 2023, 34 (2), 279– 285, DOI: 10.1021/jasms.2c00288Google ScholarThere is no corresponding record for this reference.
- 60Juetten, K. J.; Brodbelt, J. S. Top-Down Analysis of Supercharged Proteins Using Collision-, Electron-, and Photon-Based Activation Methods. J. Am. Soc. Mass Spectrom. 2023, 34 (7), 1467– 1476, DOI: 10.1021/jasms.3c00138Google ScholarThere is no corresponding record for this reference.
- 61Hyde, A. M.; Calabria, R.; Arvary, R.; Wang, X.; Klapars, A. Investigating the Underappreciated Hydrolytic Instability of 1,8-Diazabicyclo[5.4.0]undec-7-ene and Related Unsaturated Nitrogenous Bases. Org. Process Res. Dev. 2019, 23 (9), 1860– 1871, DOI: 10.1021/acs.oprd.9b00187Google ScholarThere is no corresponding record for this reference.
- 62Juetten, K. J.; Brodbelt, J. S. MS-TAFI: A Tool for the Analysis of Fragment Ions Generated from Intact Proteins. J. Proteome Res. 2023, 22 (2), 546– 550, DOI: 10.1021/acs.jproteome.2c00594Google ScholarThere is no corresponding record for this reference.
- 63Durbin, K. R.; Robey, M. T.; Voong, L. N.; Fellers, R. T.; Lutomski, C. A.; El-Baba, T. J.; Robinson, C. V.; Kelleher, N. L. ProSight Native: Defining Protein Complex Composition from Native Top-Down Mass Spectrometry Data. J. Proteome Res. 2023, 22 (8), 2660– 2668, DOI: 10.1021/acs.jproteome.3c00171Google ScholarThere is no corresponding record for this reference.
- 64Li, J.; Santambrogio, C.; Brocca, S.; Rossetti, G.; Carloni, P.; Grandori, R. Conformational effects in protein electrospray-ionization mass spectrometry. Mass Spectrom Rev. 2016, 35 (1), 111– 122, DOI: 10.1002/mas.21465Google ScholarThere is no corresponding record for this reference.
- 65Reid, G. E.; Wu, J.; Chrisman, P. A.; Wells, J. M.; McLuckey, S. A. Charge-state-dependent sequence analysis of protonated ubiquitin ions via ion trap tandem mass spectrometry. Anal. Chem. 2001, 73 (14), 3274– 3281, DOI: 10.1021/ac0101095Google ScholarThere is no corresponding record for this reference.
- 66Cobb, J. S.; Easterling, M. L.; Agar, J. N. Structural characterization of intact proteins is enhanced by prevalent fragmentation pathways rarely observed for peptides. J. Am. Soc. Mass Spectrom. 2010, 21 (6), 949– 959, DOI: 10.1016/j.jasms.2010.02.009Google ScholarThere is no corresponding record for this reference.
- 67Hellinger, J.; Brodbelt, J. S. Impact of Charge State on Characterization of Large Middle-Down Sized Peptides by Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2024, 35 (8), 1647– 1656, DOI: 10.1021/jasms.3c00405Google ScholarThere is no corresponding record for this reference.
- 68Carrera, G. V. S. M.; Jordão, N.; Santos, M. M.; da Ponte, M. N.; Branco, L. C. Reversible systems based on CO2, amino-acids and organic superbases. RSC Adv. 2015, 5 (45), 35564– 35571, DOI: 10.1039/C5RA03474DGoogle ScholarThere is no corresponding record for this reference.
- 69Bonner, J.; Talbert, L. E.; Akkawi, N.; Julian, R. R. Simplified identification of disulfide, trisulfide, and thioether pairs with 213 nm UVPD. Analyst 2018, 143 (21), 5176– 5184, DOI: 10.1039/C8AN01582AGoogle ScholarThere is no corresponding record for this reference.
- 70Macias, L. A.; Brodbelt, J. S. Investigation of Product Ions Generated by 193 nm Ultraviolet Photodissociation of Peptides and Proteins Containing Disulfide Bonds. J. Am. Soc. Mass Spectrom. 2022, 33 (7), 1315– 1324, DOI: 10.1021/jasms.2c00124Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
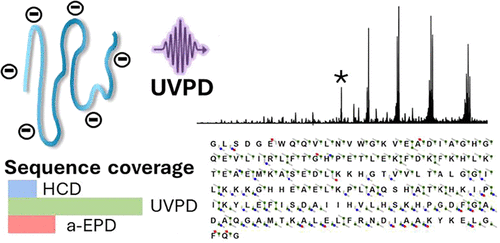
Figure 1
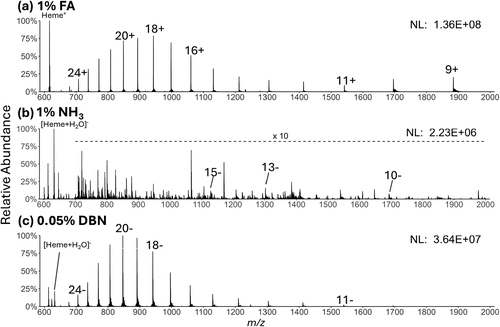
Figure 1. MS1 spectra of solutions containing 10 μM myoglobin and (a) 1% formic acid in positive mode, (b) with 1% NH3·H2O in negative mode, and (c) with 0.05% DBN in negative mode. Selected charge states of the protein and the heme are annotated.
Figure 2
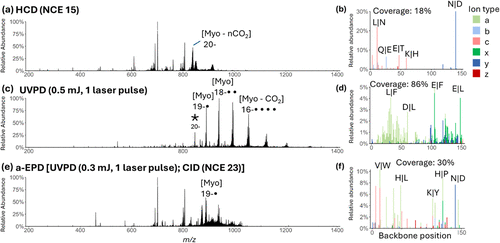
Figure 2. MS/MS spectra of myoglobin (20–, m/z 846, indicated by an asterisk in the UVPD spectrum). (a) HCD using NCE of 15. (c) UVPD using 1 laser pulse of 0.5 mJ. (e) a-EPD performed using UVPD (1 laser pulse of 0.3 mJ) followed by isolation of the charge-reduced 19−• radical ions and then CID using NCE of 23 (b), (d), (f) show sequence coverage and backbone cleavage maps based on relative intensities of fragment ions originating from different backbone cleavage sites derived from the corresponding HCD, UVPD, and a-EPD mass spectra.
Figure 3
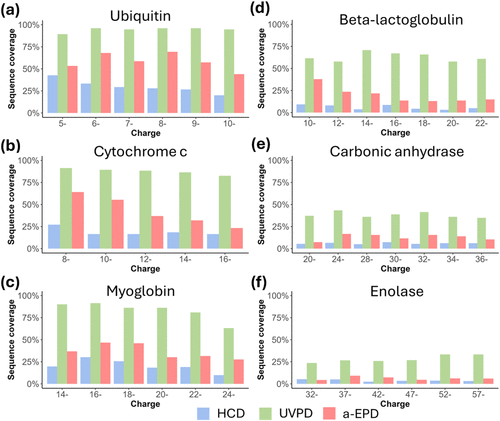
Figure 3. Sequence coverage produced by HCD, UVPD and a-EPD, as a function of precursor charge state for (a) ubiquitin, (b) myoglobin, (c) cytochrome c, (d) β-lactoglobulin, (e) carbonic anhydrase and (f) enolase.
Figure 4

Figure 4. (a) Fraction of abundance of fragment ion types generated by HCD (HCD−), UVPD (UVPD−), and a-EPD for protein anions and by UVPD for protein cations (UVPD+) with error bars showing standard deviations across six proteins and multiple charge states. (b) Violin plot showing the count of fragment ions as a function of fragment size (expressed as a percentage of the full protein length) for four MS/MS methods.
Figure 5
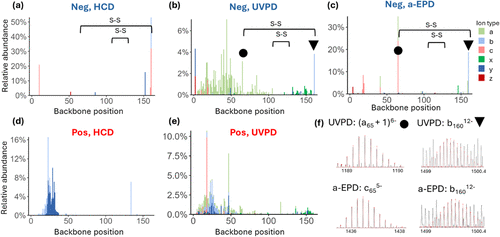
Figure 5. Backbone cleavage maps displaying relative intensities of fragment ions originating from different backbone cleavage sites of β lactoglobulin based on (a) HCD (NCE 14) of 14– charge state, (b) UVPD (1 laser pulse of 1.5 mJ) of 14– charge state, (c) a-EPD of 14– charge state [UVPD (1 laser pulse of 0.5 mJ) followed by isolation of the 13–• radical ions and then CID (NCE 20)], (d) HCD (NCE 14) of the 14+ charge state, and (e) UVPD (1 laser pulse of 1.5 mJ) of 14+ charge state. The backbone cleavage sites adjacent to the C66–C160 disulfide bond that resulted in the most abundant fragment ions are marked with circles and triangles on the backbone cleavage plots for UVPD (5b) and a-EPD (5c). (f) Isotope fitting results of the two of the most abundant fragments originating from cleavage adjacent to C66–C160 disulfide bond produced by UVPD ((a65 + 1)6– and b16012–) and a-EPD (c655– and b16012–) for the 14– charge state (all with score >0.8 in Prosight Native).
References
This article references 70 other publications.
- 1Shuken, S. R. An Introduction to Mass Spectrometry-Based Proteomics. J. Proteome Res. 2023, 22 (7), 2151– 2171, DOI: 10.1021/acs.jproteome.2c00838There is no corresponding record for this reference.
- 2Po, A.; Eyers, C. E. Top-Down Proteomics and the Challenges of True Proteoform Characterization. J. Proteome Res. 2023, 22 (12), 3663– 3675, DOI: 10.1021/acs.jproteome.3c00416There is no corresponding record for this reference.
- 3Jiang, Y.; Rex, D. A. B.; Schuster, D.; Neely, B. A.; Rosano, G. L.; Volkmar, N.; Momenzadeh, A.; Peters-Clarke, T. M.; Egbert, S. B.; Kreimer, S. Comprehensive Overview of Bottom-Up Proteomics Using Mass Spectrometry. ACS Meas Sci. Au 2024, 4 (4), 338– 417, DOI: 10.1021/acsmeasuresciau.3c00068There is no corresponding record for this reference.
- 4Chen, B.; Brown, K. A.; Lin, Z.; Ge, Y. Top-Down Proteomics: Ready for Prime Time?. Anal. Chem. 2018, 90 (1), 110– 127, DOI: 10.1021/acs.analchem.7b04747There is no corresponding record for this reference.
- 5Meyer, J. G.; Niemi, N. M.; Pagliarini, D. J.; Coon, J. J. Quantitative shotgun proteome analysis by direct infusion. Nat. Methods 2020, 17 (12), 1222– 1228, DOI: 10.1038/s41592-020-00999-zThere is no corresponding record for this reference.
- 6Lenčo, J.; Jadeja, S.; Naplekov, D. K.; Krokhin, O. V.; Khalikova, M. A.; Chocholous, P.; Urban, J.; Broeckhoven, K.; Novakova, L.; Svec, F. Reversed-Phase Liquid Chromatography of Peptides for Bottom-Up Proteomics: A Tutorial. J. Proteome Res. 2022, 21 (12), 2846– 2892, DOI: 10.1021/acs.jproteome.2c00407There is no corresponding record for this reference.
- 7Kurotani, A.; Tokmakov, A. A.; Sato, K. I.; Stefanov, V. E.; Yamada, Y.; Sakurai, T. Localization-specific distributions of protein pI in human proteome are governed by local pH and membrane charge. BMC Mol. Cell Biol. 2019, 20 (1), 36 DOI: 10.1186/s12860-019-0221-4There is no corresponding record for this reference.
- 8Penanes, P. A.; Gorshkov, V.; Ivanov, M. V.; Gorshkov, M. V.; Kjeldsen, F. Potential of Negative-Ion-Mode Proteomics: An MS1-Only Approach. J. Proteome Res. 2023, 22 (8), 2734– 2742, DOI: 10.1021/acs.jproteome.3c00307There is no corresponding record for this reference.
- 9Liigand, P.; Kaupmees, K.; Haav, K.; Liigand, J.; Leito, I.; Girod, M.; Antoine, R.; Kruve, A. Think Negative: Finding the Best Electrospray Ionization/MS Mode for Your Analyte. Anal. Chem. 2017, 89 (11), 5665– 5668, DOI: 10.1021/acs.analchem.7b00096There is no corresponding record for this reference.
- 10Leutert, M.; Entwisle, S. W.; Villen, J. Decoding Post-Translational Modification Crosstalk With Proteomics. Mol. Cell. Proteomics 2021, 20, 100129 DOI: 10.1016/j.mcpro.2021.100129There is no corresponding record for this reference.
- 11Ruhaak, L. R.; Xu, G.; Li, Q.; Goonatilleke, E.; Lebrilla, C. B. Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses. Chem. Rev. 2018, 118 (17), 7886– 7930, DOI: 10.1021/acs.chemrev.7b00732There is no corresponding record for this reference.
- 12McAlister, G. C.; Russell, J. D.; Rumachik, N. G.; Hebert, A. S.; Syka, J. E.; Geer, L. Y.; Westphall, M. S.; Pagliarini, D. J.; Coon, J. J. Analysis of the acidic proteome with negative electron-transfer dissociation mass spectrometry. Anal. Chem. 2012, 84 (6), 2875– 2882, DOI: 10.1021/ac203430uThere is no corresponding record for this reference.
- 13Robinson, M. R.; Taliaferro, J. M.; Dalby, K. N.; Brodbelt, J. S. 193 nm Ultraviolet Photodissociation Mass Spectrometry for Phosphopeptide Characterization in the Positive and Negative Ion Modes. J. Proteome Res. 2016, 15 (8), 2739– 2748, DOI: 10.1021/acs.jproteome.6b00289There is no corresponding record for this reference.
- 14Ewing, N. P.; Cassady, C. J. Dissociation of multiply charged negative ions for hirudin (54–65), fibrinopeptide B, and insulin A (oxidized). J. Am. Soc. Mass Spectrom. 2001, 12 (1), 105– 116, DOI: 10.1016/S1044-0305(00)00195-1There is no corresponding record for this reference.
- 15Bowie, J. H.; Brinkworth, C. S.; Dua, S. Collision-induced fragmentations of the (M-H)- parent anions of underivatized peptides: an aid to structure determination and some unusual negative ion cleavages. Mass Spectrom Rev. 2002, 21 (2), 87– 107, DOI: 10.1002/mas.10022There is no corresponding record for this reference.
- 16Brinkworth, C. S.; Dua, S.; McAnoy, A. M.; Bowie, J. H. Negative ion fragmentations of deprotonated peptides: backbone cleavages directed through both Asp and Glu. Rapid Commun. Mass Spectrom. 2001, 15 (20), 1965– 1973, DOI: 10.1002/rcm.457There is no corresponding record for this reference.
- 17Zuo, M.-Q.; Sun, R.-X.; Fang, R.-Q.; He, S.-M.; Dong, M.-Q. Characterization of collision-induced dissociation of deprotonated peptides of 4–16 amino acids using high-resolution mass spectrometry. Int. J. Mass Spectrom. 2019, 445, 116186 DOI: 10.1016/j.ijms.2019.116186There is no corresponding record for this reference.
- 18Chrisman, P. A.; McLuckey, S. A. Dissociations of disulfide-linked gaseous polypeptide/protein anions: ion chemistry with implications for protein identification and characterization. J. Proteome Res. 2002, 1 (6), 549– 557, DOI: 10.1021/pr025561zThere is no corresponding record for this reference.
- 19Kjeldsen, F.; Horning, O. B.; Jensen, S. S.; Giessing, A. M.; Jensen, O. N. Towards liquid chromatography time-scale peptide sequencing and characterization of post-translational modifications in the negative-ion mode using electron detachment dissociation tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19 (8), 1156– 1162, DOI: 10.1016/j.jasms.2008.04.031There is no corresponding record for this reference.
- 20Kalli, A.; Grigorean, G.; Hakansson, K. Electron induced dissociation of singly deprotonated peptides. J. Am. Soc. Mass Spectrom. 2011, 22 (12), 2209– 2221, DOI: 10.1007/s13361-011-0233-6There is no corresponding record for this reference.
- 21Yoo, H. J.; Wang, N.; Zhuang, S.; Song, H.; Hakansson, K. Negative-ion electron capture dissociation: radical-driven fragmentation of charge-increased gaseous peptide anions. J. Am. Chem. Soc. 2011, 133 (42), 16790– 16793, DOI: 10.1021/ja207736yThere is no corresponding record for this reference.
- 22Huzarska, M.; Ugalde, I.; Kaplan, D. A.; Hartmer, R.; Easterling, M. L.; Polfer, N. C. Negative electron transfer dissociation of deprotonated phosphopeptide anions: choice of radical cation reagent and competition between electron and proton transfer. Anal. Chem. 2010, 82 (7), 2873– 2878, DOI: 10.1021/ac9028592There is no corresponding record for this reference.
- 23Riley, N. M.; Rush, M. J.; Rose, C. M.; Richards, A. L.; Kwiecien, N. W.; Bailey, D. J.; Hebert, A. S.; Westphall, M. S.; Coon, J. J. The Negative Mode Proteome with Activated Ion Negative Electron Transfer Dissociation (AI-NETD). Mol. Cell. Proteomics 2015, 14 (10), 2644– 2660, DOI: 10.1074/mcp.M115.049726There is no corresponding record for this reference.
- 24Shaw, J. B.; Kaplan, D. A.; Brodbelt, J. S. Activated ion negative electron transfer dissociation of multiply charged peptide anions. Anal. Chem. 2013, 85 (9), 4721– 4728, DOI: 10.1021/ac4005315There is no corresponding record for this reference.
- 25Madsen, J. A.; Kaoud, T. S.; Dalby, K. N.; Brodbelt, J. S. 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics 2011, 11 (7), 1329– 1334, DOI: 10.1002/pmic.201000565There is no corresponding record for this reference.
- 26Greer, S. M.; Bern, M.; Becker, C.; Brodbelt, J. S. Extending Proteome Coverage by Combining MS/MS Methods and a Modified Bioinformatics Platform Adapted for Database Searching of Positive and Negative Polarity 193 nm Ultraviolet Photodissociation Mass Spectra. J. Proteome Res. 2018, 1340– 1347, DOI: 10.1021/acs.jproteome.7b00673There is no corresponding record for this reference.
- 27Shaw, J. B.; Madsen, J. A.; Xu, H.; Brodbelt, J. S. Systematic comparison of ultraviolet photodissociation and electron transfer dissociation for peptide anion characterization. J. Am. Soc. Mass Spectrom. 2012, 23 (10), 1707– 1715, DOI: 10.1007/s13361-012-0424-9There is no corresponding record for this reference.
- 28Brodbelt, J. S.; Morrison, L. J.; Santos, I. Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev. 2020, 120 (7), 3328– 3380, DOI: 10.1021/acs.chemrev.9b00440There is no corresponding record for this reference.
- 29LeBlanc, B. M.; Moreno, R. Y.; Escobar, E. E.; Venkat Ramani, M. K.; Brodbelt, J. S.; Zhang, Y. What’s all the phos about? Insights into the phosphorylation state of the RNA polymerase II C-terminal domain via mass spectrometry. RSC Chem. Biol. 2021, 2 (4), 1084– 1095, DOI: 10.1039/D1CB00083GThere is no corresponding record for this reference.
- 30Bashyal, A.; Brodbelt, J. S. Uncommon posttranslational modifications in proteomics: ADP-ribosylation, tyrosine nitration, and tyrosine sulfation. Mass Spectrom. Rev. 2024, 43 (2), 289– 326, DOI: 10.1002/mas.21811There is no corresponding record for this reference.
- 31Madsen, J. A.; Ko, B. J.; Xu, H.; Iwashkiw, J. A.; Robotham, S. A.; Shaw, J. B.; Feldman, M. F.; Brodbelt, J. S. Concurrent automated sequencing of the glycan and peptide portions of O-linked glycopeptide anions by ultraviolet photodissociation mass spectrometry. Anal. Chem. 2013, 85 (19), 9253– 9261, DOI: 10.1021/ac4021177There is no corresponding record for this reference.
- 32Robinson, M. R.; Brodbelt, J. S. Integrating Weak Anion Exchange and Ultraviolet Photodissociation Mass Spectrometry with Strategic Modulation of Peptide Basicity for the Enrichment of Sulfopeptides. Anal. Chem. 2016, 88 (22), 11037– 11045, DOI: 10.1021/acs.analchem.6b02899There is no corresponding record for this reference.
- 33Antoine, R.; Joly, L.; Tabarin, T.; Broyer, M.; Dugourd, P.; Lemoine, J. Photo-induced formation of radical anion peptides. Electron photodetachment dissociation experiments. Rapid Commun. Mass Spectrom. 2007, 21 (2), 265– 268, DOI: 10.1002/rcm.2810There is no corresponding record for this reference.
- 34Larraillet, V.; Antoine, R.; Dugourd, P.; Lemoine, J. Activated-electron photodetachment dissociation for the structural characterization of protein polyanions. Anal. Chem. 2009, 81 (20), 8410– 8416, DOI: 10.1021/ac901304dThere is no corresponding record for this reference.
- 35Cech, N. B.; Enke, C. G. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 2001, 20 (6), 362– 387, DOI: 10.1002/mas.10008There is no corresponding record for this reference.
- 36Konermann, L.; Douglas, D. J. Unfolding of proteins monitored by electrospray ionization mass spectrometry: a comparison of positive and negative ion modes. J. Am. Soc. Mass Spectrom. 1998, 9 (12), 1248– 1254, DOI: 10.1016/S1044-0305(98)00103-2There is no corresponding record for this reference.
- 37Sterling, H. J.; Daly, M. P.; Feld, G. K.; Thoren, K. L.; Kintzer, A. F.; Krantz, B. A.; Williams, E. R. Effects of supercharging reagents on noncovalent complex structure in electrospray ionization from aqueous solutions. J. Am. Soc. Mass Spectrom. 2010, 21 (10), 1762– 1774, DOI: 10.1016/j.jasms.2010.06.012There is no corresponding record for this reference.
- 38Kharlamova, A.; McLuckey, S. A. Negative electrospray droplet exposure to gaseous bases for the manipulation of protein charge state distributions. Anal. Chem. 2011, 83 (1), 431– 437, DOI: 10.1021/ac1027319There is no corresponding record for this reference.
- 39Nišavić, M.; Hozic, A.; Hamersak, Z.; Radic, M.; Butorac, A.; Duvnjak, M.; Cindric, M. High-Efficiency Microflow and Nanoflow Negative Electrospray Ionization of Peptides Induced by Gas-Phase Proton Transfer Reactions. Anal. Chem. 2017, 89 (9), 4847– 4854, DOI: 10.1021/acs.analchem.6b04466There is no corresponding record for this reference.
- 40McClory, P. J.; Hakansson, K. Corona Discharge Suppression in Negative Ion Mode Nanoelectrospray Ionization via Trifluoroethanol Addition. Anal. Chem. 2017, 89 (19), 10188– 10193, DOI: 10.1021/acs.analchem.7b01225There is no corresponding record for this reference.
- 41Stevens, A.; Abramsson, M. L.; Agasid, M. T.; Allison, T. M.; Landreh, M. Stabilization of Protein Interactions through Electrospray Additives in Negative Ion Mode Native Mass Spectrometry. Anal. Chem. 2025, 97 (20), 10738– 10744, DOI: 10.1021/acs.analchem.5c00757There is no corresponding record for this reference.
- 42Ogorzalek Loo, R. R.; Lakshmanan, R.; Loo, J. A. What protein charging (and supercharging) reveal about the mechanism of electrospray ionization. J. Am. Soc. Mass Spectrom. 2014, 25 (10), 1675– 1693, DOI: 10.1007/s13361-014-0965-1There is no corresponding record for this reference.
- 43Greer, S. M.; Cannon, J. R.; Brodbelt, J. S. Improvement of shotgun proteomics in the negative mode by carbamylation of peptides and ultraviolet photodissociation mass spectrometry. Anal. Chem. 2014, 86 (24), 12285– 12290, DOI: 10.1021/ac5035314There is no corresponding record for this reference.
- 44Le Blanc, J. C. Y.; Guevremont, R.; Siu, K. W. M. Electrospray mass spectrometry of some proteins and the aqueous solution acid/base equilibrium model in the negative ion detection mode. Int. J. Mass Spectrom. 1993, 125 (2–3), 145– 153, DOI: 10.1016/0168-1176(93)80037-FThere is no corresponding record for this reference.
- 45Ganisl, B.; Taucher, M.; Riml, C.; Breuker, K. Charge as you like! Efficient manipulation of negative ion net charge in electrospray ionization of proteins and nucleic acids. Eur. J. Mass Spectrom 2011, 17 (4), 333– 343, DOI: 10.1255/ejms.1140There is no corresponding record for this reference.
- 46Madsen, J. A.; Xu, H.; Robinson, M. R.; Horton, A. P.; Shaw, J. B.; Giles, D. K.; Kaoud, T. S.; Dalby, K. N.; Trent, M. S.; Brodbelt, J. S. High-throughput database search and large-scale negative polarity liquid chromatography-tandem mass spectrometry with ultraviolet photodissociation for complex proteomic samples. Mol. Cell. Proteomics 2013, 12 (9), 2604– 2614, DOI: 10.1074/mcp.O113.028258There is no corresponding record for this reference.
- 47Donnelly, D. P.; Rawlins, C. M.; DeHart, C. J.; Fornelli, L.; Schachner, L. F.; Lin, Z.; Lippens, J. L.; Aluri, K. C.; Sarin, R.; Chen, B. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16 (7), 587– 594, DOI: 10.1038/s41592-019-0457-0There is no corresponding record for this reference.
- 48Roberts, D. S.; Loo, J. A.; Tsybin, Y. O.; Liu, X.; Wu, S.; Chamot-Rooke, J.; Agar, J. N.; Pasa-Tolic, L.; Smith, L. M.; Ge, Y. Top-down proteomics. Nat. Rev. Methods Primers 2024, 4 (1), 38 DOI: 10.1038/s43586-024-00318-2There is no corresponding record for this reference.
- 49Larson, E. J.; Roberts, D. S.; Melby, J. A.; Buck, K. M.; Zhu, Y.; Zhou, S.; Han, L.; Zhang, Q.; Ge, Y. High-Throughput Multi-attribute Analysis of Antibody-Drug Conjugates Enabled by Trapped Ion Mobility Spectrometry and Top-Down Mass Spectrometry. Anal. Chem. 2021, 93 (29), 10013– 10021, DOI: 10.1021/acs.analchem.1c00150There is no corresponding record for this reference.
- 50Riley, N. M.; Westphall, M. S.; Coon, J. J. Activated Ion-Electron Transfer Dissociation Enables Comprehensive Top-Down Protein Fragmentation. J. Proteome Res. 2017, 16 (7), 2653– 2659, DOI: 10.1021/acs.jproteome.7b00249There is no corresponding record for this reference.
- 51Wei, B.; Zenaidee, M. A.; Lantz, C.; Williams, B. J.; Totten, S.; Ogorzalek Loo, R. R.; Loo, J. A. Top-down mass spectrometry and assigning internal fragments for determining disulfide bond positions in proteins. Analyst 2022, 148 (1), 26– 37, DOI: 10.1039/D2AN01517JThere is no corresponding record for this reference.
- 52Dunham, S. D.; Brodbelt, J. S. Enhancing Top-Down Analysis of Proteins by Combining Ultraviolet Photodissociation (UVPD), Proton-Transfer Charge Reduction (PTCR), and Gas-Phase Fractionation to Alleviate the Impact of Nondissociated Precursor Ions. J. Am. Soc. Mass Spectrom. 2024, 35 (2), 255– 265, DOI: 10.1021/jasms.3c00351There is no corresponding record for this reference.
- 53Walker, J. N.; Lam, R.; Brodbelt, J. S. Enhanced Characterization of Histones Using 193 nm Ultraviolet Photodissociation and Proton Transfer Charge Reduction. Anal. Chem. 2023, 95 (14), 5985– 5993, DOI: 10.1021/acs.analchem.2c05765There is no corresponding record for this reference.
- 54Watts, E.; Thyer, R.; Ellington, A. D.; Brodbelt, J. S. Integrated Top-Down and Bottom-Up Mass Spectrometry for Characterization of Diselenide Bridging Patterns of Synthetic Selenoproteins. Anal. Chem. 2022, 94 (32), 11175– 11184, DOI: 10.1021/acs.analchem.2c01433There is no corresponding record for this reference.
- 55Brodbelt, J. S. Deciphering combinatorial post-translational modifications by top-down mass spectrometry. Curr. Opin. Chem. Biol. 2022, 70, 102180 DOI: 10.1016/j.cbpa.2022.102180There is no corresponding record for this reference.
- 56Fornelli, L.; Srzentic, K.; Toby, T. K.; Doubleday, P. F.; Huguet, R.; Mullen, C.; Melani, R. D.; Dos Santos Seckler, H.; DeHart, C. J.; Weisbrod, C. R. Thorough Performance Evaluation of 213 nm Ultraviolet Photodissociation for Top-down Proteomics. Mol. Cell. Proteomics 2020, 19 (2), 405– 420, DOI: 10.1074/mcp.TIR119.001638There is no corresponding record for this reference.
- 57Oates, R. N.; Lieu, L. B.; Srzentic, K.; Damoc, E.; Fornelli, L. Characterization of a Monoclonal Antibody by Native and Denaturing Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2024, 35 (9), 2197– 2208, DOI: 10.1021/jasms.4c00224There is no corresponding record for this reference.
- 58Cannon, J. R.; Cammarata, M. B.; Robotham, S. A.; Cotham, V. C.; Shaw, J. B.; Fellers, R. T.; Early, B. P.; Thomas, P. M.; Kelleher, N. L.; Brodbelt, J. S. Ultraviolet photodissociation for characterization of whole proteins on a chromatographic time scale. Anal. Chem. 2014, 86 (4), 2185– 2192, DOI: 10.1021/ac403859aThere is no corresponding record for this reference.
- 59Lanzillotti, M.; Brodbelt, J. S. Comparison of Top-Down Protein Fragmentation Induced by 213 and 193 nm UVPD. J. Am. Soc. Mass Spectrom. 2023, 34 (2), 279– 285, DOI: 10.1021/jasms.2c00288There is no corresponding record for this reference.
- 60Juetten, K. J.; Brodbelt, J. S. Top-Down Analysis of Supercharged Proteins Using Collision-, Electron-, and Photon-Based Activation Methods. J. Am. Soc. Mass Spectrom. 2023, 34 (7), 1467– 1476, DOI: 10.1021/jasms.3c00138There is no corresponding record for this reference.
- 61Hyde, A. M.; Calabria, R.; Arvary, R.; Wang, X.; Klapars, A. Investigating the Underappreciated Hydrolytic Instability of 1,8-Diazabicyclo[5.4.0]undec-7-ene and Related Unsaturated Nitrogenous Bases. Org. Process Res. Dev. 2019, 23 (9), 1860– 1871, DOI: 10.1021/acs.oprd.9b00187There is no corresponding record for this reference.
- 62Juetten, K. J.; Brodbelt, J. S. MS-TAFI: A Tool for the Analysis of Fragment Ions Generated from Intact Proteins. J. Proteome Res. 2023, 22 (2), 546– 550, DOI: 10.1021/acs.jproteome.2c00594There is no corresponding record for this reference.
- 63Durbin, K. R.; Robey, M. T.; Voong, L. N.; Fellers, R. T.; Lutomski, C. A.; El-Baba, T. J.; Robinson, C. V.; Kelleher, N. L. ProSight Native: Defining Protein Complex Composition from Native Top-Down Mass Spectrometry Data. J. Proteome Res. 2023, 22 (8), 2660– 2668, DOI: 10.1021/acs.jproteome.3c00171There is no corresponding record for this reference.
- 64Li, J.; Santambrogio, C.; Brocca, S.; Rossetti, G.; Carloni, P.; Grandori, R. Conformational effects in protein electrospray-ionization mass spectrometry. Mass Spectrom Rev. 2016, 35 (1), 111– 122, DOI: 10.1002/mas.21465There is no corresponding record for this reference.
- 65Reid, G. E.; Wu, J.; Chrisman, P. A.; Wells, J. M.; McLuckey, S. A. Charge-state-dependent sequence analysis of protonated ubiquitin ions via ion trap tandem mass spectrometry. Anal. Chem. 2001, 73 (14), 3274– 3281, DOI: 10.1021/ac0101095There is no corresponding record for this reference.
- 66Cobb, J. S.; Easterling, M. L.; Agar, J. N. Structural characterization of intact proteins is enhanced by prevalent fragmentation pathways rarely observed for peptides. J. Am. Soc. Mass Spectrom. 2010, 21 (6), 949– 959, DOI: 10.1016/j.jasms.2010.02.009There is no corresponding record for this reference.
- 67Hellinger, J.; Brodbelt, J. S. Impact of Charge State on Characterization of Large Middle-Down Sized Peptides by Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2024, 35 (8), 1647– 1656, DOI: 10.1021/jasms.3c00405There is no corresponding record for this reference.
- 68Carrera, G. V. S. M.; Jordão, N.; Santos, M. M.; da Ponte, M. N.; Branco, L. C. Reversible systems based on CO2, amino-acids and organic superbases. RSC Adv. 2015, 5 (45), 35564– 35571, DOI: 10.1039/C5RA03474DThere is no corresponding record for this reference.
- 69Bonner, J.; Talbert, L. E.; Akkawi, N.; Julian, R. R. Simplified identification of disulfide, trisulfide, and thioether pairs with 213 nm UVPD. Analyst 2018, 143 (21), 5176– 5184, DOI: 10.1039/C8AN01582AThere is no corresponding record for this reference.
- 70Macias, L. A.; Brodbelt, J. S. Investigation of Product Ions Generated by 193 nm Ultraviolet Photodissociation of Peptides and Proteins Containing Disulfide Bonds. J. Am. Soc. Mass Spectrom. 2022, 33 (7), 1315– 1324, DOI: 10.1021/jasms.2c00124There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.5c00380.
(1) Mass spectra for optimization of ESI-MS in negative mode, (2) sequence maps and isotopic fitting results supporting identification of fragment ions, (3) bar graphs for sequence coverage and feature of fragment ions for varied charge states of proteins using HCD, UVPD (varied laser energy) and a-EPD, (4) bar graphs for preferential cleavage analysis of HCD, UVPD and a-EPD, and (5) mass spectra and bar graphs comparing positive and negative mode UVPD of all proteins (Figure S23) (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.