

About the Cover:
Correlating operando techniques with electrochemical analysis enables a comprehensive view of electrode evolution. Zheng et al. report an operando degradation study of ITO-PET that integrates UV–vis spectroelectrochemistry with time-resolved electrochemical impedance analysis, revealing chemical and electrochemical degradation pathways. Gemini 2.5 Flash Image was used to edit the image style.
View the article.Perspectives

Metallic Oxides and the Overlooked Role of Bandwidth
Aurland K. Watkins *- ,
Anthony K. Cheetham *- , and
Ram Seshadri *

This publication is Open Access under the license indicated. Learn More
Oxides exhibiting metallic conduction are crucial for various applications, including fuel cells, battery electrodes, resistive and magnetoresistive materials, electrocatalysts, transparent conductors, and high-temperature superconductors. Oxides that approach metallicity also play significant roles in switching applications, where the metal–insulator transition phenomenon is utilized across a range of technologies. This perspective, motivated by the question of when oxides are metallic, employs electronic structure calculations on metallic oxides to identify typical features in the electronic structure that promote metallic behavior. The critical factor of the bandwidth of the electronic energy bands near the Fermi energy is emphasized since it has been somewhat overlooked in the literature. For example, bandwidth considerations would suggest that the recently proposed phosphate “LK-99” would never be a suitable target for superconductivity. By relating the crystal structure and electronic band features obtained through density functional theory calculations, we present the general heuristic that crystals with conduction bands narrower than 1 eV (as obtained from routine electronic structure methods) are unlikely to be metallic. We further examine the origins of narrow or flat bands to distinguish between structural properties that are conducive or detrimental to physical behavior like superconductivity. This survey of representative oxide metals highlights the essential chemical and structural ingredients that contribute to extended covalent interactions and ultimately wide electronic bands. A key takeaway is that oxyanion compounds such as borates, carbonates, silicates, sulfates, nitrates, and phosphates are unlikely to exhibit metallic conduction at ambient pressure. While the focus here is on oxides, the general findings should apply across various material families, extending to conducting organic crystals, conducting polymers, and hybrid and framework materials.
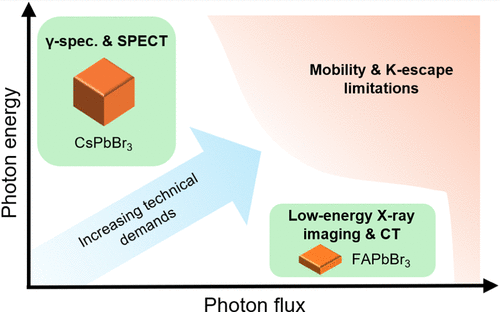
Perovskite Photon-Counting Detectors: Narrowing the Field
Thomas H. Neal - ,
Nicholas Sandor - ,
James Day - ,
Magdalena Bazalova-Carter - , and
Makhsud I. Saidaminov *
Since the pioneering demonstration of energy-resolved X-ray detection with cesium lead bromide perovskite by Mercouri Kanatzidis and coworkers in 2013, research into lead-halide perovskites for radiation detection has expanded rapidly. Among the many perovskite compositions explored, here we argue that cesium lead bromide and formamidinium lead bromide hold the strongest potential for use as photon-counting detector materials, in high- and low-photon energy applications, respectively. We justify our argument by examining the relatively low charge carrier mobility common to perovskites, which necessitates high operating biases, conditions under which many alternative compositions undergo degradation. Furthermore, we identify that the extended attenuation length associated with Pb K-shell fluorescence will likely constrain the applicability of lead-halide perovskites to two primary regimes: (1) detection of high-energy photons under low flux and low spatial resolution, as often seen in γ-ray spectroscopy; and (2) detection of low-energy X-ray photons under high flux, where reduced device thickness and small pixel size can compensate for low charge carrier mobility without the worry of excess K-escape and Compton scattering. Advancing perovskite photon-counting detectors toward practical imaging applications calls for studies under realistic photon flux conditions, focusing on parameters such as maximum count rate, charge sharing, charge cloud size, K-escape, and bias stability.
Reviews
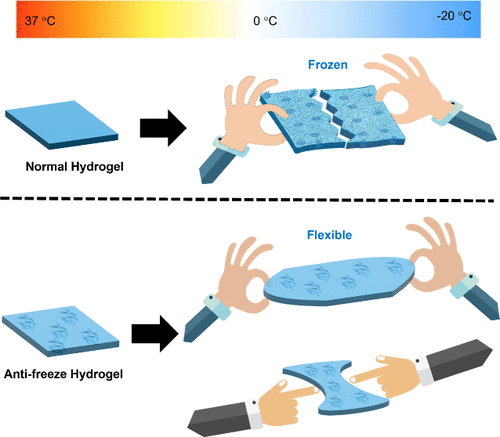
Advances in Antifreeze Hydrogel-Based Wound Dressings
Javad Esmaeili - and
Reza Jafari Aminabadi *
Wounds can occur in extreme environments, including subzero temperatures, where conventional wound dressings lose flexibility and functionality. Given these circumstances, specific wound care products that function effectively in cold environments must be developed. Recently, antifreeze hydrogels (AFHs) have garnered attention as viable options due to their ability to withstand ice crystallization, maintain biocompatibility, facilitate drug delivery, exhibit antibacterial activity, and retain flexibility. This review summarizes recent findings and provides a comprehensive overview of key advancements in AFH. Also, AFH-based wound dressings (AFHWDs) were discussed with a focus on the primary design objectives and functional aspects guiding their development. Various AFHWDs have been developed using strategies, such as the incorporation of antifreeze agents (e.g., glycerol, polyethylene glycol), the utilization of natural biomaterials (e.g., bacterial cellulose, gelatin), and the design of highly cross-linked polymer networks, each illustrating distinct antifreezing mechanisms like thermal hysteresis and ice recrystallization inhibition. This study also explores the main challenges and future scientific potential of AFHWDs. Finally, this review concludes by emphasizing the potential of alternative fabrication techniques such as 3D printing and electrospinning, and the need to explore more effective and naturally derived antifreeze agents for the development of next-generation AFHs.

Length-Controlled Synthesis of Graphene Nanoribbons
Daniel Pyle - ,
Yutong Xiang - ,
Xingchen Li - ,
Ruohai Wang - ,
Guangbin Dong *- , and
Jiangliang Yin *
Graphene nanoribbons (GNRs) have emerged as promising materials for next-generation electronic, optoelectronic, and quantum devices due to their tunable bandgaps and edge-dependent properties. A critical challenge in their integration lies in the ability to precisely control their length and ensure structural uniformity. This review highlights three major synthetic strategies developed to address this challenge: living polymerization, conventional iterative synthesis, and protecting group-aided iterative synthesis (PAIS). Living polymerization approaches enable scalable access to GNRs with narrow length distributions, although they rely on specialized monomers and catalyst design to maintain a living character. The conventional iterative synthesis strategy provides a pathway for the preparation of specific GNRs with precise length, but it is still not possible to synthesize general GNRs with a desired length or a well-defined heterogeneous monomer sequence. The PAIS strategy stands out, allowing atomic-level control over GNR length, width, edge structure, and heterojunction placement. Iterative methods offer unparalleled atomic precision and architectural flexibility but are labor-intensive and limited by solubility constraints. Each method presents complementary advantages and trade-offs. Future advancements are expected to stem from hybrid synthetic platforms, catalyst innovations, and programmable template design, ultimately enabling deterministic control over GNR structures and properties for device applications.
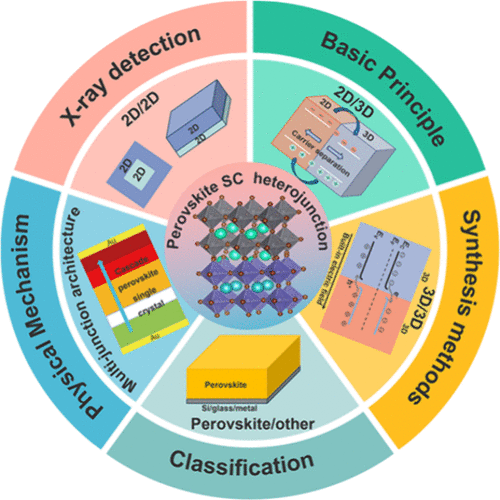
Advances of Perovskite Single-Crystal Heterojunctions for High-Performance X-ray Detectors
Hongjie Liu - ,
Wenjun Ma - ,
Jiaxin Liu - ,
Xue Sun - ,
Xutang Tao *- , and
Guodong Zhang *
Perovskite single-crystal (SC) heterojunctions have sparked great interest in enhancing the performance of optoelectronic devices due to their diverse structures and tunable compositions that benefit the low defect density and higher stability. The latest progress and future perspectives of perovskite SC heterojunctions are reviewed herein. First, we briefly introduce the fundamentals of perovskite SC heterostructures. Then, the preparation methods and the classification of perovskite SC heterostructures are discussed. Moreover, the physical mechanism and the progress in X-ray detection application of perovskite SC heterojunctions are systematically summarized. Finally, we propose a global perspective on the challenges and development of perovskite SC heterojunctions. This review summarizes the achievements of halide perovskite SC heterojunctions over the past decade, identifies their existing limitations, and offers valuable insights to guide the future development of SC heterojunctions.
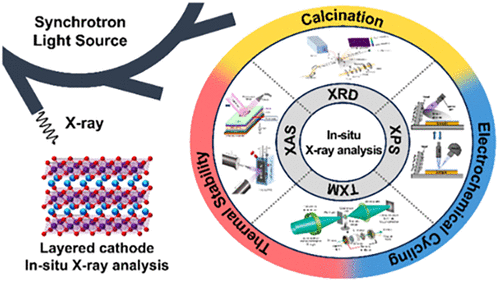
In Situ Synchrotron Characterization of Layered Oxide Cathodes for Lithium-Ion Batteries: Bridging Synthesis, Operation, and Thermal Stability
Sangbeom Kim - ,
Joon Ha Chang - ,
Beom Tak Na - ,
Sanghyeok Moon - ,
Seonho Kim - ,
Seungjun Baek - ,
Jeong-Mi Yeon - ,
Yu-Jin Kim - ,
Hyun-seung Kim *- ,
Min Wook Pin *- , and
Youngjin Kim *
This review examines synchrotron-based in situ characterization studies of layered oxide cathodes for lithium-ion batteries (LIBs) and shows that kinetic phenomena, rather than thermodynamic equilibrium, govern their behavior during synthesis, operation, and degradation. Advanced X-ray techniques reveal features including nanoscale structural domains that form substantially earlier than indicated by conventional detection methods, pronounced chemical heterogeneity within individual particles that contributes to mechanical failure, and electronic destabilization that precedes thermal decomposition. Surface–bulk divergence and intraparticle heterogeneity─often regarded as undesirable artifacts─emerge as intrinsic characteristics that strongly influence electrochemical performance. Correlative analyses of diffraction, spectroscopy, and microscopy data indicate that these materials frequently exist in nonequilibrium states in which local kinetics dominate over global thermodynamics. This review suggests a shift from equilibrium-based models toward kinetically controlled frameworks for understanding battery materials, indicating that future cathode designs might benefit from accommodating rather than eliminating these inherent heterogeneities. The mechanistic insights derived from synchrotron radiation provide guidance for developing stabilization strategies to address fundamental challenges in energy storage materials that are relevant to sustainable energy systems.
Articles

Electrochemical Instability of Flexible Indium Tin Oxide Film: A Time-Resolved Operando Study
Sijie Chen - and
Weiran Zheng *
Indium tin oxide-coated flexible polyethylene terephthalate (ITO-PET) is increasingly utilized in various electrochemical applications due to its exceptional electrical conductivity and optical transparency. However, the chemical and electrochemical instability of ITO coated on a flexible substrate poses significant challenges to maintaining long-term durability and structural integrity under harsh electrochemical conditions. In this study, we conducted a comprehensive ex situ and operando investigation of the electrical and optical degradation of ITO-PET electrodes in acidic (0.5 M H2SO4), neutral (1.0 M KCl), and alkaline (1.0 M KOH) environments by combining liquid-phase atomic force microscopy, time-resolved electrochemical impedance analysis, and time-resolved operando UV–vis spectroelectrochemistry. Our findings reveal that ITO-PET films degrade more severely than ITO glass, with degradation pathways strongly influenced by electrolyte and potential. In acidic conditions, ITO-PET undergoes rapid, catastrophic dissolution within seconds, leading to an immediate loss of both electrical conductivity and optical transparency. Neutral chloride-containing environments induce slower but significant degradation via irreversible anodic dissolution involving indium-chloride complexation, resulting in substantial morphological changes and moderate losses in conductivity and transparency. While neutral KCl solutions cause pronounced electrochemical instability, optical transparency remains relatively stable during short-term cycling. In contrast, under alkaline conditions, degradation behavior is complex, dominated by reversible Sn2+/Sn4+ redox reactions. Despite their electrochemical reversibility, these reactions progressively induce irreversible surface modifications and Sn dissolution, severely compromising optical transparency and electrical conductivity over extended cycling. By integrating real-time multimodal characterization techniques, we provide crucial insights into the degradation mechanisms of ITO-PET film, offering a framework for analyzing and optimizing its durability in practical electrochemical applications.

Atomic Layer Epitaxy of (002) κ-Ga2O3 on c-Plane Sapphire
Andy Séguret - ,
Ilyass Jellal - ,
Matthieu Weber - ,
Hervé Roussel - ,
Isabelle Gélard - ,
Laetitia Rapenne - ,
Eirini Sarigiannidou - ,
Fabrice Wilhelm - ,
Andrei Rogalev - ,
Edith Bellet-Amalric - ,
Eva Monroy - , and
Vincent Consonni *
Atomic layer epitaxy has been a powerful technique for several decades to grow epitaxial thin films of III–V and II–VI semiconducting compounds. However, the polycrystalline nature of thin films usually prevails for conducting and semiconducting binary oxides, which strongly limits their crystalline and structural quality. Here, we demonstrate the epitaxial growth of (002)-oriented κ-Ga2O3 thin films with a subnm surface roughness on c-plane sapphire by atomic layer deposition at 350 °C and using the relatively unexplored combination of triethylgallium and O3 as chemical precursors. The 16 nm-thick κ-Ga2O3 thin films exhibit an in-plane epitaxial relationship given by (060) κ-Ga2O3||(300) α-Al2O3 and (200) κ-Ga2O3||(110) α-Al2O3. The pure epitaxial κ-Ga2O3 phase is further revealed using X-ray absorption near-edge structure and X-ray linear dichroism with synchrotron radiation along with transmission electron microscopy. In particular, the (004) X-ray diffraction peak for the κ-Ga2O3 thin film grown at 350 °C is shown to have a remarkable full-width-at-half-maximum value of 0.1° following rocking curve measurements, which is comparable to the typical lowest values reported for κ-Ga2O3 thick films grown by more sophisticated epitaxial chemical and physical vapor deposition techniques. The optical bandgap energy is eventually evaluated at 4.8 eV from optical transmittance spectra. These findings show the high potential of atomic layer deposition for growing epitaxial Ga2O3 thin films at relatively low temperatures in the framework of atomic layer epitaxy, strongly challenging the well-known epitaxial physical and chemical deposition techniques.

Design Rules and Discovery of Face-Sharing Hexagonal Perovskites
M. J. Swamynadhan *- ,
Gwan Yeong Jung - ,
Pravan Omprakash - , and
Rohan Mishra *
Hexagonal perovskites with face-sharing octahedral connectivity are an underexplored class of materials. We propose quantitative design principles for stabilizing face-sharing ABX3 hexagonal perovskites based on a comparative analysis of oxides and sulfides. By mapping structural preferences across a phase space defined by an electronegativity-corrected tolerance factor and the Shannon radius of the A-site cations, we identify distinct thresholds that separate hexagonal phases from competing cubic polymorphs having corner-sharing octahedral connectivity. Our analysis reveals that sulfides differ significantly from oxides due to the increased covalency of the transition metal–sulfur bonds, which enables broader compositional flexibility. Applying these principles, we predict a set of thermodynamically formable ABO3 and ABS3 compounds that are likely to adopt face-sharing octahedral connectivity. These findings establish a predictive framework for designing hexagonal perovskites, highlighting sulfides as promising candidates for obtaining quasi-one-dimensional materials having transition-metal cations for novel ferroic phenomena.

Unlocking Lithium Superionic Conduction via Phonon Softness Descriptors: A High-Throughput Machine Learning Paradigm
Ogheneyoma Aghoghovbia - ,
Riccardo Rurali - ,
Mohammed Al-Fahdi - , and
Ming Hu *
Lithium superionic conductors (LISICONs) are pivotal for next-generation solid-state batteries, yet rational design remains challenged by poorly understood correlations between lattice dynamics (phonons) and ion transport. We unveil phonon softness as the unifying descriptor for rapid Li+ conduction through a high-throughput computational framework combining ab initio calculations, machine learning potentials (fine-tuned MACE MPA model), and molecular dynamics across 1304 dynamically stable Li materials screened from an initial pool of 8578 candidates. We demonstrate that low-frequency phonons dominate Li+ migration, with the Li+ vibrational density of states (VDOS) center and Debye frequency exhibiting strong inverse correlations with diffusion coefficients. Phonon mode-resolved analysis reveals that delocalized, collective vibrations (<2 THz) lower activation barriers by promoting in-phase host lattice coupling. Crucially, global mechanical properties─bulk modulus, shear modulus, elastic modulus, and hardness─all inversely scale with Li+ mobility, confirming that structural compliance facilitates reduced activation barriers for Li+ hopping, consistent with prior observations that lattice softness enhances ionic migration. Using ab initio molecular dynamics simulations, we confirm superionic conduction in 25 Li structures that are screened from our workflow, including halide-based and nitrogen-containing materials. Our study establishes readily accessible lattice dynamics descriptors (VDOS center, Debye frequency) and mechanical metrics as universal screening tools, accelerating the discovery of high-performance solid electrolytes for energy-dense, safe batteries.

Tuning Catalytic Activity in Nitride Perovskite Through Strain-Induced Rashba Spin Splitting Manipulation
Prajna Parimita Mohanty - ,
Showkat H. Mir - ,
Rajeev Ahuja *- , and
Sudip Chakraborty *
Rashba spin splitting is an emerging phenomenon originating from the synergistic effect of relativistic spin–orbit coupling (SOC) due to the presence of a heavy constituent element and the noncentrosymmetric crystal structure. We recently observed Rashba spin splitting in the rare nitride perovskite CeNbN3. This work explores how tuning the Rashba spin splitting strength can enhance photocatalytic water splitting and hydrogen evolution reaction (HER) activity. Based on our electronic structure calculations, we have observed the fine-tuning of Rashba spin splitting in CeNbN3 under the influence of compressive strain and the corresponding impact on HER activity. The evolution of electronic band structure, Rashba spin splitting strength, and spin texture under compressive strain corresponds well with the hydrogen adsorption free energy determined from the constructed reaction coordinate mapping of the HER mechanism. The strength of spin splitting shows a correlation with improved HER activity, which is in line with the influence of the Rashba effect.
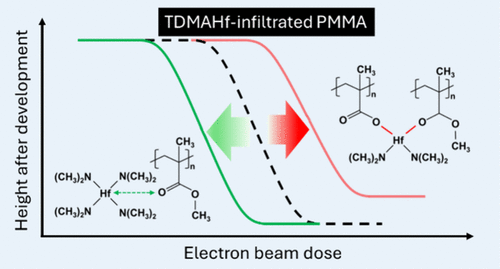
Mechanism of Vapor-Phase Infiltration of Organometallic Hf in Poly(Methyl Methacrylate) for Hybrid Resist Applications
Md Istiaque Chowdhury - ,
Xinpei Wu - ,
Won-Il Lee - ,
Peter Sun - ,
Mueed Ahmad - ,
Kim Kisslinger - ,
Jerzy T. Sadowski - ,
Samuel A. Tenney - ,
Jorge Anibal Boscoboinik - ,
Nikhil Tiwale - , and
Chang-Yong Nam *
This publication is Open Access under the license indicated. Learn More
Inorganic–organic hybrid thin films synthesized by vapor-phase infiltration (VPI) of metal oxides into organic photoresists, such as poly(methyl methacrylate) (PMMA), have recently demonstrated their utility in extreme ultraviolet lithography, critical for angstrom-era semiconductor device miniaturization. Hafnium oxide infiltration has been reported recently for this purpose, but its detailed VPI mechanism has remained largely unexplored. In this study, we investigated the VPI characteristics and mechanisms of tetrakis(dimethylamido)hafnium (TDMAHf)─the hafnium precursor predominantly used for VPI in the field─into PMMA and examined its impact on electron-beam lithography (EBL) exposure behavior. VPI was performed at temperatures ranging from 85 to 150 °C, with chemical interactions characterized using infrared reflection-absorption spectroscopy, and resist patterning performance was evaluated through EBL dose-sensitivity assessments. The results indicate that TDMAHf forms a reversible adduct with PMMA at temperatures up to 120 °C, whereas at 150 °C, covalent bond formation occurs, most likely via dealkylation that leads to acetate formation. EBL studies reveal that resist sensitivity is influenced by both infiltration temperature and developer selection, with aqueous isopropyl alcohol development demonstrating enhanced sensitivity compared to organic solvent-based development. The optimized infiltration protocol at 120 °C ensures a uniform inorganic distribution without compromising resist dissolution. These findings not only help refine hybrid resist patterning performance but also offer insights potentially applicable to the VPI of other homoleptic metal-amide organometallic VPI precursors that include TDMA ligands.
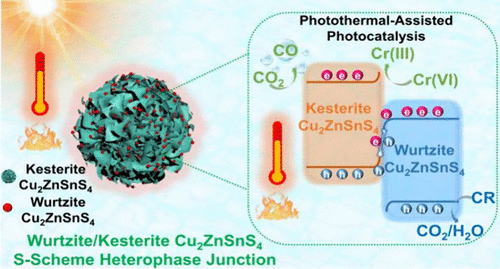
Wurtzite/Kesterite Cu2ZnSnS4 S-Scheme Heterophase Junction: A Robust Catalyst for Photothermal-Assisted Photocatalytic CO2 Reduction and Environmental Remediation
Renzhi Xiong - ,
Dongchen Duan - ,
Xiaoxue Ke - ,
Keqin Chen - ,
Yanhe Xiao - ,
Baochang Cheng - , and
Shuijin Lei *
Heterophase junction engineering offers a revolutionary strategy for constructing polymorph photocatalysts with nearly perfect interfacial matching, thereby significantly optimizing charge dynamics. Nevertheless, existing heterophase junction systems still struggle to achieve both directional charge separation and efficient utilization of the full solar spectrum. In this study, a wurtzite-phase Cu2ZnSnS4 (W-CZTS)/kesterite-phase Cu2ZnSnS4 (K-CZTS) S-scheme heterophase junction was successfully constructed through the precise regulation of the spatial assembly between W-CZTS nanoparticles and K-CZTS nanosheets. This strategic structural design achieved three key breakthroughs: (i) the formation of atomically intimate interfaces with strong built-in electric fields, (ii) the integration of superior photothermal conversion capability, and (iii) directional charge separation while preserving optimal redox potentials. The developed heterophase junction catalyst demonstrated outstanding photothermal-assisted photocatalytic performance: under UV–vis–IR illumination, the average CO yield reached 68.4 μmol g–1 h–1, which represented a 9.6-fold enhancement compared to that without IR irradiation, and 76-fold and 114-fold higher than the performance of pristine K-CZTS and W-CZTS, respectively, under UV–vis irradiation. Furthermore, the catalyst demonstrated near-complete removal efficiency for both Cr(VI) reduction and Congo red degradation. This study establishes a paradigm for the development of high-performance heterophase junction photocatalysts and provides crucial insights for advancing solar-driven environmental remediation and fuel production.
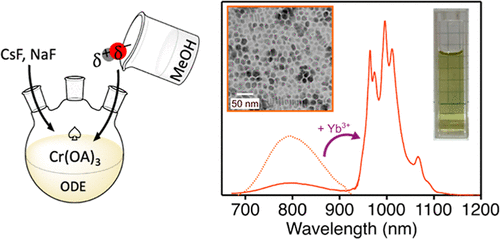
A Versatile Method for Synthesizing Colloidal Cr3+-Based Fluoride Nanocrystals: Near-IR-Emitting Cs2NaCrF6, Na3CrF6, and Yb3+-Doped Cs2NaCrF6
Eden Tzanetopoulos - and
Daniel R. Gamelin *
Colloidal fluoride nanocrystals containing luminescent rare-earth ions are powerful nanophosphors for bioimaging, optical sensing, and other photonic functions. The utility of luminescent fluoride nanocrystals could be broadened if a greater composition space could be accessed by the development of new synthetic capabilities. Here, we report a general solution-phase fluoride-salt synthesis method that allows preparation of colloidal fluoro-elpasolite and -cryolite nanocrystals, two phases that have received little attention at the nanoscale. We demonstrate that this synthetic method is compatible with various trivalent (e.g., Cr3+, Al3+, Ga3+) and monovalent (e.g., Cs+, Na+, NH4+) cations, providing access to a rich portfolio of ternary and quaternary fluoride nanocrystals. In particular, this method is used to prepare the Cr3+-based fluoro-elpasolite Cs2NaCrF6 on the nanoscale. Broadband near-infrared Cr3+ 4T2g → 4A2g emission is observed at room temperature from these nanocrystals. Under related conditions, analogous Cr3+-based cryolite nanocrystals (Na3CrF6) could also be prepared. With this method, Yb3+ was successfully doped into Cs2NaCrF6 nanocrystals at various concentrations. Cr3+ d–d transitions are found to sensitize the Yb3+ f–f luminescence at room temperature, and broad tunability of the relative photoluminescence intensities of Cr3+ and Yb3+ was achieved via composition control. The utility of this synthesis method for preparing these ternary and quaternary nanocrystals with complex and tunable compositions suggests opportunities for the development of other challenging fluoride lattices on the nanoscale using this approach.
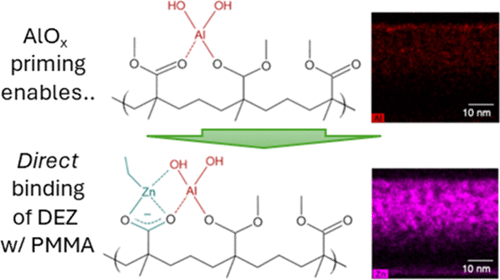
Alumina Priming-Mediated Enhanced Binding of Diethylzinc with Carbonyl Groups in Poly(Methyl Methacrylate) during Vapor-Phase Infiltration
Nikhil Tiwale *- ,
Ashwanth Subramanian - ,
Sayantani Sikder - ,
Xiaohui Qu *- ,
Guillaume Freychet - ,
Eliot Gann - ,
Cherno Jaye - ,
Kim Kisslinger - ,
Jorge Anibal Boscoboinik - , and
Chang-Yong Nam *
This publication is Open Access under the license indicated. Learn More
Vapor-phase infiltration (VPI) of inorganic materials in polymers is increasingly becoming popular for synthesizing various functional hybrid materials. While AlOx infiltration using trimethylaluminum (TMA) has been extensively studied, the mechanism of diethylzinc (DEZ)-based ZnOx infiltration, especially one that is initiated by AlOx priming, has not received much attention because highly reactive hydroxyl groups generated by AlOx-priming are expected to dominate the initial binding of DEZ, thus enabling the overall ZnOx VPI. Here, we interrogate the ZnOx infiltration mechanism in AlOx-primed poly(methyl methacrylate) (PMMA) in comparison to the control AlOx-only infiltration by utilizing a suite of complementary characterizations, including quartz crystal microbalance mass gain measurement, transmission electron microscopy, infrared reflection–absorption spectroscopy (IRRAS), and synchrotron X-ray absorption spectroscopy (XAS). The multivalent TMA precursor and associated hyperbranched AlOx network can quickly saturate the AlOx infiltration by clogging the polymer-free volume near the top. On the contrary, the ZnOx infiltration using divalent DEZ precursor, once activated via AlOx-priming, can lead to accelerated ZnOx infiltration. With the help of IRRAS, XAS, and density functional theory (DFT) simulations, we uncover that the AlOx-priming enhances the reactivity of neighboring carbonyl groups toward DEZ and opens up simultaneous reaction pathways, leading to accelerated high-fidelity infiltration of ZnOx.
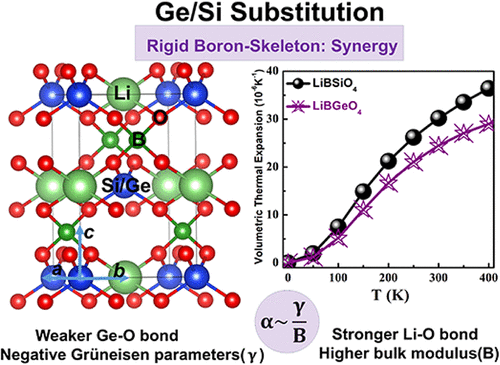
Local Bond Engineering in LiMBO4 (M = Si, Ge): Synergistic Negative Grüneisen Parameter and Bond Stiffening for Reduced Thermal Expansion
Wenjing Huang - ,
Yuanyuan Li *- , and
Dingfeng Yang *
Precise control of the thermal expansion is paramount for the stability of optical crystals in advanced photonic devices. Diamond-like borates, featuring rigid frameworks of corner-sharing [BO4] tetrahedra, are promising candidates. However, achieving ultralow or negative thermal expansion remains a significant challenge. This work demonstrates a rational strategy of local bond engineering within the LiSiBO4 prototype to dramatically suppress thermal expansion. Strategic substitution of Si4+ with larger Ge4+ yields LiGeBO4, reducing its volumetric thermal expansion coefficient from approximately 30.2 × 10–6 K–1 to 24.5 × 10–6 K–1. Structural analysis reveals that the diamond-like framework possesses exceptional rigidity, characterized by nearly invariant B–B separation within the interconnected [BO4] network, which establishes a dimensionally stable scaffold. Within this constrained scaffold, Li+ and M4+ (M = Si or Ge) cations occupy distinct interstitial sites. Crucially, the incorporation of larger Ge4+ introduces a longer, weaker Ge–O bond, inducing pronounced distortion in [GeO4] tetrahedra. This distortion generates negative Grüneisen parameters (γ), inherently counteracting lattice expansion. Concomitantly, the expanded [GeO4] tetrahedron severely compresses the spatial environment of the adjacent [LiO4] polyhedron. This spatial confinement forces the Li–O bond to contract and stiffen, significantly restricting their thermal vibrational amplitudes. The synergistic interplay between the negative γ contribution arising from the distorted [GeO4] and the bond stiffening within the spatially compressed [LiO4] collectively minimizes the overall thermal expansion of the lattice. This study establishes targeted local bond engineering within rigid, geometrically constrained frameworks as a powerful and generalizable paradigm for the design of next-generation optical materials with ultralow or potentially negative thermal expansion.
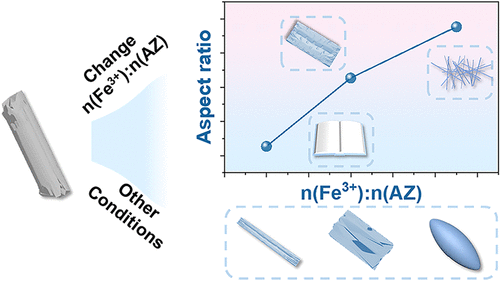
Precisely Tailoring the Architecture of Metal-Quinone Networks via a Template-Directed Coordination Assembly
Yanli Du - ,
Lulu Zhou - , and
Jing Hu *
Microarchitected materials with programmable topological configurations provide a versatile platform for enhancing functionalities and broadening applications in advanced materials science. Metal-quinone networks (MQNs) uniquely combine the inherent bioactivity of natural quinones with the structural tunability of framework-based materials, conferring integrated advantages, including high drug-loading capacity, bidirectional pH responsiveness, extensive adhesion capability, and ease of modification. However, the achievable dimensional range of MQNs through existing synthetic approaches remains limited, restricting precise control over their structural characteristics. Here, we present a strategy involving prepolymerization of natural quinones, followed by the construction of rodlike micelles and metal-coordination assembly, to achieve precise morphological control of MQNs. By systematically varying parameters including ligand-to-metal ratio, solvent types, surfactant types, and cross-linking agents, a series of MQN architectures such as book-, sheet-, thread-, spindle-, and rodlike structures were obtained. Moreover, the morphology progressively evolves from one-dimensional (1D) rod- and needle-like structures to three-dimensional (3D) rectangular blocklike forms. Notably, the rod-shaped MQNs exhibit tunable aspect ratios ranging from 3.0 to 80.0. Scanning electron microscopy (SEM) characterized solvent-induced facet-selective growth and stoichiometrically dependent anisotropic growth, thereby enabling predictable control over the geometrical shape. The findings of this work provide guiding insights into the rational construction of sophisticated structures of MQNs for potential applications.

Engineering Excited-State Dynamics in AgInS2 Quantum Dots by Gallium Incorporation and GaSy Shell Passivation
Jacquelyn Sundstrom - ,
Akshaya Chemmangat - , and
Prashant V. Kamat *
Ternary I–III–VI semiconductor quantum dots (QDs) are being explored as nontoxic alternatives to Cd- and Pb-based QDs for light-harvesting applications. Incorporation of Ga into AgInS2 reduces the number of defect states and improves its photophysical properties. Growth of a GaSy shell on a Ga-doped AgInS2 core further suppresses donor–acceptor pair (DAP states) emission and restores band-edge emission. We synthesized the core–shell architecture of AgInxGa1–xS2–GaSy QDs to make a direct comparison of the photophysical properties with those of AgInS2 and AgInxGa1–xS2 QDs. The photocatalytic activity of these QD systems was evaluated by probing electron transfer to ethyl viologen (EV2+) as an acceptor molecule. In all three cases, ultrafast electron transfer to surface-bound EV2+ occurred with rate constants on the order of ∼1011 s–1. However, the steady-state yield of the reduced product (viz., EV+•) varied, reflecting the influence of both intrinsic semiconductor properties and competing back electron transfer processes. These findings highlight how incorporation of Ga into AgInS2 improves the photophysical and photocatalytic properties of ternary semiconductor QDs and exemplifies the role of a core–shell architecture to suppress back electron transfer.

Hydrogen Bond Mediated Phase Separation of Phenolic-Based Compounds for the Preparation of Melanin-like Nanoparticles
Han Yang - ,
Yihao Gan - ,
Xinxin Han - ,
Dandan Ren - ,
Aixin Song - ,
Zhiliang Gao *- , and
Peiyu Zhang *
Melanin-like nanoparticles (MLNPs) hold great promise for biomedical applications, yet their controlled synthesis under mild conditions remains challenging. Here, we present a hydrogen bond-mediated liquid–liquid phase separation (LLPS) strategy to fabricate functional MLNPs with tunable physicochemical properties. Coacervates are formed through hydrogen bonding between hydrogen bond donor polyphenols and hydrogen bond acceptor polymers, providing a dynamic and mild environment for nanostructure formation. Leveraging this hydrogen bond-stabilized coacervate as a soft template, we synthesized monodisperse MLNPs via oxidative polymerization of coacervates. The resulting nanoparticles feature precise size control and abundant surface functionalities, enabling drug loading via electrostatic interactions, hydrogen bonding, π-π stacking, and metal-ion coordination. These multifunctional properties support diverse biomedical applications, including drug delivery, imaging, and enzyme-mimetic catalytic therapy. This work establishes a scalable and versatile platform for engineering MLNPs via hydrogen bond-driven LLPS templating, opening up opportunities for translational nanomedicine.

Rational Design of Degradable Multiarm Polymers Applicable for Photoimaging Materials
Dahye Lee - ,
Seungjun Kim - ,
Jaewon Ha - ,
Hyunseok Choi - ,
Kyuhyun Im *- , and
Myungwoong Kim *
We report the rational design and synthesis of acid-cleavable 3-arm polymers via core-first approach using a degradable reversible addition–fragmentation chain transfer (RAFT) agent, in which three chain transfer units are attached to the core through acetal bonds that are cleavable under acidic conditions. Its efficacy is confirmed through RAFT polymerization of methyl methacrylate (MMA), resulting in 3-arm PMMA with a molecular weight reduction to ≈30% of its original value after acid treatment. This approach is extended to a 3-arm terpolymer suitable for chemically amplified resists, exhibiting significantly enhanced sensitivity compared to the conventional linear terpolymer: onset energy for development decreases by ≈21% (DUV) and ≈50% (e-beam), while the energy at which development ends decreases by ≈50% for both. With DUV light, pattern formation is achieved at ≈50% lower energy than the linear terpolymer, and with e-beam, sub-100 nm pattern definition is demonstrated, which is not feasible with the linear terpolymer. The enhanced sensitivity and patternability stem from reduced molecular weight and functional group transformation induced by acid released from a photoacid generator. These findings highlight the significance of rationally designing multiarm architectures to tailor the structure and functionality of complex copolymers through an effective synthetic route, offering potential applications in advanced photoimaging materials and broader stimuli-responsive systems.
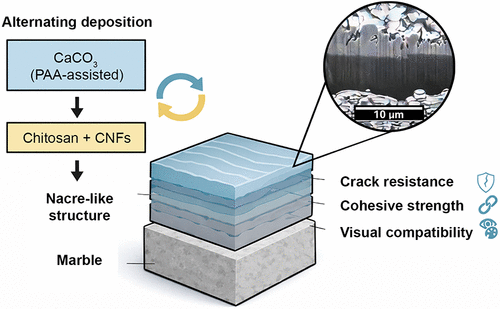
Nacre-Inspired Composite Coatings with Hierarchical Architecture for Durable Surface Protection
Aranzazu Sierra-Fernández *- ,
Diego Cortes - ,
Miguel A. Monclus - ,
Kenneth J.T. Livi - ,
Michael Kappl - ,
Stefan A.L. Weber - ,
D. Howard Fairbrother *- , and
Rafael Fort
This publication is Open Access under the license indicated. Learn More
A bioinspired multilayer coating is developed for the protection of built cultural heritage, emulating the hierarchical architecture of natural nacre. The system is fabricated through the alternating deposition of mineralized calcium carbonate (CaCO3) and organic layers composed of chitosan and cellulose nanofibrils (CNFs), with poly(acrylic acid) (PAA) acting as a mineralization-directing agent. A CO2-controlled environment promotes the formation of continuous crystalline CaCO3 layers with strong interfacial adhesion to marble substrates. The resulting composite multilayers exhibit stratified organization and mechanical properties comparable to those of the biogenic minerals. Nanoindentation and stiffness mapping reveal hardness and modulus values in the range of natural nacre, along with enhanced reinforcement with increasing numbers of multilayers. Mechanical durability under acidic conditions confirms the preservation of both structural integrity and aesthetic compatibility, with color changes remaining below perceptual thresholds (ΔEab < 5). The observed crack resistance, cohesive strength, and mechanical compatibility with the substrate highlight the effectiveness of the layered architecture for dissipating stress and inhibiting damage propagation. These results contribute to the development of an emerging class of bioinspired protective coatings that integrate mechanical resilience, chemical stability, and visual compatibility by establishing a groundwork for advanced materials tailored to the complex demands of cultural heritage conservation.

Adding Another Syngony to the K–Cd–Sb System: Synthesis, Structure, and Properties of Cubic, Clathrate-Like K3Cd12Sb10
Tori Cox - ,
Volodymyr Gvozdetskyi - ,
Nisaga Prathibhani Wanigasekara - ,
Zhen Zhang - ,
Genevieve Amobi - ,
Kaden Osborn - ,
Zeina Miari - , and
Julia V. Zaikina *
K3Cd12Sb10 was discovered using an unconventional hydride synthetic route, whereas the temperature conditions were rationalized from in situ powder X-ray diffraction. K3Cd12Sb10 can be synthesized in the single-phase form through the hydride method at relatively low temperatures (723 K). K3Cd12Sb10 melts incongruently at 794 K and has a narrow synthesizability window (691–794 K), as determined by in situ high-temperature diffraction and differential scanning calorimetry. Because of its limited thermal stability, suitable crystals for single-crystal X-ray diffraction are unavailable; the crystal structure of K3Cd12Sb10 was solved from high-resolution synchrotron powder X-ray diffraction data. K3Cd12Sb10 crystallizes in a new structure type (space group Ia3̅d, a = 18.09301(1) Å, V = 5922.87(1) Å3, Z = 8), adding another syngony for the previously reported ternary K–Cd–Sb compounds. By merging unconventional synthesis with in situ high-temperature monitoring, this study pushes the boundaries of materials discovery, revealing a clathrate-like phase with a novel structure type and hinting at vast structural diversity across other antimonide systems.

Electro-Microfluidic Exfoliation of Air-Sensitive HfTe2 into High-Quality Nanosheets and Electrically Continuous Thin Films
Mohammad Mehmandoust - ,
Yelizaveta Kudryavtseva - ,
Alina Müller - ,
Kevin Synnatschke *- , and
Michael Ruck *
This publication is Open Access under the license indicated. Learn More
The scalable production of two-dimensional (2D) materials under inert, surfactant-free conditions remains a major challenge─particularly for air-sensitive compounds such as the topological semimetal HfTe2. Here, we present a microfluidic exfoliation strategy that integrates mechanical shear with localized electrochemical delamination to produce high-quality HfTe2 nanosheets without stabilizing agents. Conducted entirely under an inert atmosphere, the process minimizes oxidation and preserves the intrinsic Dirac semimetal character of the material. The resulting dispersions enable the formation of dense, conformal thin films with clean internanosheet interfaces. Structural and spectroscopic analyses confirm that the microfluidically exfoliated nanosheets retain their crystallinity and stoichiometry, outperforming their conventionally electrochemically exfoliated counterparts. Electrical transport measurements reveal metallic conduction, indicating sufficient interflake coupling to maintain charge continuity across the film. However, the limited phase-coherence length derived from weak antilocalization and the presence of disorder-induced Parish–Littlewood-type magnetoresistance indicate that long-range quantum coherence is not preserved. These results demonstrate that microfluidic exfoliation yields electronically continuous yet structurally tunable films of air-sensitive layered semimetals, providing a scalable, surfactant-free pathway toward solution-processed, topological materials for low-power spintronic and quantum devices.
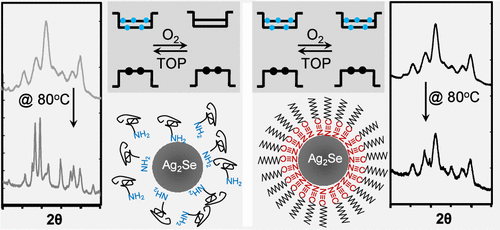
Alkylnitrile Ligands Enable Stable Phase and Electronic Structures of Monodisperse Ag2Se Nanocrystals
Zhe Wang - ,
Jie Zhu - ,
Jiongzhao Li - ,
Xudong Qian - , and
Xiaogang Peng *
Alkylnitriles, N≡CR with R as a long alkyl chain with 11–17 carbons, are introduced for synthesizing and stabilizing Ag2Se nanocrystals (NCs). Their intermediate bonding strength─stronger than alkylamines yet weaker than thiols─enables synthesis of monodisperse monoclinic Ag2Se NCs in a large size range (3–20 nm) at 120–140 °C. AgNO3 dissolved in N≡CR and Se-trioctylphosphine dissolved in NH2Ol are respectively introduced as the silver and selenium precursors, which effectively avoid formation of the Ag0 phase. Given the large excess of alkylamines in the synthesis, the native surface ligands on the as-synthesized NCs are dominated by dynamic and weak NH2Ol ligands, which can be readily replaced by relatively strong and stable N≡CR ligands at ambient temperatures. Unlike NH2Ol-coated Ag2Se NCs, N≡CR-coated ones have high phase (monoclinic) and electronic (either doped or undoped) stabilities, offering two series of Ag2Se NCs for either the short-wavelength or middle infrared window. The findings here indicate that specifically designed surface ligands not only ensure colloidal stability during synthesis and processing but also confer the desired phase and electronic stability to NCs, which may otherwise remain metastable when inappropriate ligands are used.
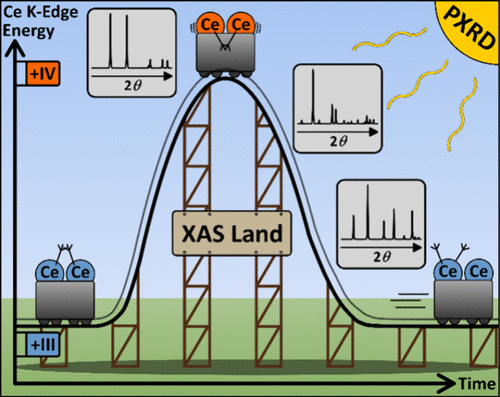
Cerium-Based Coordination Network Formation: An In Situ X-ray Absorption Spectroscopy and Powder X-ray Diffraction Study
Niklas Ruser - ,
Anastasia Yu. Molokova - ,
Kirill A. Lomachenko - ,
Zheting Chu - ,
Xiaodong Zou - ,
Felix Steinke - ,
Diletta Morelli Venturi - ,
Christoph Meier - ,
Bastian Achenbach - ,
Azat Khadiev - ,
Jonas Gosch - , and
Norbert Stock *
This publication is Open Access under the license indicated. Learn More
The Ce(NO3)3·6H2O/H2TDC/CH3COOH/CH3CN (H2TDC = 2,5-thiophenedicarboxylic acid) chemical system was studied under solvothermal reaction conditions. Four different phases that successively crystallized as a function of time were observed. Three coordination networks, [CeIV(TDC)(CH3COO)2] (1), [CeIII4(TDC)3(CH3COO)6] (2a), and [CeIII(TDC)(CH3COO)] (3), could be isolated as orange, beige, and white phase pure products, respectively, and their crystal structures were resolved from powder X-ray diffraction data. Another crystalline compound (2b) was observed in situ, which seems to be structurally related to compound 2a. Compound 2a is a metal–organic framework (MOF) with a pore size of ∼3 Å. The use of CeIII(NO3)3 as the starting material, the different colors of the products, and the crystal structures indicated a peculiar redox behavior with a Ce(III)–Ce(IV)–Ce(III) redox transformation during product formation of 1, 2a, and 3. The oxidation states of 1 and 3 were confirmed by ex situ X-ray absorption near-edge structure (XANES) measurements, and the crystallization process was followed using quasi-simultaneous in situ powder X-ray diffraction (PXRD) and X-ray absorption spectroscopy (XAS) measurements. During the reaction, the consecutive crystallization in the order 1–2b–3 was clearly observed. Linear combination fitting (LCF) of the in situ XAS data also affirmed the formation of the title compounds.
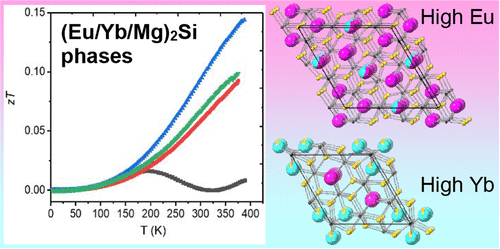
Thermoelectric and Magnetic Behavior of (Eu/Yb/Mg)2Si Zintl Phases Grown in Magnesium-Based Flux
Olufemi S. Araoyinbo - ,
Amirhossein Zareihassangheshlaghi - ,
Md Sahab Uddin - ,
Mehak Ghafoor - ,
Kaya Wei - , and
Susan E. Latturner *
Reactions of silicon with europium and ytterbium were carried out in Mg/Zn eutectic flux to synthesize complex metal silicides. Depending on the ratios of Eu and Yb reactant used, observed products were Yb2MgSi2 (when no Eu was used), Eu2Yb2.7Mg9.3Si7 (with more Yb than Eu reacted), and Eu5(Eu1–xYbx)3Mg16Si12 (with more Eu than Yb). The latter two compounds form in the Zr2Fe12P7 (P-6) and Ho5Ni19P12 (P-62m) structure types, and are charge-balanced Zintl phases. Density of states calculations show that the consistently observed composition of Eu2Yb2.7Mg9.3Si7 is electronically stabilized. Magnetic susceptibility measurements show europium and ytterbium are both divalent; highly anisotropic ferromagnetic ordering of Eu2+ moments is observed at low temperature. Thermoelectric measurements indicate that site mixing of cations lowers thermal conductivity, and that Eu6.72Yb1.28Mg15.56Zn0.44Si12 has the most promising thermoelectric behavior with a zT = 0.14 at 400 K and potential for use at high temperatures.
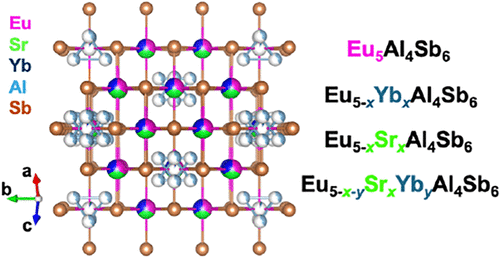
One Cation Makes a Difference: Structure–Thermoelectric Interplay in Pseudo–Rock Salt Intermetallic Eu5–xAxAl3Sb6 (A = Sr and Yb)
Luis Garay - ,
Leah Borgsmiller - ,
Duncan Zavanelli - ,
G. Jeffrey Snyder - ,
James C. Fettinger - , and
Susan M. Kauzlarich *
Polar intermetallics are an emerging class of thermoelectric materials whose electronic properties can be finely tuned by cation chemistry. Single crystals of Eu5–xYbxAl3Sb6 and Eu5–x–ySrxYbyAl3Sb6 were synthesized by flux methods and their structures determined by single-crystal X-ray diffraction, confirming monoclinic C2/m symmetry and an electron count near 3.5 e– per atom, consistent with polar intermetallic classification. The Al content in these phases can be increased from 3 to 4. A comparative study of polycrystalline synthesized Eu5Al4Sb6 and its Sr- and Yb-substituted solid solutions, along with the pseudoquinary phase Eu2.5Sr2Yb0.5Al4Sb6, is presented. Substituting Eu2+ with the more ionic Sr2+ enhances mobility and increases the magnitude of the Seebeck coefficient, while the more covalent Yb2+ drives the system metallic, lowering Seebeck values but improving zT to 0.8 at 873 K. The quinary phase further suppresses bipolar conduction, delaying the high-temperature downturn observed in both ternary solid solutions. Across all compositions, thermal conductivities remain exceptionally low (<1 W m–1 K–1), enabling promising figures of merit. These results highlight how ionic versus covalent A-site substitution can serve as a powerful lever to control scattering, band overlap, and transport in polar intermetallics, opening design pathways parallel to those of the benchmarked half-Heusler phases and PbTe.
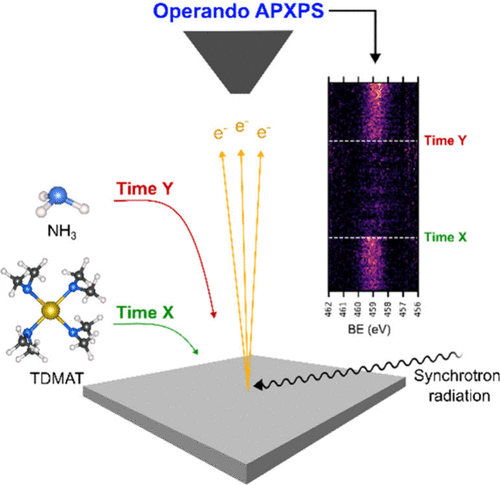
Surface Chemistry in the Initial Stages of Titanium Nitride Atomic Layer Deposition Using Operando Ambient Pressure X-ray Photoelectron Spectroscopy
Pamburayi Mpofu *- ,
Peggy Bagherzadeh Tabrizi - ,
Houyem Hafdi - ,
Premrudee Promdet - ,
Jonas Lauridsen - ,
Oscar Alm - ,
Tommy Larsson - ,
Rosemary Jones - ,
Esko Kokkonen - ,
Joachim Schnadt - , and
Henrik Pedersen *
This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Studies of the surface chemistry of the first few cycles of atomic layer deposition (ALD) using in situ and time-resolved operando techniques are attractive for realizing, understanding, and obtaining true mechanistic information during the deposition. However, the latter techniques are yet to be applied to ALD of metal nitrides. Here, we present a surface-chemistry investigation through a time-resolved ambient pressure X-ray photoelectron spectroscopy (APXPS) study of the initial growth of titanium nitride (TiN). The Ti 2p, O 1s, N 1s, C 1s, and Si 2p core-level spectra recorded at different stages during the deposition show that chemisorption occurs immediately on the silicon dioxide surface due to the interaction of tetrakis(dimethylamido)titanium(IV) (TDMAT) with the surface. A delay in nucleation on the TDMAT-terminated surface was observed during the NH3 pulse. The intensity of the Ti 2p and N 1s core levels began to increase after four ALD cycles, showing that the surface was coated with Ti and N atoms and no Si signals were observed with time. The results show that ligand exchange reactions take place before transamination reactions. This was verified using the periodic changes in the intensity and peak positions of the above-mentioned spectra and complemented by residual gas analysis using mass spectrometry. These results can provide insights into the ALD surface growth of not only TiN but also other metal nitrides.
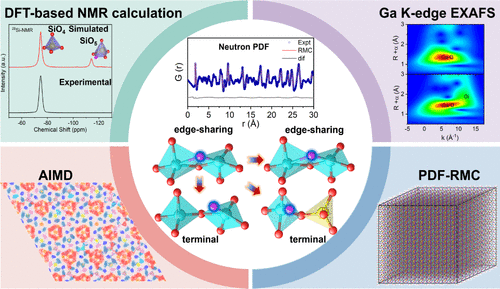
Local Rigid-Unit Design Modulates Interstitial Oxygen Coordination Geometry for Developing Langasite-Type Oxide-Ion Solid Electrolytes
Lingzhi Jiang - ,
Xiaohui Li *- ,
Cheng Li - ,
Ligang Xu - ,
Yixing Zhao - ,
Yifeng Han - ,
Shangqing Qu - ,
Qilong Gao - ,
Mingxue Tang - ,
Sihao Deng - ,
Lunhua He - ,
Junliang Sun - , and
Xiaojun Kuang *
The langasite family, featuring a three-dimensional open framework, offers exceptional structural and compositional flexibility for accommodating and transporting oxide ions. However, conventional langasite-type oxide-ion solid electrolytes (SEs) suffer from charge trapping of interstitial oxygens within the edge-sharing coordination geometry, severely limiting long-range migration. Here, we introduce a local rigid-unit design strategy to modulate interstitial oxygen coordination and unlock interstitial oxide-ion conduction in langasite La3Ga5–xGexSiO14+x/2. Incorporation of a rigid SiO4 unit and partial Ge4+ substitution stabilizes interstitial oxygen at terminal sites of GaO5 polyhedra, rather than at edge-sharing traps, enhancing migration mobility. 29Si solid-state NMR, supported by DFT calculations, and neutron PDF analysis with RMC modeling, reveal that SiO4 units preserve their intrinsic coordination geometry, while interstitial oxygens are preferentially accommodated at terminal oxygens of SiO4–GaO5 and GaO4–GaO5 linkages, and at relaxed bridging sites within edge-sharing GaO4 units, which effectively circumvent edge-sharing charge trapping and facilitate interstitial oxygen mobility. Complementary AIMD and classical MD simulations confirm the interconnected 2D long-range interstitial oxide-ion migration involving structurally distinct oxygen sites. These findings establish a design principle in which rigid structural units modulate interstitial oxygen coordination from bridging to terminal sites, providing mechanistic insight into enhancing both structural rigidity and ionic mobility in next-generation interstitial oxide-ion SEs.
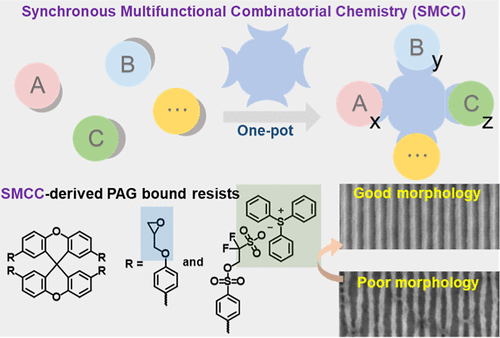
Synchronous Multifunctional Combinatorial Chemistry on a Molecular Glass Backbone─a Strategy for Developing Photoresists with Improved Lithography Performances
Yu Yan - ,
Chenfei Zhao - ,
Jingwen Hui - ,
Xinfu Zhang *- ,
Xue Zhang - ,
Yifeng Peng - ,
Pengzhong Chen - ,
Xiaojun Peng *- , and
Yi Xiao *
Although molecular photoresists exhibit higher resolution than polymer-based resists, the latter play a dominant role in lithography applications. The pivotal reason lies in the fact that multiple function integration can be readily achieved in polymer resists through one-pot copolymerization with various monomers, which is critical to enhance the overall performances; but for molecular photoresists, the functional monotonicity remains a significant drawback that needs to be overcome. Herein, a strategy named as Synchronous Multifunctional Combinatorial Chemistry (SMCC), i.e., one-pot multiple and different decorations on a molecular glass backbone, which integrates the idea of combinatorial synthesis with the approach of parallel processes analogous to copolymerization, is proposed to develop multifunction-integrated molecular photoresists. The feasibility of the SMCC strategy is exemplified by high yield syntheses of a series of diversely grafted spiroxanthene molecules via synchronous Suzuki coupling of X4Br (four bromo to be modified) with distinct phenylboronic acid/ester derivatives. Then, by the same way, an all-in-one photoresist X4EpTPS was conveniently constructed, in which both photoacid generator (PAG) triphenylsulfonium (TPS) and epoxy group (Ep, acid-driven cross-linking) were bound to a spiroxanthene core in a specified ratio. The lithography performance of X4EpTPS was studied and compared with physically blending formulation of direct adding PAG TPS-TF in X4Ep. It is confirmed that molecular level integration of multiple functions into X4EpTPS ensures more homogeneous PAG distribution during the PAB process while effectively confining photoacid diffusion in the PEB process, significantly enhancing lithographic performance. Finally, X4EpTPS achieves dense 25 nm HP L/S patterns with critical dimension 23 nm and LER 2.7 nm. Therefore, we are confident that in the future development of molecular photoresists, the strategy of SMCC is broadly extensible and applicable for performance enhancement tailored to diverse requirements.

Modulating the Photoluminescence of CsPbBr3 Nanocrystals via Cation Variation in BF4– Salts
Min-Gi Jeon - ,
Artavazd Kirakosyan - ,
Subin Yun - ,
Chang-Yeon Kim - ,
Dung Khac Nguyen - ,
Seonu Lee - ,
Huong Thi Cam Le - ,
Sunhyun Nam - , and
Jihoon Choi *
Despite metal halide perovskite nanocrystals (NCs) having shown great promise for light-emitting applications, their performance is often limited by surface defects and unstable ligand environments that promote nonradiative recombination. To overcome these challenges, the influence of countercations in tetrafluoroborate (BF4–) salts on the surface passivation and photophysical properties of CsPbBr3 NCs was systematically investigated. A series of BF4– salts with inorganic-, aromatic-, and phosphorus-based cations were examined to correlate countercation chemistry with surface reactivity. Structural analyses revealed that most BF4– salts efficiently removed metallic Pb0 defects while maintaining the phase integrity. NH4BF4 promoted the oriented attachment of nanocubes into nanowires, whereas tritylium BF4 induced the partial decomposition of BF4– into BF3 and F–, forming Pb–F bonds that stabilized the surface and reduced trap densities. In contrast, 2,4,6-triphenylpyrylium BF4 triggered a Katritzky reaction with oleylamine, leading to aggregation and Cs4PbBr6 formation. Photophysical measurements showed enhanced photoluminescence and increased trap activation energies for most BF4–-treated NCs due to suppressed nonradiative recombination. Light-emitting diodes incorporating sodium and tritylium BF4-treated NCs exhibited improved emission stability and electroluminescence. These findings highlight countercation-dependent surface chemistry as a key factor in achieving efficient defect passivation and stable perovskite optoelectronic performance.

Mechanistic Insights into the Aminolytic Decomposition of Mo(CO)6 to Form MoC1–x Nanoparticles
Brendan Ward-O’Brien - ,
Allison Forsberg - ,
Yizhen Chen - ,
Noah Malmstadt - ,
Majed S. Madani - , and
Richard L. Brutchey *
The controlled synthesis of refractory metal carbide nanoparticles under mild, solution-phase conditions remains a grand challenge in colloidal nanoparticle chemistry because the strong bonds and high formation energies of these materials make controlled nucleation and growth difficult, and, as a result, these mechanisms remain largely unexplored. Here, we provide the first mechanistic insight into the formation of α-MoC1–x nanoparticles by investigating the aminolytic decomposition of Mo(CO)6 in oleylamine (OAm) and N,N-dimethyloctadecylamine (DODA). Ex situ FT-IR studies reveal stepwise substitution of carbonyl ligands by the amine, with OAm yielding disubstituted molybdenum carbonyl species and DODA yielding only the monosubstituted species. Ex situ X-ray diffraction and in situ synchrotron small-angle X-ray scattering show that these precursors first convert into an isolable amorphous intermediate, which then rapidly crystallizes into α-MoC1–x nanoparticles. Stronger precursor-ligand complexation in OAm delays decomposition, producing rapid, synchronized nucleation and smaller nanoparticles, whereas DODA promotes earlier, more gradual decomposition and the formation of larger nanoparticles. Kinetic analysis confirms that crystallization is reaction-controlled, with intraparticle densification proceeding without significant ripening. These results highlight the critical role of solvent-dependent precursor-ligand interactions in controlling the decomposition temperature, kinetics, and final size of colloidal metal carbide nanoparticles.

Optical Magnetoelectric Effect in a Polar Ferromagnetic Two-Dimensional Organic–Inorganic Hybrid Perovskite
Po-Jung Huang *- ,
Gyeongoh Noh - ,
Shojiro Kimura - , and
Kouji Taniguchi *
Magnetoelectric effect is a coupling phenomenon between magnetism and dielectric properties that occurs in noncentrosymmetric magnetic materials. This effect can be extended to the response to electromagnetic waves, i.e., light, in materials and is referred to as the optical magnetoelectric (OME) effect. The OME effect can give rise to fascinating optical properties, such as propagation-direction-dependent light absorption, known as nonreciprocal directional dichroism. In this study, we prepared single crystals of a polar and ferromagnetic two-dimensional organic–inorganic hybrid perovskite (2D-OIHP) copper chloride, (ClBA)2CuCl4 (where ClBA+ = 4-chlorobenzylammonium ion), with sufficient size for optical measurements, and successfully detected the OME effect by measuring the difference in light absorption between opposite propagation directions. These results suggest that 2D-OIHPs are a promising class of materials for developing emergent functionalities unique to noncentrosymmetric systems.

Hierarchical Self-Assembly of Disulfide-Linked Single-Stranded DNA into Stimuli-Responsive Pods
Volkan Kilinc *- ,
Linawati Sutrisno - ,
Joel Henzie - ,
Emmanuel Picheau - ,
Yusuke Yamauchi - ,
Katsuhiko Ariga - , and
Jonathan P. Hill *
This publication is Open Access under the license indicated. Learn More
Controlling the large-scale assembly of charged biopolymers is a fundamental challenge in materials chemistry. Here, we report a chemical strategy that uses disulfide-linked single-stranded DNA (ssDNA) dimers as unique building blocks to drive the hierarchical self-assembly of functional DNA microstructures. Formed from short, random-sequence oligomers, these dimers first organize into DNA-salt composite nanobead condensates, which then serve as scaffolds for the assembly of uniform, microrod-shaped DNA condensates called DNA-pod condensates. The key innovation of this work is the material’s unique, cooperative structural transition. Upon thermal stimulation (>60 °C), dsDNA-pod condensates undergo a rapid exfoliation into an expanded ssDNA network, a process driven by significant gains in configurational entropy and the relief of electrostatic repulsion. This establishes an accessible strategy for creating stimuli-responsive DNA materials through a chemistry-driven, sequence-independent pathway. We further demonstrate that these materials act as robust host matrices for encapsulating guest molecules like doxorubicin.

Lewis Acid–Base-Driven Anisotropic Crystal Growth of Pyrochlore Pb2Ti2O5.4F1.2 with Enhanced Visible-Light H2 Evolution Activity
Gentoku Kido - ,
Hiroto Ueki - ,
Megumi Okazaki - ,
Jun Kikkawa - ,
Koji Kimoto - ,
Ryosuke Nishikubo - ,
Akinori Saeki - , and
Kazuhiko Maeda *
This publication is Open Access under the license indicated. Learn More
Mixed-anion compounds offer unique functionalities unattainable with single-anion materials, yet rational morphology control remains largely unexplored. Here, we report a Lewis acid–base-driven strategy that enables low-temperature, solution-phase morphology control of the oxyfluoride photocatalyst Pb2Ti2O5.4F1.2 (PTOF). A microwave-assisted solvothermal method with monoethanolamine (MEA) was used to tune the particle morphology and size via the precursor-solution pH, which was adjusted by the addition of formic acid or acetic acid. PTOF, an A2B2X6X′0.5-type pyrochlore with intrinsic anion vacancies (X′2, 4d site), has exposed {111} facets composed of alternating Pb-rich and Ti-rich layers. Lewis basic MEA is proposed to bind selectively to undercoordinated, strongly acidic Pb2+ sites adjacent to Ti4+ and vacancies on Ti-rich {111} facets, suppressing growth along the surface direction and stabilizing these facets, thereby driving anisotropic crystal growth and forming plate-like nanoparticles. At pH 10 (formic acid), PTOF nanoparticles (∼30 nm) with a specific surface area of 37 m2 g–1 were obtained. Compared with an analogous PTOF synthesized by a conventional solid–state reaction, the optimized sample exhibited ∼29-fold higher H2 evolution activity in an aqueous solution containing dissolved disodium ethylenediaminetetraacetate under visible-light (λ > 400 nm) with the aid of a Pt cocatalyst. Lewis acid–base-directed facet stabilization is thus shown to be a promising approach for the rational morphological design of mixed-anion oxyfluorides via solution processing.
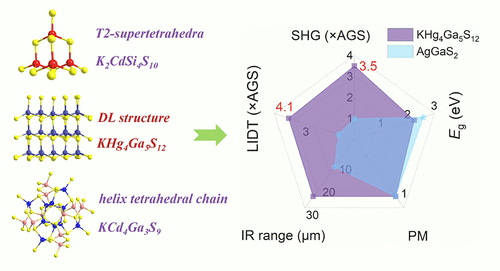
KHg4Ga5S12: A Diamond-like Tetrahedral Chalcogenide Exhibiting Giant Phase-Matching Second Harmonic Generation
Weiping Guo - ,
Chao Yang - ,
Bingxuan Li - ,
Hong-Hua Cui *- ,
Lingyun Li *- ,
Yan Yu *- ,
Zhong-Zhen Luo *- , and
Zhigang Zou
Tetrahedra-based chalcogenides constitute the most abundant type for mid-infrared (IR) nonlinear optical (NLO) crystals. Meanwhile, different connection modes of tetrahedra will determine the mid-IR NLO properties, such as birefringence (Δn) and the second harmonic generation (SHG) response. In this research, we systematically investigate the structure–property modulated relationship between tetrahedra-based compounds and mid-IR NLO properties. Herein, three compounds, KHg4Ga5S12, K2CdSi4S10, and KCd4Ga3S9, consisting of the tetrahedral units, have been successfully synthesized through a moderate-temperature solid-state reaction. Specifically, K2CdSi4S10 and KCd4Ga3S9 with the opposite polarity T2-supertetrahedra and random orientation helix tetrahedral chains exhibit a small Δn and non-phase-matching (non-PM) SHG response. Notably, KHg4Ga5S12 with the distorted diamond-like (DL) structure shows a large PM SHG response of 3.5 × AgGaS2 (AGS), moderate Δn of 0.042@2050 nm, and high laser-induced damage threshold (LIDT) of 4.1 × AGS. Therefore, the results indicate that KHg4Ga5S12 has potential for application as a high-performance mid-IR NLO crystal. The consistent arrangement and distorted DL structure can be prioritized for the design of mid-IR NLO crystals.

Dual-Fuel Driven Dissipative Self-Assembly of Gold Nanoparticles Enables Transient Conductivity
Pankaj Maity - ,
Pijush De - ,
Subhashis Ghosh - ,
Venkatesh Jha - ,
Soumen De - ,
Satyaprasad P. Senanayak *- , and
Dipak Samanta *
Inspired by the dynamic behavior of natural systems, which rely on a constant supply of energy to maintain structure and function, we have developed a synthetic platform that mimics such nonequilibrium behavior. Our system employs a chemically fueled mechanism to control the reversible assembly of gold nanoparticles in methanol. Central to this strategy is a dual-fuel approach involving 9-fluorenylmethoxycarbonyl chloride (Fmoc-Cl) and triethylamine (Et3N), both of which serve as activators, while Et3N subsequently functions as a deactivator. In this process, Fmoc groups are transiently attached to ligands on the AuNP surfaces, reducing polarity and inducing aggregation via solvophobic interactions. Meanwhile, Et3N facilitates a carbonate-forming reaction that gradually removes the hydrophobic Fmoc groups, restoring the system to its original dispersed state. A crucial design feature is that the fuel-driven attachment reaction occurs significantly faster than the cleavage, enabling precise temporal control over the assembly process across multiple cycles. Importantly, the extent of nanoparticle aggregation directly influences the electrical conductivity of the system, providing a chemically driven route to responsive, energy-dissipative nanomaterials with transient conductivity reminiscent of neuronal behavior. These findings lay the groundwork for the development of transient electronic devices, bioinspired memory, and smart materials that operate far from equilibrium–much like systems in nature.

Flame-Retardant Synergism through Chemical Intercalation for Rigid Polyurethane Foam by Using Intrinsic/inorganic Combination
Jinpeng Liu - ,
Hongmei Mu - ,
Junchao Huang *- , and
Yuhua Wang *
Rigid polyurethane foam (RPUF) is a versatile thermal and acoustic barrier material with high flammability, which raises severe safety concerns and necessitates an effective flame retardant (FR). Compared to the single FR system, organic/inorganic flame-retardant composites with the synergistic effect show better performance in both flame retardancy. While the synergistic effect is always considered among FRs and does not include the polymer matrix, it obtains limited effectiveness. Our work prepared an extender for PU foam, which contains accessible phenyl side groups that can interact with modified expandable graphite through intercalation to construct a synergistic effect between the polymer matrix and the inorganic FR. With a total additive content of 21.9%, the LOI and UL-94 ratings increased to 37% and V-0 level, respectively, which shows the greatest improvement compared to other expandable graphite systems and only induces small fluctuations in physicochemical performances. The designed FRs fulfill the major requirements of current standards on RPUF, and they also show a great practicality for future large-scale production.

Engineering Giant Strain in Bismuth Ferrite–Barium Titanate Relaxor Ferroelectrics via A-Site Driven Local Structural Disorder
Zhanpeng Li - ,
Xiaoming Shi - ,
Xianghong Zhou - ,
Yuxuan Yang - ,
Zhi Tan - ,
Chao Wu - ,
Qihang Tang - ,
Yang Zhang - ,
Haijun Wu *- ,
Ting Zheng *- ,
Shujun Zhang *- , and
Jiagang Wu *
Lead-free bismuth ferrite–barium titanate (BF–BT) relaxor ferroelectrics have emerged as promising candidates for high-strain actuator applications, yet the fundamental link between their nanoscale structure and macroscopic electromechanical performance remains elusive. This study overcomes this challenge by demonstrating that controlled A-site La3+ doping in 0.7(Bi0.95La0.05)FeO3–0.3BaTiO3 (BLF–BT) directly engineers a local structural environment characterized by chemical disorder and localized stress fields. Through local structure analysis and simulations, we reveal that La doping introduces A-site chemical heterogeneity and lattice mismatch, enhancing FeO6 octahedral distortions and local structural fluctuations. This pronounced local disorder suppresses long-range rhombohedral order, fostering a pseudocubic matrix populated by interacting randomly oriented polar nanoregions. These structural modifications create a flattened energy landscape that facilitates nearly isotropic and low-barrier polarization reorientation under an electric field. The resultant cooperative switching of these highly responsive nanodomains, the inherent lattice strain from local distortions, yields substantial unipolar strain of 0.35%, representing a 200% enhancement over undoped BF–BT. This work provides a definitive structural mechanism for giant strain in lead-free relaxors and establishes a design principle for activating large electromechanical responses through targeted local disorder.

pH Regulates Ion Dynamics in Carboxylated Mixed Conductors
Zeyuan Sun - ,
Mengting Sun - ,
Rajiv Giridharagopal - ,
Robert C. Hamburger - ,
Siyu Qin - ,
Haoxuan Li - ,
Mitchell Hausback - ,
Yulong Zheng - ,
Bohyeon Kim - ,
Heng Tan - ,
Thomas E. Gartner III*- ,
Elizabeth R. Young *- ,
Christopher J. Takacs *- ,
David S. Ginger *- , and
Elsa Reichmanis *
This publication is Open Access under the license indicated. Learn More
Coupled ionic and electronic transport underpins processes as diverse as electrochemical energy conversion, biological signaling, and soft adaptive electronics. Yet, how chemical environments such as pH modulate this coupling at the molecular scale remains poorly understood. Here, we show that the protonation state of carboxylated polythiophenes provides precise chemical control over ion dynamics, doping efficiency, solvent uptake, and mechanical response. Using a suite of multimodal operando techniques, supported by simulations, we reveal that pH dictates the balance of cation/anion uptake during electrochemical doping. Mapping across pH uncovers a quasi-nonswelling regime (≈pH 3–3.5) where charge compensation proceeds with minimal volumetric change yet pronounced stiffening. These findings establish molecular acidity as a general strategy to program ionic preference and mechanical stability, offering design principles for pH-responsive mixed conductors and soft electronic materials that couple ionic, electronic, and mechanical functionality.
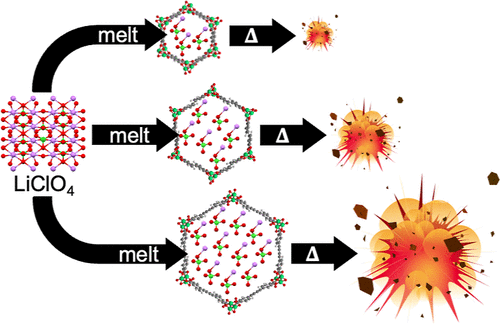
Tunable on-Demand Explosives Derived from Isoreticular Metal–Organic Framework Nanocomposites
Nicholas A. Tomalia - ,
Yulia Rakova - ,
Ashley N. Tubman - , and
Adam J. Matzger *
Energetic metal–organic frameworks (MOFs) and MOF/oxidizer composites both suffer from thermal and mechanical sensitivity. Here, we present a molecular-level design strategy that overcomes this limitation by using nonenergetic components to prepare MOF-based composite energetic materials with tunable on-demand sensitivity and energetic behavior. In this two-component approach, porous, fuel-rich MOFs serve as catalytic host frameworks that support melt-infiltration of an oxidizing salt, facilitating nanoscopic mixing of fuel (linker), catalyst (metal node), and oxidizer. Linker extension achieves isoreticular pore expansion, dictates oxidizer loading capacity, oxidizer/fuel ratio, and catalyst density. A linker designed to minimize steric bulk and incorporate high-energy functional groups establishes the linker as a tunable design feature for controlling energetic decomposition. Across a series of isoreticular MOF/oxidizer formulations, higher oxidizer/fuel ratios and lower relative metal densities lead to greater energy output and gas release while also improving thermal stability; this represents a rare synergy. This approach yields energetic nanocomposites that are insensitive to impact, friction, and electrostatic shock yet transition to thermally sensitive explosives only after heat is supplied beyond the oxidizer melting point, constituting a rational design strategy to produce safer and tunable explosives.
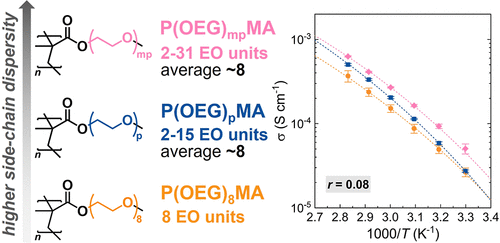
Structural Dispersity as a Determinant of Li-Ion Transport in Ethylene-Oxide-Based Graft Polymer Electrolytes
Anna Vigolo - ,
Valeria Vanoli - ,
Luca Laugeni - ,
Carlos Pavón - ,
Rossana Pasquino - ,
Edmondo M. Benetti - ,
Franca Castiglione *- , and
Francesca Lorandi *
This publication is Open Access under the license indicated. Learn More
Graft polymers with oligo(ethylene glycol) (OEG) side chains and poly(meth)acrylate backbones have been commonly studied as polymer electrolytes (PEs) owing to the ability of oligoether segments to coordinate Li+ ions. However, when poly[oligo(ethylene glycol) methyl ether methacrylate]s (P(OEG)MAs) are synthesized from commercial macromonomers, these are structurally polydisperse, as OEG segments feature a broad distribution of lengths. Herein, we investigate the influence of side-chain heterogeneity on Li-ion transport by comparing structurally polydisperse P(OEG)MAs with analogous graft polymers with homogeneous architecture, generated from discrete macromonomer feeds obtained through flash chromatography. Ionic conductivity was found to increase with increasing side-chain dispersity. For structurally polydisperse P(OEG)MAs, enhancing side-chain heterogeneity resulted in greater salt dissociation and higher ionic conductivity at relatively high salt contents. These trends are uncorrelated with differences in thermal properties, rheology, and polymer diffusivity, indicating that ion transport is not governed by overall polymer dynamics. Dispersity of side chains thus emerges as a determinant for Li-ion transport in PEs based on P(OEG)MAs. However, this effect is lost when backbone flexibility increases, i.e., when polymethacrylates are substituted with more flexible polyacrylate counterparts. By elucidating how side-chain heterogeneity and backbone flexibility affect ion transport, this work provides guidance for the rational design of graft PEs.
Dual-Mode Force-Responsive Information Encryption Enabled by Sustainable Cellulose Nanocrystal-Reinforced Waterborne Nonisocyanate Polyurethanes
Huimin Ren - ,
Qingyu Liao - ,
Ziyu Zhou - ,
Shuna Gao - ,
Ouwei Dai - ,
Xu Du - ,
Baihua Yuan *- , and
Hongbin Zhang *
Nonisocyanate polyurethanes (NIPU) have emerged as a promising alternative to conventional polyurethanes owing to their enhanced safety and sustainability across the entire life cycle. Here, we report a green waterborne NIPU elastomer toughened with cellulose nanocrystals (CNCs). In accordance with the principles of green chemistry, a tough elastomer is synthesized from renewable CNC and waste CO2 as reactants via a simple, organic solvent-free, and safe synthetic route. Furthermore, the damaged elastomer exhibits recyclability through mechanical processing. Physical cross-links among CNC, polyethylenimine, and the NIPU matrix form multiscale microdomains that act as sacrificial bonds, thereby significantly enhancing toughness. Under mechanical deformation, the elastomer exhibits reversible birefringence-induced color changes over 0–300% strain. Meanwhile, the inherent fluorescence of the nonisocyanate backbone enables the creation of UV-activated patterns that remain invisible under ambient light. By integrating these mechanochromic and fluorescent responses, the dual-mode system creates dynamic, force-dependent encryption patterns that are only revealed under concurrent UV illumination and mechanical strain, thus enabling multilevel information encryption. This work introduces a design strategy for dual-responsive, eco-friendly polyurethanes with promising applications in anticounterfeiting, flexible displays, and advanced information storage.

Scalable Fabrication of Electroluminescent Fibers with Exceptional Environmental Stability
Cong Yu - ,
Hao Yu - ,
Legeng Li - ,
Zishuo Zhang - ,
Jiaming Zheng - ,
Shaowu Pan - ,
Yingjie Zhou *- , and
Feng Yan *
Alternating current electroluminescent (ACEL) textiles show great promise for wearables, but functional limitations and unsustainable manufacturing methods limit their industrial use. Herein, we report the development of a carbon dioxide-derived poly(ionic liquid) waterborne polyurethane (CO2–WPU-IL) coating. This coating functions dualistically as an electroluminescent matrix and an encapsulation barrier for ACEL fibers, effectively eliminating interfacial defects and mitigating delamination risks. Critically, CO2-derived carbonate groups synergize with imidazolium cations, delivering superior optical transparency, robust adhesion, and potent antibacterial activity, outperforming existing encapsulation polymers. A scalable brush-coating process enables facile fabrication of ACEL textiles with uniform luminance (200 cd·m–2 at 5 V·μm–1, 1 kHz) and durability across extreme conditions (−80 to 70 °C, ultraviolet radiation, and water immersion). The integration of a 15 × 15 cm2 textile display with electronics enables dynamic pixelated lighting and programmable pattern generation through human-machine interaction. This CO2-derived platform establishes a bridge between high-performance electronics and sustainable manufacturing, charting a scalable, low-carbon-footprint pathway for next-generation interactive wearables.
Density Functional Theory–Machine Learning-Assisted Insightful Identification of Next-Generation High-Voltage Organic Dual-Ion Batteries
Poulami Paul - ,
Souvik Manna - ,
Sandeep Das - , and
Biswarup Pathak *
Organic dual-ion batteries (ODIBs) combine the sustainability of organic materials with the cost-effectiveness and ecofriendliness of dual-ion battery systems. To overcome the large material space and vast combinations of anode, cathode, and electrolyte possibilities, we developed a machine learning model to predict cell voltages for diverse p-type and n-type organic electrode materials. The model demonstrated high accuracy and reliability, validated through repeated k-fold cross-validation, density functional theory (DFT) calculations, and experimental data. Key molecular features, such as functional groups, cyclic cores, ring size, and heteroatoms, were identified as critical components. By interpreting feature contributions, we established a clear connection between the underlying molecular chemistry and predicted voltage outputs, offering insights into feature selection and design principles. This work offers practical insights for experimental researchers to identify optimal salts and organic material pairings, accelerating the development of high-performance, sustainable ODIBs. By integrating machine learning with chemistry-driven design, we provide a scalable pathway to advance next-generation battery technologies.
Additions and Corrections
Correction to “Mechanical Manipulation of Ferroelectric Domains in Molecular Ferroelectric”
K. M. Srishti Barnwal - ,
Xin Li - ,
Aryan Keshri - ,
Mohit Tanwani - ,
Yongjun Wu - ,
Zijian Hong *- , and
Sujit Das *

This publication is free to access through this site. Learn More
Correction to “PMSE in the Next 100 Years: Shaping the Future of Polymers”
Cristina Thomas *- and
Jennifer L. Schaefer *
This publication is free to access through this site. Learn More
Mastheads
Issue Editorial Masthead
This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More