

About the Cover:
Perspectives

Regulating Electronic Functionality in Lindqvist Polyoxometalates: A Molecular Approach to Tunable Metal Oxides
Gowtami Allada - ,
Pavan Kumar Matsa - ,
Kongki Gogoi - , and
Sourav Chakraborty *
Polyoxometalates (POMs) constitute a versatile family of nanoscale metal-oxo clusters characterized by well-defined architectures built from early transition metals, typically vanadium (V), molybdenum (Mo), tungsten (W), or niobium (Nb), which exhibit a range of accessible oxidation states. Lindqvist-type POMs have gained prominence within the polyoxometalate family as surface-engineerable platforms, owing to their compact topology, high structural integrity, and facile access to multiple oxidation states. Through systematic surface functionalization strategies, including heterometal substitution and organo-functionalization, clusters are tuned for precise electrochemical, optical, and catalytic properties. These structural modifications on these atomically defined platforms are correlated with ligand field effects and metal-oxo geometry to define structure–property function relationships, highlighting phenomena such as proton-coupled electron transfer, intervalence charge transfer, and bandgap modulation. This comprehensive overview aims to unify synthetic methodologies, mechanistic insights, and functional attributes, establishing Lindqvist POMs as modular components for next-generation oxide-based materials.
Articles

Reactive Strand Extension to Improve Stretchability in Semiconducting Polymers
Shine H. Huang - ,
Azalea Uva - ,
Saroj Upreti - ,
Adnan Sharif - ,
Karen Fukuda - ,
Rebecca A. Belisle - ,
Xiaodan Gu - , and
Helen Tran *
As electronics become more seamlessly integrated into our everyday lives, the demand for durable, stretchable, and electron-conducting materials will continue to grow. However, many conductive materials suffer from poor electrical performance under repeated mechanical strain, which limits their lifetime use. Inspired by developments to enhance stretchability in nonconjugated materials with covalent mechanochemistry, we explore reactive strand extension (RSE) as a strategy to mitigate poor electronic performance in conjugated polymer semiconductors under strain. Herein, we incorporated RSE into a donor–acceptor conjugated polymer by copolymerizing cinnamate dimers into the conjugated backbone and evaluated their impact on stretchability. RSE was found to improve stretchability in cross-linked conjugated polymer systems via crack onset strain measurements, atomic force microscopy, dichroic ratio measurements, and grazing-incidence wide-angle X-ray scattering. Lastly, charge carrier mobility measurements from organic field-effect transistors revealed that RSE-containing cross-linked conjugated polymers retained mobility more effectively under mechanical strain compared to unmodified conjugated polymers. Overall, our study presents an alternative strategy to improve the performance of conjugated polymers for stretchable electronics.
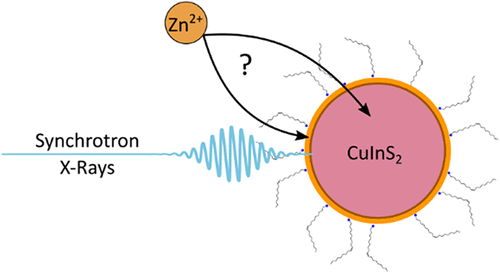
Unveiling Zn Incorporation in CuInS2 Quantum Dots: X-ray and Optical Analysis of Doping Effects, Structural Modifications, and Surface Passivation
Andrés Burgos-Caminal *- ,
Brener R. C. Vale - ,
André F. V. Fonseca - ,
Juan F. Hidalgo - ,
Elisa P. P. Collet - ,
Lázaro García - ,
Víctor Vega-Mayoral - ,
Saül Garcia-Orrit - ,
Iciar Arnay - ,
Juan Cabanillas-González - ,
Laura Simonelli - ,
Ana Flávia Nogueira - ,
Marco Antônio Schiavon - ,
Thomas J. Penfold - ,
Lazaro A. Padilha - , and
Wojciech Gawelda *

This publication is Open Access under the license indicated. Learn More
CuInS2 quantum dots (QDs) have gained significant attention owing to their remarkable broadband emission, making them desirable for various optoelectronic applications requiring efficient luminescent nanomaterials. However, maximizing radiative recombination in CuInS2 QDs necessitates minimizing intragap trap states. A common approach involves the introduction of Zn during the synthesis, which typically promotes the formation of a ZnS shell that passivates the QD surface. Despite its importance, the characterization and quantification of Zn incorporation using conventional techniques, such as optical spectroscopy or electron microscopy, remains challenging. In this study, we utilized X-ray absorption spectroscopy, in both X-ray absorption near-edge structure and extended X-ray absorption fine structure spectral ranges, to investigate Zn incorporation into CuInS2 QDs, probing at the Zn, S, and Cu K-edges. This approach allowed us to detect the formation of a ZnS surface shell, tentatively quantifying its thickness, and to distinguish between Zn as a substituent at the shell or as an interstitial defect. Additionally, we explored the dynamical properties of CuInS2 QDs using time-resolved optical spectroscopies, particularly in the presence of electron and hole acceptors (benzoquinone and phenothiazine), observing that hole transfer is highly sensitive to shell thickness. These results provide deeper insights into the Zn-induced shell.
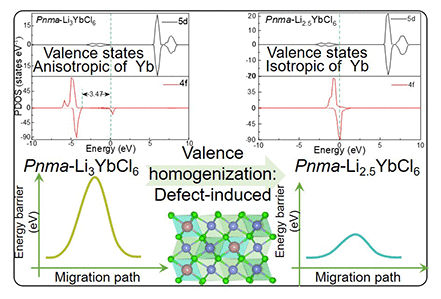
Influence of Defect-Induced Transition-Metal Valence State Changes on Ion Transport in Li3YbCl6 Solid Electrolyte
Yuan Ren *- ,
Jiahui Ye - ,
Cheng Liu - ,
Guojian Cai - , and
Chao Zhang
A unique phenomenon in high-performance solid-state electrolytes of rare-earth halides is the strong dependence of ionic conductivity on the valence state anisotropy of the rare-earth elements, which potentially impedes ion transport. To probe the presence and effects of such anisotropy, the stable crystal structures, electronic structures, ionic conductivities, and Li-migration barriers in Li3YbCl6 ionic conductors with two space groups (Pna21/Pnma) have been investigated using a first-principles approach. The change in valence state is quantified by calculating the density-of-states integral of the transition metal Yb, and ion transport is investigated using first-principles molecular dynamics. Thus, a research framework was established to investigate the relationship between valence state changes and ion conductivity in this work. The results indicate that the Yb ions exhibit anisotropic valence states and are incompletely oxidized in Pnma-Li3YbCl6. In contrast, the Yb ions in Pna21-Li3YbCl6 possess a uniform valence state and are fully oxidized. The ionic conductivity of Pna21-Li3YbCl6 is higher than that of Pnma-Li3YbCl6. This demonstrates that the homogeneity of the transition-metal ion valence state is associated with higher ionic conductivity. To regulate this valence anisotropy, we systematically introduce Li vacancies into the worse-performing Pnma-Li3YbCl6 polymorph, constructing a Pnma-Li2.5YbCl6 structure that achieves a uniform Yb valence distribution. This valence homogenization strategy significantly enhances ionic conductivity by an order of magnitude and reduces the activation energy from 0.3 to 0.1 eV. The uniform valence state mitigates the heterogeneous electronegative blocking effect of Yb during Li+ migration, facilitating more efficient ion transport. These findings highlight the critical role of transition-metal valence uniformity in optimizing the performance of halide solid electrolytes.

Boosting Anhydrous Cathode Performance in Anion Exchange Membrane Water Electrolyzers through Efficient Water Diffusion and Retention Strategies
Hao Zhang - ,
Ji Pan - ,
Yongjiang Yuan - ,
Pengda Fang - ,
Qikun Yu - ,
Tao Zhou - ,
Jinhong Shi - ,
Chengxiao Zhou - ,
Hanchi Zhang - ,
Zhenquan Chen - ,
Zhe Sun *- , and
Feng Yan *
Conventional bipolar symmetric feeding configuration in anion exchange membrane water electrolyzers (AEMWEs) induces bubble accumulation at the catalyst layer-gas diffusion layer interface, which can hinder gas transport and reduce electrolytic efficiency. Furthermore, operation under anhydrous cathode conditions creates critical durability challenges due to the insufficient water supply required for hydrogen evolution reaction. Herein, we achieve the durable operation of anhydrous-cathode electrolyzers via an efficient water diffusion and retention strategy. The sterically hindered and rotatable binaphthol configuration drives exceptional membrane water permeability. Meanwhile, the strong interactions between polyhydroxy cations and water create a coordination network, effectively preventing water loss from the catalyst surface at high temperatures. The high performance (current density of 8.32 A cm–2) and robust stability (voltage fade of only 12.5 μV h–1) have proved the feasibility of the anhydrous cathode operation mode, surpassing most bipolar symmetric feeding water electrolyzers. This strategy avoids the detrimental effects of bubble formation on catalytic efficiency and the integrity of the membrane electrode structure, providing insights for advancing cost-effective, scalable AEMWEs.
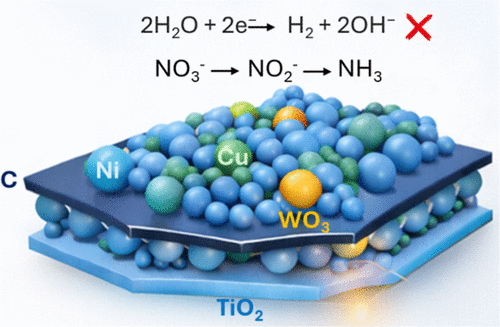
Bifunctional Catalyst Design Integrating Copper Nickel and Tungsten Trioxide on Defective Titanium Dioxide Enables Reaction Pathway Steering in Nitrate Electroreduction
Eleazar Castañeda-Morales - ,
Xochiquetzalli González-Bautista - ,
Francisco Ruiz-Zepeda - ,
Arturo Susarrey-Arce *- ,
Martha Leticia Hernández-Pichardo *- , and
Arturo Manzo-Robledo *
Metal oxide subnanometric size clusters can be “small” but “powerful” in suppressing side-reactions such as the hydrogen evolution reaction (HER), thereby improving ammonia (NH3) product during the nitrate reduction reaction (NO3-RR). This study presents the synthesis of a carbon-vulcanized (C)-defective TiO2 nanosheet (TNS) composite, modified with subnanometric WO3 clusters. It is found that among various loadings, the electrocatalyst with 3 wt % WO3 (C-3%WO3-TNS) suppresses HER. NH3 production higher than 97% is achieved by incorporating CuNi (40:60 wt %) onto C-3%WO3-TNS (Cu40Ni60/C-3%WO3-TNS), as confirmed by in situ differential electrochemical mass spectrometry (DEMS). Chemical characterizations reveal that WO3 clusters influence the Ti3+/Ti4+ ratio, thereby potentially suppressing HER. It has also been found that NH3 formation is further facilitated by Cu40Ni60, which promotes faster NO3– reduction via a multistep reaction on the C-WO3-TNS supports. The synergy between Cu40Ni60, C, WO3, and defective TNS modulates the production of H2 and NH3. This synergy can be attributed to the morphological and structural characteristics of the electrocatalyst, which indicate that Ni is positioned at specific edge sites over the C and TNS, while WO3 and Cu are well-distributed over the TNS. A mechanistic approach is proposed to explain the observed products by DEMS. This work highlights the dual potential of Cu40Ni60/C-3%WO3-TNS to suppress HER and promote NH3 synthesis, offering a promising strategy for tuning reaction pathways during NO3-RR.

Structural Design of Bismuth Telluride Nanoplates through Process Variables
Jordan Ackley - ,
Ariel E. Briggs - ,
Karthik Chinnathambi - ,
Nicholas McKibben - ,
Cadré Francis - ,
Josh Eixenberger - ,
Tony Valayil Varghese *- , and
David Estrada *
This publication is Open Access under the license indicated. Learn More
Binary pnictogen chalcogen compounds, primarily bismuth tellurides and selenides, are of great interest due to their applications in emerging quantum devices, as well as thermoelectric generators. The performance of bismuth telluride in these roles depends on its structure at the nanoscale, particularly the size, shape, and crystallinity of its nanocrystalline forms. However, current methods for controlling these features are often slow, inconsistent, or difficult to scale. Here, we demonstrate that through a solvothermal synthesis and hot injection process, precise control over the morphology of bismuth telluride nanoplates is possible with independent tuning of process variables, such as temperature and reaction time. We find that the nanoplate shape and internal porosity vary systematically with synthesis temperature and that the same morphological outcomes can be rapidly achieved at a fixed temperature by adjusting reaction duration. These results reveal that both the temperature and time can independently direct bismuth telluride morphological features, allowing for rapid, tunable synthesis strategies. Our approach offers a scalable framework, not only for bismuth telluride but also for related layered chalcogenides used in energy harvesting and quantum technologies.
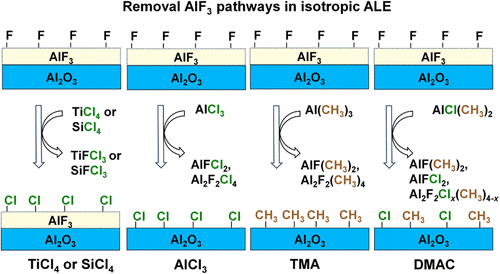
Dimer Formation as the Key Removal Pathway in the Isotropic Atomic Layer Etching of Al2O3: A First-Principles Study
Khabib Khumaini - ,
Hye-Lee Kim - ,
Taewook Nam - , and
Won-Jun Lee *
We elucidate the removal reaction mechanisms during isotropic atomic layer etching (ALE) of aluminum oxide (Al2O3) using density functional theory (DFT) calculations. Using amorphous aluminum fluoride (a-AlF3) surface models, we simulated reactions with various metal precursors: TiCl4, SiCl4, AlCl3, Al(CH3)3 (TMA), and AlCl(CH3)2 (DMAC). Our results indicate that the first two ligand-exchange steps involving TiCl4 and SiCl4 are favorable at 250 °C, releasing TiFCl3 and SiFCl3, respectively. However, the subsequent release of any Al-containing product is hindered by high activation barriers, which prevent net etching of the Al2O3 film. In contrast, we found that an alternative reaction pathway involving dimer formation is critical for successful etching processes. Although AlCl3 is ineffective via the simple ligand-exchange mechanism, it can etch the a-AlF3 layer effectively by forming a stable Al2F2Cl4 dimer product with low activation energies. The heteroleptic precursor DMAC is the most effective, offering multiple favorable reaction pathways that release various Al-containing dimers. DMAC features the lowest energy barriers, which explains its superior performance in experiments. Our calculations show that breaking the surface metal-fluoride bond is the most difficult part of the etching process, and dimer formation can offset this high-energy penalty by forming additional bonds. These theoretical results align well with experimental observations from in situ analyses, confirming that DFT is a powerful predictive tool for screening precursors and explaining reaction mechanisms in ALE processes.
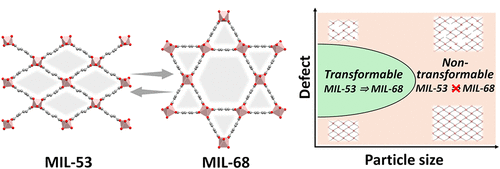
Impact of Defects and Particle Size on the Reversible Structural Transformation between Metal–Organic Frameworks
Shintaro Tanaka - ,
Yuta Kanao - ,
Takaaki Tsuruoka - ,
Kensuke Akamatsu *- , and
Yohei Takashima *
Reversible structural transformations in metal–organic frameworks (MOFs) driven by bond-switching mechanisms offer a promising approach for dynamically controlling material properties, enhancing advanced functionality, and designing smart materials. However, most reported structural transformations have been serendipitous, posing significant challenges for their deliberate design and practical application. This study focuses on the structural interconversion between MIL-53 and MIL-68, two aluminum-based MOF structural isomers, investigating the effects of framework defects and particle size on their transformation behaviors. The findings of this study provide essential insights into the mechanisms underlying MOF structural transformations and lay the groundwork for developing responsive materials with precisely controlled structural dynamics.
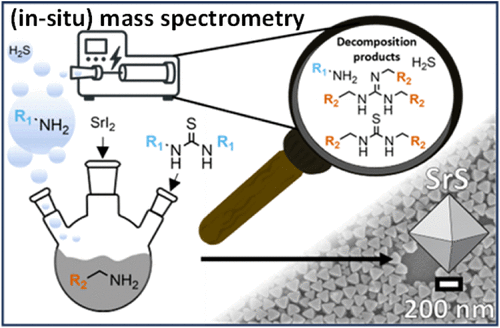
Decomposition Pathways of Thioureas in Oleylamine Control Reactive Sulfur Species in SrS Nanocrystal Synthesis
Vincent Mauritz - ,
Nico Ruhland - ,
Jonas Englhard - ,
Florian Steiger - ,
M. Eugenia Pérez-Ojeda - ,
Julien Bachmann - , and
Ryan W. Crisp *
This publication is Open Access under the license indicated. Learn More
Understanding the decomposition and reaction mechanisms of sulfur precursors used in organometallic colloidal synthesis is critical for controlling the nucleation and growth of nanocrystals. In this work, we investigate the thermal decomposition of thiourea and various N,N′-substituted thioureas in oleylamine to elucidate their distinct decomposition mechanisms, sulfur release pathways, and the implications for strontium sulfide (SrS) nanoparticle syntheses due to these decompositions. Using a combination of in situ quadrupole mass spectrometry (QMS), NMR spectroscopy, electrospray ionization mass spectrometry (ESI-MS), thermogravimetry (TGA), and DFT-calculated bond dissociation energies (BDEs), our findings reveal that substituent identity and symmetry significantly affect the decomposition onset temperatures and reaction intermediates. Unsubstituted thiourea undergoes initial isomerization with two competing subsequent fragmentation routes, releasing the gaseous products H2S and NH3, respectively. The other fragments from both routes, carbodiimide and isothiocyanic acid, react with oleylamine to form N,N′-dioleylthiourea. The N,N′-dioleylthiourea undergoes a second decomposition process, which releases H2S and the trisubstituted guanidine. Aliphatic N,N′-substituted thioureas exhibit the first fragmentation process, which releases the respective alkylamine, whereby the various chain-length substituents influence the decomposition onset temperature and the decomposition process, which releases H2S from N,N′-dioleylthiourea. Aromatic N,N′-substituted thioureas follow the same sequential decomposition mechanism, but resonance effects in N,N′-diphenylthiourea lower the decomposition temperature. These mechanistic differences are then directly correlated with the formation of strontium sulfide (SrS) nanocrystals. By tracking sulfur and amine release during synthesis, we show that the timing and concentration of reactive sulfur species, such as H2S, determine the nucleation rate, particle size, and morphology of the resulting nanocrystals. This study provides mechanistic insights into thiourea decomposition in oleylamine and establishes a direct link between the precursor structure and nanoparticle outcome. The results lay the foundation for more predictive and tunable synthesis strategies in the design of colloidal metal sulfide nanomaterials.
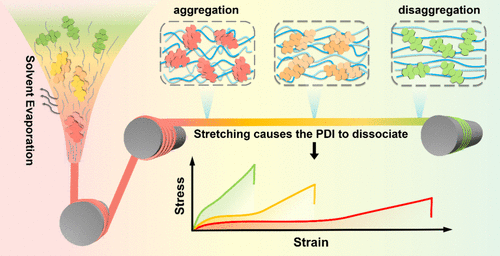
Visualization of Polymer Chain Alignment in Fiber Spinning and Hot Stretching by Noncovalent Polymer Mechanochemistry
Siqi Liu - ,
Yinfang Zhang - ,
Yifan Ge - ,
Hairui Deng - ,
Jin Wen - ,
Yangju Lin *- ,
Yinjun Chen *- , and
Meifang Zhu *
Polymer chain orientation and microstructures are crucial to achieving high performance in fibers. However, in situ detection and further control of these polymer-condensed matter structures during fibrous processing are challenging due to the high velocity of spinning and the small size of fibers. Here, we report a visual approach for monitoring polymer microstructures and chain alignment in fibers by the mechanically induced fluorescent color change of perylene diimide (PDI) mechanophores. PDI derivatives with different solubilities were synthesized and used to prepare solutions with distinct fluorescence colors, arising from varying degrees and packing of aggregation. The mechanism of distinct fluorescence further guided the synthesis of mechanoresponsive polyurethanes by embedding various contents of PDI along the polyurethane backbone. The resulting polyurethanes exhibited obvious fluorescence color change during deformation owing to the dissociation of PDI stacking, enabling real-time monitoring of microstructures in fiber processing. A fluorescent color change from red to green was observed during hot stretching, indicating the associated orientation of the polymer chains and crystalline formation. These microstructural changes, analyzed by WAXD, are quantitatively reflected in the reinforcement of fiber mechanical properties. Accordingly, the relationship between fluorescent color and fiber strength or modulus was established to enable the in situ visual detection of fiber mechanical properties during spinning. The unique mechanoresponsive mechanism of PDI mechanophores points to a novel application of noncovalent polymer mechanochemistry in the field of fiber technology.
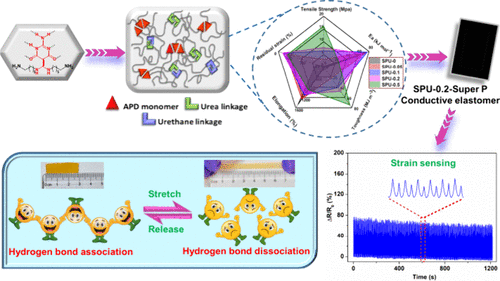
Dynamic Janus Hydrogen Bond Mimicry Unlocks Tough, Flexible Supramolecular Elastomers for Strain Sensing
Durga Lakshmi - ,
Mahendra A. Wagh - ,
Aakash Sharma - ,
Md Shafi Alam - ,
Muthamil Selvan T - ,
Arun Torris - ,
Titash Mondal *- ,
Gangadhar J. Sanjayan *- , and
Kiran Sukumaran Nair *
Flexible and wearable electronics demand stretchable sensors with polymer elastomers as key matrixes for mechanical flexibility and durability. However, despite their excellent elasticity, their limited mechanical strength remains a challenge. To address this limitation, in this study, we report the rational design of supramolecular polyurethane elastomers (SPUs) incorporating nucleobase-inspired aminopyrimidinedione with DDA-AAD (G-C mimic) reversible triple hydrogen bonds. This dual-domain architecture gives rise to a durable supramolecular network with enhanced mechanical properties, yielding elastomers that are soft, stretchable, and tough. By tuning of the density of dynamic cross-links, mechanical properties were systematically modulated. SPU-0.5 exhibited a maximum tensile strength of 16.14 MPa, representing a 67-fold strength enhancement over that of SPU-0. Although increasing the aminopyrimidinedione (APD) content reduced elongation, SPU-0.2 retained a high elongation of 1060% and showed the lowest residual strain during cyclic tests. To be of great interest, the activation energy increased with increasing hydrogen bonding content up to SPU-0.1, whereas beyond SPU-0.2 it decreased, likely due to extensive hydrogen bond formation. Furthermore, SPU-0.2-SP, a conductive variant, demonstrated a promising strain-sensing performance even after hundreds of cycles. Overall, the insights gained from this study advance the development of intelligent soft materials and lay the groundwork for next-generation flexible and wearable electronic devices.
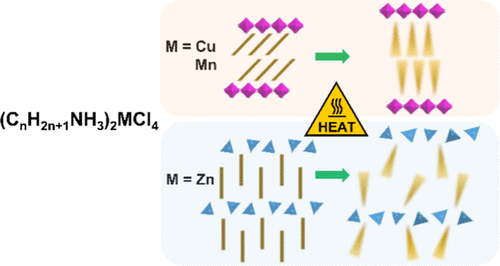
Solid–Solid Phase Change Layered Hybrid Materials for Thermal Energy Storage: Impact of the Chemical Composition and Structural Topology
Côme Archinard - ,
Ali Saad - ,
Hubert Chevreau - ,
Jean-Yves Mevellec - ,
Bernard Humbert - ,
Xavier Biquard - ,
Erik Elkaïm - ,
Kenneth Paul Marshall - ,
Dragos Constantin Stoian - ,
Wouter Van Beek - ,
Olivier Hernandez *- ,
Grégory Largiller *- , and
Thomas Devic *
Solid–solid phase change materials (SS-PCMs) are attractive candidates for thermal energy storage (TES) owing to their intrinsic shape stability, yet their widespread application remains limited by the lack of design rules linking the molecular structure to phase-transition properties. Here, we present a comprehensive study of layered hybrid chlorometallates, (CnH2n+1NH3)2MCl4 (M = Cu, Mn, Zn; n = 6–16), as tunable SS-PCMs. For that, 15 compounds (M = Cu, Mn, Zn; n = 6, 7, 12, 13, 16) were prepared, and their low-temperature (LT) forms were studied by single-crystal XRD and vibrational spectroscopies. By a multitechnique approach, involving calorimetry, temperature-dependent infrared, and Raman spectroscopies, and combined in a single synchrotron experiment temperature-dependent X-ray absorption spectroscopy (XAS), total scattering/Pair Distribution Function (PDF), and powder XRD (PXRD) analyses, we evidenced the impact of both parameters (M and n) not only on the LT structures but also on the thermal properties and on the high-temperature (HT) structures. Especially, we evidenced that although materials based on octahedrally (here Mn and Cu) and tetrahedrallly (here Zn) coordinated cations share many common features in their LT forms (alternating organic–inorganic layered structures, alkylammonium chains parallel to each other, and supramolecular organic–inorganic interactions of the same nature and strength), their HT phases strongly differ, especially at a medium range distance. This comprehensive study is not only of fundamental interest but will also help to address questions, such as the shaping and mechanical integrity of these SS-PCMs upon thermal cycling that need to be answered prior to their integration into practical devices for next-generation TES.
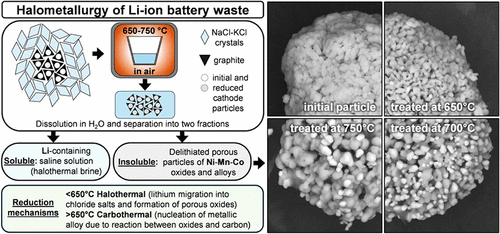
Halometallurgy: Reduction of Battery Cathode Materials under a Quasi-Inert Environment of Alkali Chloride Salts
Arseniy Bokov *- ,
Anna Shelyug - ,
Liuda Mereacre - ,
Michael Knapp - , and
Helmut Ehrenberg
This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
This study introduces halometallurgy, an approach for reducing common Li-ion cathode materials in air using a eutectic mixture of chloride salts, with direct implications for processing battery black mass containing NMC, NCA, LCO, LNMO, and LMO. In-depth analysis, including in situ XRD, SEM/EDX, and TGA-DSC, reveals that reduction in the presence of NaCl-KCl proceeds via distinct halothermal and carbothermal routes. During the halothermal stage, lithium migrates from cathode particles into the chlorides, leading to the decomposition of layered or spinel structures into a solid solution of cubic oxides. Lithium migration facilitates the melting of the salts, resulting in the encapsulation of the oxide phase and the creation of quasi-inert conditions. This enables further reduction during the carbothermal stage and promotes the nucleation of metallic crystallites. Upon washing with water, lithium predominantly remains in the saline solution, termed halothermal brine, while the insoluble fraction consists of porous transition metal oxides and graphite. Depending on cathode composition, halothermal reduction is observed at 460–640 °C, while carbothermal reduction occurs above 620–650 °C. Typical black-mass impurities, including current collectors, binders, and electrolyte residues, were also examined, demonstrating relevance for real waste streams. The proposed treatment offers a pathway toward decentralized battery recycling.
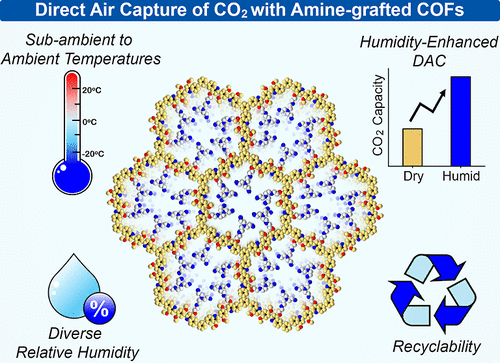
Humidity-Enhanced Direct Air Capture of Carbon Dioxide Using Amine-Grafted Covalent Organic Frameworks Under Ambient and Sub-ambient Temperatures
Arkaprabha Giri - ,
Jiaqi Zhang - ,
Xin Deng - , and
Christopher W. Jones *
This publication is Open Access under the license indicated. Learn More
The significant rise in atmospheric CO2 and its impact on accelerating climate change have triggered intense efforts to develop porous sorbents for direct air capture (DAC), a route toward carbon-neutrality. Amine-functionalized covalent organic frameworks (COFs), an emerging class of crystalline porous materials, have recently shown promising potential for DAC at ambient, indoor temperatures (25 °C). However, most of Earth’s land area has annual mean temperatures below 25 °C, accompanied by nonzero and variable relative humidity (RH). The performance of amine-grafted COFs under cold, humid conditions remains largely unexplored, even though such climates represent the majority of potential DAC deployment sites. Herein, we report a systematic investigation of a tetrahydroquinoline-linked COF covalently functionalized with diverse amines, evaluating its CO2 adsorption behavior across a broad range of ambient to sub-ambient temperatures (25 °C to −20 °C) and relative humidities (0%–70%). A unique tris(2-aminoethyl)amine-functionalized COF (ImCOF-TAEA) achieved a pseudoequilibrium capacity of 0.46 ± 0.02 mmol g–1 under dry conditions, rising ∼137% to 1.09 ± 0.09 mmol g–1 under 70% RH using 400 ppm of CO2 at 25 °C. Upon cooling to 15 °C under 70% RH, the uptake further increased to 1.25 ± 0.02 mmol g–1, showing a 205% enhancement relative to dry conditions. In situ spectroscopic analysis supports the mechanism behind the unusually high enhancement in CO2 adsorption under humid conditions. ImCOF-TAEA also demonstrates excellent recyclability under ambient/sub-ambient conditions and has modest (45 °C-65 °C) requirements for regeneration.

Silyl Hot-Injection Versus Thiocyanate Heat-Up Synthesis of Chalcohalides: Pushing the Size and Composition Envelope
Eve K. Stegner - ,
Md Riad Sarkar Pavel - ,
Anuluxan Santhiran - ,
Jack Lawton - ,
Juan-Pablo Correa-Baena - ,
Aaron J. Rossini - , and
Javier Vela *
This publication is Open Access under the license indicated. Learn More
Chalcohalide semiconductors are rapidly gaining traction as stable, biocompatible materials for energy conversion applications. While the solid-state synthesis of bulk chalcohalides is relatively well-developed, the colloidal chemistry of these materials is still in its early stages. Colloidal semiconductors are often advantageous in device fabrication due to the cost effectiveness of solution processing. Thus, we aim to increase the utility of chalcohalides in device fabrication by establishing solution phase chemistry of promising compositions. We show that silyl hot-injection is a versatile and effective method of making colloidal PnChI (Pn = Sb, Bi; Ch = S, Se) and Sn2PnS2I3 (Pn = Sb, Bi) chalcohalides of tunable sizes and compositions. Furthermore, we demonstrate the preparation of mixed-pnictide chalcohalides through direct hot-injection and/or postsynthetic cation exchange, the latter being one of the few reported instances in chalcohalides. Additionally, we use the thiocyanate heat-up approach in combination with density functional theory to study halide mixing in quaternary tin chalcohalides. By pushing the limits of each synthetic technique, we have designed more soluble chalcohalides with tunable compositions while also gaining a better understanding of the efficacy of each procedure in respect to thin film and subsequent device fabrication. In addition to size and composition tuning, silyl hot-injection can help facilitate the future development and wide-scale application of chalcohalide-based devices by expanding the selection of solution-processable chalcohalides.
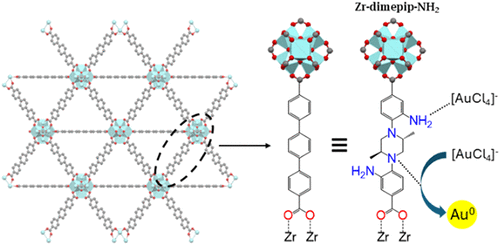
Stable Zirconium-Piperazine Metal–Organic Frameworks for Efficient Gold Recovery
Matthieu Meoli - ,
Jocelyn Roth - ,
Dragos C. Stoian - ,
Anne F. S. Belin - ,
Rosario Scopelliti - , and
Wendy L. Queen *
Gold’s importance in various industries, the ever-rising demand for electronics, and shrinking gold reserves all underscore the pressing need to develop effective gold recovery methods from waste. In this study, we systematically evaluate several large-pore Zirconium-carboxylate Metal–Organic Frameworks (Zr-MOFs) having slightly varied internal surface functionality (-H, -CH3, -NH2, and -NO2) in gold recovery from acidic, simulated e-waste solutions. We compare two Zr-MOFs constructed from oligophenylene ligands with four MOFs built from structurally analogous piperazine-based ligands, including two unreported frameworks. The piperazine-based frameworks offer a critical sustainability advantage: by replacing the central benzene core with piperazine, they eliminate the need for expensive Pd-catalyzed coupling chemistry required for oligophenylene synthesis. In this work, we demonstrate for the first time that Zr-piperazine MOFs exhibit stability in acidic conditions, significantly surpassing their oligophenylene-based counterparts. One developed Zr-piperazine MOF, functionalized with both amine and methyl groups, substantially outperforms all analogs, displaying the highest stability alongside a gold adsorption capacity of up to 1826 mg/g and rapid adsorption kinetics in a 40 ppm gold solution. X-ray Absorption Near-Edge Structure (XANES) studies reveal distinct gold adsorption mechanisms for primary versus tertiary amines. These findings establish piperazine-based building blocks as promising candidates for designing selective gold adsorbents for e-waste recovery applications.
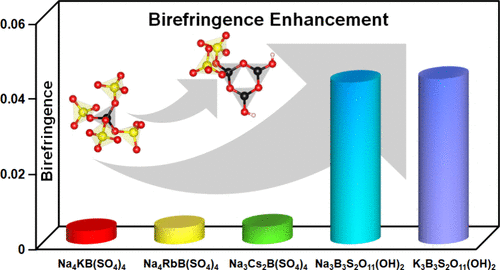
Significant Enhancement of Birefringence Resulting from the Introduction of [BO2(OH)] Groups into Borosulfates
Luyong Zhang - ,
Jianyi Zuo - ,
Guangsheng Xu - ,
Jinbin Fan - ,
Xue Bai - ,
Zhihua Yang - ,
Fangfang Zhang *- , and
Shilie Pan *
Deep-ultraviolet (DUV) birefringent crystals are essential for modern optical technologies, yet achieving a balance between large birefringence and a wide bandgap remains challenging. Although conventional borosulfates with alternating [BO4] and [SO4] tetrahedra connected by sharing corners possess a wide bandgap, they suffer from small birefringence due to weak optical anisotropy resulting from the presence of only tetrahedral groups. This work introduced advantageous “genes” [BO2(OH)] groups to modify the common [B(SO4)4] supertetrahedra for the first time and ultimately produced two borosulfates, Na3B3S2O11(OH)2 and K3B3S2O11(OH)2, striking the balance. Isolated fundamental building block (FBB) [B3O3(OH)2(SO4)2] was observed in the two compounds. Additionally, three conventional borosulfates featuring isolated [B(SO4)4] supertetrahedra, Na4KB(SO4)4, Na4RbB(SO4)4, and Na3Cs2B(SO4)4, were also obtained for comparison. Structural analysis and theoretical calculations reveal that the directional alignment of [BO2(OH)] groups significantly enhances the birefringence of the borosulfates. The calculated birefringence at 1064 nm increases by approximately 10-fold, specifically from 0.0035–0.0047 for the three conventional borosulfates to 0.043–0.044 for the two hydroxylated ones. This work demonstrates the effectiveness of employing [BO2(OH)] groups in optimizing the optical anisotropy of borosulfates, providing a design paradigm for exploring advanced DUV birefringent materials in borosulfates.

From Solid Solution to Intermetallic Compounds: Oxygen Evolution Reaction Activity Trends among Nb–Ni Phases
Büşra Sevdaroğlu *- ,
Ulrich Burkhardt - ,
Yuri Grin - , and
Iryna Antonyshyn *
This publication is Open Access under the license indicated. Learn More
The electrochemical behavior of Nb–Ni binary phases in terms of electrocatalytic activity and stability under oxygen evolution reaction (OER) conditions was investigated. The outcomes of electrochemical studies in an alkaline electrolyte, supported by extensive characterization of electrode materials before and after electrochemical experiments, were provided. OER activities using the intermetallic compounds Nb7Ni6 and NbNi3 as electrocatalyst precursors were compared with those obtained with the solid solution of NbxNi1-x (x = 0.09) and a reference Ni foil under the same conditions. While Nb7Ni6 deactivates gradually during benchmarking stability test (chronopotentiometry at current density of 10 mA cm–2 for 2 h), NbNi3 possesses remarkable OER activity and keeps it stable not only under mild conditions of benchmarking experiment but also during long-term operation at elevated current densities (50, 100, and 200 mA cm–2). To understand the difference in chemical behavior and the OER performance of Nb–Ni compounds, extensive characterization was supported by carrying out quantum chemical calculations, shedding light on the charge distribution and chemical bonding in the investigated compounds.

Ba2+ Complements Co2+ Exchange as a Reporter of Al Proximity in CHA Zeolites
Wei Ge - ,
Tania L. Class-Martínez - ,
José Rebolledo-Oyarce - ,
Alyssa McNarney - ,
Songhyun Lee - ,
Anshuman Goswami - ,
Claire T. Nimlos - ,
Ahmad Moini - ,
Subramanian Prasad - ,
Vivek Vattipalli - ,
Anthony DeBellis - ,
Sichi Li - ,
Bradley F. Chmelka - ,
Rajamani Gounder *- , and
William F. Schneider *
Co2+ exchange is commonly used as a reporter of Al pair ensembles in zeolites. We combine density functional theory (DFT) calculations, statistical models, experimental titrations, and solid-state nuclear magnetic resonance (NMR) analyses to explore the utility of other 2+ ions as alternative reporters of proximal Al ensembles in the CHA zeolite. DFT calculations suggest that Ba2+ will exchange into both eight- (8MR) and six-membered (6MR) CHA rings equally effectively, distinct from Co2+, which exchanges solely into 6MR. Simulated titration curves highlight the potential for Co2+ and Ba2+ titrations to provide complementary information about specific proximal Al site ensembles as a function of Si/Al ratio. Experiments on CHA zeolites synthesized to express different Al distributions confirm that Ba2+ uptake exceeds that of Co2+ and that this uptake can be rationalized by Ba2+ ions that titrate both 8MR and 6MR Al pair sites. Comparisons of absolute ion uptakes and two-dimensional NMR analyses of Al proximity with predictions reveal previously unrecognized differences in Al siting rules under different syntheses. These findings demonstrate that complementary titrations using divalent cations of differing ionic radii provide additional resolution on Al–Al pair ensembles and the underlying rules that govern Al distributions on zeolite frameworks.

Unlocking the Full Capacity of Iron Oxychloride as a Sustainable Mixed Anion Intercalation Material
Michael A. Spencer *- ,
Gwenaëlle Rousse - , and
Alexis Grimaud *
Mixed anion compounds offer unique opportunities to tune the redox properties of intercalation compounds. Among them, layered iron oxychloride (FeOCl) has long been envisioned as an energy-dense and sustainable alternative to layered oxides for Li–ion batteries. However, achieving full capacity has thus far remained elusive owing to detrimental solvent cointercalation typically observed in dilute liquid electrolytes and ionic liquids. In this work, we alleviate this limitation by developing a suitable electrolyte engineering approach and demonstrate that 1 Li atom can be reversibly intercalated in FeOCl. We demonstrate that solvent cointercalation can be suppressed in high-concentration electrolytes using a highly dissociating salt, lithium bis(fluorosulfonyl)imide (LiFSI), dissolved in solvents with low Li-solvent binding energy, including dimethylcarbonate and acetonitrile, achieving a capacity of 235 mAh/g and a material specific energy of 590 Wh/kg in its lithiated form. Combining X-ray absorption measurements at iron, oxygen, and chloride K-edges, we observe that the intercalation proceeds with a transition from distorted high-spin Fe3+ to low-spin Fe2+, while operando X-ray diffraction shows three successive biphasic processes associated with changes in interlayer spacing. Entropic potential measurements coupled with temperature-dependent cycling reveal a reversible cationic ordering event when half of the interlayer Li+ sites are filled. This ordering is associated with high activation energy and a slow phase-front diffusion, which does not prevent good cycling ability at room temperature even at high C-rate. Our work calls for revisiting through an electrolyte engineering approach energy dense and sustainable mixed anion materials previously discarded for their instability.

Supramolecular Assembly of Multielement Ribbon-like Structures Derived from Halide Perovskites
Heqing Zhu - ,
Cheng Zhu - ,
Yuxin Jiang - ,
Chuxi Wen - ,
Xinyu Chen - , and
Peidong Yang *
Halide perovskites are crucial materials with broad applications owing to their exceptional optoelectronic properties. Vacancy-ordered double perovskites, featuring highly tunable transition metal sites, enable controllable optoelectronic properties through multielement compositional design. In this study, we introduced 18-crown-6 into the vacancy-ordered double perovskites system and developed two-dimensional ribbon-like single crystals (18C6@K)2{PtSnTeIrRe}1Cl6 via an antisolvent supramolecular assembly method, demonstrating morphology modulation through multielement composition design. The crystals crystallize in the centrosymmetric space group P1¯. The dumbbell-shaped structural units (crown ether@A)2MX6 pack along a and b axes to form a 2-dimensional (2D) monolayer, and these monolayers further stack along the c axis to generate the ribbon-like single crystals. Energy-dispersive X-ray spectroscopy (EDX) qualitatively confirmed the uniform distribution of the five transition metals throughout the crystal, while inductively coupled plasma atomic emission spectroscopy (ICP–AES) quantitatively verified their atomic ratios. We further investigated the origin of the morphology, distinct from the previously reported cube-like single crystals with the R3¯ space group. When acetonitrile was used as the solvent, three-dimensional crystals with R3¯ symmetry were obtained, whereas dimethylformamide (DMF) was essential for forming two-dimensional ribbon-like single crystals. The essential role of DMF could be ascribed to its capability to maintain a higher concentration of the building blocks. Moreover, Ir4+ and Pt4+ cations also played critical roles in inducing the two-dimensional ribbon-like morphology. The three-element (18C6@K)2{PtSnTe}1Cl6 single crystals exhibited bright yellow emission under 375 nm laser excitation, demonstrating the tunability of optoelectronic properties of this class of material.

BODIPY-Driven π-Extended Frameworks for Efficient Photosynthesis of Hydrogen Peroxide
Shengxu Li - ,
Junyi Han - ,
Haoyong Yang - ,
Quanquan Yang - ,
Zhenhui Kou - ,
Yang Hou - ,
Qunji Xue - ,
Paolo Samorì - , and
Tao Zhang *
Hydrogen peroxide (H2O2) is an indispensable industrial feedstock and energy carrier, yet its synthesis is challenging due to the energy-intensive property and complex postpurification process. Direct photocatalytic H2O2 production from water and air offers a promising approach to addressing these problems, while obtaining efficient and robust photocatalysts has been a longstanding challenge. Herein, we describe the design and synthesis of a two-dimensional conjugated BODIPY-based framework (2D CBF) via BODIPY-mediated topologic aldol-type polycondensation. The strong solar-harvesting BODIPY units and 2D extended conjugation endow the 2D CBF with near-infrared absorption and abundant oxygen adsorption sites. Consequently, the resulting 2D CBF demonstrates a remarkable H2O2 production rate up to ∼56.8 mmol h–1 gcat–1 and benchmark apparent quantum yield of 21.9% without cocatalyst and sacrificial agents, largely superior to the BODIPY molecule. In a large-scale paradigm, the 2D CBF-based floatable photocatalysis platform (∼0.2 m2) could efficiently uptake reactant (O2) from air and produce H2O2 at a rate up to ∼15 mmol h–1 m–2 under sunlight irradiation. This work highlights the two-dimensional strategy and BODIPY building blocks for designing and synthesizing highly efficient photocatalytic platforms.

Intrinsically Weak Polarization in (4-(Aminomethyl)piperidinium) SnI4
Tianhongyi Zhao - ,
Zhenbang Dai - , and
Andrew M. Rappe *
(4AMP)SnI4 (where 4AMP is 4-(Aminomethyl)piperidinium) has recently been identified as a promising lead-free, Sn-based two-dimensional ferroelectric hybrid perovskite. However, the magnitude of its experimentally reported polarization remains controversial. In this work, we perform a detailed symmetry analysis and construct a polarization pathway originating from a manually built centrosymmetric P2/c reference structure. Our first-principles calculations reveal that the intrinsic polarization of (4AMP)SnI4 is approximately 0.97 μC/cm2, nearly an order of magnitude lower than the values reported experimentally. We attribute this discrepancy to extrinsic effects (leakage current induced by ionic migration) that can falsely enhance ferroelectric hysteresis signals. In addition, we reveal two distinct I– vacancy-mediated ion migration routes and carry out nudged elastic band calculations to evaluate the activation energy barriers of both pathways. The strong frequency dependence observed in experimental polarization hysteresis measurements supports this interpretation, pointing to a dominant contribution from ion diffusion dynamics rather than ferroelectric switching. Our findings highlight the critical importance of distinguishing intrinsic ferroelectric behavior from extrinsic artifacts in hybrid perovskites, especially those exhibiting semiconducting or leaky dielectric properties. This work not only clarifies the polarization mechanism in (4AMP)SnI4 but also provides a robust theoretical framework for evaluating ferroelectricity in emerging complex and intriguing hybrid perovskite systems.

ns2 Electron Engineering in Zero-Dimensional Metal Halides for Modulating Emission Behavior
Dhritismita Sarma - and
Arup Mahata *
The photophysical behavior of ns2 metal-based zero-dimensional (0D) halides, particularly their broad emission driven by self-trapped excitons (STEs), makes them unique and promising for light-emitting technologies. The stereochemical activity of the ns2 lone pair plays a decisive role in dictating the structural and photophysical properties of such metal halides. However, a systematic and generalized framework correlating the factors associated with ns2 electron engineering, e.g., metal identity, local coordination geometry, electronic energy level of the organic cation, and dynamical off-centering in tuning the emission characteristics, remains limited. In this work, using state-of-the-art density functional theory (DFT) and ab initio molecular dynamics (AIMD) calculations on ns2 metal (Pb2+, Sn2+, and Sb3+) bromides incorporating Cs+, aliphatic, and aromatic organic cations having octahedral, disphenoidal, and square-pyramidal coordination environments, we have studied the ground and excited-state behavior and framed a generalized structure–emission characteristic correlation. Our results demonstrate that, in higher-coordination environments, ns2 lone pair exposure primarily determines the emission behavior, with Sn2+ exhibiting more stable STE characteristics and Pb2+ remaining largely inactive. However, in lower-coordination environments, the photophysical response appears as an interplay between ns2 lone pair exposure and its coordination geometry; Sn2+, having disphenoidal coordination, displays a prominent emission characteristic compared to that of Pb2+ disphenoidal and Sb3+ square-pyramidal geometries. We find that the STE responses are largely hole-driven with a lesser role for electrons. Furthermore, our study reveals that, while A-site cation substitution has a minimal effect on ground-state hybridization, it profoundly alters excited-state behavior, where aromatic cations promote charge separation and non-STE-like excitons, whereas aliphatic cations favor STE formation. AIMD calculations further reveal that higher-coordination systems show lone pair activity through dynamic off-centering, whereas the lone pair of lower-coordination systems is stereochemically inactive to dynamic off-centering due to deviation from the optimal spatial availability of the lone pairs. Therefore, our results demonstrate that, despite the ns2 lone pair’s population at the valence band edge, ground-state treatment is insufficient to fully capture the stereochemical nature; instead, it is dictated by the excited state and dynamical treatment. These insights establish a robust atomistic framework linking the stereochemical activity, coordination geometry, and exciton localization of the lone pair, thus providing atomistic interpretation of experimentally observed trends and offering a fundamental and generalized perspective for analyzing the emission behavior and thereby providing design guidelines for engineering efficient 0D metal halide emitters.
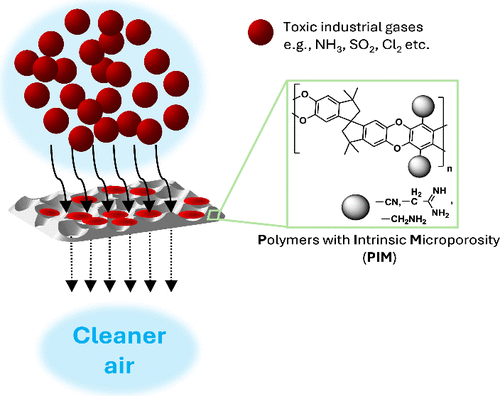
Functionalized Polymers of Intrinsic Microporosity for Toxic Chemical Filtration
Ankana Roy - ,
Brandon Blount - ,
Ryan Lively - ,
M.G. Finn - , and
Gregory W. Peterson *
Sorption or passivation of volatile toxic compounds is a critical need in industrial settings as well as in response to the use of chemical weapons. The sorption capabilities of a prototypical intrinsically microporous polymer (PIM-1) and derivatives bearing primary amine and guanidine groups were tested against five toxic industrial chemicals and three chemical warfare agents. In every case but one (ammonia under humid conditions), one or more of the organic polymers significantly outperformed standard sorbents UiO-66-NH2 (a metal–organic framework) and UFR carbon. The pattern of performance suggests a complex interplay of factors in passivation reactions, including the activity of adsorbed water, the amine nucleophilicity, and the weakly acidic nature of the guanidinium group.

Superhydrophobic Polyimides as Materials for Lunar Dust Mitigation
Giuseppina Di Stefano - ,
Rocco Di Girolamo - ,
Guido Saccone - ,
Nunzia Favaloro - ,
Fabrice Cipriani - , and
Claudio De Rosa *
This publication is Open Access under the license indicated. Learn More
The synthesis and characterization of superhydrophobic polyimides and copolyimides are presented. These polymers have been designed through a suitable choice of dianhydride and diamine monomers and incorporation of a siloxane oligomer comonomer to produce materials for mitigation of lunar dust adhesion. Lunar dust represents a major obstacle to long-duration robotic and crewed missions as its strong adhesion and abrasive nature threaten the reliability of surface systems and habitats. The synthesized polyimides and copolyimides show inherently high-performance, lightweight, and heat resistance and, in addition, exhibit superhydrophobic properties with low surface energy and adhesion properties for lunar dust regolith. The synthetic strategy is based on the use of fluorinated dianhydride and/or diamine monomers that allow incorporation of trifluoromethyl side groups within the molecular structure of the homopolyimides and incorporation of siloxane functionalities through copolymerization of diamine and dianhydride monomers with amino-terminated polydimethylsiloxane oligomers of different molecular masses. The bulky −CF3 groups contribute to decreasing the surface energy of the outermost layers of the material, reducing adhesion properties, and enhancing its hydrophobicity. Moreover, the siloxane units act as surface migrating agents as they easily migrate from the interior of the material and segregate onto the surface contributing to a further increase in hydrophobicity and a decrease of adhesion properties. These properties have been demonstrated by the high values of the water contact angles exhibited by these materials up to a remarkable value of 120°, much higher than the value of 82° measured for a well-known commercial polyimide Kapton film, without compromising outstanding thermal and mechanical properties.

Nanomorphology Control of Metallo-Supramolecular Polymers toward Photocatalytic CO2 Reduction
Arghya Ghosh - ,
Tarak Nath Das - ,
Papri Sutar - ,
Anupam Dey - ,
Sukhendu Nath - , and
Tapas Kumar Maji *
The self-assembly of small molecules with a suitable metal ion is a critical bottom-up approach for preparing solution-processable metallo-supramolecular polymers and to realize their potential as a photocatalyst. Herein, we report the design, synthesis, self-assembly, and gelation behavior of a low-molecular-weight gelator (LMWG) based on a pyrene core connected to four terpyridine units (TPY-PY) through amide linkages. TPY-PY forms an organogel (OG) in the DMSO/H2O mixed solvent with a nanofibrillar morphology. In contrast, the introduction of RuII with the LMWG resulted in a Ru-TPY-PY coordination polymer gel (CPG) with a cross-linked fibrillar nanostructure with a length of several micrometers. The Ru-TPY-PY CPG exhibited highly efficient photoreduction of CO2 to CO (yield: 10.79 mmol g–1, rate: 899.17 μmol g–1 h–1) with 90% selectivity, under visible light irradiation, and in the presence of triethylamine (TEA) as a sacrificial electron donor. Moreover, metallo-supramolecular polymers with diverse nanomorphologies, formed at different RuII to TPY-PY ratios, showed different efficiencies in the photo reduction of CO2 to CO. Among these nanostructures, 1D nanofibers exhibited enhanced photocatalytic activity compared to the nanospheres for CO2 reduction, attributed to the abundance of catalytically active sites on their surface with facile CO2 diffusion capabilities. Furthermore, femtosecond transient absorption spectroscopy, in situ DRIFTS analysis, and DFT calculation help to understand the feasibility of the electron transfer pathway, and reaction mechanism in the overall process. The “soft” processable hybrid metal–organic supramolecular polymers that integrate both the catalytic site and the light-absorbing units are a class of catalysts showing efficient CO2 photoreduction to CO.
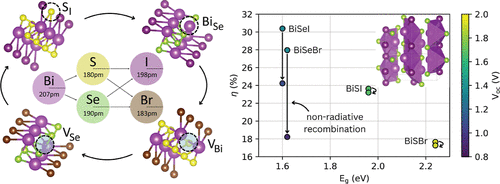
Defect-Limited Efficiency of Pnictogen Chalcohalide Solar Cells
Cibrán López - ,
Seán R. Kavanagh - ,
Pol Benítez - ,
Edgardo Saucedo - ,
Aron Walsh - ,
David O. Scanlon - , and
Claudio Cazorla *
This publication is Open Access under the license indicated. Learn More
Pnictogen chalcohalides (MChX) have recently emerged as promising nontoxic and environmentally friendly photovoltaic absorbers, combining strong light absorption coefficients with favorable low-temperature synthesis conditions. Despite these advantages and reported optimized morphologies, device efficiencies remain below 10%, far from their ideal radiative limit. To uncover the origin of these performance losses, we present a systematic and fully consistent first-principles investigation of the defect chemistry across the Bi-based chalcohalide family. Our results reveal a complex defect landscape dominated by chalcogen vacancies of low formation energy, which act as deep nonradiative recombination centers. Despite their moderate charge-carrier capture coefficients, the high equilibrium concentrations of these defects reduce the theoretical maximum efficiencies by 6% in BiSeI and by 10% in BiSeBr. In contrast, sulfur vacancies in BiSI and BiSBr are comparatively benign, presenting smaller capture coefficients due to weaker electron–phonon coupling. Interestingly, despite its huge nonradiative charge-carrier recombination rate, BiSeI presents the best conversion efficiency among all four compounds owing to its most suitable bandgap for outdoor photovoltaic applications. Our findings identify defect chemistry as a critical bottleneck in MChX solar cells and propose chalcogen-rich synthesis conditions and targeted anion substitutions as effective strategies for mitigation of detrimental vacancies.

Understanding the Effects of Tensile Strain on the Structure and Magnetism of Stoichiometric LaCoO3 Films
Daniel Russell - ,
Rebecca M. Haight - ,
Binzhi Liu - ,
Ali Barooni - ,
Allen Partin - ,
Alevtina Smekhova - ,
Florian Kronast - ,
L. Robert Baker - ,
Maryam Ghazisaeidi - ,
Jinwoo Hwang - ,
Fengyuan Yang - , and
Patrick M. Woodward *
This publication is Open Access under the license indicated. Learn More
Despite numerous reports of an insulating ferromagnetic state in epitaxial LaCoO3 thin films, no consensus has been reached on the details of ferromagnetism in these films. To better understand the origins of magnetic order in such films, stoichiometric LaCoO3 films have been deposited on SrTiO3(001) and LaAlO3(001) substrates using off-axis sputtering. This technique allows growth to occur in conditions that minimize deviations from the ideal stoichiometry. SQUID magnetometry shows that ferromagnetism is stabilized only in films grown under tensile strain on SrTiO3. The magnetic properties of these films (TC ≈ 70 K, Msat ≈ 0.3 μB/Co, and HC ≈ 5 kOe) are essentially independent of thickness, consistent with nearly uniform magnetization. At room temperature, strain induced by the SrTiO3 substrate breaks the rhombohedral symmetry of the bulk structure, leading to a–a–c0 octahedral tilting and an anisotropic distortion of the Co-centered octahedra. Low-temperature (T = 36 K) X-ray absorption spectroscopy reveals that tensile strain inherent to the SrTiO3 substrate stabilizes a substantial fraction of high- or intermediate-spin Co3+ ions, facilitating magnetic order, whereas films grown on LaAlO3 are made up nearly entirely of low-spin Co3+ ions.
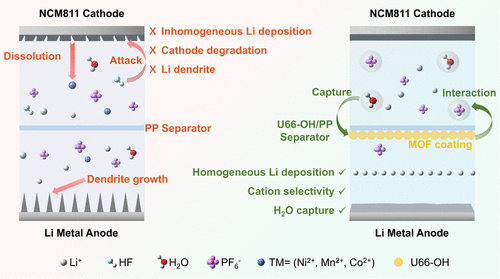
Water-Scavenging Zr-Based Metal–Organic Framework-Decorated Functional Separator for Stable Lithium Metal Batteries
Senlin Li - ,
Haitao Liu - ,
Renfei Zhao - ,
Bo Zhao - ,
Qiaozhen Sun *- , and
Gen Chen *
In lithium metal batteries (LMBs), even trace water can severely affect the battery performance and safety. Therefore, the removal of water from the electrolyte is important and necessary. In this study, the hydroxyl (−OH) functionalized metal–organic frameworks (MOFs), UiO-66-(OH)2 (noted as U66-OH) are applied as efficient and effective water scavengers in LMBs. The U66-OH crystals are pasted onto the conventional polypropylene (PP) to fabricate the U66-OH/PP separator. The results show that U66-OH can effectively capture trace water in cells, suppress the generation of acidic substances, and improve the cycling stability of the Li||NCM811 cells. Even with a 1000 ppm water-containing electrolyte, the Li||NCM811 cell equipped with a U66-OH/PP separator maintains 90% capacity retention after 300 cycles. In addition, U66-OH can effectively optimize the poor wettability of the PP separator and facilitate Li-ion transport. Consequently, the coated U66-OH layer can regulate Li+ deposition behavior and suppress the formation of lithium dendrites. The Li||Li symmetric cell employed by the U66-OH/PP separator shows stable cycling for over 750 h at 0.5 mA cm–2. This work paves a feasible route for water removal in advanced electrolytes toward high-performance LMBs.
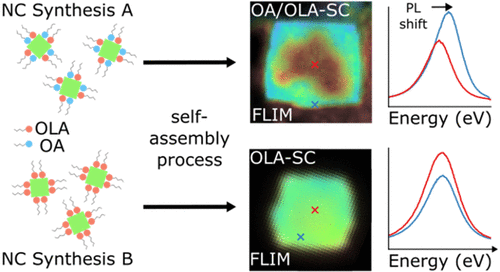
Synthesis-Dependent Fluorescence Properties of CsPbBr2Cl Supercrystals
Robert Thalwitzer - ,
Wolfgang Leis - ,
Ross E. Carter - ,
Elena Chulanova - ,
Dmitry Lapkin - ,
Gerard N. Hinsley - ,
Bihan Wang - ,
Kuan Hoon Ngoi - ,
Rustam Rysov - ,
Fabian Westermeier - ,
Ivan A. Vartanyants - ,
Ivan A. Zaluzhnyy - ,
Frank Schreiber - , and
Marcus Scheele *
This publication is Open Access under the license indicated. Learn More
We compare the fluorescence properties of CsPbBr2Cl nanocrystals, obtained via two distinct synthetic procedures and self-assembled into supercrystals using the same antisolvent crystallization technique. By spatially resolved fluorescence (lifetime) measurements we demonstrate that the optical properties of the supercrystals depend on the specific synthesis conditions of the constituting nanocrystals. Using scanning electron microscopy, small-angle X-ray scattering, and nuclear magnetic resonance spectroscopy, we find evidence that spatial fluctuations in the supercrystal fluorescence correlate with the ligand sphere of the nanocrystals. Specifically, homogeneous surface passivation of the nanocrystals leads to consistent interparticle distances and increased structural order within the supercrystals, resulting in a uniform fluorescence center wavelength and lifetime. The results of this study emphasize the importance of the relationship between crystalline structure and ligand configuration in controlling the optical properties of lead halide perovskite supercrystals.

In Situ Study of Growth Mechanism of Germanene Segregated through Ag(111) Thin Films by Raman and X-ray Photoelectron Spectroscopy
Tomo-o Terasawa *- ,
Daiki Katsube - ,
Masahiro Yano - ,
Takahiro Ozawa - ,
Yasutaka Tsuda - ,
Akitaka Yoshigoe - ,
Hidehito Asaoka - , and
Seiya Suzuki
Germanene, a honeycomb lattice of Ge atoms, has attracted attention for next-generation electronics and as a topological material. Among reported synthesis routes, the segregation method enables reproducible monolayer germanene formation on Ag(111) through simply annealing an Ag(111) thin film on a Ge(111) substrate. Despite this success, the physical origins of its monolayer selectivity and the mechanism for suppressing competing Ge phases remain unclear. Here, we investigate germanene formation via Ge segregation using in situ Raman spectroscopy and X-ray photoelectron spectroscopy to directly track Ge behavior during annealing and cooling. In situ observations revealed that annealing at 500 °C yielded no Ge-related byproducts, and the system reached a high-temperature surface equilibrium state, independent of the initial Ge amount. Cooling from this state produced a Ge-enriched surface that stabilizes the formation of monolayer germanene. In contrast, heating only to 300 °C produced three-dimensional Ge islands without Ge enrichment, followed by Ge–Ag alloy formation upon subsequent cooling. By integrating the temperature-dependent diffusion length and the process-dependent diffusion direction, we established a unified description of Ge behavior on Ag/Ge(111) substrates, in which cooling-induced Ge enrichment at the surface reproducibly stabilizes the selective formation of monolayer germanene.

Metastable Few-Layer GeS van der Waals Ferroelectrics Stabilized in SnS-GeS Heterostructures
Eli Sutter *- and
Peter Sutter
2D and layered van der Waals semiconductors have attracted interest as alternatives to conventional oxide ferroelectrics, combining attributes such as facile materials integration, distinct symmetry-breaking and polarization mechanisms, and bandgaps in the visible and near-infrared spectral region. Often, growth processes that produce high-quality ferroelectrics of a layered crystal are not easily adaptable to other materials, even within the same family. Here, we show that in such cases, a nonequilibrium ferroelectric crystal phase can be templated from one material into another across interfaces in heterostructures. Growing two types of layered SnS-GeS heterostructures, using seed crystals of equilibrium (centrosymmetric) SnS or a metastable (distorted) phase of SnS, respectively, we find that the distorted noncentrosymmetric phase can be templated from SnS to GeS. This templating effect is identified both across vertical (van der Waals) interfaces and at lateral (covalent) interfaces of the synthetic heterostructures. Furthermore, in vertically stacked regions, the GeS layers also inherit the ferroelectric stripe domain patterns from the underlying SnS seed. Heterostructures using centrosymmetric SnS seeds, on the other hand, incorporate the nonferroelectric equilibrium phase of GeS throughout. Within the studied materials system, the findings point to a rational route for obtaining ferroelectric Ge monochalcogenides. More broadly, the results indicate that interfaces in heterostructures between van der Waals crystals allow transcribing nonequilibrium crystal structures between dissimilar materials, which can be used to access distinct structures and functional properties.
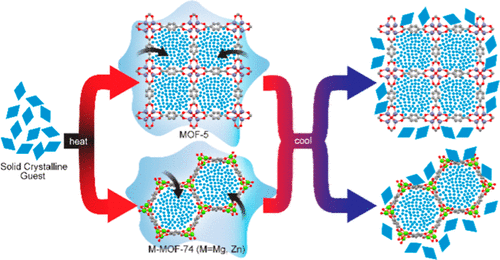
Solid Guests in Metal–Organic Frameworks: Capacity Limits and Structural Effects on Loading Kinetics
Yulia Rakova - and
Adam J. Matzger *
A method that quantifies the maximum loading of solid guests in metal–organic frameworks (MOFs) is demonstrated. The approach relies on the fact that included guests do not crystallize and quantifies the residual unincorporated guest heat of melting relative to that of the pure substance. Changes in MOF structure and metal center dramatically change inclusion rates. Whereas loading solid guests of varying melting points in MOF-5 was readily achieved at percent loadings between 31.1% and 56.7%, for Mg-MOF-74, the loadings were between 13.7% and 44.5%, with the lowest melting compounds exhibiting the greatest deviation from the theoretical maximum loading. Increasing the temperature significantly above the melting point of the lower melting guests led to dramatic incorporation improvements. The rate of inclusion was even slower in Zn-MOF-74, as demonstrated by variable temperature infrared spectroscopy. Achieving full guest loading resulted in guest@MOF composites that were near the calculated maximum loading based on pore volume considerations. This method has particular utility for making drug delivery systems and solid electrolytes.
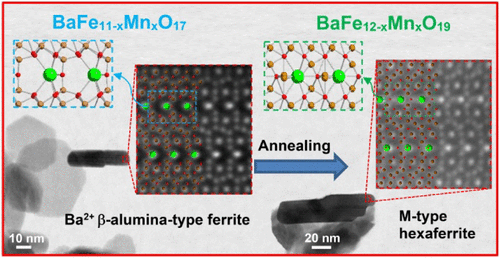
Mn-Induced Stabilization of a β-Alumina-Type Defect Structure in Barium Hexaferrite Nanoplatelets
Darko Makovec *- ,
Matic Poberžnik - ,
Janvit Teržan - ,
Tomaž Mertelj - ,
Damjan Vengust - ,
Goran Dražić - ,
Darja Lisjak - , and
Sašo Gyergyek
This publication is Open Access under the license indicated. Learn More
Hexaferrite nanoplatelets exhibit size-dependent structural variations influencing their magnetic properties. Here, we synthesized Mn-substituted barium ferrite nanoplatelets via hydrothermal methods, achieving up to ∼27% Fe substitution. Advanced STEM and Raman analyses revealed depletion of Fe(2b) trigonal lattice sites and associated oxygen vacancies, forming a β-alumina-type ferrite structure─representing the first pure Ba2+ β-ferrite analogue. First-principles modeling confirmed the thermodynamic stabilization of this defected structure at higher Mn/Fe ratios. Mn substitution reduced nanoplatelet size and suppressed magnetic properties, which were restored upon annealing at 800 °C, reverting to the M-type hexaferrite structure with expected magnetic behavior. These findings elucidate nanoscale structural adaptations induced by chemical substitution and offer insights into tailoring the magnetic properties of barium ferrite nanoplatelets through controlled synthesis and post-treatment.

Ultramicroporous Carbon Aerogel-Supported Iron Single-Atom Catalysts Toward Efficient pH-Universal Oxygen Reduction and Acidic/Alkaline Zinc–Air Batteries
Dahai Xu - ,
Haizhong Dai - ,
Jingjing Liu - ,
Jiahui Zhang - ,
Josue Pizano - ,
Shaowei Chen *- , and
Ting He *
Development of highly efficient nonprecious metal-based oxygen reduction catalysts, capable of operating within a broad pH range, has remained a great challenge in electrocatalysis. Herein, ultramicroporous carbon aerogel-supported iron single-atom catalysts (MPCA/Fe) are synthesized using a chitosan hydrogel precursor, with zinc species acting as a sacrificial template. Structural characterizations reveal that the produced ultramicropores effectively facilitate the anchoring of Fe single atoms within the carbon aerogel. The resulting MPCA/Fe composites exhibit a remarkable activity and stability toward the oxygen reduction reaction, featuring a half-wave potential of +0.93, +0.82, and +0.79 V in alkaline, neutral, and acidic media, respectively. Computational studies based on density functional theory calculations indicate that FeN4 sites embedded within the ultramicropores possess moderate *OH adsorption energy, leading to excellent catalytic performance. As MPCA/Fe also exhibits apparent electrocatalytic activity towards the oxygen evolution reaction, a zinc–air battery is assembled with the MPCA/Fe as the cathode catalyst, which delivers an open-circuit voltage (OCV) of 1.50 V and a peak power density of 240.8 mW cm–2 and excellent durability during 1600 charge–discharge cycles. When MPCA/Fe is assembled into an acid/alkali-mixed zinc–air battery, the device enables an exceptionally high OCV of 2.20 V and a discharge voltage of 2.07 V at a current density of 5 mA cm–2. Results from this study offer an effective strategy for the development of high-performance pH-universal oxygen reduction electrocatalysts.
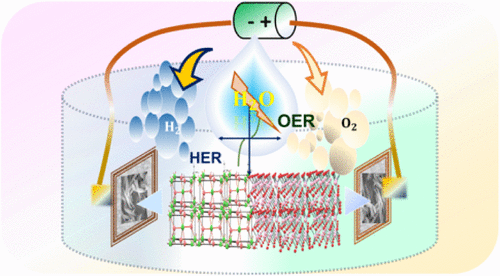
Bifunctional CrSe@FeSe2 Heterostructure with Interface Synergy and Vacancy Engineering for Enhanced Alkaline Water Splitting
Sukanya Bhattacharjee - ,
Ujjwal Phadikar - ,
Bholanath Panda - ,
Debasis Dhak - ,
Naresh Chandra Murmu - , and
Tapas Kuila *
Hydrogen energy is at the forefront of the global green energy transition, offering a clean, sustainable alternative to conventional fuels. Efficient water splitting is pivotal for large-scale hydrogen production, yet it demands highly active and durable electrocatalysts. Transition metal selenides have emerged as promising materials due to their superior electrical conductivity, tunable electronic structures, and abundant active sites. A series of CrSe-based catalysts was systematically engineered through controlled Fe incorporation and defect modulation, yielding a CrSe@FeSe2 heterostructure that synergistically enhances intrinsic catalytic activity and interfacial charge transfer. Due to the electronic coupling and the abundance of active sites introduced by vacancy engineering, it exhibited remarkable electrochemical performance in both the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER). The catalyst required an ∼220 mV overpotential to deliver a current density of 20 mA cm–2 for the OER, and 147 mV for HER in an alkaline medium to achieve the same current density. Furthermore, the material demonstrated excellent long-term durability for 40 h, delivering high current densities of 600 mA cm–2 (OER) and 1A cm–2 (HER) in chronoamperometric tests. When used as both an anode and cathode in a two-electrode configuration, the CrSe@FeSe2-based electrolyzer achieved sustained water-splitting performance at 100 mA cm–2 for 40 h with negligible potential degradation. This work highlights the efficacy of vacancy engineering and heterostructure design in optimizing selenium-based transition-metal catalysts for high-performance, durable electrochemical water splitting.

A Facile Method to Create a High-Performance Electrochromic Material: Electrografting of Bis-Terpyridine-Iron Motif onto Extended Indium Tin Oxide Support
Salma Jadali - ,
Iraklii I. Ebralidze - ,
E. Bradley Easton *- , and
Olena V. Zenkina *
Fe(II)–bis(terpyridine) units were covalently embedded on the extended surface of the indium tin oxide (ITO) support, and oligomeric molecular wires derived from these units were subsequently grown using a diazonium electrografting under ambient temperature and pressure. The resulting smart material exhibits excellent electrochromic performance, changing its optical properties in response to external voltage, both in a liquid electrolyte (3-electrode cell) and when assembled into a solid-state electrochromic device (2-electrode cell). Electrografting molecular architectures within interparticle pores results in dense molecular packing, as confirmed by surface coverage measurements and the presence of π–π* satellites in both the C 1s and N 1s X-ray photoelectron spectra. Electrografting is more time-efficient than the conventional layer-by-layer growth of coordination-based molecular assemblies. Moreover, it eliminates the need for a separate surface templating layer, resulting in enhanced conductivity through the molecular wire. Thus, the electron transfer rate of the developed material is in par with that of monolayer-based materials. The electrochromic devices were assembled by incorporating the material and surface-enhanced ITO as electrodes through a lithium gel electrolyte and a Nafion layer. In-operando measured optical properties demonstrate that the device exhibits extremely high cycling stability, notable coloration efficiency, and short switching times.
Additions and Corrections
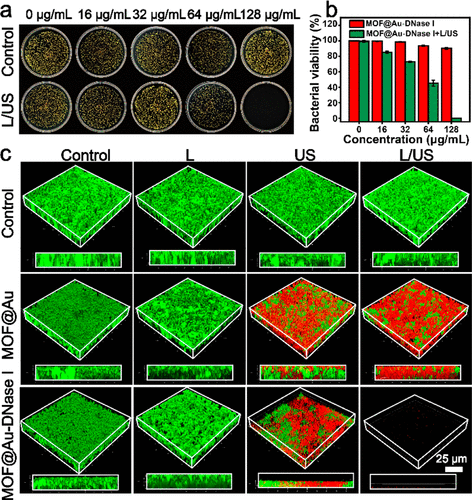
Correction to “Near-Infrared Light-Propelled MOF@Au Nanomotors for Enhanced Penetration and Sonodynamic Therapy of Bacterial Biofilms”
Wei Guo - ,
Yanmin Wang - ,
Kai Zhang *- ,
Xiaoguang Dai - ,
Zhuangzhuang Qiao - ,
Zhiwen Liu - ,
Bingran Yu - ,
Nana Zhao *- , and
Fu-Jian Xu *

This publication is free to access through this site. Learn More
Correction to “Polymorphism and Phase Control in Dion–Jacobson 2D 3-(Aminomethyl)piperidinium-Based Metal Iodide Perovskites”
Jared D. Fletcher - ,
Marios Zacharias - ,
Shoshanna Peifer - ,
Jin Hou - ,
Anastasia D. Pournara - ,
Aditya D. Mohite - ,
Richard D. Schaller - ,
Jacky Even - ,
Claudine Katan - , and
Mercouri G. Kanatzidis *
This publication is free to access through this site. Learn More
Mastheads
Issue Editorial Masthead
This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More