

About the Cover:
Thermophysical and Thermochemical Properties
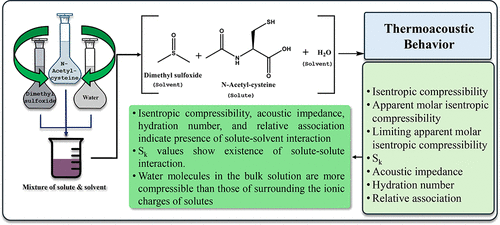
Investigation on the Molecular Interaction of N-Acetylcysteine in Aqueous Dimethyl Sulfoxide Systems and Thermoacoustic Properties at Various Temperatures
Mehidi H. Khan - and
Mohammad A. Yousuf *
The potential molecular interactions among N-acetyl-l-cysteine (NAC), water (H2O), dimethyl sulfoxide (DMSO), and aqueous dimethyl sulfoxide (DMSO-H2O) were investigated from the experimental speed of sound (c) and density values (ρ) over the temperature range from 298.15 to 318.15 K and at atmospheric pressure. The acoustic parameters, viz., isentropic compressibility (κs), apparent molar isentropic compressibility (κs,ϕ), limiting apparent molar isentropic compressibility (κs,ϕ0), Sk, acoustic impedance (Z), hydration number (NH), and relative association (RA), were determined using experimental sound velocities. The results were interpreted in terms of possible solute–solute and solute–solvent interactions. Negative κs,ϕ values identifying molecules of water in the bulk solution seemed to be more compressible than those surrounding the ionic charges of the solute. κs, κs,ϕ0, and Z values indicate the presence of solute–solvent interaction, whereas Sk values specify that solute–solute interaction is also present in the studied system.
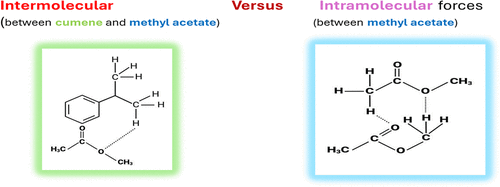
Comparative Evaluation of Thermophysical and Acoustical Properties of Binary Mixtures of Cumene + Methyl Acetate and Cumene + Ethyl Acetate over the Range of Molar Compositions and Temperatures from 298.15 to 318.15 K: Experimental Measurements and Application of Deviation Modeling
Padmanaban Radhakrishnan - ,
Gayathri Ahobilam - ,
Seema Kapoor - ,
Gopalan Anantha Iyengar *- ,
Dong-Eun Lee - ,
Venkatramanan Kannan *- ,
Vaithinathan Karthikeyan - ,
Dani Samer Assi - , and
Vellaisamy A. L. Roy
The viscosity (η), activation energy (Ea), density (ρ), ultrasonic velocity (U), and refractive index (nD) were determined for binary liquid mixtures of cumene (CM, an alkylbenzene) with ethyl acetate (EA) or methyl acetate (MA) over different mole fractions of CM (0.08–0.87) in a wide temperature range (298.15–313.15 K). The increasing trend in η with the mole fraction of CM was correlated with the strengthening of the molecular interactions between the constituent components. Various theoretical models were utilized to predict the deviation in the properties of the binary mixtures over the compositions. Typically, the Kandoll and Monroe models predicted lower values for η, while models like Gruenberg and Nissan suggested higher values for the CM + MA binary mixture. Likewise, the usefulness of the models in predicting other properties is detailed. The average percentage of deviation (APD) was used to predict the most suitable model for η, U, and nD. The various acoustical properties and excess parameters were calculated and interpreted in terms of charge transfer, dipole–dipole, and dipole-induced dipole interactions. The results suggested that the presence of both aryl and aliphatic moieties in CM contributed to the intermolecular interactions with nearly nonpolar (EA/MA) molecules.
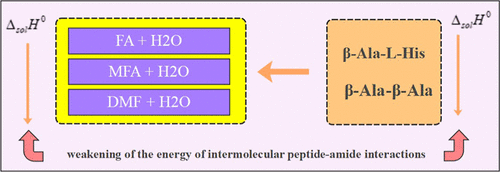
Study of the Thermochemical Aspects of the Dissolution and Solvation of Certain β-Alanyl-Dipeptides in Aqueous Solutions of Formamides at a Temperature of T = 298.15 K
Valeriy I. Smirnov *
This paper presents, for the first time, the standard values of the enthalpies of β-alanyl-l-histidine (β-Ala-L-His) dissolution (ΔsolH0) in the binary solvents water + (formamide, N-methylformamide, and N,N-dimethylformamide) at T = 298.15 K, obtained calorimetrically. Various parameters such as solvation enthalpies, (ΔsolvH0), transfer enthalpies, (ΔtrH0) and the enthalpic coefficients of pairwise interactions (hxy) were calculated from these values. The obtained data were compared with similar data for β-alanyl-β–alanine (β-Ala-β-Ala). A similar character of solvation of both peptides in mixtures of the same name has been established. This is evidenced by the linear relationship between the enthalpic coefficients of the pairwise interactions of both peptides. The solvation of peptides is weakened in the series of mixed solvents (H2O + FA) < (H2O + NMF) < (H2O + DMF), which is associated with an increase in the hydrophobic properties of the formamides and an increase in the intermolecular interactions in the mixtures themselves in the same order. However, β-Ala-L-His is more strongly solvated than β-Ala-β-Ala. This is due to the presence of specific solvation centers in the side chain of β-Ala-L-His.
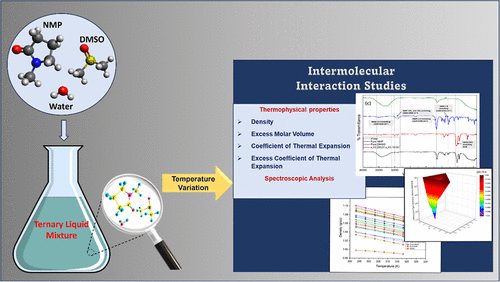
Intermolecular Interactions in a Ternary Mixture of Industrial Solvents (N-Methyl-2-Pyrrolidone + Dimethyl Sulfoxide + Water) Using Spectroscopic and Volumetric Analysis
Praseeda P. Nair - ,
Eshita Raghunath Kakodkar - ,
Suvanjan Bhattacharya - , and
Ranjan Dey *
This study investigates the thermodynamic and molecular interaction properties of the N-Methyl-2-pyrrolidone (NMP), Dimethyl sulfoxide (DMSO), and water ternary system across a range of compositions and temperatures (293.15–318.15 K). Densities were measured for 19 different compositions, from which excess molar volumes (VmE) and excess coefficients of thermal expansion (αPE) were calculated to quantify deviations from an ideal mixing behavior. All compositions exhibited negative VmE values, indicating a significant volume contraction due to strong intermolecular interactions. Fourier-transform infrared (FTIR) spectroscopy confirmed the presence of enhanced hydrogen-bonding networks between water and the organic solvents. Temperature dependence studies revealed that while thermal energy partially disrupts these interactions, the structured domains persist throughout the temperature range studied. The positive αPE values indicate that the mixture’s thermal expansion exceeds predictions based on ideal mixing, highlighting the system’s sensitivity to thermal perturbation. These findings provide valuable insights for optimizing NMP–DMSO–water mixtures in applications, including chemical synthesis, pharmaceutical formulations, polymer processing, and extraction technologies.

Binary Mixtures of n-Alkylbenzenes and Pentadecane: Densities, Speeds of Sound, and Viscosities within the Range of 288.15 and 333.15 K and at 0.1 MPa
Dianne J. Luning Prak *- and
Jim S. Cowart

This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
The physical properties of mixtures of alkanes and aromatic compounds can aid in the understanding and modeling of their combustion in engines. Herein, densities, viscosities, and speeds of sound of binary mixtures of n-alkylbenzenes with pentadecane and their corresponding excess molar volumes (VmE), excess speeds of sound (cE), excess isentropic compressibilities (ΚsE), and viscosity deviations (Δη) are reported. In general, mixture densities, viscosities, and speeds of sound increased monotonically with changing mole fractions, except for the speeds of sound of toluene, ethylbenzene, propylbenzene, and butylbenzene mixtures. These properties can be used for fuel modeling purposes. VmE’s, Δη’s, cE’s, and KsE’s ranged from 0.04 to 0.56 cm·mol–1, −0.14 to 0.09 mPa·s, −14.8 to 2.5 m·s–1, and −2.2 to 18.1 TPa–1, respectively. Increasing the n-alkylbenzene size (up to decylbenzene) caused the equimolar mixture’s VmE’s, Δη’s, and KsE’s to decrease and cE’s to increase at 293.15 K. The properties of smaller n-alkylbenzene molecules were more affected by the aromatic group. In comparison with n-alkylcyclohexane/pentadecane mixtures, n-alkylbenzene/pentadecane mixture Δη trends were similar, but VmE’s, cE’s, and KsE’s trends were different, suggesting that molecular interactions of the benzyl and cyclohexyl groups affect volume and compressibility more than they affect viscosity.
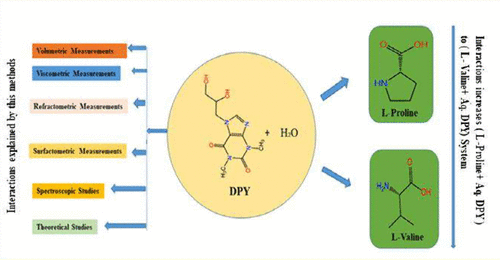
Physiochemical Investigation of Different Interactions of Some Essential Amino Acids Prevailing in Aqueous Media of a Biologically Potent Drug Molecule at Various Temperature Optimized by DFT
Anindita Poddar - ,
Biplab Rajbanshi - ,
Sukdev Majumder - ,
Debadrita Roy - ,
Subhajit Debnath - ,
Ayesha Hossain - ,
Subhankar Choudhury - ,
Akashdeep Jaiswal - ,
Modhusudan Mondal - ,
Biraj Kumar Barman - , and
Mahendra Nath Roy *
The comparative molecular interaction between a biologically potent drug molecule, dyphylline, and two essential amino acids, l-proline and l-valine, has been studied in an aqueous medium using physicochemical methodologies at 303.15, 308.15, and 313.15 K at 0.1 MPa atmospheric pressure. The methodologies engaged herein (density, refractive index, viscosity, and surface tension) recognized the presence of the molecular interaction of dyphylline with the amino acids. Different parameters like “apparent molar volumes” (ϕv), “partial molar volumes at infinite dilution” (ϕv0), “limiting apparent molar expansibilities” (ϕE0), transfer properties (Δtrφv0), “isobaric thermal expansion coefficient” (α), viscosity B-coefficients, hydration number (nH), and thermodynamic parameters (Δμ10≠, Δμ20≠, TΔS20≠, and ΔH20≠) of viscous flow obtained from density and viscosity measurements are also used for molecular interaction determination. 1H and 13C NMR spectroscopic study shows significant evidence for the presence of the solute–cosolvent interaction in solution. The interaction between the molecules has been discussed in the context of the structure-breaking/structure-making ability of the molecules in solution. The thermodynamic background of their interaction in aqueous solution has also been explored with the help of density and viscosity measurements. DFT studies construct the theoretical basis of the interaction between the components in solution.
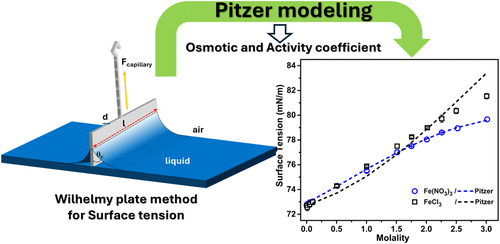
Thermodynamic Modeling of Osmotic Coefficients and Surface Tension of the Ferric Chloride–Water and Ferric Nitrate–Water Systems Using the Pitzer Model
Mouad Arrad *- ,
Biswajit Biswas - ,
Ka Chon Ng - , and
Heather C. Allen
A comprehensive thermodynamic model for aqueous solutions of ferric nitrate and ferric chloride was presented. The temperature dependency of the Pitzer parameters was determined based on the available literature data. New surface tension measurements were carried out in this work for the aqueous solution of ferric nitrate and ferric chloride at 293 K. The model was able to reproduce accurately the thermodynamic properties such as the osmotic coefficient and the activity coefficient. The model was extended to predict the surface tension of these electrolytes with good agreement to the measured values of surface tension.
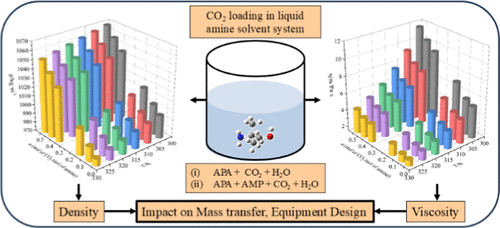
Physicochemical Characteristics for CO2-Loaded Aqueous Bis(3-aminopropyl)amine and Its Mixture with 2-Amino-2-methyl-1-propanol
Mehul Darji - ,
Chetna Shukla - ,
Sukanta K. Dash *- , and
Kalisadhan Mukherjee *
Removal of CO2 from industrial exhaust gas is vital and is commonly achieved by using chemical absorption with newly formulated solvents. The density and viscosity of carbonated solutions are critical physicochemical properties for selecting an efficient solvent in the CO2 absorption processes. In this work, aqueous bis(3-aminopropyl)amine (APA) and its mixture with 2-amino-2-methyl-1-propanol (AMP) are considered to be promising absorbents for the CO2 capture process. New experimental density and viscosity data for CO2-loaded APA and APA + AMP mixtures were determined over a temperature range of 303.25–328.25 K and different CO2 concentrations pertaining to the gas absorption condition. The density and viscosity of the CO2-loaded APA and APA-AMP solvent systems were correlated using thermodynamics models, with parameters determined for different aqueous carbonated solvent compositions across the studied temperature range. The modeling results show that the percentage average absolute deviations (% AADs) between the experimental and model results of density were 0.03 for the ternary system (APA + H2O + CO2) and 0.13 for the quaternary system (APA + AMP + H2O + CO2). The % AADs between experimental and model viscosity data are 0.88 and 2.97, respectively. The obtained results provide valuable insights into the development of amine-based solvents for efficient CO2 capture applications.
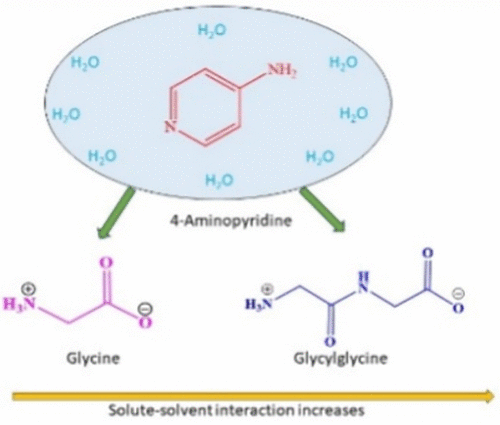
Unraveling Glycine and Glycylglycine Dynamics in Aqueous 4-AP Solutions: A Combined Experimental and DFT Study Across Temperatures
Priyanka Roy - ,
Modhusudan Mondal - ,
Doli Roy - ,
Ayesha Hossain - ,
Kangkan Mallick - ,
Debadrita Roy - ,
Mantu Dey - ,
Agnisha Ghosh - ,
Subhankar Choudhury - , and
Mahendra Nath Roy *
In the present investigation, aqueous solutions of 4-aminopyridine (4-AP) at molalities of 0.001, 0.003, and 0.005 mol·kg–1 were utilized as solvent media to explore the physicochemical properties of the amino acids (AAs) glycine and glycylglycine across a temperature span of 293.15 to 313.15 K under ambient pressure. Experimental determinations of density and viscosity facilitated the evaluation of critical thermodynamic and transport parameters, including apparent molar volumes (Vϕ), limiting partial molar volumes (Vϕ0), transfer volumes (ΔtrVϕ0), Jones–Dole viscosity B-coefficients, and the activation free energies for viscous flow (Δμ10# and Δμ20#), interpreted through the framework of Transition State Theory. The obtained data indicated stronger intensification of solute–solvent interactions with increasing concentrations of 4-AP, consistent with the Co-sphere Overlap Model. Both glycine and glycylglycine were found to exhibit structure-disrupting effects on the aqueous medium in the presence of 4-AP, suggesting a perturbation of the inherent water structure. Complementary fluorescence spectroscopic analysis, through systematic variation in AA concentration, enabled the estimation of association constants, while 1H NMR spectroscopy provided insights into the presence of hydrophobic interactions between 4-AP and the AAs. These experimental insights were further substantiated through computational modeling, offering a coherent and molecular-level perspective on the interaction dynamics governing these systems.
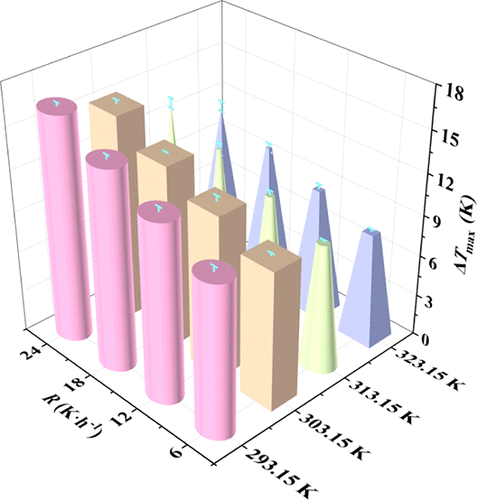
Effect of Stirring Paddle Geometry on Nucleation and Growth Crystal Size of Ammonium Sulfate Crystals
Jihao Xiong - ,
Jian Han - ,
Huixin Zhang - ,
Kaili Liu - ,
Xuan Yin - ,
Suzhen Zhang - , and
Jianxin Chen *
Stirring paddle selection is a critical part of the design and optimization of the crystallization processes, directly influencing crystal nucleation and crystal quality. This work investigates the effect of two different stirring paddles, the propeller and anchor paddle, on the properties of the metastable zone and the nucleation kinetics of ammonium sulfate. The experiments were conducted by determining the metastable zone widths with two different types of stirring paddles and deriving the nucleation kinetics parameters from a dual analytical point of view by using the self-consistent Nývlt equation with the classical 3D nucleation theory. It is found that the nucleation mechanism under the action of both stirring devices is progressive nucleation, and the interfacial energy increases with decreasing saturation temperature. The anchor paddle system exhibits higher nucleation rates at the same chemical potential difference. In addition, crystal products prepared with the propeller were significantly larger in size than the anchor paddle, and crystal growth was more regular. This study provides a theoretical basis for the selection of stirring paddles in the industrial crystallization process.

Activity Coefficients of HCl in Solutions Related to “Tris” Buffers in Artificial Seawater. II. HCl + NaCl + TrisHCl + H2O, and Tris Buffer + NaCl + H2O, to High Ionic Strength and from 5 to 40 °C
Igor Maksimov *- ,
Toshiaki Asakai - ,
Yuya Hibino - , and
Simon L. Clegg *
This publication is Open Access under the license indicated. Learn More
The substance Tris (2-amino-2-hydroxymethyl-1,3-propanediol, CAS 77-86-1), and its protonated form TrisH+, are used in the preparation of ‘total’ pH buffers in artificial seawater media. The development of a chemical speciation model of the buffer solutions, using the Pitzer equations to calculate solute activity coefficients, is desirable in order to quantify the effects of composition change, convert the total pH to other scales, and address metrological requirements for traceability to the International System of Units. Here, in the second of a series of studies, we present Harned cell measurements of potentials and mean activity coefficients of HCl in solutions containing HCl, NaCl, and TrisHCl for ionic strengths from 1.0 to 5.5 mol kg–1 and from 5 to 40 °C. The results at 25 °C are consistent with those of the literature studies of the two end-member solutions (aqueous HCl + NaCl, and HCl + TrisHCl). We also present results of measurements of buffer solutions containing equimolal Tris and TrisHCl (hence TrisH+), and NaCl, at ionic strengths of 0.2, 1.0, and 4.0 mol kg–1 at the same temperatures. These are compared with literature data for Tris buffers in an artificial seawater medium. Aspects of the development of a Pitzer model for these solutions are discussed.
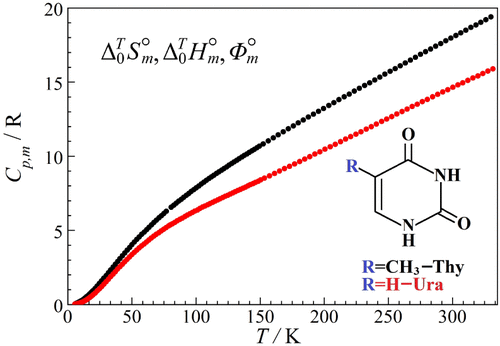
Low-Temperature Thermodynamic Properties of Pyrimidine Bases: Thymine and Uracil
Michael A. Bespyatov *
This study reports precise heat capacity data for crystalline thymine (C5H6N2O2; CAS Number: 65-71-4; fraction purity: 0.999) and uracil (C4H4N2O2; CAS Number: 66-22-8; fraction purity: 0.999) over the temperature range of 6 to 330 K. Adiabatic calorimetry was used to perform the measurements. The calorimeter was validated by measuring the heat capacity of reference standards (copper and benzoic acid). The experimental heat capacity data were fitted using Einstein-Planck functions and extrapolated to 0 K following the Debye law. The data enabled the calculation of thermodynamic functions (entropy, enthalpy increment, and reduced Gibbs energy) for thymine and uracil between 0 and 330 K.
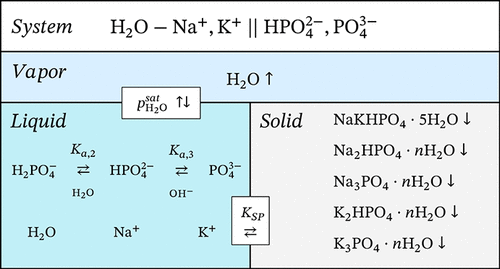
Thermodynamic Modeling of the H2O–Na+, K+ || HPO42–, PO43– System
Artem A. Novikov *- ,
Nikita Kovalenko - , and
Irina A. Uspenskaya
A comprehensive thermodynamic model for aqueous solutions containing sodium and potassium monohydrogen phosphates has been developed based on the Pitzer–Simonson–Clegg formalism for excess properties, the Helgeson–Kirkham–Flowers equation of state for standard-state properties, and the IAPWS formulations for pure water. The model accounts explicitly for acid–base equilibria among H2PO4–, HPO42–, and PO43– species and includes all necessary ion–ion and ion–neutral interactions to accurately describe solution properties over a wide temperature range. Thermodynamic parameters were evaluated using critically selected literature data on osmotic coefficients, water activity, solubility, enthalpies of dilution, and heat capacities. Solubility products of relevant solid phases, including multiple hydrates of Na2HPO4 and K2HPO4, were parametrized. The resulting model reproduces experimental phase equilibria and thermodynamic properties with high accuracy from the crystallization to the boiling point of saturated solutions. The model forms a foundation for the future inclusion of dihydrogen phosphate species and enables the consistent prediction of multicomponent phosphate solution behavior across wide concentration and temperature ranges.
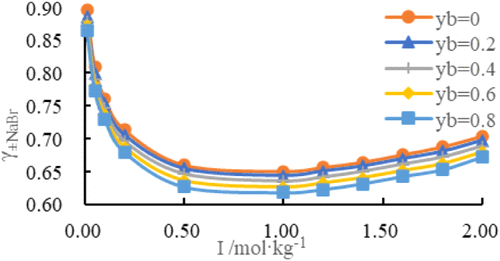
Measurements of the Mean Activity Coefficients of NaBr in NaBr–SrBr2–H2O Ternary Solutions at 318.15 K and Applications in Prediction of Phase Equilibria
Rong Yang - ,
Shi-Hua Sang *- ,
Yan Feng - , and
Xiao-Tian Tan
The mean activity coefficients of NaBr in the NaBr–SrBr2–H2O ternary solution at 318.15 K were investigated by using the cell potential method. Sodium- and bromide-selective electrodes were used to measure the cell potentials of the NaBr–H2O solutions. The standard cell potential (E0) and electrode response slope (κ) were derived from these measurements. Subsequently, the mean activity coefficients of NaBr in the NaBr–SrBr2–H2O system were determined for a total ionic strength range of 0.01 to 2.00 mol·kg– 1. The ionic strength fraction of SrBr2 (yb) was set to 0.0, 0.2, 0.4, 0.6, or 0.8. Using mean activity coefficents and the Pitzer model, we used a programming solver to fit the Pitzer ion interaction parameters θNa+,Sr2+ and ψNa+,Sr2+,Br−. The mean activity coefficients of SrBr2, the osmotic coefficient Φ, the water activity aw, and the excess Gibbs free energy GE of the mixed solution were calculated. Additionally, the solubility of the ternary system was modeled using the Pitzer model. The predicted values were compared with experimental data at 308.2 and 323 K, confirming the reliability of the modeled results and the good adaptability of the mixed ion interaction parameters for solubility prediction.
Vapor-Liquid Equilibria and Supercritical Fluid Equilibria
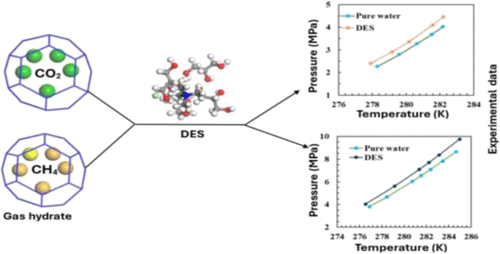
Phase Equilibria of Carbon Dioxide and Methane Gas Hydrates in the Presence of Tetramethylammonium Chloride and Tetrapropylammonium Bromide-Based Deep Eutectic Solvents
Njabulo Mziwandile Zulu *- ,
Hamed Hashemi - , and
Kaniki Tumba
This publication is Open Access under the license indicated. Learn More
The effects of four deep eutectic solvents (DESs) on the phase equilibrium conditions of CO2 and CH4 gas hydrates were investigated. The mixtures of tetramethylammonium chloride (TMAC) and tetrapropylammonium bromide (TPAB) as hydrogen-bond acceptors with glycerol and ethylene glycol (EG) as hydrogen-bond donors were used to formulate the DESs. The combinations of TMAC/glycerol, TMAC/EG, TPAB/glycerol, and TPAB/EG were all made at a 1:3 molar ratio. The concentrations of DESs in the aqueous solutions were 2 and 4 wt %. The hydrate dissociation conditions for CO2 and CH4 systems were measured using an isochoric pressure-search method in the temperature ranges of (277.85 to 282.97) K and (276.03 to 285.15) K, respectively, and pressure ranges of (2.03 to 5.28) MPa and (3.71 to 10.39) MPa, respectively. All the studied DESs demonstrated the thermodynamic gas hydrate inhibition effect on CO2 and CH4 hydrate formation. The gas hydrate inhibition ability ranking from the highest to the lowest was found to be in the order of: TMAC/glycerol > TMAC/EG > TPAB/glycerol > TPAB/EG. This result proved that DESs have great potential to be effective green thermodynamic hydrate inhibitors.
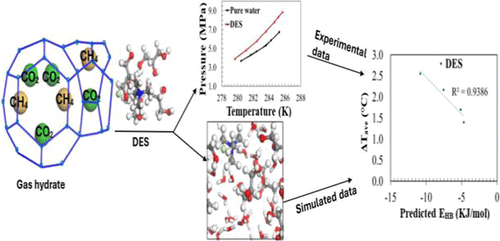
Thermodynamic Inhibition of CO2–CH4 Gas Hydrates by DESs: Experimental and Computational Study
Njabulo Mziwandile Zulu *- ,
Hamed Hashemi - ,
Kaniki Tumba - , and
Victoria T. Adeleke
This publication is Open Access under the license indicated. Learn More
This study presents an experimental and computational investigation of thermodynamic hydrate inhibition behavior of four deep eutectic solvents (DESs) on binary mixed CO2–CH4 gas hydrates. The mixtures of hydrogen bond acceptors, tetramethylammonium chloride, and tetrapropylammonium bromide with hydrogen bond donors, glycerol, and ethylene glycol were used to prepare the DESs. The hydrate liquid–vapor equilibrium data for studied systems were measured using an isochoric pressure-search method within the temperature and pressure ranges of (280.51–285.20) K and (3.86–9.48) MPa, respectively. All the investigated DESs inhibited CO2–CH4 hydrates, with the inhibition effect increasing with the DES concentration. The experimental results were validated using a computational approach through the analysis of sigma profile data and hydrogen bonding energies obtained for the investigated DESs. The order of the obtained sigma profile results and hydrogen bonding energies was consistent with the experimental results, thereby validating the inhibition ability of the investigated DESs. The hydrogen bonding energies that were obtained in this study correlated well with the inhibition ability of the studied DESs, and this proved the reliability of the computational approach used. Furthermore, this computational approach was successfully used as the prescreening tool to predict the inhibition ability of theoretically formulated DESs without experimental data.

Vapor–Liquid Equilibrium of Toluene/Heptane and Benzene/Cyclohexane Systems with Biobased Cyrene: Experimental Data and Thermodynamic Modeling
Yaqiong Zhao - ,
Hongwei Kang - ,
Yuming Tu - ,
Qunsheng Li - ,
Chencan Du *- , and
Zhongqi Ren *
Separation of hydrocarbon mixtures is essential for the efficient utilization of resources in the chemical industry. In this study, we investigated the vapor–liquid equilibrium (VLE) behavior of two binary systems, toluene + heptane and benzene + cyclohexane, and examined the application of the biobased solvent, Cyrene, as an extractant in the ternary systems. The vapor–liquid-phase equilibrium data were correlated by a nonrandom two-liquid (NRTL) model, and the results showed that the addition of Cyrene significantly increased the relative volatility of the two binary systems up to 3, and the deviation of all systems was less than 5%. Compared with conventional organic solvents such as SUL, DMSO, NMP, and DMF, Cyrene not only demonstrated good separation performance but also offered significant advantages in terms of environmental sustainability. The results suggest that Cyrene has great potential as a green alternative for sustainable hydrocarbon separation processes.
Liquid-Liquid Equilibria and Vapor-Liquid-Liquid Equilibria
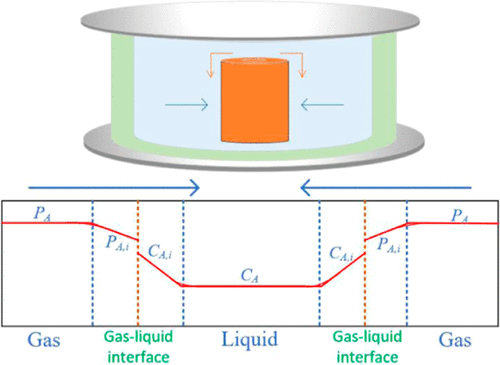
Mass Transfer Kinetics and Thermodynamics Evaluation of 1-Amino-2-propanol/Sulfolane Biphasic Solution for CO2 Absorption
Guangjie Chen - ,
Guangying Chen *- ,
Ge Gao - ,
Li Sze Lai *- ,
Swee Pin Yeap - ,
Wee Horng Tay - , and
Helei Liu
The utilization of biphasic absorbents for CO2 capture can substantially reduce energy consumption. This study investigated the mass transfer kinetics and thermodynamics of the phase separation absorption process using 1-amino-2-propanol (MIPA)/sulfolane (TMS) biphasic solvents. 13C NMR spectroscopy was employed to analyze the species in different CO2-loaded solutions. The direction and extent of species transfer during the absorption process were elucidated and the phase separation mechanism was explored. CO2 absorption kinetics were studied in a wetted-wall column, and the physicochemical properties and the absorption reaction kinetics data under different experimental conditions were obtained. Results showed the overall mass transfer coefficient (KG) was 1.6 times higher than 30 wt % MEA and twice that of DETA/TMS solution. NMR and kinetic data analysis revealed that phase separation is mainly contributing to the deterioration of the mass transfer process in biphasic solution, reducing the liquid-film mass transfer coefficient (KL) by 63.2% during the phase separation stage. Furthermore, the desorption heats of different biphasic solvents with identical amine concentrations were measured using a microcalorimeter. Findings indicated that TMS did not alter the thermodynamic properties of the solution. Since only the CO2-rich phase is regenerated, the energy consumption is significantly reduced compared to that of MEA.
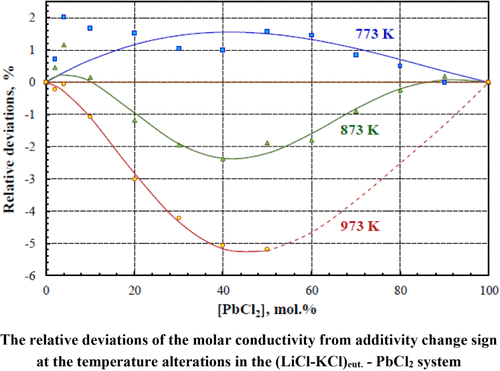
Electrical Conductivity of (LiCl–KCl)eut.–PbCl2 Molten Mixtures
Alexander Salyulev - and
Alexei Potapov *
The electrical conductivity of molten (LiCl–KCl)eut.–PbCl2 has been measured from the liquidus temperature up to 901–994 K over the entire concentration range. Literature data on the (LiCl–KCl)–PbCl2 density were recalculated to approximate them for our eutectic system. Using the calculated density values, the (LiCl–KCl)eut.–PbCl2 molar conductivity was calculated. The specific electrical conductivity of molten (LiCl–KCl)eut.–PbCl2 was found to decrease with PbCl2 addition, while the molar conductivity increases. The molten PbCl2 molar electrical conductivity is greater than the LiCl–KCl eutectic conductivity. For all molten mixtures, the ln Λ vs. 1/T dependence has a nonlinear form (Λ─molar conductivity, T─absolute temperature). At 773 K, the molar conductivity has positive deviations from additive values. The deviations shift in the negative direction and increase as the temperature grows (−5.3% at 973 K). This indicates a change in the predominant mechanism of electricity transfer as the temperature increases. Using breakpoints, the liquidus line of this system has been built. The activation energy of molar electrical conductivity decreases up to the PbCl2 concentration of 30–40%, and then increases. The results are discussed by considering the available data on the structure of the melts.
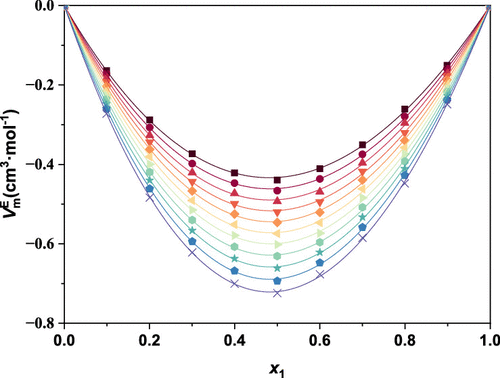
Densities and Viscosities for Four Binary Systems of Tetrahydrotricyclopentadiene with n-Dodecane, Methylcyclohexane, Decalin, or 1,2,3,4-Tetrahydronaphthalene at T = (293.15 to 343.15) K
Peilun Wang - ,
Xiwei Ye - ,
Yitong Dai - ,
Ji Mi - ,
Wenjun Fang *- ,
Pengfei Jiang *- , and
Yongsheng Guo
In order to achieve a comprehensive understanding of the properties of tetrahydrotricyclopentadiene (THTCPD) as a potential candidate for high-energy-density hydrocarbon fuels, the density and viscosity of binary mixtures comprising THTCPD and four representative hydrocarbons─n-dodecane, methylcyclohexane, decalin, and 1,2,3,4-tetrahydronaphthalene─were experimentally determined under conditions of T = (293.15 to 343.15) K and atmospheric pressure p = 0.1 MPa. At a constant temperature, both density and viscosity exhibited continuous increases with the rising molar fraction of THTCPD in the binary systems. Furthermore, the excess molar volume (VmE) and viscosity deviation (Δη) of these binary systems were calculated and subsequently fitted by using the Redlich–Kister equation. With the exception of the THTCPD + 1,2,3,4-tetrahydronaphthalene system, the VmE values for all other systems were negative, with their absolute values increasing as the temperature rose. The Δη values for all systems were also negative, and their absolute values decreased significantly with increasing temperature. These findings are interpreted in terms of intermolecular interactions and structural effects, providing critical reference data for the development of high-energy-density hydrocarbon fuels.
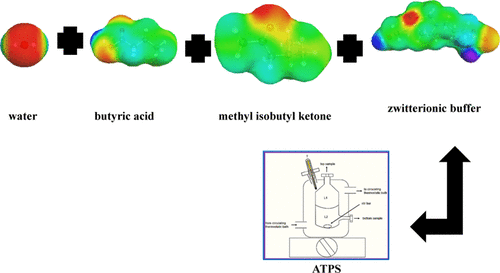
Separation of Butyric Acid with Methyl Isobutyl Ketone and Zwitterionic Buffers (EPPS/MOPSO/HEPES/TAPS) Based on Liquid Liquid Equilibrium Experiments and a Quantum Chemistry Approach
Ming-Chen Lin - ,
Saidah Altway *- ,
Farah Meutia - , and
Ardila Hayu Tiwikrama *
This publication is Open Access under the license indicated. Learn More
The aim of this study is to examine the ATPS consisting of butyric acid, water, MIBK, and a biological buffer at T = 293.15 K and atmospheric pressure. The zwitterionic buffer effect as a green auxiliary agent was also investigated using four different biological buffers, i.e., N-[tris (hydroxymethyl)methyl]-3-aminopropanesulfonic acid (TAPS), 2-hydroxy-3-(morpholin-4-yl)propane-1-sulfonic acid (MOPSO), 4-(2-hydroxyethyl)-1-piperazine propanesulfonic acid (EPPS), and 2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid (HEPES). Based on the experimental findings, the performance of extraction followed the sequence HEPES > MOPSO > EPPS > TAPS, which also indicated the effectiveness of buffering-out. Two thermodynamic models, the nonrandom two-liquid (NRTL) and the universal quasi-chemical (UNIQUAC), successfully correlated the experimental ATPS LLE data. The enthalpies of mixing were used to predict the sigma profile and energy interactions of butyric acid, water, MIBK, and the zwitterionic buffer. The molecular interaction via excess enthalpies of water and butyric acid was greater than that of MIBK and butyric acid. In addition, quantum chemical calculations were performed to study the interactions between MIBK–water and MIBK–butyric acid via the reduced density gradient (RDG). The results showed that the separation of butyric acid with MIBK from an aqueous solution was dominated by hydrogen bonding and vdW forces.
Toward Evaluation of Diethylene Glycol-Based Deep Eutectic Solvents for the Separation of Benzene and n-Hexane Using Pseudoternary Liquid-Liquid Equilibrium Data
Salal Hasan Khudaida - ,
Jiarou Liu - ,
Mohamed Tarek Ahmed - ,
Yu-Cheng Chang - , and
Ardila Hayu Tiwikrama *
This publication is Open Access under the license indicated. Learn More
The pseudoternary systems of deep eutectic solvents (DESs) has attracted interest in separation technology due to its novel physicochemical properties. Two DESs of [ChCl][DEG] with molar ratios of 1:4 (DES1) and 1:8 (DES2) were prepared. The pseudoternary systems of n-hexane + benzene + DES1 and n-hexane + benzene + DES2 measured the tie lines of liquid–liquid equilibrium (LLE) data at T = 298.15–318.15 K and at atmospheric pressure. The assumption of DESs as the pseudopure component was performed to investigate pseudoternary LLE, which implies that the DESs remain unaltered in a single phase. DES2 was found to substantially increase the selectivity (S) values for the separation of benzene and n-hexane. The Othmer–Tobias and Bachman correlations were used to evaluate the reliability of the experimental tie-line data. The NRTL model shows a good agreement of the calculated and experimental values of n-hexane + benzene + DES1 with the root-mean-square deviation of 0.0076. The pseudoternary system of n-hexane + benzene + DES2 shows a good agreement with the UNIQUAC model with the RMSD of 0.0095. To verify the tie-line consistency, GM/RT was used to topologically analyze the Gibbs mixing energy.
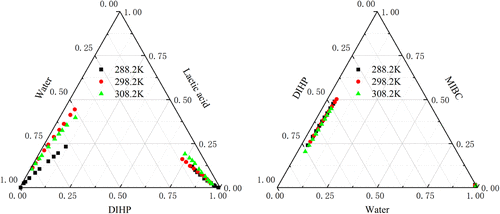
Analysis of Solvation Effects in Lactic Acid Ester-Containing Systems at 288.2–308.2 K: Liquid–Liquid Equilibrium Experiments, Thermodynamic and Quantum Chemical Studies, and Interaction Mechanism Analysis
Shaolong Dong - ,
Lu Chen - ,
Xuqiang Li - ,
Yujie Zhen - ,
Erkang Li - ,
Siyu Zheng - ,
Jingli Sun - ,
Houchun Yan - , and
Yingmin Yu *
In the traditional lactic acid ester synthesis process, lactic acid and 4-methyl-2-pentanol (MIBC) under certain temperature and acidic conditions undergo an esterification reaction to produce 2-hydroxypropanoic acid-1,3-dimethylbutyl ester (DIHP) and water, forming a complex system containing lactic acid, MIBC, DIHP, and water. To investigate the mutual solubility of the substances in the system, the ternary liquid–liquid equilibrium (LLE) data for DIHP + lactic acid + water and water + MIBC + DIHP were determined at 288.2–308.2 K and 101.3 kPa in this paper. The measured experimental data were correlated with nonrandom two-liquid (NRTL) and universal quasi-chemical (UNIQUAC) models to obtain the binary interaction parameters between substances. In addition, parameter rationality was evaluated by using the GUI-MATLAB tool, and the conclusions indicated that these parameters were reliable. This will provide critical and reliable fundamental data points for constructing a more complete thermodynamic model that predicts the phase behavior of the quaternary system. Finally, this paper reveals the interaction mechanism between the substances at the microscopic level by combining the experimental results with quantum chemical calculations, which, in turn, reflect the solvation effects between the substances.
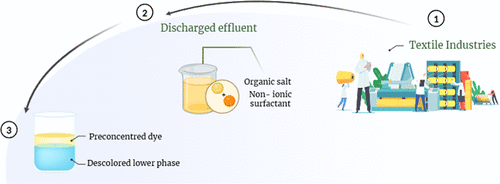
Phase Behavior, Dye Extraction, and Potential for Pretreating Textile Effluents in Aqueous Two-Phase Systems of Nonionic Surfactants (Tween 20 or Tween 80) and Organic Salts (Sodium Citrate or Sodium Tartrate)
Renata Aparecida Fideles - ,
Lohayne Ligya Barbosa Silva Nascimento - ,
Matheus Torres Duarte Figueiredo - ,
Amanda Luise Pereira Murta - ,
Melany Alejandra Ruiz Lopez - ,
Aparecida Barbosa Mageste - , and
Gabriel Max Dias Ferreira *
This publication is Open Access under the license indicated. Learn More
Aqueous two-phase systems (ATPSs) based on nonionic surfactant (Tween 20 or Tween 80) and organic salt (sodium citrate or sodium tartrate) are proposed as alternatives for dye extraction and pretreatment of textile effluents. The newly studied ATPSs exhibited an upper phase rich in surfactant and a lower phase rich in salt. An increase in temperature expanded the biphasic region, altered the phase compositions, and increased the tie-line length. Systems containing citrate exhibited larger biphasic regions due to its higher hydration capacity, while Tween 80 promoted greater phase separation than Tween 20, attributed to its higher hydrophobicity. Partitioning studies with rhodamine B and methyl orange showed preferential dye transference to the surfactant-rich phase, with extraction percentages above 98.7% and 99.2%, respectively. When applied to a textile effluent, the formation of ATPSs using the effluent as solvent led to an expanded immiscibility region in tartrate-based systems. Additionally, color removal efficiency remained above 91.52%, regardless of overall system composition or phase mass ratio, demonstrating the practical feasibility of using these ATPSs for the pretreatment of textile effluents. To the best of our knowledge, this is the first study to evaluate the effect of textile effluent composition on both the formation and dye extraction performance of Tween-based ATPSs.
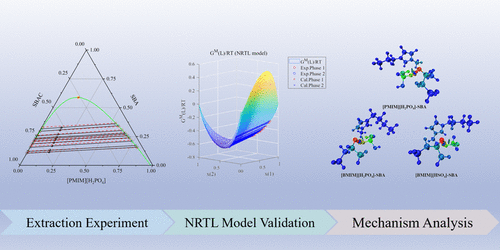
Experimental Investigation and Mechanism Analysis for the Separation of sec-Butanol and sec-Butyl Acetate System via Liquid–Liquid Extraction with Ionic Liquids
Jingya Hou - ,
Yutong Wang *- ,
Yu Sheng - ,
Yulin Li - ,
Sixuan Wang - ,
Hua Xin - ,
Qinqin Zhang - , and
Zhigang Zhang *
Efficient separation of sec-butanol (SBA) and sec-butyl acetate (SBAC) is important for the sustainability of methyl ethyl ketone (MEK) and its subsequent solvent and pharmaceutical chemical processes. However, SBA and SBAC form a binary azeotrope, which complicates the separation. In this work, three ionic liquids: 1-propyl-3-methylimidazolium dihydrogen phosphate ([PMIM][H2PO4]), 1-butyl-3-methylimidazolium dihydrogen phosphate ([BMIM][H2PO4]), and 1-butyl-3-methylimidazolium hydrosulfate ([BMIM][HSO4]) are screened and employed to separate SBAC and SBA azeotropic mixture using liquid–liquid extraction. Ternary liquid–liquid equilibrium (LLE) data at 303.15 K and atmospheric pressure are determined with distribution coefficients and selectivity evaluated, and data reliability is confirmed via Othmer-Tobias and Hand correlations. The experimental LLE data are correlated by the nonrandom two-liquids (NRTL) model, with the consistency of NRTL model parameters verified through the GMcal_TieLinesLL. The underlying separation mechanism is comprehensively elucidated through QC calculations, with ESP mapping, bond length and interaction energy, IGMH analysis, and QTAIM topological analysis, demonstrating that the interaction between ionic liquids (ILs) and SBA is stronger than the ILs and SBAC, among which hydrogen bond plays a dominant role. The feasibility of industrial-scale application is confirmed by a process flowsheet simulated and optimized with Aspen Plus.

Liquid-Liquid Equilibrium Measurement and Modeling of Methyl 2-Hydroxyisobutyrate, Water, and Different Extractants at 303.2 K
He Ma - ,
Houchun Yan *- ,
Menghao Du - ,
Yingmin Yu - , and
Qingsong Li *
To separate methyl 2-hydroxyisobutyrate from an aqueous solution, alkyl alcohols were selected as extractants. The liquid–liquid equilibrium data for the system {water + methyl 2-hydroxyisobutyrate + extractants (1-hexanol, 1-heptanol, 1-octanol, and 1-nonanol)} were determined at a temperature of 303.2 K. The partition coefficients and separation factors were calculated to assess the effectiveness of the extraction process. Additionally, the data were correlated using the NRTL and UNIQUAC models, with parameter regression conducted accordingly. The GMcal_TieLinesLL tool was utilized to evaluate the regression model parameters, and the results conformed to the Gibbs stability criterion. The present study further investigates the separation mechanism through analyses of σ-profile, deformation charge density, interaction energies, and Reduced Density Gradient (RDG) analysis. Ultimately, it was concluded that 1-nonanol exhibited the most effective extraction performance.

Recovery of Mannosylerythritol Lipids Using Alcohol/Salt Aqueous Two-Phase Extraction
Mellisa Z. Ncube - ,
George M. Teke - ,
Eugéne van Rensburg - , and
Robert W. M. Pott *
This publication is Open Access under the license indicated. Learn More
Mannosylerythritol lipids (MELs) are biosurfactants with applications in cosmetics, pharmaceuticals, and bioremediation. However, large-scale production using oil substrates complicates downstream recovery, resulting in low yields and high production costs. This study investigates aqueous two-phase extraction (ATPE) as an alternative recovery method. Alcohol/salt aqueous two-phase systems (ATPS) were evaluated using ethanol, 1-propanol, and 2-propanol in combination with various anions (phosphate, sulfate, citrate, and tartrate), while varying pH, temperature, and salt concentrations. The 1-propanol/sulfate system achieved the highest MEL recovery of 83.4% in the alcohol-rich top phase. The addition of 2% (w/w) NaCl improved recovery to 86.4%, while 8% (w/w) NaCl decreased MEL recovery. Since U. maydis coproduces cellobiose lipids (CBLs), we demonstrate that an ethanol/tartrate system could effectively separate MELs from CBLs. Oleic acid, a key contaminant, was found to preferentially partition into the top phase, indicating the necessity for further purification. To validate the system, MELs produced by Ustilago maydis were recovered using the 1-propanol/sulfate ATPS, yielding 87.28% recovery. This study is the first to demonstrate ATPE for MEL recovery and separation from coproducts such as CBLs, offering a promising and scalable alternative to conventional purification methods in industrial biosurfactant production.

Liquid–Liquid Equilibrium Investigations and Thermodynamic Modeling of Ternary Mixtures (Propylene Carbonate + Toluene or Phenol + n-Hexane or n-Octane) at 293.2 K
Sina Shekarsaraee *- ,
Zeynab Mohammadi - , and
Niyayesh Narimani Sabegh
The liquid–liquid equilibria (LLE) of four ternary systems (propylene carbonate (PC) + toluene or phenol + n-hexane or n-octane) were studied at 293.2 K and 101.3 kPa. The study aimed to assess the effectiveness of PC in extracting nonpolar and polar aromatic compounds from alkanes with molar weights similar to gasoline components. Tie-line data and mutual solubilities were determined using refractive index measurements, while cloud point titration was employed to define binary phase regions. Systems with toluene exhibited Type I LLE behavior, while those with phenol showed Type II behavior. Experimental tie-lines were correlated with NRTL and UNIQUAC models, yielding rmsd values below 1%, indicating excellent model accuracy. NRTL binary interaction parameters were validated via MATLAB GUI tools, and activity coefficients were calculated. Toluene exhibited negative deviations from Raoult’s law in both phases, whereas phenol showed negative deviations at low phenol concentrations, with positive deviations observed in the alkane phase. Distribution coefficients and selectivities confirmed the strong extraction ability of PC, particularly for phenol, which exhibited values significantly higher than those of toluene, highlighting the greater separation efficiency for polar aromatic compounds.
Solid-Solid Equilibria and Solid-Fluid Equilibria

Comprehensive Physicochemical Investigation of the Water-Soluble Adduct of C60 with l-Methionine (C60(C5H11NO2S)3): Important Data for Further Applications
Konstantin N. Semenov *- ,
Ali Mlhem - ,
Alexander V. Akentiev - ,
Dmitry A. Nerukh - ,
Natalia V. Petukhova - ,
Ilnaz T. Rakipov - ,
Kirill V. Timoshchuk - ,
Gleb O. Iurev - ,
Andrey V. Petrov - ,
Igor V. Murin - ,
Nikolay A. Charykov - ,
Olga S. Vezo - ,
Anastasia V. Penkova - ,
Dilafruz K. Kholmurodova - ,
Jasur A. Rizaev - ,
Aziz S. Kubaev - , and
Vladimir V. Sharoyko *
This work is devoted to the study of the physicochemical properties of the water-soluble adduct fullerene C60-l-methionine. The adduct was characterized using 13C solid-state NMR spectroscopy, IR spectroscopy, elemental analysis, UV/vis spectroscopy, and HPLC. The measured physicochemical properties included density, viscosity, refraction, electrical conductivity, speed of sound, surface properties of aqueous solutions, nanoparticle size distribution in water, the molecular dynamics simulation of the association of C60-Met molecules in water and isotonic saline (0.15 M NaCl solutions), the study of solubility in binary C60-Met–H2O and ternary C60-Met–NaCl–H2O systems, as well as their distribution in the n-octan-1-ol–water system.
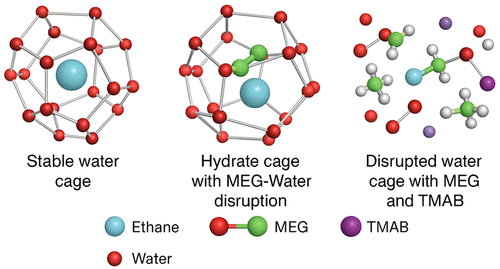
Ethane Hydrate Phase Equilibria in WATER, MEG, and TMAB Semiclathrate System
Dariush Mardomi - ,
Mehdi Ardjmand *- ,
Hassan Pahlavanzadeh *- , and
Ferial Nosratinia
The oil and gas industries can benefit from the potential of storing clathrate hydrate gas by managing it with precise knowledge of thermodynamic conditions or preventing its formation in gas transmission lines. In this study, despite the assurance of the thermodynamic inhibition property of MEG on the formation of ethane hydrate, the effect of TMAB on the stability or decomposition of ethane hydrate in the presence of water and MEG was investigated for the first time across six different experimental systems with combinations of 1, 2, 3, and 5 wt % of TMAB from the group of quaternary ammonium salts. The reactor pressure in each system ranged from 0.5 to 3.5 MPa. The minimum temperature of the experiments for maximum clathrate hydrate formation was determined to be 272 K. According to the obtained data, the shift of the equilibrium diagram of ethane hydrate from 1 to 5 wt % changes in favor of the inhibition of stability and inhibition performance. The maximum inhibition of TMAB on ethane hydrate was observed in the presence of MEG and water in combinations close to stoichiometric ratios.
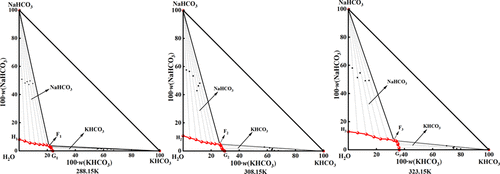
Measurements and Thermodynamic Model on the Solid–Liquid Phase Equilibria of the Ternary System KHCO3–NaHCO3–H2O at Multitemperature (288.15, 308.15, and 323.15 K)
Xiang Zhang - ,
Shi-Hua Sang *- ,
Chun-Tao Hu - , and
Ling-Xuan Wang
The thermodynamic solid–liquid phase equilibria of the ternary system KHCO3–NaHCO3–H2O at 288.15, 308.15, and 323.15 K were investigated using the isothermal dissolution equilibrium method. The phase diagrams of the ternary system KHCO3–NaHCO3–H2O are simple, containing one invariant point, two solubility curves, and two crystallization fields (corresponding to KHCO3 and NaHCO3), with no double salts or solid solutions formed. As the temperature increases, the crystallization fields of both KHCO3 and NaHCO3 expand, but the crystallization field of NaHCO3 always remains larger than that of KHCO3. This suggests that in this saturated ternary system, NaHCO3 is more prone to crystallize out. Based on the solid–liquid phase equilibrium experiments, the mixed salt Pitzer parameters (ΨK+, Na+, HCO3–) for the ternary system KHCO3–NaHCO3–H2O at 308.15 and 323.15 K were fitted and calculated. The Pitzer model was further employed to theoretically predict the solid–liquid phase equilibria of this ternary system at 288.15, 308.15, and 323.15 K. The modeling and experimental phase diagrams are in good agreement. This result validates the applicability and accuracy of the Pitzer model and the fitted mixed salt parameters for this ternary system.
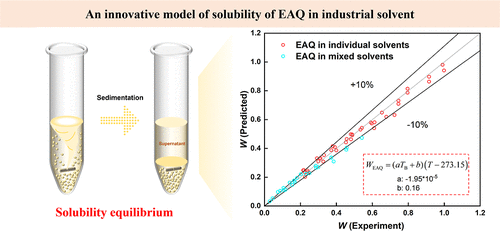
Modeling of the Solubility of 2-Ethyl or 2-tert-pentyl Anthraquinone in Organic Solvents
Junjie Wang - ,
Lin Sheng - ,
Yuyang Xing - ,
Jian Deng - , and
Guangsheng Luo *
Hydrogen peroxide production via the Reidl-Pfleiderer process critically depends on the solubility of alkylated anthraquinones in the working solvents. Existing solubility prediction models suffer from limitations such as complex parametrization and poor interpretability. To address this, this study develops a boiling point correlation model (BPCM) that predicts 2-ethylanthraquinone (EAQ) solubility using solvent boiling points (TB) and temperature (T) between 303.15 and 343.15 K. Experimental solubility data for EAQ and 2-tert-pentylanthraquinone (TPAQ, an isomer mixture of 2-tert-pentylanthraquinone and 2-s-pentylanthraquinone with a mass ratio of 3:1) in aromatic solvents (toluene, xylenes, and trimethylbenzene) and tris(2-ethylhexyl) phosphate (TOP) were measured systematically via dynamic equilibrium methods. The BPCM achieved average relative deviations (ARD) of <5% for pure solvents and mixing solvents, surpassing Wilson/NRTL models in simplicity. Furthermore, a universal correlation bridged TPAQ to EAQ solubility (ARD < 1%), enabling direct TPAQ estimation from EAQ data. This work reduces experimental workloads by >90% while maintaining industrial-grade accuracy for EAQ and TPAQ solubility in aromatic and phosphate-based systems.
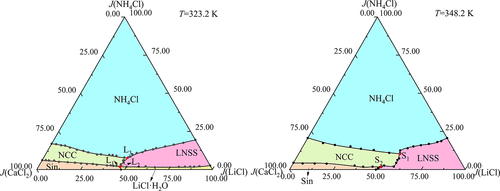
Solid–Liquid Phase Equilibria of Quaternary System Li+, NH4+, Ca2+//Cl––H2O at 323.2 and 348.2 K
Shuang Wang - ,
Jinniu Chen - ,
Jiubo Liu - ,
Jiantuan Jia - ,
Shuai Chen - ,
Changhao Wu - , and
Xudong Yu *
The solid–liquid phase equilibria of the quaternary system Li+, NH4+, Ca2+//Cl––H2O at 323.2 and 348.2 K was investigated using isothermal dissolution method, targeting deep brines rich in ammonium and calcium. Equilibrium liquid-phase composition and density were determined. The solid solution (NH4Cl)x(LiCl·H2O)1–x was confirmed by XRD and TG-DSC. The results show that the quaternary system Li+, NH4+, Ca2+//Cl––H2O at 323.2 and 348.2 K exhibits complex behavior involving the formation of solid solution (NH4Cl)x(LiCl·H2O)1–x and double salt (2NH4Cl·CaCl2·3H2O). The phase diagram of the system at 323.2 K consists of three invariant points, seven univariate curves, and five crystalline phase regions (NH4Cl, LiCl·H2O, 2NH4Cl·CaCl2·3H2O, (NH4Cl)x(LiCl·H2O)1–x, and CaCl2·2H2O). At 348.2 K, it has two invariant points, five univariant curves, and four crystalline regions (NH4Cl, 2NH4Cl·CaCl2·3H2O, (NH4Cl)x(LiCl·H2O)1–x, and CaCl2·2H2O). Comparative analysis of multitemperature phase diagrams of the quaternary system Li+, NH4+, Ca2+//Cl––H2O at 298.2, 323.2, and 348.2 K demonstrates that the crystalline phase region of LiCl·H2O decreases with the increase of temperature. Notably, at 348.2 K, lithium chloride exclusively exists within the (NH4Cl)x(LiCl·H2O)1–x. This thermal behavior suggests that cooling crystallization could be effectively employed for lithium extraction from such brines.
Mastheads
Issue Editorial Masthead

This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More