

About the Cover:
Reviews
Deep Eutectic Solvents as Sustainable Media in Maleic Anhydride Esterification: Potential and Application Analysis
Yaosen Wang - ,
Qiuyan Ding *- ,
Shun Liu - ,
Hong Li *- , and
Xin Gao *
Maleate esters, particularly dimethyl maleate (DMM), are pivotal platform chemicals and key intermediates in the synthesis of polymers, plasticizers, and fine chemicals. However, the conventional production via maleic anhydride (MAH) esterification is often constrained by equilibrium limitations and the use of harsh, nonrecyclable acid catalysts. In this context, deep eutectic solvents (DESs) have recently emerged as sustainable and versatile alternatives in esterification reaction, owing to tunable acidity, negligible volatility, and outstanding environmental compatibility. We systematically summarize the three synergistic roles of DESs: (i) as solvents, enhancing reactant miscibility; (ii) as catalysts, providing tunable acidity for efficient carbonyl activation; and (iii) as reactive extractants, separating byproduct water in situ to drive reactions. This intrinsic multifunctionality enables significant process intensification by coupling reaction and separation into a single unit. Based on the current research status of DMM production, the feasibility, advantages, and potential process strategies for applying DESs to DMM synthesis are thoroughly discussed. Finally, this review concludes by outlining current challenges and future research directions for MAH esterification, emphasizing the need for rational design of DESs. The findings collectively underscore the potential of DESs to revolutionize the sustainable production of maleate esters.
Research on Deep Eutectic Solvents for Fuel Desulfurization: Methods, Mechanisms, and Emerging Trends
Siqi Zhu - ,
Sitong Jin - ,
Congfei Yao *- ,
Jitong Xue - ,
Qiuyu Chen - , and
Zheng Liu
With increasing environmental concerns and stricter sulfur-emission regulations, efficient removal of sulfur-containing compounds from fuels has become a critical issue. Traditional hydrodesulfurization (HDS) faces challenges, including high energy consumption, limited efficiency for refractory sulfur compounds, and the need for severe operating conditions. Therefore, alternative desulfurization methods using novel solvents and green materials have gained interest. Among these, deep eutectic solvents (DESs) are particularly promising due to their tunable physicochemical properties, low toxicity, biodegradability, and excellent extraction capabilities. Unlike previous reviews that primarily emphasize experimental performance comparisons of DESs, this review provides a mechanism- and computation-driven perspective on DES-based fuel desulfurization, with particular emphasis on mechanistic elucidation through the integration of experimental investigations and multiscale computational approaches across different desulfurization technologies. It first introduces the definition, classification, and key physicochemical properties of DESs and their influence on desulfurization performance. Experimental and computational approaches were used to elucidate desulfurization mechanisms, including multiscale simulations and advanced characterization techniques. Finally, emerging research trends in DESs research, such as the design of composite systems, multifunctional development, and strategies for green and sustainable applications, are discussed.
Thermophysical and Thermochemical Properties
Refractive Index Measured at 589 nm from 283.15 to 358.15 K to Evaluate Temperature Invariance of Berthelot, Gladstone–Dale, Lorentz–Lorenz, and Eykman Equations
Adriana A. Rivolta - ,
Yanet Rodriguez Herrero *- ,
Natalia Montoya Sánchez - , and
Arno de Klerk
Molar refractivity is temperature independent, and its relationship to molar volume is described by a refractive index function. Four refractive index functions describing the relationships between molar refractivity, refractive index, and molar volume were evaluated: Berthelot, Gladstone–Dale, Lorentz–Lorenz, and Eykman. Experimental measurements of refractive index (n, 0.000001 readability) and density (ρ, 0.001 kg·m–3 readability) were obtained for pure compounds at five temperatures, from 283.15 to 358.15 K at atmospheric pressure, with nine replicates per temperature condition. In total, 72 compounds from the n-alkanes, 1-alkenes, 1-alkynes, cyclic compounds, alkyl aromatics, 1-alcohols, carboxylic acids, and those containing sulfur were included in the study. Using the experimental data as input, the variation in molar refractivity with temperature was calculated using each of the four refractive index functions. It was found that the Eykman refractive index function, (n2 −1)/(n + 0.4), resulted in the lowest temperature-dependent variation in molar refractivity. The rank order of performance of the refractive index functions was consistent for all groups of compounds, with Eykman being the best (0.03% relative variability in molar refractivity with temperature), followed by Gladstone–Dale, Lorentz–Lorenz, and Berthelot.
Thermodynamic Description of Aqueous Mixtures of New Antifreezes
Elena N. Tsurko *- ,
Tobias Lange - ,
Kerstin Betz - ,
Johannes Stauber - ,
Simon Schwarz - ,
Werner Kunz - , and
Rainer Müller

This publication is Open Access under the license indicated. Learn More
Combustion engines for motor vehicles will still play a substantial role for many years further. As a consequence of the power increase leading to high local surface temperatures of up to 260 °C in a modern engine, the requirements on coolants regarding stability and ability to maintain the dissipation of heat are also increased. In search for possible alternatives to currently applied freezing point depressants (coolants) and to create an overview about antifreezes in use, thermodynamic properties, namely, freezing points, densities, viscosities, specific isobaric heat capacity, boiling point curve data, and osmotic coefficient values were measured, generalized, and systemized with interpolated data from the literature for aqueous solutions of four molecule classes studied in wide temperature (from −45 to 95 °C) and composition ranges and are given as a database. The nonaqueous components are diols: 1,2-propanediol and 1,3-butanediol; glycols: ethylene glycol, diethylene glycol, triethylene glycol, and tetraethylene glycol; glycerols: glycerol and diglycerol; and carbonic acid salts: potassium acetate, sodium propionate, and potassium propionate. Conclusions have been drawn about the applicability of liquids with the best flow behavior and thermal properties as antifreezes, which fulfill the technical requirements but also those of sustainable chemistry including low toxicity, efficiency, and cheapness.
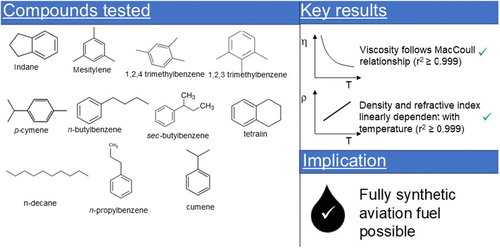
Viscosity and Density of C9–C10 Aromatics at 223.15–323.15 K Relevant to Aviation Turbine Fuel
Abhijith Vivek - ,
Stefanía Betancur Marquez - , and
Arno de Klerk *
There are likely to be differences in the aromatic composition of fully formulated aviation turbine fuels compared to petroleum-derived fuels. In this study, the viscosity and density of a range of C9–C10 aromatic compounds were measured from 223.15 to 323.15 at 10 K intervals. It was of interest to determine whether the MacCoull kinematic viscosity–temperature relationship in ASTM D341 was generally valid for all aromatic compounds tested. As an internal check, the refractive index at 589 nm was measured over the temperature range 283.15–323.15 at 5 K intervals and is also reported. All measurements were performed on air-saturated compounds of known purity at a near-atmospheric pressure. It was found that the measured data for all compounds was described by the MacCoull relationship with correlation coefficients r2 ≥ 0.996. Liquid density changed linearly with temperature, with r2 ≥ 0.999, even in data sets including measurements near the freezing point temperature. Refractive index changed linearly with temperature, r2 ≥ 0.999, and temperature invariant molar refractivity calculated using the Eykman relationship had a relative standard deviation of ≤0.04%.

Volumetric Properties of Amino Acids in Aqueous Protic Ionic Liquid Solutions: Probing Molecular Interactions
Jaspreet Kaur - and
Vickramjeet Singh *
Volumetric properties of amino acids (glycine and l-valine) in aqueous solutions of protic ionic liquids (PIL) (diethylammonium acetate [DEAAc] and ethanolammonium acetate [EAAc]) were investigated at different temperatures. The apparent molar volume (V2,ϕ), infinite dilution partial molar volume (V2°), infinite dilution partial molar volume of transfer (ΔtV2°), partial molar compression at infinite dilution (Ks,2°), and partial molar isentropic compression of transfer at infinite dilution (ΔtKs,2°) were determined from density/sound velocity data to investigate the nature of interactions between amino acid and cosolute (PIL) in water. Partial molar expansibilities (E°ϕ), Hepler’s constant ((∂2V2° /∂T2)P), and hydration number (NH) were also derived to assess the solvation behavior. Transfer volumes for both amino acids in [EAAc] solutions were positive, whereas positive and negative transfer volumes were observed for l-valine, and only negative transfer volumes were observed for glycine in the presence of [DEAAc]. Positive transfer volumes indicate the dominance of hydrophilic interactions among ions of PIL and charged functional groups (amino or carboxylic) of amino acids. Hydrophobic interactions contributed toward negative transfer volumes. The volumetric analysis offers insight into specific interactions between the ions of PILs and amino acids, helping to elucidate molecular forces that contribute to solute−solvent behavior and stabilization mechanisms in such systems.
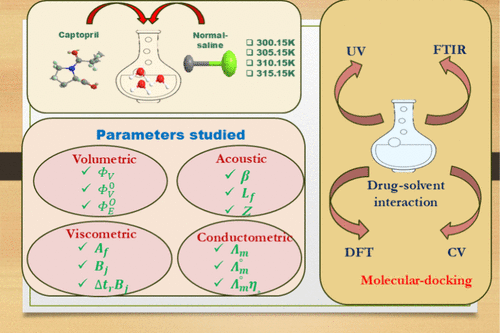
Physicochemical, Spectroscopic, and Computational Insights into the Interactional Behavior of the Antihypertensive Drug Captopril in Aqueous and Normal Saline Systems
Sunaina Sharma - ,
Parveen Kumar - ,
Palak Ahir - ,
Vishal Thakur - ,
Palak Verma - ,
Inesh Kumar - , and
Sunil Kumar *
Captopril (CAP) is an antihypertensive drug commonly prescribed for hypertension and heart failure. In current research, the molecular interactions of CAP were examined in water and normal saline at various concentrations (0.001–0.010) mol·kg–1 and temperatures (295.15–315.15) K. To address this objective, we measured the densities (ρ), ultrasonic velocities (c), specific conductance (κ), and viscosities (η) of CAP in water and normal saline solvent systems. The experimentally determined data were used to evaluate various volumetric, acoustic, conductometric, and viscometric parameters. The outcomes obtained from these parameters were estimated in terms of drug-solvent and drug–drug interactions. The findings of physicochemical studies reveal strong binding affinity and structural organization behavior of CAP in water/normal saline, and these findings were supported by UV–visible and FTIR studies. The DFT studies were also carried out to evaluate various quantum parameters and visualize FMOs and ESPS of CAP and NaCl in water. The ADMET profile of CAP and molecular docking with the targeted protein 2YDM were studied to evaluate their pharmacokinetic behavior and binding efficacy. This investigation can help the scientific community to optimize drug design and drug delivery, leading to improved biological efficacy in the pharmaceutical and healthcare sectors.

Thermophysical Properties and Quantum Chemical Calculations of 3-Aminopropyltris(trimethylsiloxy)silane, 3-[1,3,3,3-Tetramethyl-1-[(trimethylsilyl)oxy]-1-disiloxanyl]-1-propanamine, and 1,1,1,3,5,5,5-Heptamethyl-3-N-2-(aminoethyl)-3-aminopropyltrisiloxane
Yukai Wu - ,
Dan Liao - ,
Chenchen Li - ,
Dan Cao - ,
Hong Dong *- , and
Chuan Wu *
This study systematically investigated the synthesis, thermophysical properties, and electronic characteristics of three novel trimethylsiloxy (TMS)-functionalized amino silane coupling agents: 3-aminopropyltris(trimethylsiloxy)silane (AP-trisTMS), 3-[1,3,3,3-tetramethyl-1-[(trimethylsilyl)oxy]-1-disiloxanyl]-1-propanamine (AP-diTMS), and 1,1,1,3,5,5,5-heptamethyl-3-N-2-(aminoethyl)-3-aminopropyltrisiloxane (AEAP-diTMS). By precise design of the molecules through partial replacement of alkoxy groups with TMS, the hydrogen bonding ability, hydrophobicity, and thermal stability of these compounds were adjusted. Based on experimental results such as density, viscosity, and vapor pressure, combined with density functional theory calculations of electronic properties (the analysis of electrostatic potential energy of molecular surfaces, frontier molecular orbitals, simplified density gradient functional analysis, and localized orbital locator scatter plots), the molecular characteristics are comprehensively discussed. This research not only deepens our understanding of these compounds but also highlights their potential as high-performance materials.
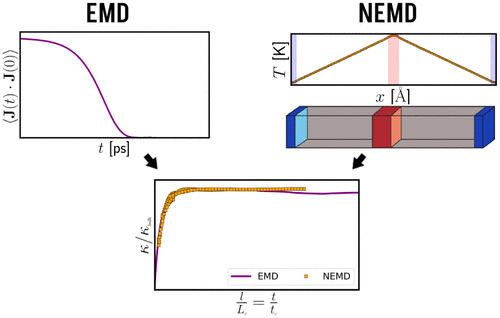
Consistency of Equilibrium and Nonequilibrium Molecular Dynamics to Assess Thermal Conductivity
Nikolas Ferreira de Souza - ,
Luís Fernando Mercier Franco *- , and
Benoit Coasne *
Accurate evaluation of thermal conductivity using molecular simulation can be challenging depending on the system’s dynamical behavior. Two computational strategies are commonly employed in this field: equilibrium molecular dynamics (EMD) and nonequilibrium molecular dynamics (NEMD). The EMD approach, which relies on the Green–Kubo formalism, requires extensive sampling of the heat flux time autocorrelation function. The NEMD approach, which relies on the direct implementation of Fourier’s law, requires obeying the linear regime and checking for any finite size effects. Here, using a united atom model for methane and a flexible zeolite framework, we discuss the fundamental aspects of such methods by analyzing the convergence in time and/or in size. We show that both approaches can be rationalized by invoking characteristic time and length scales, tc and Lc. In an equivalent manner as the convergence in EMD approach is achieved when the simulation time t ≫ tc, the convergence in NEMD methods requires system sizes L ≫ Lc (in the latter case, smaller simulation boxes lead to finite size effects due to phonon scattering at the temperature gradient boundaries). Notably, EMD and NEMD data collapse on the same curve when rescaled through these parameters.

Volumetric Properties of Aromatic Nitrogen Compounds and Their Binary Mixtures at Various Temperatures
Deepthi Jayachandran Sreekala - ,
Anantharaj Ramalingam *- ,
Pachimatla Rajesh - ,
Gayathri Mahavishnu - , and
Balachandar Vijayakumar
In this study, the densities of pyrrole (1) + pyridine (2), pyridine (1) + quinoline (2), pyrrole (1) + quinoline (2), indoline (1) + quinoline (2), indoline (1) + pyridine (2), and indoline (1) + pyrrole (2) were measured over the entire composition range at temperatures T = 298.15–343.15 K and pressure p = 101.305 kPa. The data were used to calculate excess molar volume (VmE), partial molar volume (V̅i), excess partial molar volume (V¯iE), apparent molar volume (Vφ,i), and the isobaric thermal expansion (αp). Excess molar volumes (VmE) were fitted to a Redlich–Kister equation to evaluate the deviation from ideal mixing behavior. It was noted that indoline (1) + quinoline (2) has a negative (VmE) deviation from ideality. Whereas the pyridine (1) + quinoline (2), pyrrole (1) + quinoline (2), indoline (1) + pyridine (2), and indoline (1) + pyrrole (2) mixtures showed positive excess molar volume deviations from ideality due to dissimilar structures. The FT-IR spectra of all the studied binary mixtures showed the presence of N–H bonding within the molecules. Finally, the sigma profile and the sigma potential of individual compounds were generated and analyzed.
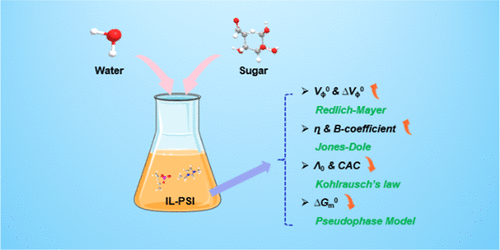
Thermodynamic Properties and Synergistic Interactions in the IL-PSI–Sugar–Water System: Hydrogen-Bond Network Enhancement and Microstructural Strengthening
Shasha Gong - ,
Yingqiu Wu - ,
Jingjing Sun - ,
Junfeng Wang *- , and
Yi Nie
This work examines the thermophysical properties, (density, volumetric properties, electrical conductivity, viscosity) of the 1-ethyl-3-methylimidazolium diethyl phosphate (IL-PSI)–sugar–water system at 298.15 K. Experimental density (ρ) and viscosity (η) data were analyzed using the Redlich–Mayer and Jones–Dole equations to obtain the apparent molar volume (VΦ) and the viscosity B-coefficient, respectively. Kohlrausch’s law was used to determine the limiting molar conductivity (Λ0), and the Walden product (Λ0η0) was subsequently calculated. Critical aggregation concentrations were identified through conductometric measurements, and the related aggregation thermodynamics were quantified via a pseudophase model. Through this thermodynamic analysis, solute–solute and solute–solvent interactions were probed to shed light on the microscopic organization of these systems.

Experimental Thermophysical Properties and Temperature Dependence of Four Non-Ionic Deep Eutectic Solvents
Jingwen Wang - ,
Fanjing Wei - ,
Qingqing Zhang *- ,
Shaofu Li - ,
Lijie Guan - ,
Hao Qin *- , and
Zhiwen Qi
Deep eutectic solvents (DESs) have emerged as promising alternatives to conventional solvents due to their tunable properties and potential environmental benefits. While most studies focus on ionic DESs, nonionic DESs remain less explored. In this work, four nonionic deep eutectic solvents (DESs) are prepared using diethylene glycol dimethyl ether or isoquinoline as hydrogen-bond acceptors (HBA) and cyclohexanecarboxylic acid, nonanoic acid, or 1-naphthylamine as hydrogen-bond donors (HBD) at specific molar ratios. Their fundamental thermophysical properties, including density, viscosity, surface tension, electrical conductivity, and melting point, are systematically measured over the temperature range of 303.15–343.15 K. The temperature dependence of all measured properties is quantitatively analyzed using appropriate empirical or Arrhenius-type correlations. The results provide insights into the influence of molecular composition on nonionic DES behavior, expanding the experimental database and highlighting their potential as low-viscosity, tunable, and versatile solvents for chemical engineering applications.
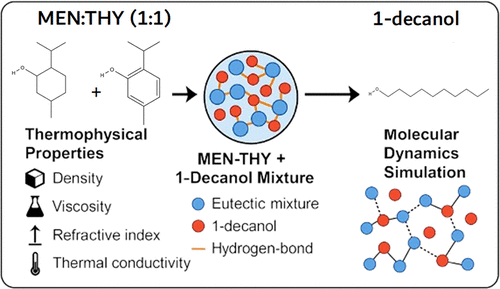
Liquid Structuring and Dynamics in Hydrophobic Deep Eutectic Mixtures of MEN/THY and 1-Decanol
Rafael Alcalde - ,
Cristina Benito - ,
Mert Atilhan - ,
Jose L. Trenzado *- , and
Santiago Aparicio *
This work considers the liquid structuring and dynamic behavior of the hydrophobic deep eutectic solvent composed of menthol and thymol (MEN/THY, 1:1 molar ratio) when mixed with 1-decanol over the entire composition range. The system, although ternary in nature, is treated as a binary mixture between the preformed eutectic solvent and 1-decanol in order to isolate the mixing process and elucidate the underlying molecular interactions governing the transition from DES-rich to alcohol-rich domains. A comprehensive experimental study was conducted including measurements of density, viscosity, refractive index, thermal conductivity, and pH, complemented by Raman spectroscopy to probe microscopic organization and intermolecular forces. Thermophysical data were analyzed through polynomial and excess property correlations to assess the evolution of volume, cohesive energy, and free volume upon mixing. Molecular dynamics simulations were further employed to rationalize the macroscopic observations, providing atomistic insights into hydrogen-bond topology, dispersion interactions, and dynamic rearrangements within the liquid. The combined experimental–computational framework establishes a coherent molecular interpretation of hydrophobic DES–alcohol mixtures, advancing the understanding of how associative and dispersive forces determine liquid structuring, dynamics, and potential solvent design strategies.
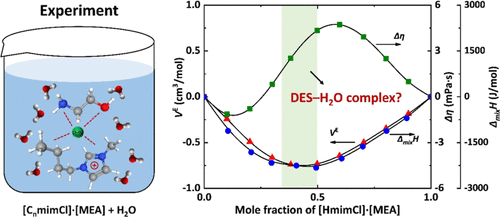
Thermodynamic Study of Amine-Based Deep Eutectic Solvents with H2O
Zhida Zuo - ,
Yusi Shen - ,
Linghong Lu - ,
Yudan Zhu - ,
Xiaohua Lu *- , and
Xiaoyan Ji *
This publication is Open Access under the license indicated. Learn More
The densities and viscosities of ([CnmimCl][MEA] + H2O) systems (n = 2, 4, 6) were measured over 288.15–323.15 K, and the enthalpies of mixing were determined at 298.15 and 308.15 K. Density data showed nonmonotonic composition dependence with extrema near xDES ≈ 0.4–0.5, and negative excess molar volumes indicated enhanced molecular packing. Viscosity increased sharply at low DES content and more gradually toward pure DESs, exhibiting S-shape deviation profiles that reflect structural rearrangements in both DES-rich and H2O-rich regions. Negative enthalpies of mixing confirmed exothermic mixing due to strong DES–H2O interactions, and the data were well correlated by the NRTL model. The coincidence of extrema in density and enthalpy of mixing, together with viscosity transitions, suggests the formation of complexes among the DES constituents and H2O, probably leading to compact ternary DES-like microstructures. Further comparative analysis revealed that H2O addition reorganizes the hydrogen-bond network, forming extensive MEA–H2O associations. These findings offer molecular-level insights for the rational design of DES-based solvents for CO2 capture and related separation processes.
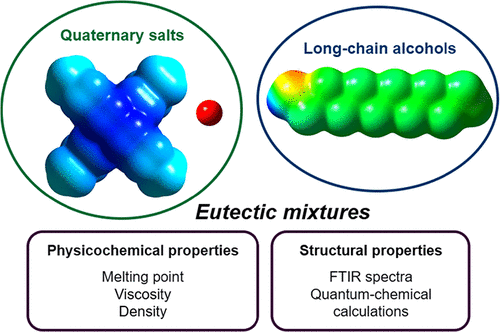
Experimental and Computational Studies of Hydrophobic Eutectic Mixtures Based on Tetrabutyl Ammonium and Phosphonium Salts and Alcohols
Dmitriy M. Makarov *- ,
Yuliya A. Fadeeva - ,
Michael A. Krestyaninov - , and
Arkadiy M. Kolker
Hydrophobic eutectic mixtures of tetrabutylammonium bromide (TBABr) or tetrabutylphosphonium bromide (TBPBr) with 1-decanol or 1-dodecanol were investigated using a combination of thermal, rheological, spectroscopic, and quantum-chemical methods. Phase diagrams demonstrate that TBPBr-based mixtures exhibit pronounced negative deviation from ideal behavior and form eutectic minima at a 1:1 composition, while TBABr-based systems behave nearly ideally. Viscosity and density were measured over the temperature range of 293.15 to 343.15 K. FTIR spectroscopy indicates that the addition of small amounts of salt perturbs the hydrogen-bond network of the alcohols, while higher salt fractions partially restore it. Quantum-chemical calculations and QTAIM analysis show that TBPBr forms stronger O–H···Br hydrogen bonds and more compact ion-pair structures than TBABr, consistent with the enhanced intermolecular association and the higher viscosity observed experimentally. The physicochemical properties of these hydrophobic eutectic mixtures arise from the cumulative effect of multiple weak interactions, primarily hydrogen bonding and dispersion forces, rather than a single dominant interaction. These results advance the molecular-level understanding required to design hydrophobic eutectic solvents with tailored properties.
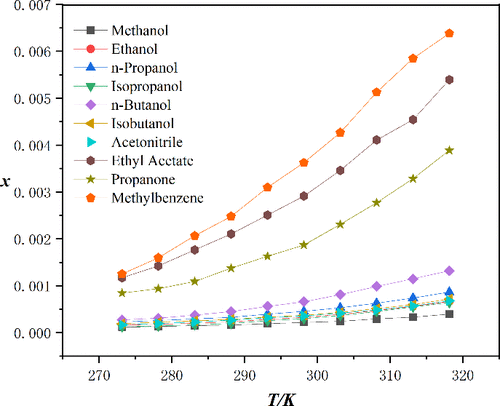
Solubility Determination and Thermodynamic Analysis of Clioquinol in Ten Pure Solvents and Three Binary Solvents at 273.15 to 318.15 K
Jun Xiong - ,
Yuyang Zhang - ,
Yiyang Yu - ,
Wenge Yang *- , and
Yonghong Hu *
The solubility of clioquinol in ten different pure solvents─specifically methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, acetonitrile, ethyl acetate, toluene, and acetone─as well as in three binary mixtures containing methanol combined with acetone, ethyl acetate, or toluene was determined using the static equilibrium method combined with HPLC. The temperature range under ambient pressure was from 273.15 to 318.15 K. Data revealed that clioquinol demonstrated maximum solubility in toluene and minimum in methanol as the temperature rose. In binary solvent systems, clioquinol solubility increased with the molar ratio of the more effective solvent. The solubility figures obtained from pure solvents were correlated using the Modified Apelblat model, the λh model, and the CNIBS/R-K model, while the Jouyban–Acree model and the SUN model were applied for the binary solvent mixtures. Thermodynamic model accuracy was verified through relative average deviation (RAD) and root-mean-square deviation (RMSD) calculations of the fitting outcomes. Finally, the KAT-LSER model was utilized to examine how solvent characteristics impact pure solvent solubility.
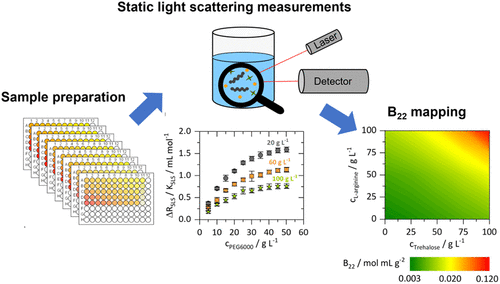
Enhanced Insights into Macromolecular Interactions: High-Throughput B22 Mapping Using Static Light Scattering
Marcel Pillath - and
Christoph Brandenbusch *
Static light scattering (SLS) is a widely used technique for characterizing size, molecular weight, and interactions of macromolecules such as polymers and proteins. In pharmaceutical formulation design, protein–protein interactions (PPI) are commonly assessed by the second osmotic virial coefficient (B22) derived from SLS data. While conventional methods like the composition-gradient multi-angle light scattering (CG-MALS) yield highly accurate B22-values, they are limited by lengthy processing times and substantial material requirements. Within this work we developed a novel high-throughput method for rapid generation of experimental SLS data using automated liquid handling and plate-based SLS measurements. Validation of this method was performed through direct comparison with SLS data and B22-values obtained via CG-MALS. To demonstrate its applicability, B22 maps of PEG6000 and dextran in the presence of trehalose and l-arginine in phosphate buffer were generated. These maps reliably captured the influence of varying excipient concentrations on macromolecular interactions, except in cases with minimal B22 changes, as observed with dextran and l-arginine. However, for applications requiring extensive SLS data but do not require the highest level of sensitivity, the high-throughput method is especially well-suited. A potential future application could be its extension to biological systems, thereby supporting pharmaceutical formulation development.
Solubility Determination of 2-Phenyl-4H-benzo[h]chromen-4-one in Mixed Solvents and Its Correlation with Thermodynamic Models
Yuhao Cai - ,
Xianlang Chen - ,
Tongyang Song - ,
Liang Cai - , and
Rongrong Li *
The solubility of 2-Phenyl-4H-benzo[h]chromen-4-one was measured in mixed solvents of ethyl acetate with n-propanol, n-butanol, or isobutanol using the isothermal saturation method at atmospheric pressure (101.3 kPa), across a temperature range of 278.15 to 318.15 K. Experimental data were well correlated by both the modified Apelblat model and the λh equation, with all correlation coefficients (R2) exceeding 0.99. According to the van’t Hoff equation, the apparent thermodynamic parameters (ΔsolH°, ΔsolS°, ΔsolG°) of the dissolution process were determined. Results demonstrate that the solubility of 2-Phenyl-4H-benzo[h]chromen-4-one increases significantly with temperature in all solvent systems studied. Furthermore, the positive values of ΔsolH°, ΔsolS°, and ΔsolG° indicate an endothermic, entropy-driven, and nonspontaneous dissolution process under the experimental conditions. These comprehensive findings provide essential guidance for optimizing the crystallization of 2-Phenyl-4H-benzo[h]chromen-4-one, facilitating the rational selection of solvent composition and operating temperature. This work also establishes a thermodynamic foundation for designing efficient antisolvent crystallization strategies.
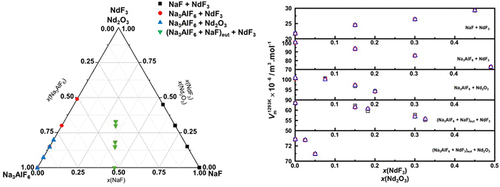
NdF3 and Nd2O3 Effect on the Physicochemical and Volume Properties of Cryolite-Based Melts and the NaF + NdF3 Systems
Jarmila Mlynáriková *- and
František Šimko
This study investigates the physicochemical and volume properties of neodymium-containing cryolite-based melts and the NaF + NdF3 system. Densities were measured using the Archimedean method, showing a linear decrease with increasing temperature and an increase with the NdF3 or Nd2O3 content. Calculated molar and partial molar volumes indicate local expansion upon addition of neodymium compounds. However, the partial molar volumes of NdF3 at infinite dilution are lower than those of Nd2O3, due to stronger Nd3+–F– interactions and more compact local structures in fluoride melts. In cryolite-based melts, the [AlF6]3– network enhances packing, resulting in higher partial molar volumes for Nd2O3. Direct calculations are in good agreement with predictive procedures (1A and 1B), validating the data set. These results provide a detailed thermophysical description of Nd-containing melts, offering a valuable basis for modeling and optimizing industrial processes for neodymium separation and refining.
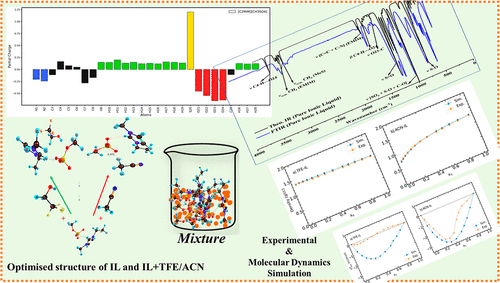
Thermophysical Characteristics, Molecular Interactions, and Molecular Modeling of 1-Ethyl-3-methylimidazolium Methylsulfate ([C2MIM][CH3SO4]) with Organic Solvents in Binary Mixtures
Malik Raihan Ahmad - ,
Riyazuddeen *- ,
Amey S. Thorat - ,
Ashutosh Kumar Verma - ,
Jindal K. Shah *- , and
Mohammad Jane Alam
The thermophysical properties, density (ρ), speed of sound (u), and dynamic viscosity (η) of pure 1-ethyl-3-methylimidazolium methylsulfate [C2MIM][CH3SO4], 2,2,2-trifluoroethanol (TFE), and acetonitrile (ACN), as well as their binary mixtures ([C2MIM][CH3SO4] + TFE/ACN), have been measured at 1 atm pressure over a temperature range of (298.15–318.15) K and across the entire mole fraction composition. Excess molar volumes (VE), deviations in isentropic compressibilities (Δκs), and viscosity deviations (Δη) have been derived from the experimental data and correlated with the Redlich–Kister equation. The Prigogine–Flory–Patterson theory has been applied to VE values and showed good agreement with the experimental findings. The FT-IR study is used to probe inter- and intramolecular interactions in mixtures and to investigate their nonideal behavior. Molecular dynamics simulations have been performed to calculate the properties, density, self-diffusion coefficient, and radial distribution function, g(r), at 308.15 K across the entire mole fractions. Furthermore, density functional theory (DFT/B3LYP-D3) calculations have been carried out with dispersion correction to examine the interactions in pure ionic liquid and IL–solvent mixtures. The reactivity and interactions are analyzed with the help of HOMO–LUMO gap, interaction energy, noncovalent interaction (NCI) plot, radial distribution gradient (RDG), simulated IR, and calculated reactivity descriptors.

Physicochemical and Electrochemical Properties of ZnCl2/Propan-1,3-diol Deep Eutectic Solvent for Zinc-Ion Battery Electrolytes
Jing Li - ,
Yifan Huang - ,
Yaping Sun - ,
Cheng Lian - ,
Haiping Su *- ,
Jingkun Li *- , and
Honglai Liu
As an environmentally benign solvent system, deep eutectic solvents (DESs) are of great significance in the field of sustainable development. This work pioneers a novel application of ZnCl2/propan-1,3-diol (1,3-PDO) DES as an environmentally friendly, low-cost, and safe electrolyte for zinc-ion batteries. The viscosity (η), conductivity (κ), and surface tension (γ) of DESs, and their correlations with ZnCl2 concentration, temperature, and hydrogen bonding strength are investigated. Furthermore, DES with a proper ZnCl2:1,3-PDO ratio exhibits a broadened operating temperature range and enhanced cycle life, owing to its well-balanced η, κ, and γ, as well as excellent thermal and chemical stability. The Zn||Zn symmetric cell with the ZnCl2/1,3-PDO (1:10) electrolyte achieves a cycle life of over 5000 h, demonstrating outstanding practical potential. This study provides important theoretical and data support for the application of ZnCl2/1,3-PDO DES as an electrolyte in zinc-ion batteries.
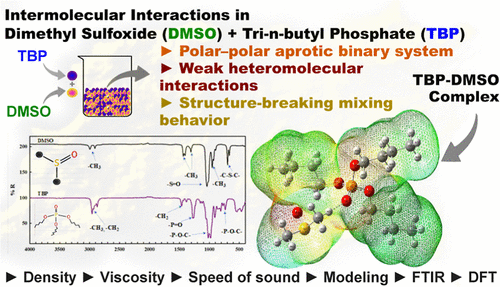
Intermolecular Interactions and Thermophysical Modeling of Dimethyl Sulfoxide + Tri-n-butyl Phosphate Mixtures
M. Nur Hossain *- ,
M. Mehedi Hasan Rocky *- ,
M. Masum Billah - ,
Ariel Hernández - ,
Irin Hossain - ,
Hiroshi Hasegawa - , and
Shamim Akhtar *
Density, viscosity, and speed of sound were measured for dimethyl sulfoxide (DMSO) + tri-n-butyl phosphate (TBP) mixtures at atmospheric pressure (0.10 MPa) over the full composition range and temperatures from 298.15 to 323.15 K. The mixtures exhibit pronounced structure-breaking behavior, with excess molar volumes reaching positive maxima of 1.05–1.07 cm3·mol–1 near a TBP mole fraction of ≈0.45. Viscosity deviations show positive peaks between 0.9 and 1.7 mPa·s, while isentropic compressibility deviations remain positive across all temperatures. Negative speed of sound deviations, reaching −300 m·s–1, further indicate weakened intermolecular networks compared to the pure components. FTIR spectra reveal composition-dependent shifts of 5–12 cm–1 in the S═O and P═O stretching bands, consistent with weak hydrogen bonding and dipole–dipole interactions. Density functional theory (DFT) calculations support formation of a stabilized TBP–DMSO adduct, showing short intermolecular contacts, charge redistribution, and reduced HOMO–LUMO gaps, consistent with enhanced polarity upon mixing. Experimental data were correlated using the Jouyban–Acree model, while the PC-SAFT equation of state reproduced liquid densities with an overall average relative deviation (ARD) of 0.08%. In addition, new interaction parameters for the (CH2)3PO–DMSO pair were computed and applied in group-contribution-based predictive models (UNIFAC-VISCO and UNIFAC-THERMO), yielding viscosity prediction ARDs of 2.7% and 2.4%, respectively.
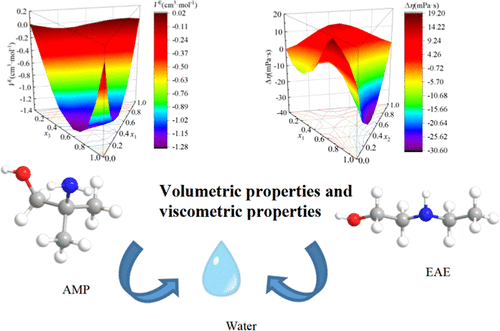
Densities and Viscosities of 2-Amino-2-methyl-1-propanol, 2-(Ethylamino)ethanol, and Water Solutions
Zhen Chang - ,
Yaxuan Zhen - ,
Chunying Zhu *- ,
Taotao Fu - , and
Youguang Ma
The densities and dynamic viscosities of pure components, binary solutions and ternary solutions of 2-amino-2-methyl-1-propanol (AMP), 2-ethylaminoethanol (EAE) and water were systematically measured at (293.15–323.15) K. Based on experimental findings, calculations were made for the excess molar volume, thermal expansion coefficient, dynamic viscosity deviation, and solution viscous activation energy to assess intermolecular interactions. The negative excess molar volumes of binary and ternary aqueous solutions indicate significant hydrogen bonding interaction and a filling effect within the solutions. The negative excess molar volumes of binary and ternary aqueous solutions indicate significant hydrogen bonding interactions and a filling effect within the solutions. For the AMP–EAE system, the hydrogen bonding interaction and filling effect are weakened due to the similar volume and steric hindrance effect.
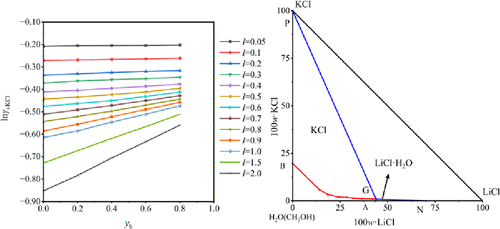
Activity Coefficients and Phase Equilibria in the LiCl–KCl–CH3OH–H2O Mixed System at 298.2 K
Qing-Shuang Wang - ,
Shi-Hua Sang *- ,
Yun-Yun Gao - ,
Kuang-Yi Zhu - ,
Zhen-Hua Feng - , and
Yang Tang
The Qaidam Basin of China abounds in salt lake resources containing lithium, potassium, and boron. Lithium precipitation mother liquor is generated during lithium carbonate production. To investigate solution thermodynamic properties of lithium salts and support its utilization, the thermodynamic activity coefficients and phase equilibria of the LiCl–KCl–CH3OH–H2O mixed solvent system at 298.2 K are studied. The mean activity coefficients of KCl in the KCl−CH3OH−H2O and the LiCl−KCl−CH3OH−H2O mixed solvent system are measured using cell potentials and the Nernst equation. Via multiple linear regression and nonlinear programming fitting, the Pitzer single-salt parameters of LiCl (β(0), β(1), and CΦ) and ion interaction parameters (θK,Li and ψK,Li,Cl) at 298.2 K are obtained, and then mean activity coefficients of LiCl, osmotic coefficients, water activities, and excess Gibbs free energies via the Pitzer model are calculated accordingly. Moreover, the mixed solvent system’s phase equilibria via isothermal dissolution equilibrium are also determined. Solubility data modeling with these parameters agrees well with experimental results, validating the model’s efficacy in predicting lithium–potassium salt phase behavior in methanol-containing mixed solvents.

Experimental and Computational Assessment of Propyl Propionate with 2-Alkanols (C3–C7): Density, Viscosity, and DFT Analysis
Mohammad Almasi - ,
Adel Noubigh - ,
Nasim Rahmani-Ivriq - , and
Razieh Sadat Neyband *
Densities and viscosities were measured for five binary mixtures of propyl propionate (PP) with 2-alkanols ranging from 2-propanol to 2-heptanol over the full composition range at temperatures from 293.15 to 323.15 K. All systems exhibited positive excess molar volumes, with the extent of expansion increasing with both temperature and the length of the alkanol alkyl chain. This behavior is consistent with structure-breaking effects resulting from disruption of self-association in the pure alkanols. Viscosity deviations were negative throughout and increased in magnitude with alkanol chain length, indicating that the mixtures exhibit weaker intermolecular interactions relative to those present in the corresponding pure components. A detailed understanding of hydrogen bonding in complex liquid mixtures is crucial for optimizing process behavior and enhancing predictive thermodynamic models. Experimental measurements elucidate the macroscopic thermodynamic consequences of these interactions, while complementary density functional theory (DFT) calculations provide molecular-level insight into the hydrogen-bonding networks present in propyl propionate +2-alkanol systems. To further refine this perspective, computational studies employing the M05–2X functional with the 6–311++G** basis set were conducted, enabling a more comprehensive characterization of the intermolecular hydrogen-bonding patterns within these mixtures.
Vapor-Liquid Equilibria and Supercritical Fluid Equilibria
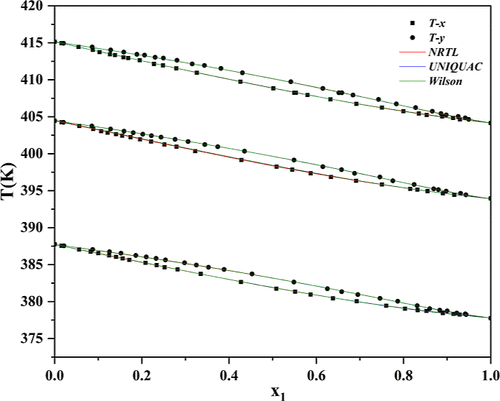
Isobaric Vapor–Liquid Equilibrium of Binary Mixtures of Ethylene Glycol Diacetate and 1,2-Butanediol Diacetate at 5.00, 10.00, and 15.00 kPa
Yulong Li - ,
Fengzhi Li - ,
Yang Xiao - ,
Wenyu Ma - ,
Yan Wang *- , and
Wangfeng Cai *
The isobaric vapor–liquid equilibrium (VLE) data for the binary system of ethylene glycol diacetate (EGDA) and 1,2-butanediol diacetate (1,2-BDDA) were measured at 5.00, 10.00, and 15.00 kPa using a modified Rose–Williams equilibrium still. The thermodynamic consistency of the experimental data was confirmed by Fredenslund and Van Ness tests. The measured data were correlated with the nonrandom two-liquid (NRTL), universal quasi-chemical, and Wilson activity-coefficient models, and the corresponding binary interaction parameters were obtained through regression. The accuracy of each model was evaluated using the root-mean-square deviations (RMSDs) of the vapor-phase mole fraction (y1) and equilibrium temperature (T). All three models satisfactorily reproduced the experimental results, with the NRTL model showing the best agreement. The reliable VLE data and correlations reported in this work provide essential thermodynamic information for the modeling and design of separation processes involving EGDA and 1,2-BDDA.
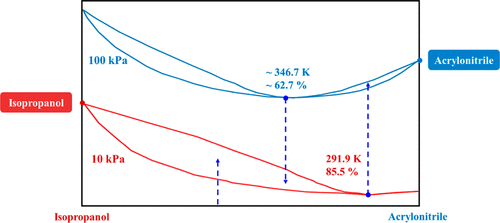
Vapor Liquid Equilibrium for the Binary Systems Involving Acrylonitrile and Isopropanol at 10.0, 50.0, 100.0 kPa
Yan-Yang Wu *- ,
Xin-Wei Pei - ,
Xue Li - ,
Cheng-Hao Xing - ,
Bin Wu - ,
Kui Chen - , and
Li-Jun Ji
Isobaric vapor liquid equilibrium (VLE) data for the acrylonitrile-isopropyl alcohol binary system were measured at 10.0, 50.0, and 100.0 kPa. It showed that the system forms a minimum-boiling azeotrope at each of the three studied pressures with different compositions and boiling temperatures. The experimental data were correlated with the NRTL, Wilson, and UNIQUAC activity coefficient models, and their binary interaction parameters were obtained correspondingly. Thermodynamic consistency was verified using both the Fredenslund method and Redlich–Kister area test. According to the maximum absolute deviation, root-mean-square deviation, and average absolute deviation, all three models provided satisfactory correlations with the experimental data. In view of the differences in the azeotropic composition and VLE behavior of acrylonitrile-isopropyl alcohol at 10.0 and 100.0 kPa, pressure-swing distillation could be used to separate the components. This work provided a crucial thermodynamic basis for the design and optimization of distillation processes for separating the acrylonitrile and isopropanol mixtures.

Vapor–Liquid Equilibrium Measurement of Limonene + Citronellal, Limonene + β-Citronellol, and Citronellal + β-Citronellol Binary Systems in Kaffir Lime Essential Oil at Vacuum Pressures
Putri Amalia Devianda - ,
Anisa Fatma Aulia - , and
K Kuswandi *
The isobaric vapor–liquid equilibrium data for limonene + citronellal, limonene + β-citronellol, and citronellal + β-citronellol binary systems at vacuum pressures of 20 and 40 kPa were obtained experimentally by using a simple static ebulliometer with a modified Glass Othmer-Still type. The apparatus was previously validated by using pure solutions at various pressures. Liquid and vapor phase samples were analyzed using gas chromatography to obtain the composition. The experimental results were tested for thermodynamic consistency using the L.W. Wisniak and Fredenslund method. The data were correlated using Wilson, NRTL, and UNIQUAC models to obtain the binary interaction parameters. The minimum average absolute deviations (AADs) T for the limonene + citronellal binary system at 20 kPa are 0.1 K, 0.1 K, and 0.1 K, and those at 40 kPa are 0.2 K, 0.5 K, and 0.2 K. However, the AADs T for the limonene + β-citronellol binary system at 20 kPa are 0.3 K, 0.3 K, and 0.3 K, and those at 40 kPa are 0.4 K, 0.5 K, and 0.4 K. In addition, the AADs T for the citronellal + β-citronellol binary system at 20 kPa are 0.1 K, 0.1 K, and 0.1 K, and those at 40 kPa are 0.3 K, 0.3 K, and 0.3 K.
Liquid-Liquid Equilibria and Vapor-Liquid-Liquid Equilibria
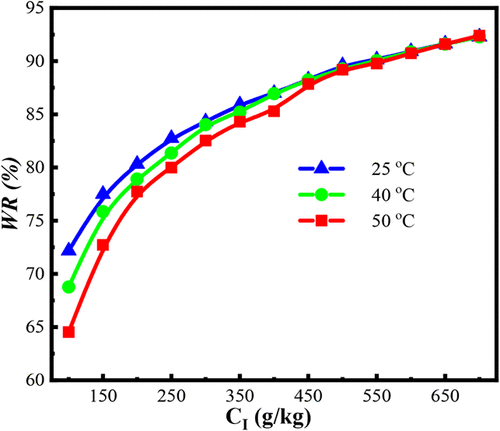
Dewatering of Butanol Biofuel by a Highly Soluble Carbohydrate
Yuanxin Zhao *
Biobutanol is a superior biofuel alternative, but its recovery from acetone-butanol-ethanol (ABE) fermentation is inefficient due to azeotrope formation and high energy costs. This study investigates fructose-induced sugaring-out for phase separation and breaking the azeotrope, optimizing ABE recovery by analyzing the effects of sugar concentration and temperature. The results demonstrate efficient phase separation, with butanol recovery reaching 93.1%, acetone recovery at 66.0%, ethanol recovery at 59.1%, and water removal at 92.3%. Increasing fructose concentration enhances water exclusion and phase separation, while temperature variations play a secondary role compared to fructose concentration, only affecting separation efficiency at lower sugar concentrations. The findings highlight the dominant role of fructose-induced sugaring-out in phase formation and its potential for optimizing biofuel recovery. This method offers a highly efficient and tunable strategy for solvent recovery and water removal in biorefinery applications.

Liquid–Liquid Equilibrium Determination and Correlation for Ethylbenzene and n-Butanol with Deep Eutectic Solvents
Weizhuo Li - ,
Jiaqi Song - ,
Longwei Cao - ,
Jun Gao - ,
Dongmei Xu *- ,
Lianzheng Zhang - , and
Yinglong Wang
Separation of the azeotropic mixture of ethylbenzene (EB) and n-butanol (n-BuOH) is of critical importance, since EB can be catalytically converted from renewable n-BuOH, yet it remains challenging by conventional distillation. Three deep eutectic solvents (DESs) were formulated wherein 1,4-butanediol served as the hydrogen bond donor, paired with choline chloride (ChCl), l-lactic acid, and dl-menthol acting as hydrogen bond acceptors (HBAs). The liquid–liquid equilibrium (LLE) data were acquired for the pseudoternary mixtures (EB + n-BuOH + DESs) at 298.15 K and 101.3 kPa. The extraction performance was quantified based on the distribution coefficient and selectivity. The results showed that DES (ChCl: 1,4-butanediol, 1:2, molar ratio) indicated its strong potential as an efficient and eco-friendly extractant. The NRTL thermodynamic model was employed for the purpose of correlating the collected LLE data, resulting in lower values of root-mean-square deviation (RMSD), confirming its reliability for modeling the pseudoternary mixtures.
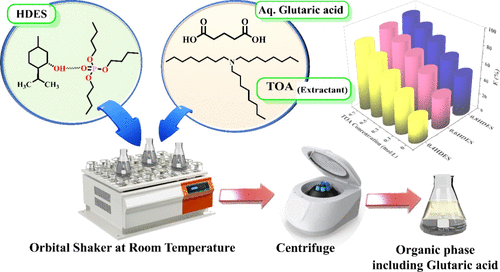
Reactive Extraction of Glutaric Acid from Aqueous Solutions Using Tri-n-octylamine in Deep Eutectic Solvents
Ashwini S. Thakre - ,
Kamleshwar L. Patle - ,
Diwakar Z. Shende - , and
Kailas L. Wasewar *
Glutaric acid, a value-added dicarboxylic acid with wide applications in food, polymers, pharmaceuticals, and speciality chemicals, is typically produced via fermentation and requires efficient downstream recovery for process viability. In this study, the recovery of glutaric acid via reactive extraction with deep eutectic solvents (DESs) was investigated to intensify the process sustainably. DES was synthesized by combining dl-menthol as the hydrogen bond donor (HBD) and tributyl phosphate (TBP) as the hydrogen bond acceptor (HBA), with tri-n-octylamine (TOA) as the extractant. DES was characterized using FTIR and NMR to investigate structural stability. The effects of operational parameters, including the initial acid concentration, amine concentration, solvent composition, and temperature, were systematically evaluated at 298 K and at atmospheric pressure. Key separation parameters, such as distribution coefficients and loading factors, confirmed the effectiveness of the process. This work establishes a scalable,enviornment-friendly approach fro glutaric acid recovery.The highest reactive extraction efficiency (92.85%) and distribution coefficient (12.99) were observed at the maximum TOA concentration (0.77 mol/L) and at 0.8XT DES (XT represents the mole fraction of TBP in the TBP-TOA DES).
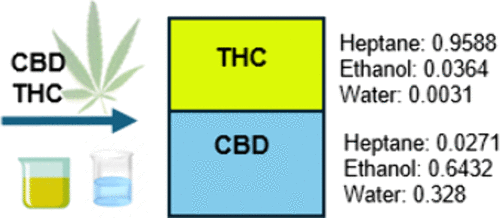
Liquid–Liquid Equilibrium of the System: Water + Ethanol + Heptane + Δ9-Tetrahydrocannabinol and Cannabidiol
Ada Carolina Gallo-Molina *- ,
Víctor Alfonso Roa-Gómez - ,
César Augusto Sánchez-Correa - , and
Iván Darío Gil-Chaves
In the development of novel medical cannabis products, several technologies have been proposed, including Centrifugal Partition Chromatography. This technique enables the separation of compounds through multiple liquid–liquid extraction processes that occur inside the equipment. The biphasic solvent system used for the isolation of Δ9-tetrahydrocannabinol (Δ9-THC) from cannabidiol (CBD) enriched matrices consists of n-heptane, ethanol, and water. In this study, experimental data on the liquid–liquid equilibrium (LLE) of a system comprising water, ethanol, n-heptane, Δ9-THC, and CBD at 282.15, 285.15, and 288.15 K at 75.11 kPa are presented. To fit the LLE data, the nonrandom-two-liquid model was applied using an in-house MATLAB application. Binary interaction parameters for the multicomponent system were calculated and are also reported in this study. This research contributes to the generation of new knowledge by (i) providing binary interaction parameters between cannabinoids and the selected solvents and (ii) offering a detailed analysis of liquid–liquid equilibria in cannabinoid separation processes, with valuable implications for future applications in the field. The parameter estimations were shown to be thermodynamically consistent through both graphical and numerical analysis.
Liquid–Liquid Equilibrium Data for 2-Nonanone or 3-Octanone with Acetic Acid and Water at 293.2–333.2 K and 0.1 MPa
Nivaar Brijmohan *- and
Kuveneshan Moodley
It is important to study the separation of acetic acid from water via solvent extraction, as acetic acid is used in the production of vinegar, the synthesis of numerous acetic acid derivatives, and specific applications in the pharmaceutical industry. Identifying better solvents is an area of ongoing research as solvent choice affects process economics and environmental impact as well as health and safety. In this work, liquid–liquid equilibrium measurements were performed for the ternary systems water + acetic acid + 2-nonanone and water + acetic acid + 3-octanone at 293.2, 313.2, and 333.2 K and 0.1 MPa. The purpose of this study was to determine the selectivity and capacity of 2-nonanone and 3-octanone in order to ascertain their effectiveness as potential extraction solvents for the water/acetic acid mixture. A double-walled glass cell was used to perform the experimental measurements, with the equilibrium compositions determined by using gas chromatography. The results indicate that 2-nonanone and 3-octanone offer comparative extraction performance with solvents, such as toluene and ethyl acetate. The data were successfully modeled using the NRTL and UNIQUAC activity coefficient models.

Predicting the Solubilization of Hydrocarbons in Aqueous Surfactant Solutions
Milan Völkel - and
Gabriele Sadowski *
This publication is Open Access under the license indicated. Learn More
Surfactants are widely employed across numerous applications due to their amphiphilic nature, which drives aggregate formation and enables the solubilization of hydrophobic compounds in aqueous solutions once the critical micelle concentration (CMC) is exceeded. In this work, we present a holistic modeling approach that considers both aggregation equilibria as well as phase equilibria in ternary systems of water, hydrophobic solutes (“oil”), and surfactants. For that purpose, PC-SAFT is incorporated into a novel mixed-aggregate formation model. Micellar solubilization effects are described by accounting for the formation of mixed oil-surfactant aggregates. Consequently, our model is based on a continuous distribution along all possible compositions and sizes of the mixed aggregates while calculating their equilibrium geometries. Our approach, for the first time, enables quasi-quantitative predictions of liquid–liquid equilibria in water–oil–surfactant systems. It accurately captures the sharp increase in oil solubility at surfactant concentrations just above the CMC and captures the influence of surfactant polarity on the maximum solubilization capacity for various hydrophobic solutes in very good agreement with experiments. Our methodology is validated for polyethoxylated alkyl ethers (CiEj) as surfactants and for alkanes and alkenes as example oil components, with the predicted binodal curves closely matching experimental LLE data.
Liquid–Liquid Equilibria of Toluene–Alkanol Azeotrope Mixtures Using a Deep Eutectic Solvent: Measurement and Thermodynamic Modeling
Mohammad Alizadeh Sarami - ,
Ali Ghanadzadeh Gilani *- ,
Neda Gilani - , and
Behnaz Mohammadi Khanghah
Ternary liquid–liquid equilibrium (LLE) data were experimentally determined and systematically compared for the systems containing binary azeotrope mixtures {toluene + alkanols (from C1 to C5)} and a deep eutectic solvent (DES) at 298.2 K and 101 kPa. The prepared DES was a binary mixture of choline chloride and ethylene glycol (DES; 1:4 mol ratio). The DES was stable during the LLE experiments and was used to remove a series of short chain n-alcohols (from C1 to C5) from an aromatic hydrocarbon (toluene) through LLE experiments. The equilibrium data of the studied systems and extraction capacity of the DES for each system was compared and interpreted. The local composition model of the NRTL was correlated with the measured equilibrium data, and the model parameters were regressed. The reliability of the obtained model parameters was confirmed by topological analysis associated with the Gibbs tangent law and stability test. Evaluation of the extraction capacity of the studied systems was carried out through the solute distribution coefficient and selectivity data. The prepared DES lead to different selectivity values for the studied systems. The solute distribution coefficient values were found to be different for the ternary systems containing methanol, ethanol, 1-propanol, 1-butanol, and 1-pentanol.
Green Auxiliary Buffering Agent Effect on the Separation of Acetic Acid from Aqueous Solution: Phase Equilibrium Measurements and Modeling Study
Saidah Altway *- ,
Nadza Basyuni Ramli - ,
Melvin Iga Maulidia - ,
Eva Oktavia Ningrum - , and
Danawati Hari Prajitno
Wastes produced by the biological oxidation of solid waste materials frequently contain diluted acetic acid solutions. Therefore, separating acetic acid from a mixture of several aqueous components remains difficult. In this study, the liquid–liquid extraction method was used to conduct an experimental separation of acetic acid from its aqueous solution using 1-heptanol as a solvent with the addition of biological buffer (HEPES/MOPS/EPPS) as a green auxiliary agent at 303.15 K and atmospheric pressure (0.1 MPa). The biological buffer was added in place of salt, which can boost the distribution coefficient and separation factor. The performance of acetic acid extraction using 1-heptanol as a solvent can be determined based on the distribution coefficient and separation factor. The buffering out strength sequence was MOPS > HEPES > EPPS, with separation factor and distribution coefficient values up to 507 and 14.1, respectively. The experimental results on liquid–liquid equilibrium for the 1-heptanol + acetic acid + water + biological buffer systems were found to be consistent using the Hand equation. Furthermore, the experimental data for liquid–liquid equilibrium can be well correlated by the NRTL and UNIQUAC models, with the RMSD values for both models in all systems of less than 1%.
Solid-Solid Equilibria and Solid-Fluid Equilibria

Solubility of Organic Photoelectric Material Intermediate 9,9-Dimethylfluorene in 15 Single Solvents: Solvent Effect Analysis, Molecular Simulation, and Model Correlation
Jintong Zhang - ,
Xingyu Liu - ,
Weikun Tang - ,
Yan Chen - ,
Pingping Jiang - ,
Bowen Pi - ,
Wenbo Zhang - , and
Peng Wang *
9,9-Dimethylfluorene is a core intermediate underpinning the development of modern organic electroluminescent (OLED) display technology. Its unique molecular structure and physicochemical properties make it a key basic raw material for the synthesis of OLED materials. These materials are widely applied in mainstream display devices such as smartphones and televisions. Therefore, the dissolution behavior of 9,9-dimethylfluorene in individual solvents holds certain research value. The solubility of 9,9-dimethylfluorene was measured using a static gravimetric method. The measurements were conducted in 15 individual solvents, which encompass ethanol, n-butanol, methanol, iso-propanol, iso-butanol, iso-pentanol, acetone, n-pentanol, acetonitrile, 2-butanone, n-propanol, methyl acetate, sec-butanol, propyl acetate, and ethyl acetate. The solubility increased with temperature in all 15 solvents. At 298.15 K, it was the lowest in methanol (0.009091 mol/mol) and the highest in 2-butanone (0.2575 mol/mol). A comprehensive analysis of cohesive energy density, hydrogen bonding, polarity, and Hansen solubility parameters was conducted. Dispersion forces were revealed to be the key factors governing the dissolution behavior of the substance. Additionally, among the four model fittings, the Apelblat model exhibited the highest degree of fitting. Molecular simulations were employed to systematically elucidate the internal interactions within 9,9-dimethylfluorene. The simulations included the analysis of molecular electrostatic potential (MEP) surfaces and the calculation of interaction energies.
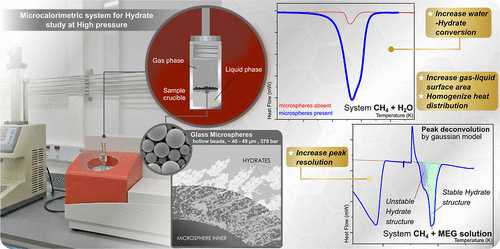
Determination of Hydrate Thermodynamic Equilibrium Data Using μDSC with Resolution Enhancement Employing Glass Microspheres
Isabelle Rodrigues de Oliveira - ,
Rafael Cavalcante dos Santos - ,
Adriana Teixeira - ,
Amaro Gomes Barreto Jr.- ,
Frederico Wanderley Tavares - , and
Ingrid Azevedo de Oliveira *
This publication is Open Access under the license indicated. Learn More
Accurate determination of hydrate equilibrium properties is crucial for developing offshore oil and gas strategies and enabling technological applications in gas storage and transportation. This study presents a multicycle calorimetric method using glass microspheres to enhance resolution for determining hydrate equilibrium properties in CH4 + H2O systems, both uninhibited and inhibited with 10% or 30% MEG. In MEG-inhibited systems, merged peak clusters required Gaussian deconvolution to extract properties. Glass microspheres enhance calorimetric determination by improving signal resolution via increased surface area and induced nucleation, reducing experimental time. For noninhibited systems, Tonset is 294.3 K (300 bar) and 286.0 K (100 bar) with a 0.02% deviation. For inhibited systems, Tpeak is 285.0 K (100 bar, 10% MEG) and 276.4 K (100 bar, 30% MEG), and a deviation of up to 0.1%. In strongly MEG-inhibited systems, microspheres enable effective separation of overlapping dissociation peaks, making Gaussian deconvolution essential for accurate identification of equilibrium states. While the methodology also shows potential for measuring dissociation enthalpies in uninhibited systems (64 ± 2 kJ·mol–1), values diverged from references in MEG-inhibited systems (30 ± 2 kJ·mol–1, 10% MEG and 10.3 ± 0.6 kJ·mol–1, 30% MEG), indicating the inhibitor's high concentration influences the dissociation process itself.
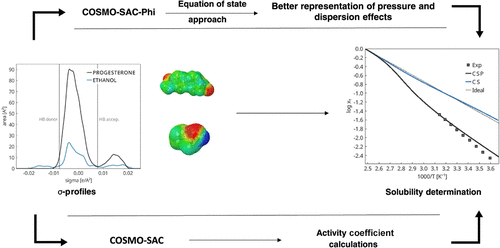
Prediction of Progesterone Solubility by Modern Quasi-Chemical and Classical Group Contribution Models
Enzo B. Paiva - ,
Edgar T. de Souza Jr.- ,
Paula B. Staudt - , and
Rafael de P. Soares *
This publication is Open Access under the license indicated. Learn More
Progesterone is a hydrophobic steroid hormone that plays a crucial role in human health. Its limited aqueous solubility presents a significant challenge for both pharmaceutical applications and academic research, showing the importance of accurately predicting its solubility behavior in different solvents. This study assesses the performance of the modern quasi-chemical equation of state known as COSMO-SAC-Phi (CSP), in comparison with its underlying COSMO-SAC (CS) activity coefficient model when modeling solid–liquid equilibrium of progesterone in 14 different solvents. Pure compound parameters employed in CSP calculations were obtained from the vapor pressure and liquid volume data of each pure compound. No binary parameters were adjusted. The results were compared with experimental solid–liquid equilibrium data collected from the literature. As a reference, the classical UNIFAC (Do) group contribution method was also used. The CSP model generally provided more accurate predictions of phase equilibrium, captured solubility trends among similar solvents, and reproduced the correct deviations from ideality for most systems, whereas the CS model was often less accurate in these aspects. An intermediate performance was observed for UNIFAC (Do). The mean absolute deviation in log10 units from experimental solubility data highlights the advantage of the equation-of-state approach, yielding an average value of 0.26 for CSP compared to 0.60 for the underlying activity model and 0.37 for UNIFAC (Do). Nevertheless, CS should be sufficiently accurate for the preliminary screening of new solvent alternatives.
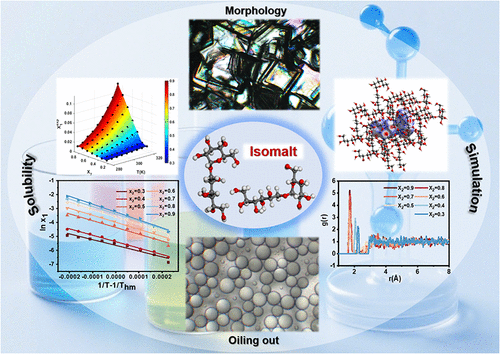
Liquid–Liquid Phase Separation and Solubility of Isomalt in Five Binary Solvents: Hansen Solubility Parameters, Molecular Dynamics Simulations and Thermodynamic Properties at 283.15–323.15 K
Yingchen Wang *- ,
Mingting Yuan - ,
Yimin Jia - ,
Mei Ma - , and
Qiushuo Yu *
The mole fraction solubility of Isomalt in five binary solvent systems (water + methanol, ethanol, 1-propanol, isopropanol, tert-butanol) was determined via the gravimetric method at atmospheric pressure and 283.15–323.15 K. Solubility increased with rising temperature and water molar fraction, following the order: water + methanol > water + ethanol > water +1-propanol > water + isopropanol > water + tert-butanol, with solubility curve intersections indicating varying temperature sensitivities. Liquid–liquid phase separation occurred in water +1-propanol/isopropanol/tert-butanol at high water mole fractions. Four thermodynamic models (modified Apelblat, van’t Hoff, λh, Jouyban–Acree van’t Hoff) yielded excellent fits (ARD < 10%, RMSD < 0.1%), with the modified Apelblat model being best correlation. Hansen solubility parameter analysis showed solubility was governed by hydrogen-bonding parameters (Δδh) and total solubility parameter (Δδ̅). Hirshfeld surface analysis (HS) and radial distribution function (RDF) simulations confirmed hydrogen bonding as the dominant solute–solvent/solvent–solvent interaction. Thermodynamic calculations based on the van’t Hoff equation confirmed that the dissolution process is endothermic and entropy-driven. These findings provide comprehensive solubility data and mechanistic insights, supporting future research and applications of isomalt in alcoholic solutions.
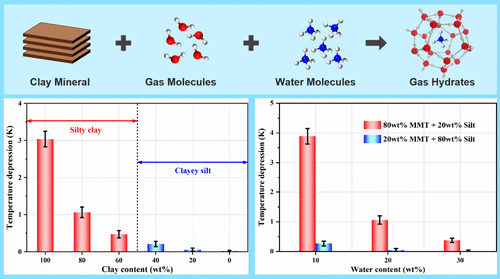
Phase Equilibrium Condition of Methane Hydrates in Clayey-Silty Sediments: Effects of Clay and Water Content
Dajiang Zhu - ,
Kui Zhang *- ,
Xiaodong Yang - ,
Yafeng Lu - ,
Lin Zhang - ,
Jiejing Nie - ,
Naiyan Zhang - , and
Bin Yang
Over 90% of natural gas hydrates (NGHs) on Earth occur in fine-grained clayey-silty sediments, which also serve as ideal sites for hydrate-based CO2 geological sequestration. However, hydrate phase equilibria in such sediments remain poorly understood, hindering advancements of NGH exploitation and carbon sequestration technologies. Here, we conducted a series of experiments to investigate the phase behavior of methane hydrates in representative clayey-silty sediments (montmorillonite and silt) with varying clay and water contents and measured the hydrate dissociation conditions via a stepwise heating method. The results demonstrate that the hydrate dissociation temperature depression increases exponentially with rising clay content and decreasing water content. This leads to a more pronounced dissociation temperature shift in silty clays (clay content >50 wt %) than in clayey silts (clay content <50 wt %). Specifically, at a water content of 20 wt %, the hydrate dissociation temperature depression in silty clays (80 wt % montmorillonite and 20 wt % silt) is as high as 1.5 K on average relative to bulk hydrates, whereas that in clayey silts (20 wt % montmorillonite and 80 wt % silt) remains below 0.3 K. Furthermore, compared to clayey silts, the hydrate dissociation temperature depression in silty clays exhibits a stronger dependence on water content. These findings highlight the pivotal role of clay and water content in regulating hydrate stability within geological systems.
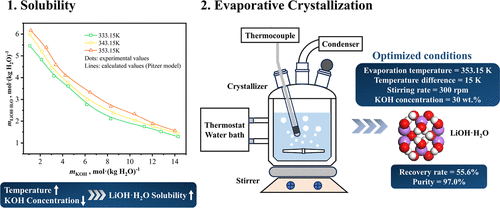
Solubility of LiOH·H2O in KOH Solutions from 333.15 to 353.15 K: Pitzer Modeling and Optimized Evaporative Crystallization for Lithium Recovery
Yu-Fei Zhao - ,
Zhuo-Fan Chen - ,
You-Fa Jiang - ,
Ying Yang *- , and
Cheng-Lin Liu *
Lithium hydroxide monohydrate (LiOH·H2O) is a critical alkaline lithium salt for battery applications. The solubility behavior of LiOH·H2O in KOH solutions and its evaporative crystallization process were systematically investigated in this study. Solubility was measured from 333.15 to 353.15 K, and a Pitzer model was developed for this system, demonstrating an excellent correlation with the experimental data. In the LiOH–KOH–H2O ternary system, LiOH·H2O solubility increased with temperature but decreased with KOH concentration at fixed temperatures. During evaporative crystallization experiments, the effects of mixing intensity of the solution, heat-transfer temperature difference, evaporation temperature, and mother-liquor KOH concentration on both the purity of LiOH·H2O and lithium recovery rate were systematically examined. Optimized conditions (evaporation temperature: 343.15 K, heat-transfer temperature difference: 15.0 K, stirring rate: 300 rpm, and KOH concentration in mother liquor: 30 wt %) yielded a lithium recovery rate of 55.6% and product purity of 98.3%. This study provides thermodynamic insights and process guidelines for the efficient separation and purification of lithium hydroxide from alkaline solutions.
Experimental Investigation and PC-SAFT Modeling of Etodolac Solubility in Deep Eutectic Solvents and Aqueous Solutions
Bin Liu - ,
Yewei Ding - ,
Haomin Wu - ,
Xiaojie Hu - ,
Zheng Zhang - , and
Yuanhui Ji *
The measurement and modeling of drug solubility are crucial for guiding solvent selection in the pharmaceutical formulation development process. Here, the solubility of etodolac was determined in six deep eutectic solvents (DES1–DES6) based on choline chloride as the hydrogen bond acceptor combined with different hydrogen bond donors (polyols and carboxylic acids) and their corresponding hydrogen bond donor (HBD1–HBD6) aqueous solutions, under conditions of 293.15–333.15 K and 101.3 kPa. All HBD and DES systems exhibited a significant solubilizing effect. Among them, the propionic acid system showed the most pronounced enhancement in the solubility of etodolac. Powder X-ray diffraction (PXRD) results confirmed that the crystal form of etodolac remained stable throughout the experiments, ensuring the reliability of the solubility data. The solubility data were further modeled using the Perturbed-Chain Statistical Associating Fluid Theory (PC-SAFT) equation of state, combined with solid–liquid equilibrium theory. The results showed good agreement between the calculated and experimental values, with most ARD values below 10%, and the minimum value reaching only 0.564%, indicating that the PC-SAFT model reliably predicts the solubility behavior of etodolac in various solvent systems and serves as a powerful theoretical tool for drug research and development.
Phase Equilibria in o-Xylene–Di-(2-ethylhexyl)phosphoric Acid–Lanthanum (Praseodymium, Terbium, Lutetium) Di-(2-ethylhexyl)phosphate Systems at 298.15 K
Mariia D. Kaplina - ,
Anatoly S. Arkhipin - ,
Aleksandr V. Nesterov - ,
Andrei A. Eliseev - ,
Svetlana V. Kurdakova *- , and
Irina A. Uspenskaya
This study investigates the phase equilibria in the systems containing o-xylene, di-(2-ethylhexyl)phosphoric acid (D2EHPA), and lanthanum, praseodymium, terbium, and lutetium di-(2-ethylhexyl)phosphates at 298.15 K. The research provides insight into the solubility of these rare-earth element complexes in organic solvents. The study outlines an approximate homogeneity boundary for the solutions of lanthanum, praseodymium, terbium, and lutetium di-(2-ethylhexyl)phosphates in D2EHPA and the o-xylene–D2EHPA mixture at 298.15 K. The study demonstrates a decreasing stability region for the solution of o-xylene–D2EHPA–rare earth elements (La, Pr, Tb, Lu) di-(2-ethylhexyl)phosphates systems from La to Lu. The reported density of “liquid” praseodymium di-(2-ethylhexyl)phosphate (PrA3) at 298.15 K, which is not available in the literature, is optimized based on experimental data, which are obtained using the Redlich–Kister polynomial equation. The interaction parameters of o-xylene–D2EHPA, along with the optimized density value of PrA3, enable a satisfactory description of the densities in o-xylene–D2EHPA–PrA3 solutions. These results have relevance for various industrial applications, particularly in the field of rare earth element extraction and separation processes.

Solid–Liquid Phase Equilibrium in the Quaternary System H3BO3 + CsCl + RbCl + H2O at 298.15 K and Identification of a Solid Solution Phase 2H3BO3·3(Cs,Rb)Cl
Ziyu Zhuang *- ,
Manman Zhang - ,
Dandan Gao - ,
Wolfgang Voigt - ,
Dongdong Li - , and
Haitao Feng *
Previously, the formation of a stable adduct 2H3BO3·3CsCl (BC) was observed in CsCl(aq) but not in RbCl(aq). The phase equilibria of the H3BO3 + RbCl + CsCl + H2O system were studied to assess the potential for selective separation of Cs from Rb. Solubility isotherms were determined experimentally. Appropriate liquid composition ranges were preselected using modeling with ISLEC’s equilibrium software. Schreinemakers’ wet solid method, XRD, Raman and IR spectra, Roozeboom’s plots, and SEM-EDS were applied to identify the four phase regions H3BO3, 2H3BO3·3(Cs,Rb)Cl (Bss), (Rb,Cs)Cl, and (Cs,Rb)Cl. Notably, in the presence of RbCl, the BC phase incorporates Rb+, forming a substitutional solid solution, Bss, which inhibits selective separation of CsCl. Roozeboom’s plot analysis revealed that RbCl exhibits higher solubility in the Bss phase compared to the (Cs,Rb)Cl solid solution. The separation factor βCs/Rb between the solid Bss phase and the corresponding solution was 2.0, significantly lower than that of (Cs,Rb)Cl (βCs/Rb = 9.5) in the ternary system without H3BO3. This indicates a stronger coordination between CsCl and H3BO3 relative to that of RbCl in the ternary system without H3BO3. The above understanding provides new strategies designing novel B–OH compounds to enhance Cs/Rb separation efficiency in aqueous systems.
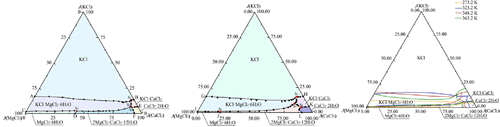
Solid–Liquid Equilibria (SLE) of Aqueous Quaternary System K+, Mg2+, Ca2+// Cl–-H2O at 323.2 and 348.2 K
Xudong Yu *- ,
Tong Pan *- ,
Jiantuan Jia - ,
Jiubo Liu - ,
Jinniu Chen - ,
Qi Li - , and
Zongde Ma
Compared with salt lake brine, deep brine features high temperature and high calcium content, which alters potassium’s phase equilibria behavior. Accordingly, the phase equilibria of the quaternary system K+, Mg2+, Ca2+//Cl–-H2O at 323.2 and 348.2 K were investigated using the isothermal dissolution method. The phase diagrams at both temperatures consist of four quaternary invariant points, nine univariate curves, and six crystallization regions. Three double salts chlorocalcite (KCl·CaCl2), carnallite (KCl·MgCl2·6H2O), and tachyhydrite (2MgCl2·CaCl2·12H2O) were identified. In this system, potassium crystallizes as KCl, KCl·MgCl2·6H2O, and KCl·CaCl2, with KCl having the largest crystallization region, followed by KCl·MgCl2·6H2O and then KCl·CaCl2. A multitemperature comparison (298.2 to 348.2 K) of K+, Mg2+, Ca2+//Cl–-H2O reveals that at 298.2 K, potassium crystallizes only as KCl and KCl·MgCl2·6H2O, and the crystallization phase region of KCl has the largest region, which is favorable for the separation and extraction of potassium. When at 323.2 and 348.2 K, in addition to KCl and KCl·MgCl2·6H2O, the double-salt KCl·CaCl2 also forms, and the crystallization region of KCl·CaCl2 increases with increasing temperature, which may affect the stability of potassium extraction raw materials such as KCl and KCl·MgCl2·6H2O.
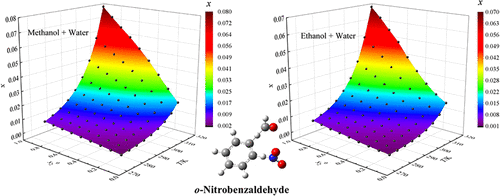
Measurement and Correlation of Solubility of o-Nitrobenzaldehyde in Four Binary Solvents from 273.15 to 313.15 K
Xinhui Chen - ,
Dongmei Zhang - ,
Shaochuan Bai - ,
Panpan Li - ,
Qi Yu - ,
Xia Jiang *- , and
Guan Wang *
The mole fraction solubility of o-nitrobenzaldehyde was measured in four binary aqueous–alcoholic solvent systems (methanol–water, ethanol–water, n-propanol–water, and isopropanol–water) by the laser dynamic method over the temperature range of 273.15–313.15 K. The results showed that the solubility of o-nitrobenzaldehyde increases with both rising temperature and increasing mole fraction of the alcoholic component. The solubility data were correlated using the modified Apelblat model, the Yaws model, the Van’t Hoff–Jouyban–Acree model, and the Wilson model. Among these, the modified Apelblat model provided significantly superior correlation results compared to the other models. The calculated Hansen solubility parameters indicated that hydrogen bonding energy plays a predominant role in the mixing process.
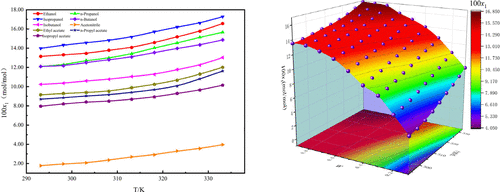
Solubility Measurement, Correlation, and Solvent Effect of 3,5-Dihydroxybenzoic Acid in Nine Pure Solvents and Binary Solvent Mixture (Ethanol + Acetonitrile) at Temperatures from 293.15 to 333.15 K
Pengqi Hou - ,
Shaolan Zhuang - ,
Yire Ma - ,
Jibin Song - ,
Hongkang Zhao *- , and
Qunsheng Li *
The solubility of 3,5-dihydroxybenzoic acid (3,5-DHBA) in nine pure organic solvents and in one binary mixture (ethanol + acetonitrile) was measured gravimetrically at ambient pressure over the temperature range of 298.15–333.15 K. The mole fraction solubility increased with rising temperature in all systems studied. In pure solvents, solubility followed this order: isopropanol > ethanol > n-propanol > n-butanol > isobutanol > ethyl acetate > n-propyl acetate > isopropyl acetate > acetonitrile. In the ethanol + acetonitrile mixture, solubility increased continuously with higher ethanol mass fraction.Experimental data were correlated using three semiempirical equations (van’t Hoff, modified Apelblat, and λh) and three activity coefficient models (NRTL, Wilson, and UNIQUAC). The modified Apelblat equation yielded the best correlation, with average relative deviations (ARD) below 0.60% across all systems. Dissolution in pure solvents was analyzed using the “like-dissolves-like” principle and Hansen solubility parameters. Solvent effects were quantitatively assessed via the KAT-LSER model, where multiple linear regression showed that solubility was mainly governed by π* (41.83%) and δH (30.69%), with lesser contributions from β (21.80%) and α (5.68%). These solubility data provide a valuable reference for designing and optimizing industrial crystallization processes for 3,5-DHBA.
Mastheads
Issue Editorial Masthead

This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More