

About the Cover:
Reviews
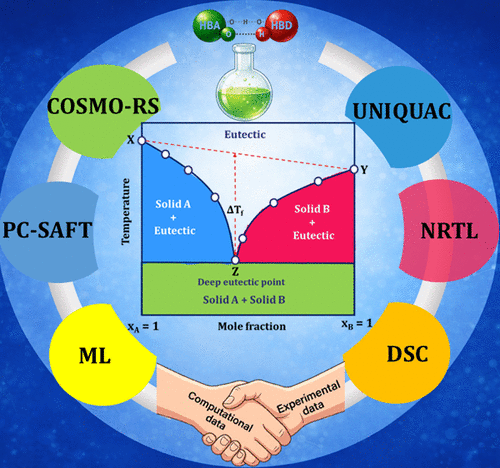
Revisiting the Definition, Solid–Liquid Equilibria, and Thermodynamic Nonideality of Deep Eutectic Solvents
Shaibuna Machingal - and
Ramesh L. Gardas *
Since their introduction, deep eutectic solvents (DESs) have been widely used, often without a rigorous or consistent definition. This has led to misuse of the term, where DESs are applied without considering their underlying thermodynamics, resulting in misclassification and studies that overlook essential physicochemical behaviors. This review aims to provide a comprehensive understanding of DESs by examining their definitions, thermodynamic properties, phase behavior, and modeling approaches through representative examples. First, common misconceptions regarding DES definitions are addressed, followed by a clear, thermodynamically grounded definition. Next, the types of components capable of providing the nonideal thermodynamic interactions necessary for DES formation are outlined. The review then emphasizes the importance of constructing solid–liquid equilibrium (SLE) phase diagrams for accurately defining DESs, discussing both experimental and thermodynamic modeling methods for determining these diagrams. Recent studies on SLE determination of DESs using various approaches are also summarized. Finally, the effects of water on DES stability and other challenges are discussed, along with perspectives for future research. Overall, DES research holds great promise, and this review supports the rational design, selection, and modeling of DESs, which are essential for efficient and practical process development.
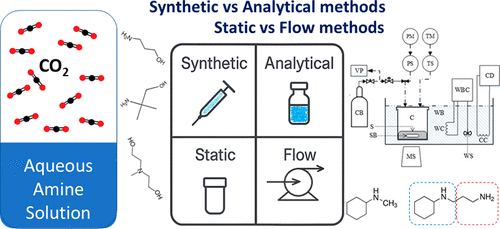
Experimental Techniques for Measuring the CO2 Solubility in Aqueous Amine Solutions
Giannis Kontos - and
Ioannis Tsivintzelis *
Data on CO2 solubility in aqueous amine solutions are crucial for designing efficient CO2 absorption processes. This review provides a comprehensive discussion of the experimental methodologies used for measuring CO2 solubility in aqueous amine systems. Initially, the variations of the equilibrium cells, the main component in every experimental setup used for phase equilibrium measurements, appeared in literature studies, are discussed. The experimental methods are categorized, first, based on the synthetic or analytical approach for the determination of phases’ compositions (synthetic and analytical methods) and, second, based on the presence or not of a flowing phase (static and flow systems), respectively. In every case, the main advantages and disadvantages are discussed.
Thermophysical and Thermochemical Properties
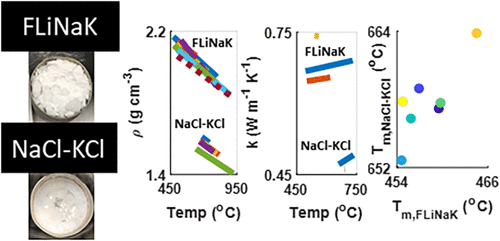
Round Robin Measurements of Molten Salt Properties for LiF-NaF-KF (FLiNaK) and NaCl-KCl Mixtures
Troy Munro - ,
Randall Chiu - ,
Melissa A. Rose - ,
Ondrej Benes - ,
Miroslav Boca - ,
D. Nathanael Gardner - ,
Amanda Leong - ,
Sara Mastromarino - ,
Kim L. Pamplin - ,
Markus H. A. Piro - ,
Mouna Saoudi - ,
Juliano Schorne-Pinto - ,
Christian Michael Sclafani - ,
Anna L. Smith - ,
Nathan D. Smith - ,
Dino Sulejmanovic - ,
Allison M. Berry - ,
Renkun Chen - ,
Ka Man Chung - ,
Christa Dahman - ,
Kent Detrick - ,
Tianshi Feng - ,
J. A. Ocádiz Flores - ,
Ryan Gallagher - ,
Levi Gardner - ,
Xiaofeng Guo - ,
J. Matthew Jackson - ,
Toni Karlsson - ,
Peter Kasper - ,
Hojong Kim - ,
Logan McIlwain - ,
Matthew Memmott - ,
Brian Merritt - ,
Marisa J. Monreal - ,
Kentaro Oishi - ,
Liana Orlovskaya - ,
S. Scott Parker - ,
Andrew A. Prudil - ,
Aaron D. Robison - ,
Sean Scott - ,
Raul C. Romero III- ,
John Vlieland - ,
Michael E. Woods - ,
Jinsuo Zhang - ,
Theodore M. Besmann - , and
Raluca O. Scarlat *
The development, operation, and regulation of nuclear reactors that utilize molten salts as fuel or as heat transfer media require knowledge of the thermal properties of the salt systems and quantification of the corresponding uncertainties. Knowledge of molten salt properties is also necessary for applications in material synthesis, processing, separations, solar thermal power generation, and energy storage. A round robin was conducted with national laboratory and university participants from twenty-one laboratories in five countries to compare property measurements, to better understand uncertainties, and to identify possible best practices. Two salt mixtures, each from a common batch, were distributed to participants for evaluation: equimolar NaCl-KCl and 45.0LiF-13.7NaF-41.3KF mol % (FLiNaK). Measurements were performed to determine the major constituent composition, oxygen content, density, thermal expansivity, melting point, and thermal conductivity. Error analysis was performed on each measurement for uncertainty quantification for each type of property that was explored. The resulting discussion of the methodologies used in this work is meant to lay the groundwork for the development of standard methods and reference materials for future high-temperature property measurements on halide melts.

Thermodynamic Properties of Carbohydrate (d-Glucose, d-Fructose, Sucrose, and Xylitol), Choline Chloride, and Water Mixtures: Experimental Investigations and Modeling Approaches
Renata C. Gaioto - ,
Mariana C. G. C. Dias - ,
Papa M. Ndiaye - ,
Luciana Igarashi-Mafra - , and
Marcos R. Mafra *

This publication is Open Access under the license indicated. Learn More
Carbohydrates are biomolecules with different applications and play a fundamental role in the development of biotechnology and industry. In this work, experimental analyses of density, viscosity, and speed of sound in ternary solutions composed of carbohydrates (d-fructose, d-glucose, sucrose, or xylitol), choline chloride (ChCl), and water were conducted in the temperature range of 278.15 to 323.15 K at atmospheric pressure. Apparent molar volumes (Vϕ0) and apparent molar compressibility (κs,ϕ0) at infinite dilution and B-coefficient were calculated from the experimental data to obtain the transfer parameters (ΔVϕ0, Δks,ϕ0, and ΔB) for the carbohydrates. The limiting apparent molar expansibility (Eϕ0), the isobaric expansion coefficient (αp), and Hepler’s relation were also determined to study the effects caused by temperature. These parameters were chosen to understand the behavior of carbohydrates in the presence of ChCl in an aqueous medium, aiming to elucidate mixing effects resulting from intermolecular interactions in solutions. The results were interpreted in terms of saccharide-ChCl-water interactions.

Understanding the Density and Viscosity Behavior of Thymol–Fatty Acid-Based DES: A Combined Experimental and Simulation Approach
Nouman Rafique - ,
Ludmila Baldan do Rosario - ,
Kalil Bernardino - , and
Oliver Järvik *
This publication is Open Access under the license indicated. Learn More
This study aims to understand the thermodynamic properties of hydrophobic deep eutectic solvents (DESs) comprising thymol (THY) and the carboxylic acids decanoic acid (C10), undecanoic acid (C11), and dodecanoic acid (C12), utilizing experimental measurements alongside molecular dynamics (MD) simulations. To understand the influence of composition and thermal conditions on the macroscopic properties of the DESs, density (ρ) and viscosity (η) measurements were taken at 293.15 to 353.15 K and 293.15 to 323.15 K, respectively, at mole fractions of THY (XTHY) ranging from 0.1 to 0.9. MD simulations were carried out to elucidate the hydrogen bonding in the hydrophobic DES mixtures to complement the experimental work. The hydrogen bonds between thymol and the acid molecules were computed for a few compositions, enabling the characterization of interactions and bonding. The insights advanced the understanding of the structure–property relationships of DESs and reinforced the rational design of the solvents for thermal energy storage materials and environmentally friendly solvent systems.

Density, Viscosity, Electrical Conductivity, and Specific Heat Capacity of Dicationic IL-Based Deep Eutectic Solvents
Yingying Zhang *- ,
Ying Li - ,
Xuzhao Yang *- ,
Ruixue Zhang - ,
Daming Wu - ,
Jingli Han - ,
Yakun Li - ,
Jianqiang Zhang - ,
Cong Liu - ,
Xiaoning Liu - , and
Jingxiang Hu
In this study, six pure deep eutectic solvents (DESs) were prepared using dicationic ionic liquids ([C2(MIM)2Br2] or [C4(MIM)2Br2]) as hydrogen bond acceptors and ethylene glycol (EG) as the hydrogen bond donor, with HBA/HBD molar ratios of 1:4, 1:5, and 1:6. The melting points of the synthesized ILs, DESs, and EG were determined. The density, viscosity, electrical conductivity, and specific heat capacity of these DESs were measured over the temperature range of 288.15∼323.15 K, and the thermal expansion coefficients were derived. Results show that the density decreases with temperature, while the viscosity, electrical conductivity, and specific heat capacity increase. The density and specific heat capacity correlate well with the empirical models, and the Vogel–Fulcher–Tammann equation accurately describes the viscosity and electrical conductivity behavior. With an increase of the HBD/HBA molar ratio, the density and viscosity decrease, while the electrical conductivity and specific heat capacity increase. Due to the interplay between chain length and molecular flexibility, the density and viscosity follow the trend of [C2(MIM)2Br2]/EG < [C4(MIM)2Br2]/EG, and the electrical conductivity and specific heat capacity follow the trend of [C4(MIM)2Br2]/EG < [C2(MIM)2Br2]/EG. These properties were compared with those of [C3(MIM)2Br2]/EG. This study supports the potential application of dicationic IL-based DESs in extraction.
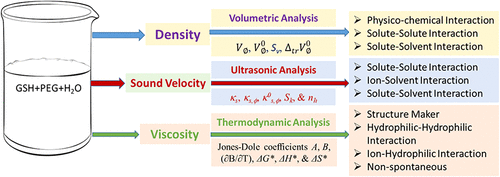
Thermodynamic and Acoustic Mapping of Glutathione–Poly(ethylene glycol) Interactions in Water: A Volumetric, Ultrasonic, and Viscometric Investigation
Mehidi H. Khan - ,
Md Mizanur Rahman Badal - , and
Mohammad A. Yousuf *
This study systematically investigates the volumetric, ultrasonic, and viscometric properties of glutathione (GSH) and poly(ethylene glycol) (PEG) in aqueous solutions (0.01 to 0.05 mol·kg–1) across a temperature range of 293.15 to 318.15 K. To elucidate molecular interactions, density measurements were used to derive key volumetric parameters, including the limiting apparent molar volume (VØ0) and transfer volume (ΔtrVØ0). Results for GSH in lower concentrations of PEG (0.01 to 0.03 m) indicated a predominance of solute–solute interactions, evidenced by increasing VØ0 and positive experimental slopes (Sv). Conversely, higher PEG concentrations (0.04 to 0.05 m) suggested stronger solute–solvent interactions. Ultrasonic data, including isentropic compressibility (Ks), apparent molar isentropic compressibility (Ks,Ø), and hydration numbers (nh), corroborated the coexistence of these competing forces. Viscometric analysis via Jones–Dole coefficients (A, B) and the temperature derivative (∂B/∂T) identified both solutes as structure makers within the aqueous medium. Finally, thermodynamic activation parameters, namely, Gibbs free energy of activation (ΔG*), enthalpy of activation (ΔH*), and entropy of activation (ΔS*) were calculated to provide a comprehensive profile of the viscous flow and molecular mechanism. These findings offer critical insights into the existing molecular interaction and stability of GSH in the presence of PEG.

A Combined Experimental and PC-SAFT Modeling Investigation on the High-Pressure Density of Ionic Liquids with CO2
Lucas H. G. de Medeiros - ,
Moacir F. L. da Costa - ,
Jean-Patrick Bazile - ,
Filipe X. Feitosa - ,
Hosiberto B. de Sant’Ana - , and
Jean-Luc Daridon *
In this study, the density of four binary mixtures of ionic liquids (ILs) with carbon dioxide (CO2) was measured at high pressure by using a vibrating-tube densimeter. The ionic liquids triethylsulfonium bis(trifluoromethylsulfonyl) imide [S222][(CF3SO2)2N], 1,2-dimethyl-3-propylimidazolium bis(trifluoromethylsulfonyl) imide [C3C1C1Im][(CF3SO2)2N], 1-methyl-1-propylpyrrolidinium bis(trifluoromethylsulfonyl) imide [C3C1pyr][(CF3SO2)2N], and 1-butyl-2,3-dimethylimidazolium bis(trifluoromethylsulfonyl) imide [C4C1C1Im][(CF3SO2)2N] were selected because of their different cations and the common anion. Measurements were performed at a pressure range of P = (10.0 to 70.0) MPa, temperatures from 303.15 to 313.15 K, and CO2 mole fractions from 0.2 to 0.7. From these results, the excess molar volume was calculated and presented negative values under almost all composition range, indicating a volume contraction behavior, despite the small values. Finally, the experimental density results were used to estimate the parameters of a Tammann–Tait-type equation and PC-SAFT equation of state. In addition, the regressed parameters were used to obtain the isothermal compressibility kT.
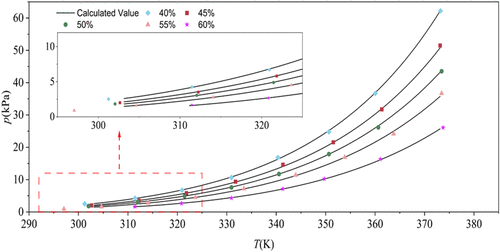
Vapor Pressure, Density, Viscosity, Specific Heat Capacity, Dissolution Enthalpy, and Specific Enthalpy of the LiCl–[BMIM]Cl/H2O Working Pair
Xiaoran Lv - ,
Muqun Li - ,
YuFan Yang - ,
Muran He - ,
Chunting Zhou - , and
Chunhuan Luo *
Imidazolium-based ionic liquids have been reported to enhance the performance of conventional absorption working pairs; however, the effect of 1-butyl-3-methylimidazolium chloride ([BMIM]Cl) on the LiCl/H2O system remains unclear. In this study, a novel working pair, LiCl–[BMIM]Cl/H2O, was proposed, and the optimal mass ratio between LiCl and [BMIM]Cl was systematically investigated. Based on the optimal mass ratio, the vapor pressure, density, viscosity, specific heat capacity, enthalpy of dissolution, and specific enthalpy were measured and calculated over a temperature range of 303.15–373.15 K and a concentration range of 40–60%. Considering both crystallization temperature and application cost, the optimal LiCl-to-[BMIM]Cl mass ratio was determined to be 1:1. The measured data show good agreement with fitted data using the least-squares fitting method. The results of this study provide essential thermophysical property data for evaluating the feasibility of LiCl–[BMIM]Cl/H2O as a working pair in absorption heat pump systems.
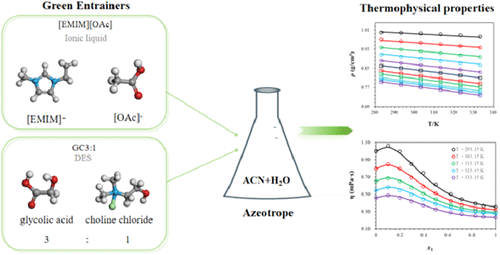
Experimental Determination and Correlation of Density and Viscosity for the Acetonitrile–Water Azeotropic System Containing [EMIM][OAc] and Glycolic Acid/Choline Chloride Deep Eutectic Solvent at Atmospheric Pressure
Yayimei Gao - ,
Yicang Guo - ,
Jiashu Li - ,
Leyi Li - ,
Yingjie Liu - ,
Qing Ye - , and
Jinlong Li *
To enhance the separation efficiency of the acetonitrile–water azeotrope, the densities and viscosities of the azeotropic mixture with two entrainers─1-ethyl-3-methylimidazolium acetate ([EMIM][OAc], an ionic liquid) and a deep eutectic solvent (DES, denoted as GC3:1) prepared by glycolic acid and choline chloride at a molar ratio of 3:1─were experimentally determined at atmospheric pressure. Density measurements were performed over a wide composition range at temperatures from 283.15 to 333.15 K, while viscosity measurements were carried out at 293.15–333.15 K under the same composition range. Thermodynamic parameters including excess molar volume were calculated from the experimental density data. The experimental density and viscosity data were well correlated using empirical linear models, with high correlation coefficients. The addition of either entrainer significantly increased both the density and viscosity of the acetonitrile–water azeotrope. This work provides essential thermophysical data and thermodynamic parameters for acetonitrile recovery via extractive distillation, which offers valuable guidance for entrainer screening and process optimization in industrial applications.
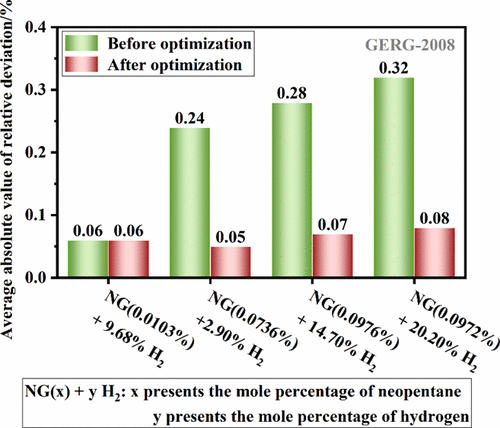
Density Data and Calculation Methods for Hydrogen-Blended Natural Gas Containing Neopentane
Qingjin Lin *- ,
Pu Zhang - ,
Yong Chen - ,
Li Zhou - ,
Bowen Sheng *- , and
Maoqiong Gong
Blending hydrogen into mature natural gas pipelines for mixed transportation is a relatively fast way to achieve large-scale and low-cost hydrogen energy transportation. The density of hydrogen-blended natural gas has a significant impact on process optimization, metrological calibration, and pipeline design. Our previous work involved experimental density measurements for hydrogen-blended natural gas mixtures representative of a Chinese pipeline gas (without neopentane). This study, as the second part of our investigation, aims to determine the density of hydrogen blended with a typical Chinese natural gas containing neopentane using the same high-accuracy single-sinker densimeter. The data cover a range of mole fractions of H2 (3–20%), and temperatures (253.15–350.15 K), with pressures up to 10 MPa. The relatively high neopentane concentration leads to degradation in the predictive performance of the GERG-2008 and AGA8-DC92 equations of state for the studied hydrogen-blended natural gas, with the maximum relative deviation reaching approximately 0.5%. Based on GERG-2008, a systematic evaluation of the neopentane assignment rule was conducted, and a calculation method suitable for relatively high neopentane concentrations (up to 0.1%) is proposed.
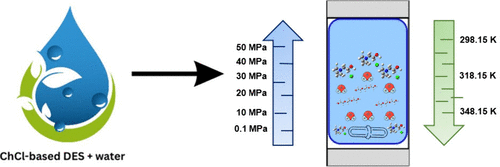
PVT Properties of Choline Chloride-Based Deep Eutectic Solvents with Polyols and Water under Extreme Conditions
Salal Hasan Khudaida - ,
Kao-Jing Yu - ,
Miriana Kfoury - ,
Ardila Hayu Tiwikrama *- , and
Sophie Fourmentin *
In this study, the PVT properties of choline chloride-based deep eutectic solvents (DESs) with two polyols were measured using a high-pressure densitometer from 298.15 to 348.15 K and up to 50 MPa at two different molar ratios and various water content. The high-pressure densities experimental data were used to determine the excess molar volumes and isothermal compressibilities, which were also evaluated using the modified Redlich–Kister model. The binary interactions between DESs and water were calculated using the FOV and Schotte equations of state (EOS). Results show that the density of DESs decreases as the temperature increases and increases as the pressure and the DES mole fraction increase. The isothermal compressibility of the DES–water mixture increases with increasing temperature and decreases with pressure. Deviations in the correlation of densities with the Tait equation are less than 0.09%. The excess molar volumes were calculated using a modified Redlich–Kister equation with deviations between 0.01 and 0.1. The deviations in the binary interactions between DES and water are less than 0.2% and 0.29% for the FOV and Schotte EOS, respectively. These findings suggest that choline chloride has strong interactions with water, reducing densities over a wide range of temperatures and pressures.
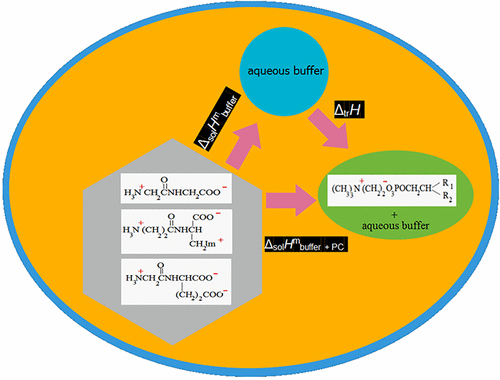
Thermochemical Characterization of the Interaction of the Zwitterionic Surfactant with Zwitterionic, Cationic, and Anionic Peptides: Binding of Glycyl-Glycine, β-Alanyl-l-Histidine, and Glycyl-l-Glutamic Acid to Phosphatidylcholine in a Buffered Saline
Vladimir P. Barannikov - ,
Valeriy I. Smirnov *- , and
Marina S. Kurbatova
The enthalpies of dissolution of three peptides, glycyl-glycine (GlyGly), β-alanyl-l-histidine (β-AlaHis), and glycyl-l-glutamic acid (GlyGlu), in buffered saline containing micellar-type aggregates of l-α-phosphatidylcholine (PC) were measured calorimetrically. The thermochemical characteristics of the interaction of peptides with PC micellar-type aggregates in a buffered saline are found from the calorimetric data as the enthalpies of transfer of peptides from a buffer to (buffer + PC) solution. The exothermic effect of interaction with zwitterionic surfactant aggregates was observed to weaken in the series: β-AlaHis cations > GlyGlu anions > GlyGly zwitterions. The effect of peptide charge on the binding characteristics is more pronounced in the case of zwitterionic aggregates than in the case of charged micelles. The observed differences are related to the different binding modes of peptides to micellar-type aggregates and the absence of the effect of counterions in zwitterionic surfactants.
Vapor-Liquid Equilibria and Supercritical Fluid Equilibria

Isobaric Vapor–Liquid Equilibrium and Intermolecular Interactions in Benzene + Ethanol + Ionic Liquid Ternary Systems
Wenxiu Li - ,
Songyan Chen - ,
Man Yang - ,
Jiawei Zhao - ,
Haoxia Wang - ,
Hongfan Guo - ,
Jungang Fan - , and
Tao Zhang *
The separation of the benzene + ethanol azeotrope is of great significance for environmental protection and resource conservation. 650 ionic liquids composed of 26 cations and 25 anions were screened using the COnductor-like Screening MOdel for Realistic Solvents thermX (COSMOthermX) software (version C2.1, release 01.11) with BP_TZVPD_FINE_C30_1601 parametrization as the entrainer for the separation of benzene + ethanol azeotrope. Tetraoctylammonium acetate ([N8,8,8,8][OAC]), tetrabutylphosphonium acetate ([P4,4,4,4][OAC]), and tributylethylphosphonium acetate ([P4,4,4,2][OAc]) were chosen. To compare the separation capabilities of the three ionic liquids, the isobaric vapor–liquid equilibrium (VLE) data of the benzene + ethanol mixture containing ionic liquids were measured under atmospheric pressure (101.3 kPa). The experimental data were correlated using the nonrandom two-liquid (NRTL) model. The order of the separation capacity of the ionic liquids is [P4,4,4,2][OAc] > [P4,4,4,4][OAc] > [N8,8,8,8][OAC] on the basis of the NRTL equation. The mechanisms of separation were analyzed by means of Sigma profiles (σ profiles), interaction energies (ΔE), electrostatic potentials (ESP), and interaction region indicators (IRI). Fourier-transform infrared (FT-IR) spectroscopy was used to confirm the presence of molecular interactions between ionic liquids and ethanol.
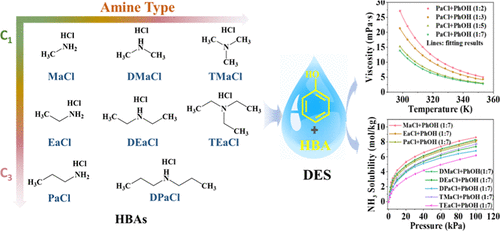
Physicochemical and Structure–Property Relationship of Alkylamine Hydrochlorides-Phenol Deep Eutectic Solvents for NH3 Absorption
Lu Zheng - ,
Saisai Ju - ,
Siqi Fang - ,
Jiayin Zhang - ,
Hongwei Zhang - ,
Zhenping Cai - ,
Lin Teng *- ,
Kuan Huang *- , and
Lilong Jiang
This work elucidates the structure–property relationships of alkylamine hydrochloride-based deep eutectic solvents (DESs) for efficient NH3 capture from industrial gas streams. Through systematic variation of hydrogen bond acceptors (HBAs) including amine type (primary/secondary/tertiary) and alkyl chain length (C1–C3). Three key structural parameters governing absorption performance: (1) amine substitution state (primary > secondary > tertiary), (2) alkyl chain length (C1 > C2 > C3), and (3) HBA: HBD molar ratio (1:7 > 1:5 > 1:3 > 1:2). Primary amine-based DESs achieve optimal performance, exhibiting exceptional NH3 capacities (8.32 mol/kg at 313.2 K, 101.4 kPa) coupled with low viscosities (9.6–13.9 mPa·s). Spectroscopic and computational studies reveal a cooperative dual-site absorption mechanism involving both phenolic –OH and ammonium protons of alkylamine hydrochlorides. The optimized solvents display outstanding NH3/CO2 selectivity (>170) and cycling stability (>88% retention after 5 cycles) compared to many reported absorbents, while comprehensive physicochemical characterization provides critical data for industrial implementation, such as viscosity, density, and thermal stability. These findings establish molecular design principles for NH3 capture solvents that simultaneously address capacity, selectivity, and transport property requirements.
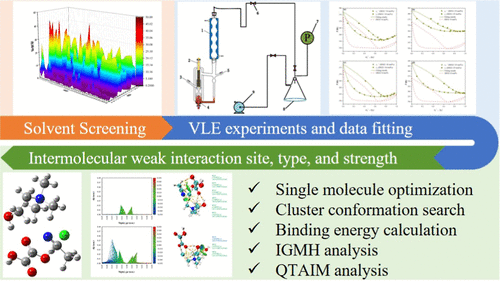
Recovery of Acetonitrile from Wastewater Using Green Low-Toxicity Deep Eutectic Solvents as Extractive Distillation Entrainers
Renting Li - ,
Sen Li - ,
Yan Cheng - ,
Jing Yang - ,
Zhenyu Zhang - ,
Haigang Liu - ,
Zhanhua Ma - ,
Jun Li *- , and
Lanyi Sun *
Acetonitrile (ACN) is an important organic compound and versatile chemical intermediate. Recovering ACN from pharmaceutical waste liquids can reduce production costs and mitigate environmental hazards. At atmospheric pressure, ACN forms an azeotrope with water, and extractive distillation is one of the most effective separation methods. In this study, the conductor-like screening model–segment activity coefficient (COSMO-SAC) was employed to identify deep eutectic solvents (DESs) prepared using choline chloride (ChCl) as the hydrogen bond acceptor and oxalic acid (OA) and glycolic acid (GA) as hydrogen bond donors for separating the ACN–water azeotrope. Vapor–liquid equilibrium (VLE) experiments demonstrate that adding 25 mol % DESs significantly enhances the relative volatility of ACN to water. The nonrandom two-liquid (NRTL) model was used to correlate the experimental data with satisfactory accuracy. Quantum chemical calculations were performed to analyze the types, sites, and strengths of weak interactions between DESs and azeotropic components. Results confirm that interactions between DESs and water are stronger than those with ACN, thereby reducing the activity coefficient of water in the liquid phase and enabling azeotrope separation.
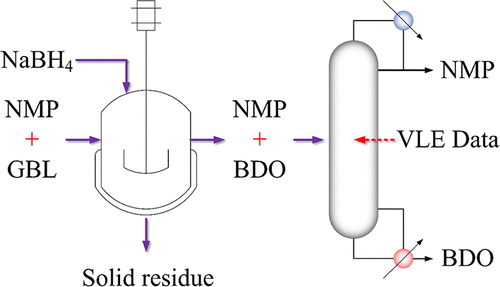
Isobaric Vapor–Liquid Equilibrium for the N-Methyl-2-pyrrolidone + 1,4-Butanediol Binary System at 40.0, 20.0, and 10.0 kPa
Li Sun - ,
Hua Zhou - ,
Xue E Wu - , and
Liming Che *
N-Methyl-2-pyrrolidone (NMP) is a critical solvent in diverse industrial applications, particularly in the microelectronics and lithium-ion battery sectors, where high purity is paramount. A promising purification strategy involves the reactive conversion of its persistent impurity, γ-butyrolactone (GBL), into 1,4-butanediol (BDO). To provide essential design data for the subsequent distillation-based separation of NMP + BDO mixture, isobaric vapor–liquid equilibrium (VLE) data were experimentally measured at reduced pressures of 40.0, 20.0, and 10.0 kPa using a modified Othmer still. The experimental VLE data underwent rigorous thermodynamic consistency verification using both the Fredenslund’s test and Van Ness’s point-to-point test. Subsequently, these data were successfully correlated with the Wilson, NRTL, and UNIQUAC models. All three models demonstrated an excellent fit to the experimental data. Furthermore, the thermodynamic consistency of the regressed binary interaction parameters was confirmed via a graphical analysis of the Gibbs energy of mixing. These comprehensive VLE data and validated model parameters are crucial for the accurate design, simulation, and optimization of the NMP purification process.
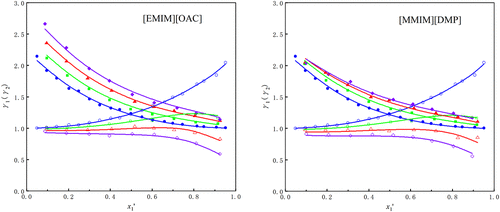
Isobaric Vapor–Liquid Equilibrium Experiment and Thermodynamic Model Correlation of Methyl Butyrate + n-Propanol with Different Ionic Liquids at 101.3 kPa
Zuobin Li - ,
Gaoyang Ma - ,
Shaolan Zhuang - ,
Sentao Yuan - ,
Qunsheng Li *- , and
Hongkang Zhao *
The separation of methyl butyrate (MB) and n-propanol (NPA) is of significant industrial importance for resource recovery in transesterification processes, biofuel formulation, and solvent recycling. However, the formation of a minimum-boiling azeotrope at atmospheric pressure poses a challenge for conventional distillation. In this study, 1-ethyl-3-methylimidazolium acetate ([EMIM][OAC]) and 1,3-dimethylimidazolium dimethyl phosphate ([MMIM][DMP]) were selected as entrainers to facilitate the separation process. The vapor–liquid equilibrium (VLE) of the binary system MB (1) + NPA (2), as well as ternary systems MB (1) + NPA (2) + [EMIM][OAC] (3) and MB (1) + NPA (2) + [MMIM][DMP] (3), was investigated at atmospheric pressure. The experimental data were correlated by using the NRTL, Wilson, and UNIQUAC thermodynamic models. Among them, the NRTL model was found to be more suitable for the studied systems. The results show that both ionic liquids effectively eliminate the azeotropic point, with [EMIM][OAC] demonstrating a slightly superior separation performance compared to that of [MMIM][DMP]. The average relative deviations (ARDs) calculated by the NRTL model were 1.87% for the binary system, while those for the ternary systems were 3.16% and 3.65%, respectively, demonstrating that this model can satisfactorily describe the phase equilibrium behavior of the system.
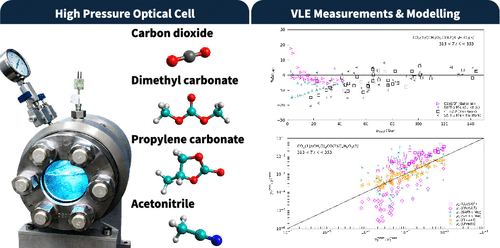
Vapor–Liquid Equilibrium Measurements of Binary and Ternary Systems of Carbon Dioxide, Dimethyl Carbonate, Acetonitrile, and Propylene Carbonate
Bomin Kim - ,
Dongho Yoo - ,
Soomin Gee - , and
Tae Jun Yoon *
Vapor–liquid equilibria (VLE) of three binary (CO2/dimethyl carbonate, CO2/propylene carbonate, and CO2/acetonitrile) and ternary mixtures (CO2/dimethyl carbonate/propylene carbonate, CO2/dimethyl carbonate/acetonitrile, and CO2/propylene carbonate/acetonitrile) were measured at 313, 333, and 353 K and at pressures up to approximately 200 bar using a variable-volume view cell coupled with gas chromatography (GC) analysis. These measurement results and data collected from earlier works were used to model the phase behavior of these mixtures. Two thermodynamic models were applied to represent the bubble-pressure (pbubl) and vapor-phase composition data (yi): a cubic equation-of-state/excess Gibbs free energy (CEoS/GE) model and the statistical associating fluid theory-γ Mie group-contribution (SAFT-γ Mie) equation of state. For the yi data, the Chrastil correlation was also adopted to test the performance of thermodynamic EoSs. The CEoS/GE model provided a satisfactory correlation of the pressure–composition data, while the SAFT-γ Mie model exhibited good performance in representing ternary data when unlike-interaction parameters were properly adjusted based on binary VLE data.
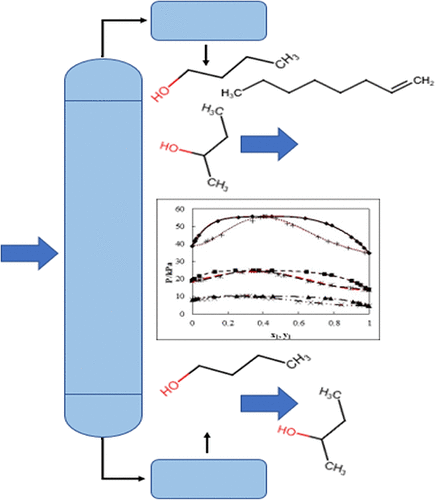
Subatmospheric P-T-x-y Measurements and Modeling for the Butan-1-ol/Butan-2-ol + Oct-1-ene Systems in the Range of T = (323.2–363.2) K
Kuveneshan Moodley *
The phase behaviors of alkene-oxygenate mixtures are highly nonideal and not well studied in the literature. To expand the available literature for these systems, isothermal vapor–liquid equilibrium (VLE) data were measured for the butan-1-ol/butan-2-ol + oct-1-ene systems using the dynamic circulation method. The measurements were conducted at three temperatures in the subatmospheric range (T = 333.2, 343.2, 363.2) K, and complex azeotropic behavior was observed. The data were fit using the γ-Φ formulation to the NRTL and UNIQUAC activity coefficient models combined with the virial equation of state using parameters from the Hayden-O’Connell correlation. A temperature-dependent activity coefficient model parameter term was used. The NRTL model combination generally resulted in a superior fit of the VLE data when considering deviations in pressure and vapor composition. This is attributed to the model strength and the additional regressed nonrandomness parameter. The VLE data were found to be thermodynamically consistent according to the Area, Point, and Infinite dilution tests.
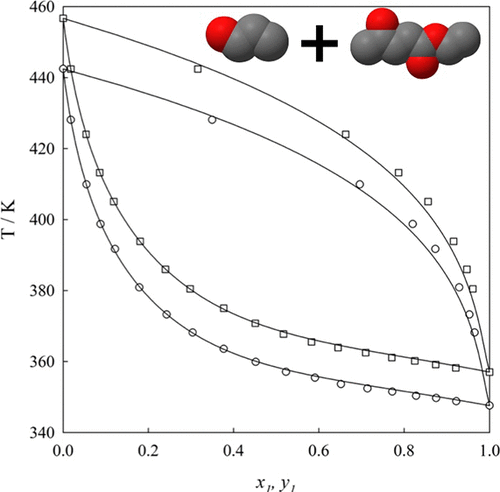
Vapor–Liquid Equilibria, Liquid Dynamic Viscosity, and Surface Tension of the 1-Propanol and Ethyl Levulinate Binary Mixture
Marcela Cartes - and
Andrés Mejía *
The thermodynamic characterization of a potential new alternative biofuel formed from 1-propanol and ethyl levulinate is reported. This characterization is based on experimental determinations of vapor–liquid equilibria (VLE), liquid mass density, liquid dynamic viscosity, and surface tension. Specifically, the VLE are reported at 40 and 60 kPa and over the temperature range from 347 to 456 K. Liquid dynamic viscosity and surface tension for the mixture are measured at 298.15 and 101.30 kPa. The experimental VLE show nonazeotropic behavior, with a positive deviation from Raoult’s law. The VLE data are thermodynamically consistent and are well correlated by classical activity coefficient models (i.e., NRTL, Wilson, and UNIQUAC), where the NRTL model shows lower deviations. Liquid mass density decreases as the mole fraction of 1-propanol increases. Viscosimetry shows that the liquid dynamic viscosity negative deviates from linear behavior, decreases with increasing mole fraction of 1-propanol, x1, reaching a minimum at x1 = 0.69, and then increases. This behavior is correlated using four parameters of the Myers–Scott expansion. Tensiometry results indicate that surface tension exhibits a negative deviation from its linear behavior, decreases as the mole fraction of 1-propanol increases, and is well-correlated by four parameters of the Redlich–Kister polynomial.
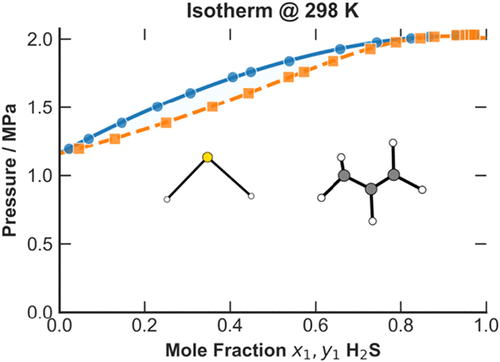
Vapor–Liquid Equilibrium of the Hydrogen Sulfide (H2S)–Propylene (C3H6) Binary System: Experimental and Modeling Study
Alain Valtz - ,
Ismail Lemrabti - ,
Christophe Coquelet *- , and
Antonin Chapoy
The study of the phase behavior of the binary system hydrogen sulfide (H2S)–propylene (C3H6) is necessary for the optimization of gas sweetening processes and petrochemical streams. This study presents new isothermal vapor–liquid equilibrium (VLE) measurements for this system at 278.21, 298.12, 323.06, and 348.13 K, at pressures up to 5.8 MPa. The data were obtained using a precise static-analytic method with two magnetic capillary samplers (ROLSI(R)) for phase analysis by gas chromatography. The measurement uncertainties are u(T)= 0.02 K for temperature, u(P)= 0.0009 MPa for pressure, and u(x,y) = 0.001 for molar compositions. To model this data, a ϕ–ϕ approach utilizing the translated consistent Peng–Robinson (tc-PR) equation of state was used. For the liquid phase, we compared the classical van der Waals mixing rules against the Wong-Sandler mixing rules coupled with the NRTL model. Subsequently, a multiparametric equation of state was utilized to extend the analysis. After optimizing the parameters of each model by fitting them to experimental data, the final models accurately describe the phase behavior of the system. Their reliability and suitability for industrial process design and simulation are thereby demonstrated.
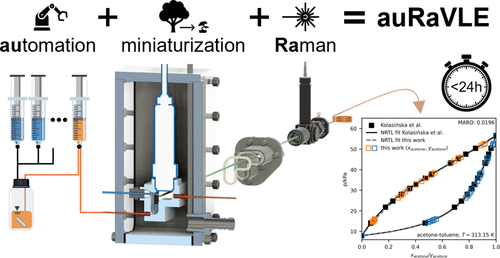
An Automated Milliliter-Scale Setup for Vapor–Liquid Equilibrium Measurements with Raman Spectroscopy: Validation with Binary Mixtures Ranging from 2 to 98 kPa and 278–343 K
Christoph Busch - ,
Marvin Kasterke - ,
Thorsten Brands - ,
André Bardow - , and
Carsten Flake *
This publication is Open Access under the license indicated. Learn More
Vapor–liquid equilibrium (VLE) data are limited due to the resource-intensive nature of conventional measurements. Overcoming this limitation calls for compact VLE measurement setups that allow for rapid data generation. In this paper, we present the automated Raman VLE setup (auRaVLE), an automated milliliter-scale VLE setup, using Raman spectroscopy. Miniaturization accelerates equilibration through improved heat and mass transfer, while reducing sample consumption. Raman spectroscopy enables fast, sampling-free in-situ composition analysis in both phases. Automated sample preparation and experiments mitigate common sources of error and allow 24/7 data generation. We validate our setup with vapor pressure measurements of acetone, ethanol, heptane, and toluene over a range of 278.2–343.1 K and 2.2–97.5 kPa. Furthermore, we measure binary VLE (pTxy) for acetone-heptane (313.15 K), acetone-toluene (313.15 K), and acetone-ethanol (328.15 K), which we validate with literature data and consistency tests. With the auRaVLE setup, we can automatically measure binary VLE in under 24 h, thereby significantly accelerating data output to support rapid process design while minimizing sample consumption.
Liquid-Liquid Equilibria and Vapor-Liquid-Liquid Equilibria
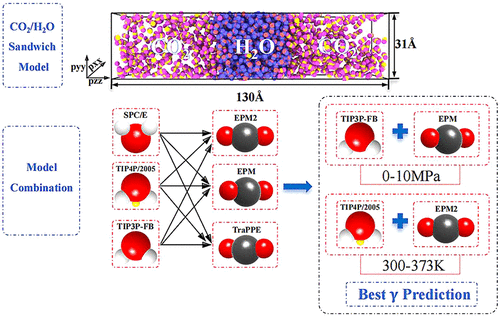
Comparing Molecular Models for CO2/H2O Interfacial Tension over 0–10 MPa and 300–373 K
Junchao Xu *- ,
Mingliang Wang - ,
Fan Li - ,
Li Lv - , and
Huaqiang Chu *
This paper comprehensively compares the performance of different CO2/H2O force fields in predicting interfacial properties. The comparison includes three CO2 molecular models (all rigid) and three H2O molecular models (all rigid). Results show that the TIP4P/2005 model has the smallest deviation in predicting pure water surface tension (γ0). The TIP3P-FB + EPM model combination yields the lowest overall deviation for interfacial tension (γ) prediction over 0–10 MPa at 300 K. Meanwhile, the TIP4P/2005 + EPM2 model combination yields the lowest overall deviation for γ prediction over 300–373 K at 2 MPa. The accuracy of γ is more dependent on the water model used, with the TIP3P-FB water model having the highest weight. These results may be helpful for selecting the appropriate force field to further investigate the kinetics and interfacial properties of CO2/H2O systems.

A Glycerol-Based Deep Eutectic Solvent as an Effective Extractant for the Separation of Alcohols from Esters: A Liquid-Liquid Equilibrium Study
Artemiy Samarov - ,
Natalia Volodina *- ,
Igor Prikhodko - ,
Georgii Misikov - , and
Alexander M. Toikka
This study investigated the use of a deep eutectic solvent (DES) for separating azeotropic mixtures of alcohol–ester systems. The DES was composed of choline chloride and glycerol. Experimental liquid–liquid equilibrium (LLE) data were acquired for several pseudoternary systems. The studied systems consisted of an alcohol (ethanol, n-propanol, or n-butanol), an ester (n-propyl acetate, n-butyl acetate, ethyl propionate, n-propyl propionate, ethyl acetate, or n-butyl propionate), and the DES as the solvent. The experimental measurements were conducted at 293.15 and 313.15 K and 101.3 kPa. A comparative analysis revealed structure–property relationships, demonstrating how separation performance is affected by the alkyl chain length of both the alcohol and the ester. The efficiency of the separation was evaluated by calculating selectivity and distribution coefficients from the determined tie-line data. This work offers important findings on the viability of using a choline chloride-based DES as an effective agent for breaking azeotropes in alcohol–ester mixtures.
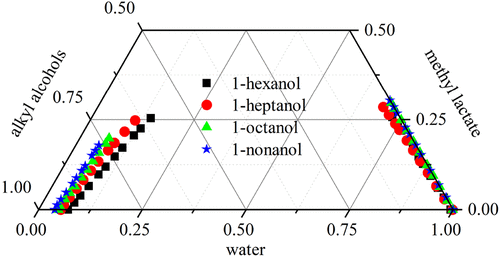
Liquid–Liquid Equilibrium Behavior and Intermolecular Interactions of Water, Methyl Lactate, and Alkyl Alcohols (C6∼C9)
Houchun Yan *- ,
Yue Wang - ,
Xuqiang Li - ,
He Ma - ,
Shaolong Dong - , and
Qingsong Li *
This work explores the liquid–liquid equilibrium (LLE) of a water + methyl lactate (ML) + alkyl alcohol system. The LLE data were acquired for water + ML + (1-hexanol, 1-heptanol, 1-octanol, and 1-nonanol) at a temperature of 303.2 K and a pressure of 101.3 kPa. The system’s equilibrium behavior was characterized by determining water and ML distribution coefficients across the two phases and calculating the separation factor. Moreover, activity coefficient models were utilized to regress the experimental LLE data, with binary interaction parameters subsequently derived. The model parameters were validated with the GMcal_TieLinesLL tool, confirming adherence to Gibbs stability criteria. Additionally, the study encompassed an analysis of intermolecular interactions within the system, employing the σ profile, deformed charge density, interaction energy, and reduced density gradient (RDG) analysis to elucidate the interactions.

Vapor–Liquid Equilibrium of Methyl Acetate + Ethanol with 1-Butyl-3-methylimidazolium Dibutyl Phosphate and 1-Ethyl-3-methylimidazolium Diethyl Phosphate at 101.3 kPa
Niangeng Wu - ,
Gaoyang Ma - ,
Zhe Xue - ,
Hao Liu - ,
Yongbo Chen - ,
Qunsheng Li - ,
Hongkang Zhao *- , and
Xuefeng Feng *
This study explores the separation of the ethanol–methyl acetate azeotrope using ionic liquids as entrainers. Vapor–liquid equilibrium (VLE) data for the binary system and ternary systems containing 1-butyl-3-methylimidazolium dibutyl phosphate ([BMIM][DBP]) and 1-ethyl-3-methylimidazolium diethyl phosphate ([EMIM][DEP]) were measured at 101.3 kPa. Both ionic liquids effectively eliminated azeotropic behavior, with [EMIM][DEP] showing superior performance across the full composition range, attributed to stronger hydrogen bond basicity and reduced unfavorable interactions. Comparison with literature data for [EMIM][OAc], [BMIM][OAc], [EMIM][MeSO3], and DMSO confirmed that ionic liquids generally outperform DMSO. For [BMIM]+-based systems, separation efficiency is as follows: [EMIM][OAc] ≈ [BMIM][OAc] > [BMIM][DBP] > [EMIM][MeSO3] > DMSO. For [EMIM]+-based systems, [EMIM][DEP] provides higher relative volatility in ethanol-rich regions, highlighting the selective capability of the [DEP]− anion. The VLE data were well correlated by the NRTL model, with overall average relative deviations below 5%, and thermodynamic consistency tests confirmed data reliability. These findings indicate that [DEP]−-based ionic liquids are promising entrainers for high-purity methyl acetate production via extractive distillation.
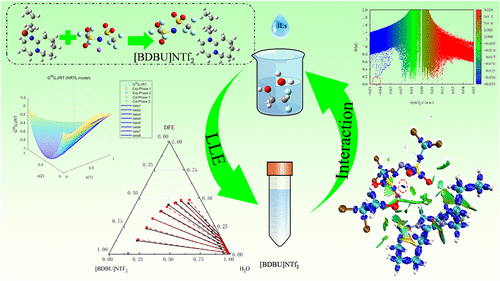
Liquid-Liquid Equilibrium for Extraction of 2,2-Difluoroethanol and Water with Ionic Liquids
Ying Qin - ,
Le Cao - ,
Dongmei Xu - ,
Lingling Sun - ,
Jun Gao *- , and
Yinglong Wang
2,2-Difluoroethanol (DFE) is an eco-friendly intermediate, which is employed to produce pharmaceuticals and fluoropolymers. During industrial manufacturing and practical use, an aqueous solution of DFE is usually generated. Owing to the formation of an azeotropic mixture of water and DFE, it is infeasible to separate such an azeotrope by conventional distillation due to its inherent thermodynamic limitations. Thus, extraction is considered for the separation of water and DFE. Three hydrophobic ionic liquids (ILs) {8-butyl-1,8-diazabicyclo [5,4,0]-7-undecene bis (trifluoromethyl sulfonyl) imide ([BDBU]NTf2), 1-butyl-1-methylmorpholinium bis (trifluoromethyl sulfonyl) imide ([BMMor]NTf2), and 1-butylpyridinium bis (trifluoromethyl sulfonyl) imide ([Bpy]NTf2)} were screened using the COSMO-SAC model for separating the azeotropic mixture. The liquid–liquid equilibrium data were ascertained for three mixtures (water + DFE + ILs). The computed data for partition ratio and selectivity of the extractants were employed for assessing their extraction capabilities. The NRTL equation was utilized to regress the obtained experimental data. The consistency of the fitting parameters was further verified. Finally, the intermolecular interactions between DFE/water and the ILs were analyzed and discussed from various perspectives by quantum chemistry calculations, providing a basis for designing rational IL structures to separate the azeotrope.
Solid-Solid Equilibria and Solid-Fluid Equilibria
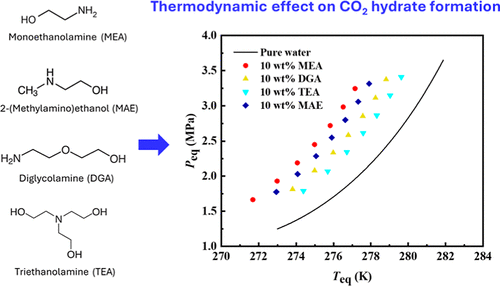
Experimental Investigation of the Thermodynamic Effects of Monoethanolamine, Diglycolamine, Triethanolamine, and 2-(Methylamino)ethanol on CO2 Hydrate Formation
Salal Hasan Khudaida - ,
Wei-Shan Chen - , and
Chie-Shaan Su *
This publication is Open Access under the license indicated. Learn More
Controlling the formation of carbon dioxide (CO2) hydrate is crucial for CO2 capture, transportation, and storage. Understanding the effects of additives on CO2 hydrate formation is essential for selecting suitable chemicals to ensure flow assurance. This study presents new experimental data on the effects of alkanolamine additives: monoethanolamine (MEA), diglycolamine (DGA), triethanolamine (TEA), and 2-(methylamino)ethanol (MAE) on CO2 hydrate formation. Equilibrium dissociation temperatures and pressures were measured using isochoric and temperature-cycling methods over a temperature range of 272–281 K, pressure range of 1.7–3.4 MPa, and additive concentrations of 5 and 10 wt %. All additives thermodynamically inhibited CO2 hydrate formation, with MEA exhibiting the strongest inhibitory effect. Compared to other thermodynamic hydrate inhibitors (THIs), including amines, alcohols, ionic liquids, and alcohol amine mixtures reported in the literature, MEA demonstrated the highest inhibition strength. Additionally, the Clausius–Clapeyron equation was used to speculate on CO2 hydrate structures, suggesting that the sI CO2 hydrate is generated in the presence of each additive. Furthermore, the Østergaard–Masoudi–Tohidi–Danesh–Todd (ØMTDT) equation was employed to effectively correlate the equilibrium dissociation data of CO2 hydrates in the presence of these additives.
Solubility Estimation and Thermodynamic Modeling of Vanillin in Ecologically Friendly Monosolvents and Binary Systems
Kosuru Ravi Kumar - and
Bankupalli Satyavathi *
The solid–liquid equilibrium data of vanillin is estimated in ecologically friendly two monosolvents, acetonitrile and methanol, along with five binary systems, isopropanol + water, ethanol + water, ethylene glycol + water, acetonitrile + water, and methanol + water, in the temperature range of 288.15–323.15 K with varying mole fractions of binary mixtures at an atmospheric pressure of 94.47 kPa. For better comparison, solubility estimation was also carried out in pure solvents (water, isopropanol, ethanol, ethylene glycol), which are already reported in the literature, and it was found that there is very little deviation between the experimental and literature data. It was observed that methanol exhibited the highest solubility in monosolvent, followed by acetonitrile, ethylene glycol, ethanol, isopropanol, water, and its corresponding binary system. The solubility of vanillin increases with increases in temperature and mole fraction, as expected. Thermodynamic modeling was carried out using five different models, and the experimental data are in good agreement with the modified Apelblat model for most of the systems, apart from the acetonitrile + water binary system.
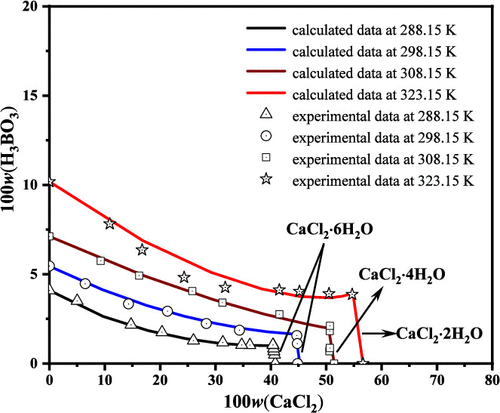
Solubility Measurement and Thermodynamic Model of Solid–Liquid Phase Equilibrium in the Ternary System CaCl2-H3BO3-H2O at T = (288.15 and 308.15) K and P = 0.1 MPa
Jiaxin Han - ,
Yunqing Wang - ,
Jiazheng Qin - ,
Jia Xu - ,
Caixiong Quan - ,
Dan Li *- ,
Yong Ma - , and
Lingzong Meng *
The solid and liquid equilibria in the ternary CaCl2-H3BO3-H2O system at 288.15 and 308.15 K were investigated using the dissolution equilibrium method. The phase diagrams of this system at two temperatures consist of two single-salt crystallization regions corresponding to CaCl2·6H2O and H3BO3 at 288.15 K, CaCl2·4H2O and H3BO3 at 308.15 K, two univariant solubility curves, and one invariant point. A comparison of the phase diagrams at four temperatures shows that the solubilities of H3BO3 and calcium chloride salts increase as the temperature increases. The concentrations of different boron species were calculated with the pH data and the concentration of total boron in the solution. The predominant boron species in the solution is H3BO3 with the mole fraction no less than 0.99. Combining the Pitzer binary parameters of CaCl2, the temperature dependence equations for Pitzer parameters and dissolution equilibrium constant of H3BO3 from 288.15 to 323.15 K were obtained by fitting the solubilities in the ternary system on the basis of Pitzer model. The calculated solubility data at the four temperatures with the Pitzer model agree with the experimental data.

Solubility Measurement, Data Correlation, and Solvent Effect of 4-Methoxycinnamic Acid in Pure Solvents and in Three Binary Solvent Mixtures from 293.15 to 333.15 K
Shaolan Zhuang - ,
Zuobin Li - ,
Yire Ma - ,
Hongkang Zhao - , and
Qunsheng Li *
4-Methoxycinnamic acid (P-MAC) is an important pharmaceutical intermediate. Investigating the solubility of P-MAC in organic solvents is crucial to understanding its crystallization and separation processes. This study employed the gravimetric method to determine the solubility of P-MAC in ten pure solvents (methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, methyl acetate, ethyl acetate, propyl acetate, and dimethylformamide (DMF)) and three binary solvent mixtures (ethanol and ethyl acetate, ethanol and n-propanol, and ethyl acetate and propyl acetate) at temperatures ranging from 293.15 to 333.15 K under atmospheric pressure. The solubility of P-MAC was found to increase with the temperature in all pure and binary solvent systems investigated. P-MAC was observed to be more soluble in aprotic solvents than in protic ones. Furthermore, a correlation analysis of solubility data using various thermodynamic models revealed that the Yaws equation exhibited the best correlation with the experimental data in pure solvent systems. In binary solvent mixtures, the Jouyban-Acree-Van’t Hoff equation had the best correlation with experimental data. Analysis based on the KAT-LSER model indicated that π* and δH significantly influence the dissolution process. The thermodynamic properties of the mixture (ΔGmix, ΔHmix, and ΔSmix) indicate that the mixing process of P-MAC is endothermic, spontaneous, and driven by entropy.
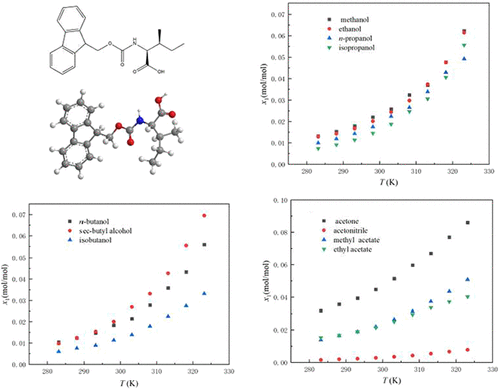
Fmoc-l-Isoleucine Solubility in 12 Monosolvents: Solvent Effects, Molecular Simulation, Thermodynamic Modeling, and Comparison of Structurally Similar Substances
Wei Gao - ,
Weikun Tang - ,
Haolin Yang - ,
Chenyan Li - ,
Yubo Wang - ,
Xinping Hu - ,
Yibo Wang - ,
Peng Wang *- , and
Yanxia Ge *
At present, there is a lack of comprehensive solubility data and thermodynamic analysis, which limits the optimization of crystallization and purification process of Fmoc-l-isoleucine. In this study, the solubility of Fmoc-l-isoleucine in 12 pure solvents in the temperature range of 283.15–325.15 K was measured, and it was found that it was positively correlated with temperature. The solubility in acetone is the highest (8.604 × 10–2 mol/mol at 323.15 K), and the solubility in water is the lowest. The thermodynamic correlation of the four semiempirical models shows that the Yaws model has the best fitting degree, and the average relative deviation (100ARD) is only 1.31%. Molecular simulations (HS, MEPS, and IRI) quantitatively confirmed that the hydrogen bonding between the carboxyl/ester group of the solute and the active site of the solvent is the main driving force of the dissolution process. This study could provide a key thermodynamic basis for industrial process design.
Mastheads
Issue Editorial Masthead

This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More