

About the Cover:
In the cell, miRNA-loaded hAgo2 scans and binds target mRNAs, directing them for degradation via the RNA-Induced Silencing Complex (RISC) in the cytoplasm.
View the article.Application Notes
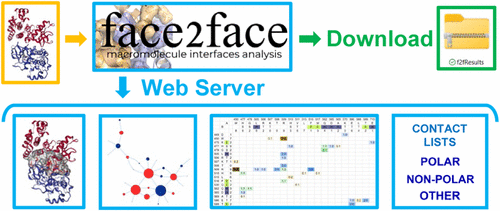
Fast, Comprehensive, and User Customizable Macromolecule Interface Analysis with FACE2FACE
Patrizio Di Micco *- ,
Mario Incarnato - ,
Gianmarco Pascarella - ,
Allegra Via - , and
Veronica Morea *

This publication is Open Access under the license indicated. Learn More
Structural analysis of interfaces in macromolecular complexes is crucial to unveiling the mechanisms underlying molecular recognition. While several valuable computational tools exist for interface analysis, many web-based tools have limitations in input types, analysis comprehensiveness, or output customization, and there remains a need for an immediately accessible solution requiring no software installation, especially for users with limited computational skills. We have developed FACE2FACE, a user-friendly, fast, and comprehensive tool available as a web server for macromolecule interface analysis. FACE2FACE analyzes interfaces between proteins, nucleic acids, and other biological macromolecules or small molecules, providing extensive information that can be instantly visualized on the server interface and easily downloaded. The downloaded materials comprise files in formats that can be easily parsed and imported in spreadsheet applications as customizable contact maps and scripts to quickly visualize interface features in widely used applications such as PyMol and ChimeraX. Examples of FACE2FACE contributions to research projects are described.
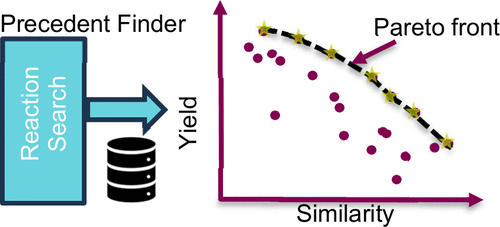
Precedent Finder: Locating Pareto-Optimal Reactions
Christoph A. Bauer *- ,
Thierry Kogej - ,
Samuel Genheden - , and
Per-Ola Norrby
We present Precedent Finder, a cheminformatics search tool for locating relevant reaction information in chemical reaction databases. Precedent Finder is a multiobjective optimization, in that it retrieves Pareto-optimal data points. We choose two different axes of reaction similarity, yield, and date as the initial parameters and present the results for different searches as well as how we use the tool to support the work of synthetic chemists. Precedent Finder can highlight successful catalytic systems for query reactions and thus help optimize synthetic routes to complex pharmaceutical substances.
Perspectives

In Search of Beautiful Molecules: A Perspective on Generative Modeling for Drug Design
Remco L. van den Broek - ,
Shivam Patel - ,
Gerard J. P. van Westen - ,
Willem Jespers *- , and
Woody Sherman *
This publication is Open Access under the license indicated. Learn More
Generative modeling with artificial intelligence (GenAI) offers an emerging approach to discover novel, efficacious, and safe drugs by enabling the systematic exploration of chemical space and to design molecules that are synthesizable while also having desirable drug properties. However, despite rapid progress in other industries, GenAI has yet to demonstrate clear and consistent value in prospective drug discovery applications. In this Perspective, we argue that the ultimate goal of generative chemistry is not just to generate “new” or “interesting” molecules, but to generate “beautiful” molecules─those that are therapeutically aligned with the program objectives and bring value beyond traditional approaches. We focus on five essential considerations for the successful applications of GenAI for drug discovery (GADD): 1) chemical synthesizability (accounting for time/cost constraints); 2) favorable ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties; 3) desirable target-specific binding to modulate the biological mechanism of interest; 4) the construction of appropriate multiparameter optimization (MPO) functions to drive the GenAI toward the project objectives; and 5) human feedback from experienced drug hunters. Interestingly, defining the beauty of a molecule in a drug discovery program is not always obvious, being context-dependent as data emerge and priorities shift, making the role of expert human input indispensable. While MPO frameworks using complex desirability functions or Pareto optimization can help operationalize multifaceted project objectives, they cannot yet fully capture the nuanced judgment of experienced drug hunters. Reinforcement learning with human feedback (RLHF) offers a path to guide the GenAI toward therapeutically aligned molecules, just as RLHF played a pivotal role in training large language models (LLMs) like ChatGPT, especially in aligning the model’s behavior with human expectations. While not responsible for the model’s base knowledge, RLHF is essential in shaping how the model responds. In addition to RLHF, future progress in GADD will depend on better property prediction models and explainable systems that provide insights to expert drug hunters. “Beauty is in the eyes of the beholder”─for drug discovery, beauty is judged by experienced drug hunters and clinical success.
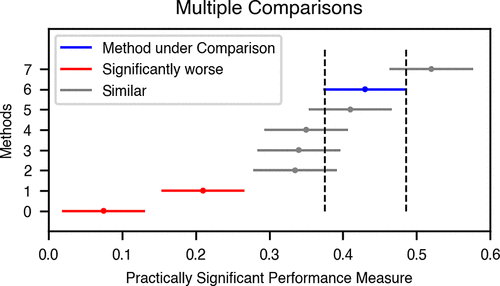
Practically Significant Method Comparison Protocols for Machine Learning in Small Molecule Drug Discovery
Jeremy R. Ash - ,
Cas Wognum *- ,
Raquel Rodríguez-Pérez - ,
Matteo Aldeghi - ,
Alan C. Cheng - ,
Djork-Arné Clevert - ,
Ola Engkvist - ,
Cheng Fang - ,
Daniel J. Price - ,
Jacqueline M. Hughes-Oliver - , and
W. Patrick Walters

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Machine Learning (ML) methods that relate molecular structure to properties are frequently proposed as in silico surrogates for expensive or time-consuming experiments. In small molecule drug discovery, such methods inform high-stakes decisions like compound synthesis and in vivo studies. This application lies at the intersection of multiple scientific disciplines. When comparing new ML methods to baseline or state-of-the-art approaches, statistically rigorous method comparison protocols and domain-appropriate performance metrics are essential to ensure replicability and ultimately the adoption of ML in small molecule drug discovery. This paper proposes a set of guidelines to incentivize rigorous and domain-appropriate techniques for method comparison tailored to small molecule property modeling. These guidelines, accompanied by annotated examples using open-source software tools, lay a foundation for robust ML benchmarking and thus the development of more impactful methods.
Machine Learning and Deep Learning

3D Spatial Learning for Adsorption Energy Prediction in Multi-Temporal Solution Systems: The MTSS Data Set and a GCN-Based Network
Lanqi Li - ,
Rui Luo - ,
Xiaolu Chen - ,
Huapeng Wei - ,
Wenming Zhang - ,
Qiang Lu - ,
Weiming Dong - ,
Jianmei Lu *- ,
Bing Zhang *- , and
Fan Tang *
Existing methods for adsorption energy prediction primarily focus on individual molecules or static molecular pairs, lacking the capabilities to model the diverse spatial configurations found in complex solution systems. While traditional data sets are static, dynamic systems explore a vast conformational space over time. This paper introduces the Multi-Temporal Solution System (MTSS) data set containing 500,000 temporally resolved configurations (3D atomic coordinates + adsorption energy labels) across five solvents. To address solution-level interactions (solute–solvent/solvent–solvent), we propose SEP-Net─a dual-channel graph network integrating rotational-invariant geometric learning and molecular SMILES embeddings. Experimental validation shows SEP-Net achieves an MAE of 211.02 kJ/mol on known solvents and 507.37 kJ/mol on unseen solvents, surpassing MLP (3827.33 vs 507.37 kJ/mol on ACE solvent). This work establishes new benchmarks in system-level adsorption prediction through geometric deep learning.

PhyCysID: Plant Cystatin Protein Prediction by an Artificial Intelligence Approach
Sadaf Aqil - ,
Isabel C. Cadavid - ,
Nureyev F. Rodrigues - ,
Natalia Balbinott - ,
Geancarlo Zanatta - , and
Rogerio Margis *
This publication is Open Access under the license indicated. Learn More
Phytocystatins are proteinaceous inhibitors found in plants that competitively target various classes of cysteine proteinases, including papain-like enzymes, cathepsins, and legumains. Based on structural characteristics and gene organization, phytocystatins can be classified into four subtypes: intronless (I1 and I2), intron-containing (IwI), and multidomain cystatins containing more than one inhibitory region (II). This work presents PhyCysID, a dedicated web server designed for the rapid classification of phytocystatin subtypes. PhyCysID uses a set of 21 features derived from amino acid composition, in combination with 15 distinct machine learning algorithms, to classify phytocystatin sequences into one of the four subtypes. Initially, the input sequence is analyzed to verify if it comprises a true phytocystatin sequence. If so, the input sequence is further analyzed using a specialized classification pipeline called PhyCysID 12M, which integrates 12 machine learning models to assign it to one of the four defined phytocystatin classes. As a case study, a curated dataset of phytocystatin sequences from the UniProt database was used to evaluate the algorithm’s performance. The PhyCysID web server enables rapid classification of both individual and batch-submitted sequences in less than 15 s, providing high-throughput analysis for an accurate identification of phytocystatin class and function. PhyCysID is freely available at https://www.ufrgs.br/labec/phycysid.

Leveraging Language Model, Crystal Structure Prediction and First-Principles Calculation for Material Design
Lei Zhang *- ,
Ben Ni - ,
Kaiyang Xu - ,
Yiru Huang - ,
Qingfang Li - , and
Lifeng Liu *
Large language models (LLMs) have demonstrated transformative potential for materials discovery in condensed matter systems, but their full utility requires both broader application scenarios and integration with ab initio crystal structure prediction (CSP), density functional theory (DFT) methods and domain knowledge to benefit future inverse material design. Here, we develop an integrated computational framework combining language model-guided materials screening with genetic algorithm (GA) and graph neural network (GNN)-based CSP methods to predict new photovoltaic material. This LLM + CSP + DFT approach successfully identifies a previously overlooked oxide material with unexpected photovoltaic potential. Through transformer-based vector similarity analysis coupled with unsupervised clustering and first-principles calculations, we demonstrate that this material exhibits a direct band gap and high theoretical efficiencies that are suitable for photovoltaic application. Our work highlights a hierarchical computational inverse design pipeline that can efficiently navigate the material space to identify nonintuitive functional materials with tailored optoelectronic properties.
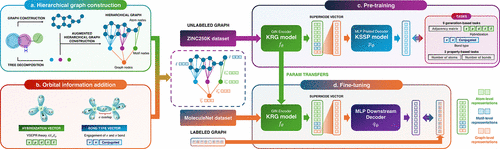
KGG: Knowledge-Guided Graph Self-Supervised Learning to Enhance Molecular Property Predictions
Van-Thinh To - ,
Phuoc-Chung Van Nguyen - ,
Gia-Bao Truong - ,
Tuyet-Minh Phan - ,
Tieu-Long Phan *- ,
Rolf Fagerberg - ,
Peter F. Stadler - , and
Tuyen Ngoc Truong *
This publication is Open Access under the license indicated. Learn More
Molecular property prediction has become essential in accelerating advancements in drug discovery and materials science. Graph Neural Networks have recently demonstrated remarkable success in molecular representation learning; however, their broader adoption is impeded by two significant challenges: (1) data scarcity and constrained model generalization due to the expensive and time-consuming task of acquiring labeled data and (2) inadequate initial node and edge features that fail to incorporate comprehensive chemical domain knowledge, notably orbital information. To address these limitations, we introduce a Knowledge-Guided Graph (KGG) framework employing self-supervised learning to pretrain models using orbital-level features in order to mitigate reliance on extensive labeled data sets. In addition, we propose novel representations for atomic hybridization and bond types that explicitly consider orbital engagement. Our pretraining strategy is cost efficient, utilizing approximately 250,000 molecules from the ZINC15 data set, in contrast to contemporary approaches that typically require between two and ten million molecules, consequently reducing the risk of potential data contamination. Extensive evaluations on diverse downstream molecular property data sets demonstrate that our method significantly outperforms state-of-the-art baselines. Complementary analyses, including t-SNE visualizations and comparisons with traditional molecular fingerprints, further validate the effectiveness and robustness of our proposed KGG approach. The key advantages of KGG are its data efficiency and architectural versatility, driven by orbital-informed representations. By distilling essential chemical knowledge from modest corpora, it avoids extensive pretraining and excels in low-data fine-tuning, providing a robust and chemically meaningful foundation for diverse GNN architectures.
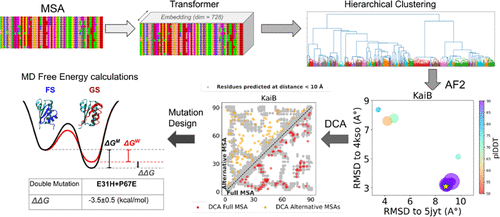
Evolutionary Constraints Guide AlphaFold2 in Predicting Alternative Conformations and Inform Rational Mutation Design
Valerio Piomponi *- ,
Alberto Cazzaniga - , and
Francesca Cuturello *
This publication is Open Access under the license indicated. Learn More
Investigating structural variability is essential for understanding protein biological functions. Although AlphaFold2 accurately predicts static structures, it fails to capture the full spectrum of functional states. Recent methods have used AlphaFold2 to generate diverse structural ensembles, but they offer limited interpretability and overlook the evolutionary signals underlying the predictions. In this work, we enhance the generation of conformational ensembles and identify sequence patterns that influence the alternative fold predictions for several protein families. Building on prior research that clustered multiple sequence alignments to predict fold-switching states, we introduce a refined clustering strategy that integrates protein language model representations with hierarchical clustering, overcoming limitations of density-based methods. Our strategy effectively identifies high-confidence alternative conformations and generates abundant sequence ensembles, providing a robust framework for applying direct coupling analysis (DCA). Through DCA, we uncover key coevolutionary signals within the clustered alignments, leveraging them to design mutations that stabilize specific conformations, which we validate using alchemical free energy calculations from molecular dynamics. Notably, our method extends beyond fold-switching, effectively capturing a variety of conformational changes.
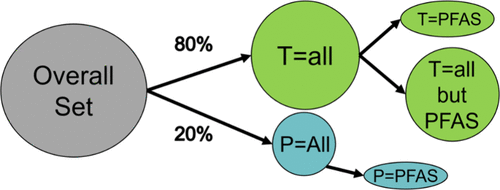
Physicochemical Property Models for Poly- and Perfluorinated Alkyl Substances and Other Chemical Classes
Todd M. Martin *- ,
Landon R. Batts - ,
Nathaniel Charest - ,
Charles N. Lowe - ,
Gabriel Sinclair - , and
Antony J. Williams
To assess environmental fate, transport, and exposure for PFAS (per- and polyfluoroalkyl substances), predictive models are needed to fill experimental data gaps for physicochemical properties. In this work, quantitative structure–property relationship (QSPR) models for octanol–water partition coefficient, water solubility, vapor pressure, boiling point, melting point, and Henry’s law constant are presented. Over 200,000 experimental property value records were extracted from publicly available data sources. Global models generated from data for diverse chemical classes resulted in more accurate property value predictions for PFAS than local models generated from a PFAS-only data set, with an average 11% reduction in mean absolute error (MAE). The global models across all property endpoints achieved strong performance on test data (R2 = 0.76–0.89 for all chemical classes). The test set mean absolute error for PFAS was about 33% higher than the value for all chemicals in the test set (when averaged over the six data sets). The new global models yielded superior PFAS prediction statistics relative to those for existing Toxicity Estimation Software Tool (T.E.S.T) models, with an average 13% reduction in MAE. A nearest neighbor-based measure of model applicability domain (AD) was shown to exclude poor predictions while maintaining a relatively high fraction (∼95%) of chemicals inside the AD. In addition, most test set PFAS are outside the AD when the model was generated without PFAS in the training set.
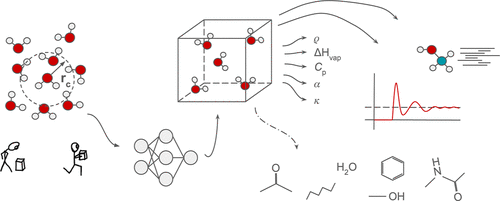
Transferable Neural Network Potentials and Condensed Phase Properties
Anna Katharina Picha - ,
Marcus Wieder - , and
Stefan Boresch *
This publication is Open Access under the license indicated. Learn More
Transferable neural network potentials (NNP) are undergoing rapid development. Many practical applications of NNPs focus on single molecules; e.g., using NNPs as a fast replacement for quantum chemical methods for dihedral angle scans in force field development. Similarly, the reference data on which most transferable NNPs have been trained are single molecule properties. As NNPs are beginning to be used to simulate more complex systems, such as solute–solvent simulations, the question arises whether the current generation of transferable NNPs is accurate enough to reproduce condensed phase properties, which in most cases are outside the training domain of the models. Here we present a first analysis of how well two transferable NNPs (ANI-2x, MACE-OFF23(S/M)) perform in reproducing properties such as density, heat of vaporization, heat capacity, and isothermal compressibility of several pure liquids (water, methanol, acetone, benzene, n-hexane at room temperature, and N-methylacetamide at 100 C). In addition, we examine selected radial distribution functions and the self-diffusion constant. We find specific weaknesses for each of the models, and seemingly small flaws lead to poor performance when applied to condensed phase simulations. The varied outcomes observed with the machine learning potentials suggest that, currently, selecting an architecture or model for all-NNP simulations of real-world applications requires careful consideration and testing.

Predicting HOMO–LUMO Gaps Using Hartree–Fock Calculated Data and Machine Learning Models
Md Mehedi Hasan - ,
Omid Tarkhaneh - ,
Sharene D. Bungay - ,
Raymond A. Poirier - , and
Shahidul M. Islam *
The calculation of the highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) gap for chemical molecules is computationally intensive using quantum mechanics (QM) methods, while experimental determination is often costly and time-consuming. Machine Learning (ML) offers a cost-effective and rapid alternative, enabling efficient predictions of HOMO–LUMO gap values across large data sets without the need for extensive QM computations or experiments. ML models facilitate the screening of diverse molecules, providing valuable insights into complex chemical spaces and integrating seamlessly into high-throughput workflows to prioritize candidates for experimental validation. In this study, we leveraged a data set of HOMO–LUMO gap values for small molecules obtained through Hartree–Fock (HF) calculations and developed ML models to predict HOMO–LUMO energy gaps for organic molecules. Molecular descriptors generated from Simplified Molecular Input Line Entry System (SMILES) representations using RDKit were used as input features to train various regression-based ML models. The data set included 46,717 small molecules with carbon chain number ranging from 1 to 8. Among the tested models, LightGBM regressor, Bidirectional LSTM, CatBoost regressor, and Multilayer Perceptron (MLP) achieved mean absolute error (MAE) values below 0.25 eV. Further improvement was achieved by creating a weighted ensemble model combining the LightGBM regressor, Bidirectional LSTM, and MLP, resulting in a prediction accuracy with an MAE of 0.1660 eV. This ensemble model outperformed others across various data sets, with the LightGBM regressor showing better performance for predicting the HOMO–LUMO gap of saturated linear molecules. SHAP analysis was conducted which identified 20 molecular descriptors critical for accurate predictions. Additionally, the models were empirically adapted to estimate experimental HOMO–LUMO gap values for both small and large molecules (up to carbon number 50), demonstrating their versatility and practical applicability.
Chemical Information

Can Reasoning Power Significantly Improve the Knowledge of Large Language Models for Chemistry?─Based on Conversations with LLMs
Dong-Xu Cui - ,
Shi-Yu Long - ,
Yi-Xuan Tang - ,
Yue Zhao - , and
Qiao Li *
This study presents a systematic evaluation of five reasoning-enhanced Large Language Models (LLMs)─Deepseek-R1–0528, OpenAI-o4 mini, Gemini-2.5-pro, doubao-seed-1.6-thinking, and qwen-max-latest─across nine key chemistry tasks. By comparing these models with traditional LLMs and established computational tools, we systematically investigate the influence of reasoning capabilities and prompt engineering on chemical cognition. The results demonstrate that reasoning-enabled LLMs achieve significant performance improvements in fundamental tasks and that, in most cases, overly complex prompts are not beneficial for these models. However, domain-specific limitations persist; for instance, all five models exhibited structural inaccuracies in CIF file generation (such as incorrect bond topologies). Notably, while reasoning frameworks enhance logical coherence, they do not fundamentally resolve challenges in stereochemical identification or the recognition of rare symmetry groups. In essence, the spatial recognition capabilities of current Large Language Models remain insufficient. These findings underscore the necessity of developing domain-optimized training paradigms to bridge the gap between general reasoning capabilities and specialized chemical applications.
Computational Chemistry
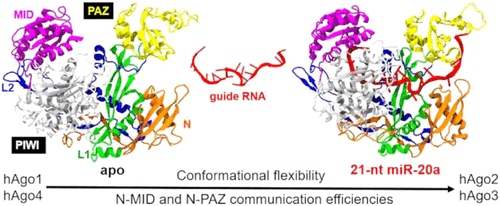
Conformational Dynamics of hAgo2 Silencing: Decoding Functional Divergence across Human Argonaute Paralogs
Antonella Paladino *- ,
Andrea Catte - ,
Jorge Franco - ,
Elisabetta Moroni - , and
Silvia Rinaldi *
This publication is Open Access under the license indicated. Learn More
RNA interference (RNAi) is a key mechanism for controlling gene expression, with Argonaute (Ago) proteins serving as core effectors of the RNA-induced silencing complex (RISC). By loading small noncoding RNAs, Agos target complementary messanger RNAs (mRNAs), leading to their direct catalytic cleavage or the activation of translational repression. Among the four human Ago isoforms (hAgo1–4), only hAgo2 exhibits catalytic activity, a feature not fully explained by structural differences alone. This study explores the structural and functional distinctions among hAgo isoforms, both in their unbound and bound states, using miRNA-20a as a model system. Microsecond-scale molecular dynamics (MD) simulations reveal insightful differences in structural flexibility and plasticity. Catalytically active hAgo2 demonstrates enhanced conformational dynamics, enabling essential structural transitions for efficient RNA silencing. Conversely, hAgo4 exhibits a more rigid conformation, consistent with its reduced catalytic activity. These findings suggest that human isoforms employ a conformational selection mechanism, where the interplay between structural rigidity and flexibility fine-tunes their functional roles. The isoform-specific dynamics unveiled in this study illuminate the functional specialization of human Ago isoforms, providing critical insights into their distinct role in RNA silencing. This understanding opens new possibilities for therapeutic innovation by modulating Ago-mediated pathways in an isoform-specific manner.
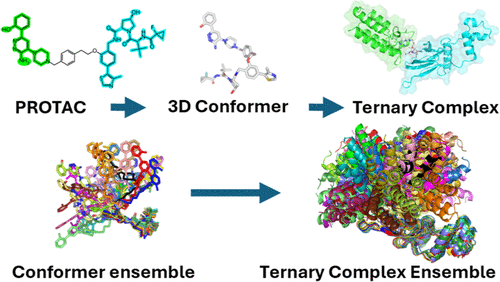
Data-Driven Generation of Conformational Ensembles and Ternary Complexes for PROTAC and Other Chimera Systems
Fabio Montisci - ,
Laura Friggeri - ,
Kepa K. Burusco-Goni - ,
Patrick McCabe - ,
Bojana Popovic - , and
Jason C. Cole *
This publication is Open Access under the license indicated. Learn More
We present the protolysis-targeting chimera (PROTAC) Conformer Generator, a fast and knowledge-based tool for generating robust conformational ensembles of PROTACs and other chimeric degraders. The modeling protocol integrates conformer generation, rigid-body ternary complex (TC) assembly, and conformational sampling strategies that address the inherent flexibility and complexity of these molecules. Each modeled TC is evaluated using a clash-score and a surface-score, designed to prioritize sterically and geometrically plausible models with favorable protein surface interactions. The protocol was validated using experimentally determined PROTAC-mediated TC structures from the Protein Data Bank and “PROTAC-like” structures from the Cambridge Structural Database, demonstrating accuracy across diverse systems. The results show that the PROTAC Conformer Generator can reliably reproduce experimental conformations and generate simple TC models that recapitulate the relative orientations between E3 ubiquitin ligase and the protein of interest as observed in protein crystal structures. This robust validation supports the method’s reliability and establishes a reference framework for degrader modeling studies. The PROTAC Conformer Generator provides a structured and validated workflow for modeling and assessing degrader conformations and ternary complexes, enabling rapid ensemble generation and downstream integration into relevant early stage drug design pipelines.
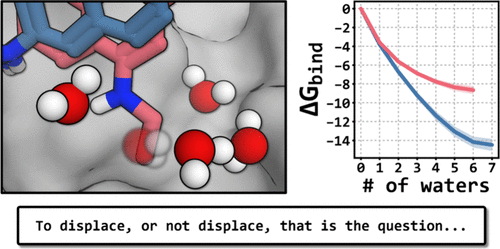
Decoding BCL6 Inhibitors: Computational Insights into the Impact of Water Networks on Potency
Daniella E. Hares - ,
Andrea Scarpino - ,
Michael S. Bodnarchuk *- , and
Swen Hoelder *
This publication is Open Access under the license indicated. Learn More
Water molecules in the binding site can have a critical role in small molecule binding to proteins and are an important consideration in structure-based drug design. Water networks have additional complexity as displacing one water molecule has subsequent effects on the remaining network. Modification of a lead compound that disrupts a water network can have beneficial or detrimental impacts on potency and this outcome is impossible to determine experimentally without time-consuming synthesis of the new compound. Computational methods are ideally suited to study the interplay between ligand optimization and water displacement by predicting the effect of structural changes on both the activity of the compound and the stability of neighboring water molecules. We used Grand Canonical Monte Carlo (GCMC) simulations and alchemical free energy calculations to retrospectively study a series of B-cell Lymphoma 6 (BCL6) inhibitors that sequentially displaced water molecules from a network. The methods were used to rationalize the structure–activity relationship of the compounds by quantifying the individual contributions to the binding affinity from the changes in the water network and new interactions with the protein. GCMC simulations are well-suited for studying water networks in the binding site and were able to reproduce 94% of the experimentally observed water sites from the crystal structures in a subpocket of BCL6. Using the BCL6 project as an example, we show the power of these computational methods to study water networks and how they can provide insights that are able to guide drug discovery projects.
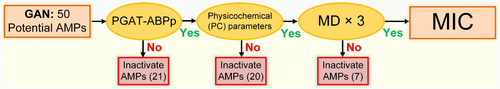
From AI-Driven Sequence Generation to Molecular Simulation: A Comprehensive Framework for Antimicrobial Peptide Discovery
Chunsuo Tian - ,
Yuelei Hao - ,
Haohao Fu *- ,
Xueguang Shao *- , and
Wensheng Cai *
Antimicrobial Peptides (AMPs) are a promising strategy to address bacterial resistance, yet only a limited number have advanced to clinical trials. Recent advances in deep learning provide new opportunities for AMP design. Here, we propose an integrated computational framework combining deep learning with molecular simulation to systematically design and screen novel AMPs. Employing a naïve character-string-based generative adversarial network (GAN), we generated 50 candidate sequences, which were preliminarily screened by the antibacterial peptide discriminative network PGAT-ABPp along with key physicochemical parameters. This screening identified 9 potential functional AMPs. Subsequent molecular dynamics simulations revealed that two peptides can induce water pore formation in bacterial membranes within a limited simulation period, suggesting their potential antibacterial activity. These two peptides were synthesized and tested in vitro, demonstrating efficacy against both Gram-negative (E. coli) and Gram-positive (S. aureus) bacteria, thus confirming their clinical potential. This study not only discovered two novel AMPs but also established a cost-effective design strategy, highlighting the broad applicability of this approach for AMP discovery.

Chemical Space Exploration with Artificial “Mindless” Molecules
Thomas Gasevic - ,
Marcel Müller - ,
Jonathan Schöps - ,
Stephanie Lanius - ,
Jan Hermann - ,
Stefan Grimme - , and
Andreas Hansen *
We introduce MindlessGen, a Python-based generator for creating chemically diverse, “mindless” molecules through random atomic placement and subsequent geometry optimization. Using this framework, we constructed the MB2061 benchmark set, containing 2061 molecules with high-level PNO-LCCSD(T)-F12 reference data for H2-promoted decomposition reactions. This set provides a challenging benchmark for testing, validating, and training density functional approximations (DFAs), semiempirical methods, force fields, and machine learning potentials using molecular structures beyond conventional chemical space. For DFAs, we initially hypothesized that highly parametrized functionals might perform poorly on this set. However, no consistent relationship between the fitting strategy and accuracy was observed. A clear Jacob’s ladder trend emerges, with ωB97X-2 achieving the lowest mean absolute error (MAE) of 8.4 kcal·mol–1 and r2SCAN-3c offering a robust cost-efficient alternative (19.6 kcal·mol–1). Furthermore, we discuss the performance of selected semiempirical methods and contemporary machine-learning interatomic potentials.
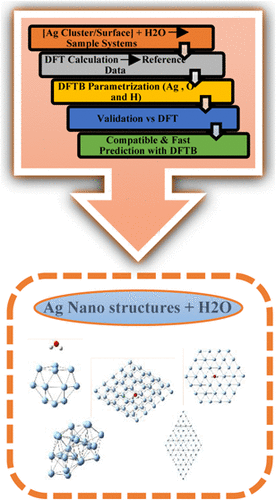
Exhaustive DFTB Parameterization and Its Implementation for the Exploration of Ag Nanostructures + H2O Complexes
Paria Fallahi - and
Hossein Farrokhpour *
To describe Ag–H2O hybrid systems, a new SCC-DFTB parameterization is introduced by generating Ag-X, O-X, and H-X (X = Ag, O, H) pair parameters using the density functional-based tight binding (DFTB) module in Materials Studio 2020. We verify the accuracy of the parameters designated as DFTB-AgOH by juxtaposing them with the outcomes of DFT-DMOL3 and DFTB-HYB for several Ag systems, including clusters (n = 2, 4, 6, 11, 17, 22), monolayer surfaces (n = 9, 16, 25, 37, 49), a bilayer (Ag50), and Ag–H2O complexes. The new parameters align closely with DFT-DMOL3 for morphology, energy, and electronic properties. They also outperform DFTB-HYB, which frequently produces anomalous surface structures. DFTB-AgOH effectively optimizes extensive Ag surfaces and forecasts stable configurations for Ag–H2O systems, closely resembling the findings of DFT. DFTB-AgOH and DFT-DMOL3 both predict analogous adsorption sites for water molecules on silver nanostructures. Nevertheless, the hydrogen orientation is frequently flipped, indicating that this parameterization lacks precision. We employ two linear scaling equations, Y = −0.096 X – 12.998 (adsorption energy) and Y = −0.143 X – 15.383 (interaction energy), to correlate DFTB-AgOH data with DFT-DMOL3 values. This enables us to identify trends and assess the compatibility of the two systems across various sizes. The activation energies for water dissociation from DFTB-AgOH and DFT-DMOL3 are in strong agreement. Molecular dynamics simulations indicate that the dynamic behavior forecasted by DFTB-AgOH aligns more closely with DFT-DMOL3 than with DFTB-HYB, particularly for temporal variations.
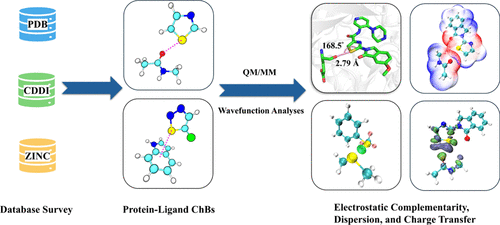
Characterization of Protein–Ligand Chalcogen Bonds: Insights from Database Survey and Quantum Mechanics Calculations
Wenhao Cai - ,
Ziyue Li - ,
Wangchen Zhou - ,
Hongli Chen - ,
Yungen Xu *- ,
Qihua Zhu *- , and
Yi Zou *
Chalcogen bonds (ChBs) are essential noncovalent interactions in biological systems and drug molecules, playing a key role in stabilizing protein structures and determining the bound conformations of drugs. While previous studies on ChBs in biological systems have primarily focused on interactions between residues and within drug molecules, systematic investigations into the characteristics of ChBs between proteins and ligands remain limited. This study systematically explores ChBs between proteins and sulfur-containing ligands using comprehensive database analyses and precise quantum mechanics (QM) calculations. The findings reveal the persistent prevalence of sulfur throughout the drug development process, underscoring the importance of considering sulfur’s role in fragment-based drug design. A survey of the Protein Data Bank (PDB) identified specific preferences in distance, angle, and backbone/side chain interactions for ChBs within protein–ligand complexes, offering valuable insights for structure-based drug design. Furthermore, QM calculations demonstrate that protein–ligand ChBs are characterized by electrostatic complementarity, dispersion, and inter/intramolecular charge transfer. This systematic investigation substantially advances our understanding of protein–ligand ChBs in biological systems.
Computational Biochemistry
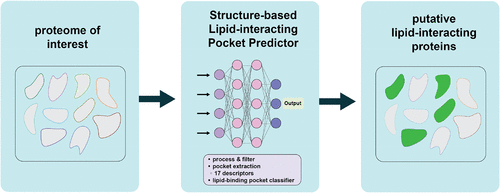
A Machine Learning Model for the Proteome-Wide Prediction of Lipid-Interacting Proteins
Jonathan Chiu-Chun Chou - ,
Poulami Chatterjee - ,
Cassandra M. Decosto - , and
Laura M. K. Dassama *
This publication is Open Access under the license indicated. Learn More
Lipids are essential metabolites that play critical roles in multiple cellular pathways. Like many primary metabolites, mutations that disrupt lipid synthesis can be lethal. Proteins involved in lipid synthesis, trafficking, and modification, are targets for therapeutic intervention in infectious disease and metabolic disorders. The ability to rapidly detect these proteins can accelerate their evaluation as targets for deranged lipid pathologies. However, it remains challenging to identify lipid binding motifs in proteins because the rules that govern protein engagement with specific lipids are poorly understood. As such, new bioinformatic tools that reveal conserved features in lipid binding proteins are necessary. Here, we present Structure-based Lipid-interacting Pocket Predictor (SLiPP), an algorithm that leverages machine learning to detect protein cavities capable of binding to lipids in protein structures. SLiPP uses a Random Forest classifier and operates at scale to predict lipid binding pockets with an accuracy of 96.8% and an F1 score of 86.9% when testing against a set of 8,380 pockets embedded within proteins. Our analyses revealed that the algorithm relies on hydrophobicity-related features to distinguish lipid binding pockets from those that bind to other ligands. SLiPP is fast and does not require substantial computational resources. Use of the algorithm to detect lipid binding proteins in various proteomes produced hits annotated or verified as bona fide lipid binding proteins. Additionally, SLiPP identified many new putative lipid binders in well studied proteomes. Because of its ability to identify novel lipid binding proteins, SLiPP can spur the discovery of new and “targetable” lipid-sensitive pathways.
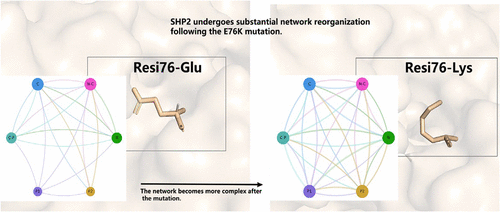
E76K Mutation Promotes SHP2 Activation by Rewiring Allosteric Networks That Drives Conformational Transitions
Derui Zhao - ,
Mengting Liu - ,
Hui Duan - ,
Junyao Zhu - ,
Liquan Yang *- , and
Peng Sang *
The E76K mutation in protein tyrosine phosphatase (PTP) SHP2 is a recurrent driver of developmental disorders and cancers, yet the mechanism by which this single-site substitution promotes persistent activation remains elusive. Here, we combine path-based conformational sampling, unbiased molecular dynamics (MD) simulations, Markov state models (MSMs), and neural relational inference (NRI) to elucidate how E76K reshapes the activation landscape and regulatory architecture of SHP2. Using a minimum-action trajectory derived from experimentally determined closed and open structures, we generated representative transition intermediates to guide the unbiased MD simulations. This strategy captured a thermodynamically relevant ensemble spanning the full activation process. MSMs analyses revealed that E76K flattens the energy landscape, stabilizes activation-prone conformations, and accelerates conformational transitions into catalytically competent states. NRI further uncovered a rewiring of allosteric communication networks, characterized by increased interdomain coupling and an elevated centrality of key relay residues. Shortest-path and temporal analyses revealed that the E76K-induced network remodeling precedes and facilitates domain opening, linking topological reorganization to structural activation. Together, our study provides a dynamic and mechanistic framework for understanding SHP2 activation by oncogenic mutation and illustrates the power of integrating ensemble modeling with interpretable network inference to dissect allosteric regulation.
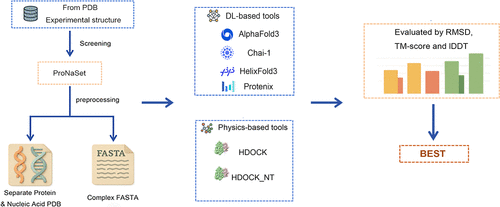
Precision in Predicting Protein–Nucleic Acid Complexes: Establishing a Benchmark Data Set and Comparative Metrics
Huizi Cui - ,
Yuxuan Wang - ,
Yu Fu - ,
Xiangyu Yu - ,
Wannan Li - ,
Feng Lin *- , and
Weiwei Han *
Protein–nucleic acid interactions are fundamental to biological processes and biotechnology, yet their computational prediction lags behind protein structure or protein–protein interaction modeling. This study introduces ProNASet, a benchmark data set of 100 experimentally resolved protein–nucleic acid complex structures, alongside a multidimensional evaluation framework using root mean square deviation (RMSD), TM-score, and local distance difference test (LDDT) metrics. We systematically evaluated four deep learning (DL) algorithms (AlphaFold3, Chai-1, HelixFold3, and Protenix) and two physically driven docking methods (HDOCK and HDOCK_NT). Our analysis revealed that physically driven methods significantly outperform current DL approaches in predicting protein–nucleic acid complex structures. The template-less HDOCK_NT demonstrated the highest success rate at 74.5% (using thresholds RMSD <2 Å, TM-score >0.9, and LDDT >0.6), compared to 63.8% for template docking and only 34.0% for the best-performing DL method, AlphaFold3. These results underscore the substantial need for improvement in DL methods for this specific task. The ProNASet benchmark provides a standardized testing platform, highlights intrinsic shortcomings in current DL models for capturing protein–nucleic acid interaction features, and guides the development of next-generation computational tools crucial for advancing genome editing and synthetic biology.

PAZ Domain Pivoting is the Rate-Limiting Step for Target DNA Recognition in the Middle Region of Thermus thermophilus Argonaute
Jinchu Liu - ,
Kun Xi - , and
Lizhe Zhu *
Thermus thermophilus Argonaute (TtAgo) is a DNA-guided programmable endonuclease with emerging applications in genome engineering, yet the rate-determining dynamic mechanisms governing its transition from guide-target hybridization to catalytic activation remain unresolved. Here, we employ molecular dynamics simulations and the Traveling-salesman-based Automated Path Searching (TAPS) approach to dissect the target DNA recognition in the middle region (nt 9–12) of TtAgo. We designed two paths to tackle this problem: one assumed that coordination of the target DNA backbone occurs before base-pairing between the target and guide DNA; the other hypothesized a concerted transition without preferred order between backbone-coordination and base-pairing. While the first path exhibits two high energy barriers (12.45 and 14.12 kcal/mol), the second path is featured by a single rate limiting barrier (12.56 kcal/mol) and therefore more probable to occur. Crucially, the flexible PAZ domain in both scenarios dominates the three rate limiting barrier steps driving bidirectional cavity modulation through pivoting motions. These findings underscore the PAZ domain’s indispensable role in manipulating DNA recognition in the middle region, offering mechanistic insights for engineering high-efficiency Argonaute variants by targeting domain plasticity.
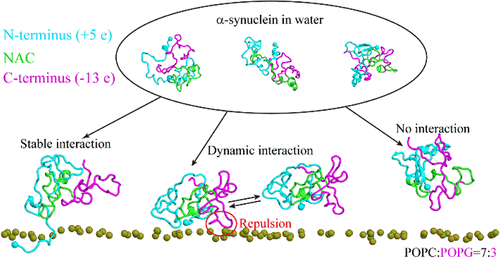
Molecular Dynamics Simulations Reveal Conformational Determinants of the Dynamic Association between α-Synuclein and Membranes
Jiahui Huang - and
Cong Guo *
Interaction of α-synuclein with membranes is associated with normal cellular functions and the etiology of neurodegenerative diseases. Structural characterization of the membrane-bound α-synuclein is key to understanding the interaction mechanism. However, it represents a significant challenge because the intrinsically disordered nature of α-synuclein leads to a multitude of membrane-binding modes and highly dynamic conformations at the membrane surface. The present work investigated the binding of α-synuclein to a mixed POPC/POPG bilayer and provided atomic-level characterization of the protein–membrane complex based on extensive molecular dynamics simulations. The binding process is triggered by the adsorption of lysine residues to the negatively charged PG headgroups and results in differential binding modes stemming from the balance of heterogeneous intramolecular contacts of α-synuclein and its interaction with the membrane. The membrane-binding residues are primarily located in the first nine residues and the five imperfect KTKEGV repeats in the N-terminus. Network analysis of intramolecular interactions identifies the interaction between the N-terminus and C-terminus as the major interference factor in membrane binding. Repeats 1, 3, and 5 which are less engaged in intramolecular contacts display higher membrane-binding propensities, whereas Repeat 4 is the least membrane-bound due to strong interactions with Repeat 3, Repeat 5, and the C-terminus. Our results reveal crucial intramolecular interactions governing the membrane binding of α-synuclein and would enlighten the development of therapeutic strategies targeting the α-synuclein–membrane interaction.
Pharmaceutical Modeling
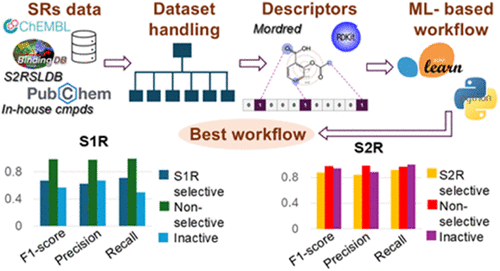
Prediction of Activity and Selectivity Profiles of Sigma Receptor Ligands Using Machine Learning Approaches
Lisa Lombardo - ,
Verena Battisti - ,
Thierry Langer - ,
Rosaria Gitto - , and
Laura De Luca *
This publication is Open Access under the license indicated. Learn More
Sigma (σ) receptors (SRs) have emerged as important therapeutic targets due to their roles in various biological pathways. They are classified into two subtypes: S1R, primarily distributed in the central nervous system and related to neuroprotection and neurodegenerative diseases, and S2R mainly expressed in cancer cells and associated with cell proliferation and apoptosis, as well as in neurons. Although S1R and S2R exhibit structural differences in receptor architecture and assembly, they share similar binding site features and ligand recognition mechanisms. This similarity underscores the importance of identifying selective ligands for therapeutic design, especially given the distinct physiological functions of these receptors. In this project, we developed three distinct machine learning (ML) approaches based on classification, regression, and multiclassification models to predict the activity and selectivity profiles of SR ligands. High-quality data sets were curated from public and in-house source; in turn, the data sets were systematically organized and processed for each workflow. Models were built using molecular descriptors and fingerprints, including Mordred, RDKit, ECFP4, ECFP6, and MACCS keys, and trained with various ML algorithms such as extra trees, random forest, support vector machine, k-nearest neighbors, and XGBoost. Rigorous nested and classical 5-fold cross-validation protocols were applied for model selection and validation. At the end, identification of the best workflow was performed by an external validation procedure. Among the workflows, the one-step multiclassification approach, based on extra trees combined with Mordred descriptors, showed the best predictive performance in external validation, offering a robust tool for the identification of selective S1R and S2R ligands.
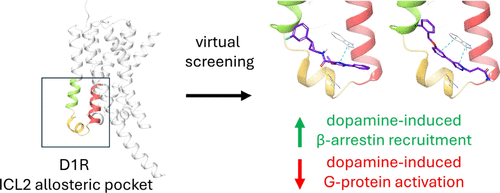
Virtual Compound Screening for Discovery of Dopamine D1 Receptor Biased Allosteric Modulators
Yang Zhou - ,
William C. Wetsel - ,
Steven H. Olson *- , and
Lawrence S. Barak *
This publication is Open Access under the license indicated. Learn More
The dopamine D1 receptor (D1R) is a therapeutic target for a variety of central nervous system disorders including Parkinson’s disease (PD). Challenges thus arise in the development of safer D1R therapies in limiting off-target drug activity. This issue is particularly relevant to PD therapy, where L-DOPA has been the “gold standard” drug for decades despite a problematic side-effect profile. Recent studies of G-protein and β-arrestin functionally selective signaling offer new strategies for developing superior D1R orthosteric and allosteric compounds with fewer side effects. We designed a desktop-computer drug-screening platform to examine large virtual chemical libraries for allosteric compounds binding D1R intracellular loop 2 (ICL2) determinants. Two structurally distinct hits were strong enhancers of dopamine-induced β-arrestin recruitment and inhibitors of dopamine-induced G-protein activation. The lead candidate DUSBI-A3 was highly selective for D1R over closely related dopamine receptors when assessed by β-arrestin activation, providing proof-of-concept for pursuing D1R selective, biased compounds in the treatment of PD.
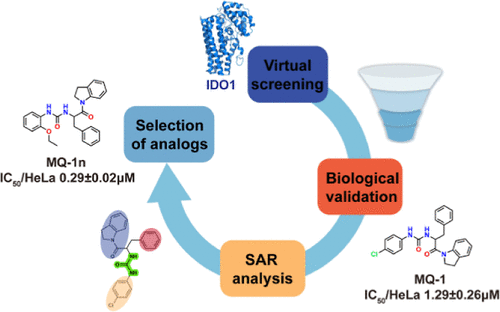
Identification of a Novel Core Structure of Apo-Ido1 Inhibitors Through Virtual Screening and Preliminary Hit Optimization
Yekui Yin - ,
Meiqi He - ,
Jianda Yue - ,
Yaqi Li - ,
Jiuxi Peng - ,
Xiao Luo - ,
Zhenyu Wang - ,
Xiao He - ,
Songping Liang - ,
Zhonghua Liu *- , and
Ying Wang *
Indoleamine 2,3-dioxygenase 1 (IDO1) is a heme-containing enzyme considered as a potential therapeutic target for neurodegenerative diseases and cancer. However, the further development of traditional IDO1 inhibitors has been hindered by their limited clinical efficacy. Recently, type IV apo-IDO1 inhibitors offer a new strategy for developing IDO1 inhibitors due to their highly selective and durable inhibition. In this study, we developed a virtual screening (VS) workflow to identify novel apo-IDO1 inhibitors. A hit compound MQ-1 (IC50 = 1.29 μM) was identified by molecular docking and binding pose metadynamics (BPMD). Biological evaluations confirmed that MQ-1 selectively targets apo-IDO1 and disrupts heme binding. To optimize the structure of MQ-1, free energy landscape was constructed, and the dissociation mechanism was explored by random accelerated molecular dynamics and self-organizing maps. Finally, several MQ-1 analogs with improved inhibitory activity were discovered, such as MQ-1a (IC50 = 1.03 μM), MQ-1e (IC50 = 0.81 μM), and MQ-1n (IC50 = 0.29 μM). The established VS workflow effectively applied to IDO1 and can also be applied to similar targets. The novel apo-IDO1 inhibitor core structure provides a starting point for potential antitumor drug development.
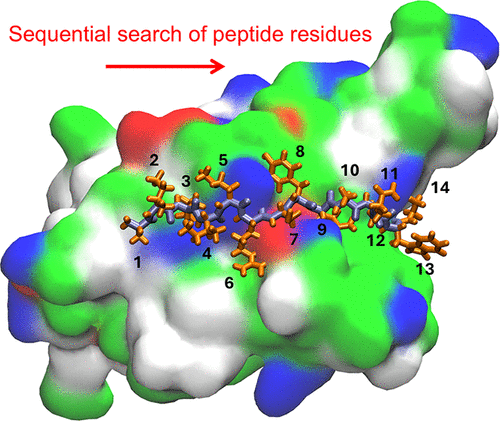
On-the-Fly Sequential Design of Simple Peptides
Francesco Coppola - and
Petr Král *
We introduce a simple and efficient computational methodology capable of designing peptides lacking higher order structures. The method is based on the sequential modification of residues in a simple peptide attached to a substrate of interest. The peptide design can start at an arbitrary point of the substrate and proceed into a direction chosen spontaneously or guided by an external potential. The decision about using a certain residue is based on its binding free energy to the substrate, as evaluated in molecular dynamics simulations. Here, this approach is tested on the design of peptides binding to spike proteins in SARS-CoV-2. The methodology can be easily modified according to actual needs and extended to other molecules.
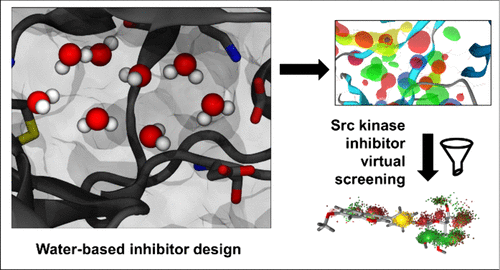
Water-Based Pharmacophore Modeling in Kinase Inhibitor Design: A Case Study on Fyn and Lyn Protein Kinases
Martin Ljubič - ,
Marija Sollner Dolenc - ,
Jure Borišek *- , and
Andrej Perdih *
This publication is Open Access under the license indicated. Learn More
Water-based pharmacophore modeling is an emerging approach in inhibitor design that leverages the dynamics of explicit water molecules within ligand-free, water-filled binding sites to derive 3D pharmacophores for virtual screening. In this study, we assess the potential of this strategy through a case study targeting the ATP binding sites of Fyn and Lyn protein kinases─members of the Src family that have been less explored in anticancer drug discovery compared to other family members. Molecular dynamics simulations of multiple kinase structures were used to generate and validate several water-derived pharmacophores, which were subsequently employed to screen chemically diverse libraries of compounds. Two active compounds were identified in biochemical assays: a flavonoid-like molecule with low-micromolar inhibitory activity and a weaker inhibitor from the library of nature-inspired synthetic compounds. Structural analysis via molecular docking and simulations revealed that key predicted interactions, particularly with the hinge region and the ATP binding pocket, were retained in the bound states of these hits. However, interactions with more flexible regions, such as the N-terminal lobe and activation loop, were less consistently captured. These findings outline both the strengths and challenges of using water-based pharmacophores: while effective at modeling conserved core interactions, they may miss peripheral contacts governed by protein flexibility. Incorporating ligand information where available may help address this challenge. Overall, water-based pharmacophore modeling presents a promising ligand-independent strategy for identifying novel chemotypes and exploring undercharged chemical and conformational space in kinases as well as other therapeutically relevant targets.
Bioinformatics

Multiview Deep Learning Framework for Precise Prediction of Transcription Factor Binding Sites
Yiben Lin - ,
Huiliang Luo - ,
Liang Yan - ,
Changmiao Wang - ,
Yao Li *- , and
Ruiquan Ge *
Transcription factors (TFs) are essential proteins that regulate gene expression by specifically binding to transcription factor binding sites (TFBSs) within DNA sequences. Their ability to precisely control the transcription process is crucial for understanding gene regulatory networks, uncovering disease mechanisms, and designing synthetic biology tools. Accurate TFBS prediction, therefore, holds significant importance in advancing these areas of research. While machine learning methods, particularly deep learning approaches, have achieved notable progress in TFBS prediction in recent years, several challenges persist. These include modeling the intricate structural features of the DNA double helix, capturing long-range dependencies within sequences and integrating diverse biological data sources. To address these issues, we propose an innovative solution known as multiview deep learning for Transcription Factor Binding Prediction (MDNet-TFP), which leverages multiple views of DNA sequences─including different representational forms and diverse processing strategies─to enhance prediction capabilities. Specifically, our framework introduces a bidirectional reverse complement module (BiRC-Mamba) that effectively accounts for the bidirectional and reverse complement properties characteristic of DNA sequences. Furthermore, we developed a multiscale convolutional recurrent attention network (MCRAN) that extracts both structural and functional DNA features across multiple dimensions while integrating information from various biological data sets. These advancements allow our model to outperform existing methods across 165 ChIP-seq data sets, achieving an average ACC of 88.13% (±0.47), an ROC-AUC of 93.72% (±0.15), and a PR-AUC of 93.40% (±0.21). The model not only excels with this specific data set but also maintains its high performance across a wider array of 690 ChIP-seq data sets. To further validate the model’s effectiveness, we employ motif visualization techniques. This approach reveals that the regions receiving high attention from our model align with known transcription factor binding motifs, offering valuable biological insights. Additionally, this correspondence substantiates the model’s ability to generalize and interpret complex genomic data effectively. By addressing critical limitations in the field, MDNet-TFP offers a promising new avenue for advancing research in transcriptional regulation and biomedical applications.
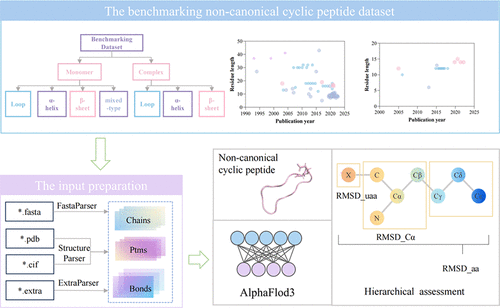
AlphaFold3 for Noncanonical Cyclic Peptide Modeling: Hierarchical Benchmarking Reveals Accuracy and Practical Guidelines
Chengyun Zhang - ,
Wentong Wang - ,
Ning Zhu - ,
Zhigang Cao - ,
Yaling Wu - ,
Qingyi Mao - ,
Cheng Zhu - ,
Chenhao Zhang - ,
Jingjing Guo - , and
Hongliang Duan *
Despite the revolutionary impact of AlphaFold3 on structural biology, this model’s capability in predicting noncanonical cyclic peptides remains unexplored. Given the clinical significance of cyclic peptides containing unnatural residues as a therapeutic modality, we present the first systematic evaluation of AlphaFold3 for this class of molecules. To facilitate benchmarking, we developed an automated input pipeline to streamline AlphaFold3 predictions for cyclic peptides. Our study aims to (1) quantify the hierarchical accuracy (all atoms, Cα atoms, and atoms of unnatural residue levels) of AlphaFold3 in predicting both noncanonical cyclic peptide monomers and complexes, (2) assess the reliability of AlphaFold3's confidence metrics, (3) evaluate the influence of multiple sequence alignment and structural templates, and (4) identify systematic biases in AlphaFold3's predictions. Based on these analyses, we provide practical guidelines for applying AlphaFold3 in cyclic peptide structure prediction to facilitate the related research of bioactive cyclic peptides.

An Empirical Biasing Force Constant to Minimize Overfitting in Cryo-EM Flexible Fitting Refinement
Daisuke Matsuoka - ,
Yuji Sugita - , and
Takaharu Mori *
This publication is Open Access under the license indicated. Learn More
Reliable modeling of protein structures from a cryo-EM density map is one of the central issues in structural biology. Typically, the constructed model is refined using a flexible fitting method combined with molecular dynamics, where a biasing potential is introduced to guide the protein structure toward the density map. However, the appropriate force constant for the biasing potential is generally unknown a priori. Here, we propose an empirical force constant that enables flexible fitting refinement with minimal overfitting. The rule is derived from systematic flexible fitting calculations performed on 29 selected systems (map resolution = 3.0–6.8 Å) using a range of force constants from weak to strong values. The refined structures are evaluated based on the MolProbity score and secondary-structure-forming tendencies. Our analysis shows that in most systems, the MolProbity score increases monotonically as the force constant increases. In addition, α-helix and β-strand tend to collapse beyond a certain force constant depending on the system size or number of fitting atoms (Natom). Based on these findings, we propose that a suitable choice for the biasing force constant in the cross-correlation coefficient-based flexible fitting refinement is 3Natom kcal/mol. This provides a practical guideline for an initial selection of the force constant, aiding in the search for a more suitable value and serving as a useful default parameter in flexible fitting protocols to achieve reliable models with minimal overfitting.
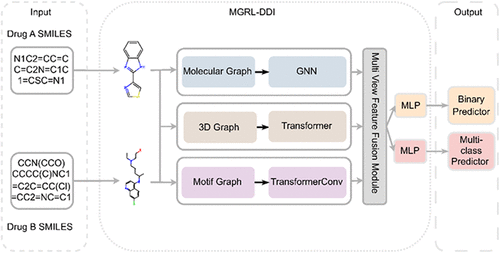
MGRL-DDI: Multiview Graph Representation Learning for Accurate Drug–Drug Interaction Prediction
Peng Xiong - ,
Hu Chen - ,
Jiaxu Zhou - ,
Yuni Zeng *- , and
Qi Dai *
Drug–drug interactions (DDIs) present a significant challenge in clinical practice, as they may lead to adverse reactions, diminished therapeutic efficacy, and serious risks to patient safety. However, most existing methods depend on single-view representations of drug molecules or substructures, which limits their capacity to capture the diverse and complex nature of drug properties. To overcome this limitation, we propose MGRL-DDI, a multiview graph representation learning framework that comprehensively models drug structures from three complementary perspectives: Three-dimensional (3D) molecular graphs, motif graphs, and molecular graphs. Specifically, the 3D graph captures the spatial and topological configuration of drug molecules, the motif graph encodes biologically meaningful substructures and their interactions, and the molecular graph reflects local atomic connectivity. To effectively integrate information across these structural dimensions, we introduce a multiview fusion module. Extensive experiments conducted on multiple real-world data sets demonstrate that MGRL-DDI consistently outperforms most advanced methods in both warm-start and cold-start scenarios, underscoring the advantages of multiview structural modeling for DDI prediction.

Accelerating Prediction of Antiviral Peptides Using Genetic Algorithm-Based Weighted Multiperspective Descriptors with Self-Normalized Deep Networks
Shahid Akbar - ,
Ali Raza - ,
Quan Zou - ,
Wajdi Alghamdi - ,
Xiaorui Kang - ,
Hashim Ali - , and
Ximei Luo *
The accurate prediction of antiviral peptides (AVPs) plays a crucial role in accelerating the development of peptide-based therapeutics. Despite extensive production of antiviral medications, viral diseases remain a major human health concern. AVPs have emerged as potential candidates for the development of novel antiviral drugs. However, the available traditional methods are labor-intensive, expensive, and cannot provide a deeper structural and contextual understanding of the peptide sequences. To address these problems, we propose a novel deep computational model, TargetAVP-DeepCaps, for the precise prediction of AVPs. In this model, multiple innovative feature representation strategies were presented by encoding the input peptides using a pretrained ProtGPT2 model for contextual embeddings. On the other hand, sequence-to-image transformations are performed using SMR and RECM matrices. Additionally, the produced 2D images were locally decomposed using the CLBP approach to obtain the SMR-CLBP and RECM-CLBP descriptors. A differential evolution mechanism was applied to form a weighted-feature-based multiperspective vector. The optimal features were selected using a hybrid MRMD + SFLA feature selection approach. Finally, a novel self-normalized capsule network (Sn-CapsNet) model was developed to achieve a superior predictive accuracy of 97.36%, outperforming the available predictors by approximately 12% with an area under the curve (AUC) of 0.98. To ensure the generalization of the TargetAVP-DeepCaps model, our training achieved an approximately 8% higher prediction than previous models using an independent data set. The demonstrated effectiveness and robustness of TargetAVP-DeepCaps provide an advanced therapeutic tool for understanding peptide mechanisms and related applications in drug discovery.
Mastheads
Issue Editorial Masthead

This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More