

About the Cover:
Venturing beyond the traditional flatland of kinase binders (The image was generated using Adobe Firefly (8.2.0) and edited with Adobe Photoshop (26.8.0)
View the article.Viewpoints

Blind Challenges Let Us See the Path Forward for Predictive Models
John D. Chodera - ,
W. Patrick Walters *- ,
Sriram Kosuri - , and
James S. Fraser

This publication is Open Access under the license indicated. Learn More
The rapid proliferation of AI/ML models in drug discovery heralds an era of extraordinary progress but also raises urgent questions about whether the true predictive performance is as good as advertised. On-target prediction models often benefit from high-resolution structural or atomistic representations that capture the subtleties of binding affinity and pose. In contrast, off-target and ADMET liabilities have typically relied on more implicit representations of molecular interactions. Retrospective benchmarks often provide a misleading picture of how successful these diverse representations are at predicting properties, and the community lacks standardized, prospective comparisons. Blind challenges, such as the OpenADMET × ASAP × PolarisHub Challenge featured in this issue, are crucial for realistically evaluating progress, encouraging iterations, and directing collective efforts toward major accuracy barriers. With ongoing investment in large-scale, open data creation, and community-led challenges, predictive modeling is poised to rapidly transform drug discovery by enabling accurate, multiparameter optimization.
Application Notes
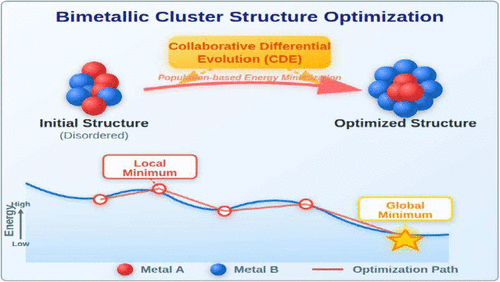
C++ Toolkit for Bimetallic Cluster Structure Optimization Using Collaborative Differential Evolution
Xiaomin Wu - ,
Miao He - , and
Yousi Lin *
Global optimization of bimetallic and monometallic cluster structures remains computationally challenging, particularly due to the rapid increase in homotops with system size and compositional complexity. To address this issue, we present a Collaborative Differential Evolution (CDE) algorithm featuring a multisubpopulation collaborative architecture specifically designed for efficient structure prediction of diverse nanocluster systems. The framework integrates three functionally specialized subpopulations for exploration, exploitation, and balance along with adaptive operations tailored for metallic nanoclusters. This algorithm is implemented as a user-friendly online C++ toolkit. We demonstrate the versatility and robustness of our approach through comprehensive structural optimization across three distinct case studies: Pt–Pd and Cu–Au bimetallic clusters, as well as monometallic Pt clusters. The CDE algorithm consistently achieves 50–100% faster convergence and superior stability compared to conventional methods across all tested systems, establishing itself as a robust and generalizable tool for accelerating the discovery of stable configurations in diverse cluster materials.

DFDD: A Cloud-Ready Tool for Distance-Guided Fully Dynamic Docking in Host–Guest Complexation
Kowit Hengphasatporn *- ,
Lian Duan - ,
Ryuhei Harada - , and
Yasuteru Shigeta
This publication is Open Access under the license indicated. Learn More
Fully dynamic sampling of host–guest inclusion remains difficult because conventional docking and conventional molecular dynamics simulations can sample inclusion, but crystal-like binding is typically stochastic and difficult to reproduce. Here, we introduce DFDD (Distance-Guided Fully Dynamic Docking), a cloud-ready implementation of the LB-PaCS-MD framework designed to capture inclusion processes via unbiased molecular dynamics in explicit solvent. DFDD automates system setup, parameter generation, iterative short-cycle MD sampling, and trajectory analysis within a single workflow that runs on Google Colab without any installation. Progress toward complexation is guided only by the host–guest center-of-mass distance, allowing force-free exploration of insertion pathways and enabling the recovery of both stable and transient binding modes. Using β-cyclodextrin as a representative host, DFDD reproduces experimentally observed inclusion geometries within minutes and reveals intermediate states along the insertion route. Optional coupling with pKaNET-Cloud enables pH-aware, stereochemically consistent ligand protonation states prior to simulation, supporting robust host–guest modeling. This Application Note provides a transparent and accessible platform for efficient host–guest complexation studies. The DFDD framework is publicly available at https://github.com/nyelidl/DFDD.
Reviews
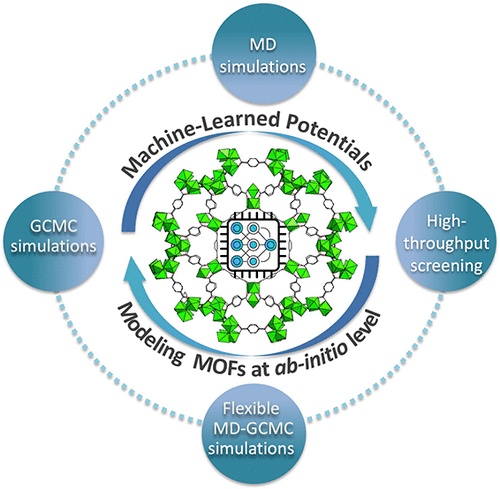
Transforming MOF Modeling with Machine-Learned Potentials: Progress and Perspectives
Omer Tayfuroglu *- and
Seda Keskin *
Machine-learned potentials (MLPs) have emerged as transformative tools for modeling metal–organic frameworks (MOFs), bridging the accuracy of quantum mechanics with the efficiency required for large-scale molecular simulations. By learning the potential energy surface directly from quantum-mechanical reference data, MLPs enable a unified description of the complex nature of MOFs and their interactions with guest molecules across multiple length and time scales. Recent developments have demonstrated the capability of MLPs to model intrinsic MOF properties such as lattice dynamics, thermal expansion, and mechanical response, as well as to describe adsorption thermodynamics, diffusion, and cooperative host–guest behavior in flexible frameworks. Developing reliable and transferable MLPs for MOFs remains a significant challenge due to the vast chemical and structural diversity of MOFs and the complexity of sampling guest-framework configurations. The lack of openly shared, standardized, and user-friendly MLP implementations also limits their broader adoption. This review focuses on the current progress in MLP-based modeling of MOFs, highlighting methodological advances, data-generation strategies, and active-learning protocols, while outlining the key challenges and future directions for developing transferable, accessible, and universal MLPs for the predictive design and discovery of MOFs.
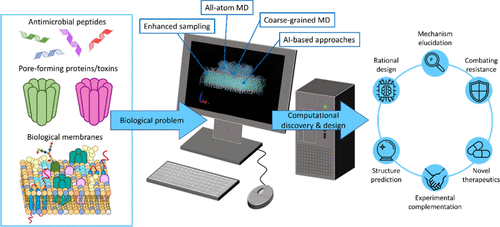
Current Status of Molecular Dynamics Simulations of Membrane Permeabilization by Antimicrobial Peptides and Pore-Forming Proteins: A Review
Sofia Cresca - ,
Jure Borišek *- ,
Alessandra Magistrato *- , and
Igor Križaj *
Biological membranes are crucial for cellular integrity and function, but their selective permeability can be compromised by various peptides and proteins, such as antimicrobial peptides (AMPs) and pore-forming proteins/toxins (PFPs/PFTs). These molecules induce membrane permeabilization through diverse mechanisms, ranging from the formation of well-defined pores to more nuanced disruptions of the lipid bilayer. Understanding molecular mechanisms underlying membrane integrity disruption is vital for developing novel tools to be applied in medicine, biotechnology, and agriculture. However, due to their transient and dynamic nature, characterizing membrane-disrupting mechanisms is a significant experimental challenge. In silico methods, particularly all-atom and coarse-grained molecular dynamics (MD) simulations, are an indispensable tool to complement and enrich experimental studies, and can offer detailed insights into peptide/protein–membrane interactions, insertion, oligomerization, and pore formation. This review provides a comprehensive overview of the structural and mechanistic diversity of AMPs and PFPs, highlighting representative case studies and discussing key challenges emerging from MD simulations.
Machine Learning and Deep Learning
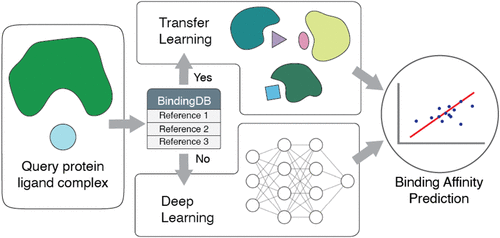
More Accurate Binding Affinity Prediction Using Protein Homology and Ligand-Based Transfer Learning
Justin Purnomo - ,
Caitlin Kim - ,
Kunyang Sun - ,
Yingze Wang - , and
Teresa Head-Gordon *
Accurate and rapid prediction of protein–ligand binding affinities is critical for drug discovery, particularly when evaluating large chemical libraries or new drug molecules from high-throughput generative models. We present UCBbind, a hybrid framework that combines a similarity-based transfer module with a deep-learning-based prediction module, to efficiently estimate binding affinities of small molecules to target proteins. For each query protein–ligand pair, UCBbind transfers experimental data from highly similar reference pairs when available and applies the prediction module when no sufficiently similar reference exists. We benchmarked UCBbind on multiple datasets, including the CASF-2016 set, the HiQBind dataset post 2020, and the COVID Moonshot database. Our results show that UCBbind achieves state-of-the-art predictive performance, particularly for test entries with high similarity to well-characterized reference proteins and ligands, and can support downstream tasks such as binding site prediction and binder/nonbinder classification.

Chemical Feature Engineering and Defect-Aware Structural Fingerprint Representations for Complex Defects in 2D Materials
Cheewawut Na Talang - ,
Aniwat Kesorn - ,
Chanaprom Cholsuk - ,
Tobias Vogl - ,
Rutchapon Hunkao - ,
Asawin Sinsarp - ,
Sujin Suwanna *- , and
Suraphong Yuma *
This publication is Open Access under the license indicated. Learn More
Designing descriptors for multiple defects in two-dimensional materials is challenging due to the diverse local atomic environments created by different defect types and arrangements. Existing physics-informed descriptors struggle to distinguish distinct defect configurations with identical composition, while deep learning models, though powerful, require large data sets and are less interpretable. In this work, we address this limitation by engineering chemical descriptors and constructing structural features from nearest-neighbor distributions provided by the classical force-field-inspired descriptors (CFID). We show that our engineering method, combined with defect-aware structural features derived from the Hellinger distance, even excluding the full distribution features, improves data point discrimination in high-dimensional feature space while reducing the number of features by 50%. In predicting formation energy per defect site, this extended feature set balances reliance on a few dominant features, enhancing model interpretation and generalization at the cost of a marginal 10% increase in prediction error compared to baseline descriptors. This generalization capability is empirically validated on an external out-of-distribution data set of bulk hBN defects, where our model exhibits lower uncertainty and superior stability within the applicable physical domain (− 1 < Ef < 5 eV). However, predicting a highly complex and nonlinear target, such as the HOMO–LUMO gap, remains challenging, as none of our extensions outperform the baseline. This physics-informed approach offers an interpretable and computationally efficient alternative to deep learning models, providing new insights into defect representations in 2D materials and serving as a tool for the high-throughput prescreening of stable defect candidates prior to expensive first-principles calculations.
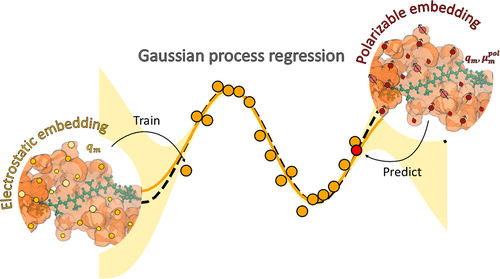
Machine-Learning Framework for Excitation Energies of Chromophores in Polarizable Environments
Chris John - ,
Edoardo Cignoni - ,
Lorenzo Cupellini *- , and
Benedetta Mennucci *
Excited states of embedded chromophores are highly influenced by their interaction with the environment. Herein, we present a machine-learning (ML) framework capable of predicting the different environmental contributions to excitation energies of chromophores in a polarizable embedding. Our ML models are built in a hierarchical structure to capture both the effect of ground-state polarization and the response of the polarizable environment to the electronic transition. With the use of the right descriptors, the models trained on the quantum mechanics/molecular mechanics (QM/MM) calculations in a nonpolarizable environment are able to successfully predict the effects of a polarizable environment on excitation energies. The ML models are applied to three chromophores present in light-harvesting complexes (chlorophyll a, chlorophyll b, and lutein) and are used to reproduce the excitonic structure of a multichromophoric system unseen in the training set to a level of accuracy offered by a polarizable QM/MM calculation, while taking a fraction of its time.
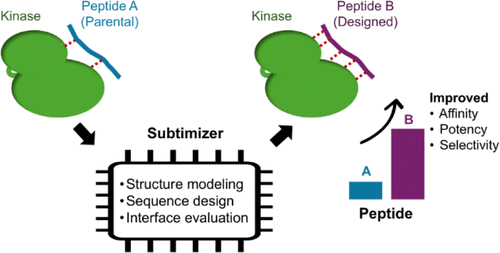
Subtimizer: Computational Workflow for Structure-Guided Design of Potent and Selective Kinase Peptide Substrates
Abeeb A. Yekeen - ,
Cynthia J. Meyer - ,
Melissa McCoy - ,
Bruce Posner - , and
Kenneth D. Westover *
Kinases are pivotal cell signaling regulators and prominent drug targets. Short peptide substrates are widely used in kinase activity assays essential for investigating kinase biology and drug discovery. However, designing substrates with high activity and specificity remains challenging. Here, we present Subtimizer (substrate optimizer), a streamlined computational pipeline for structure-guided kinase peptide substrate design using AlphaFold-Multimer for structure modeling, ProteinMPNN for sequence design, and AlphaFold2-based interface evaluation. Applied to five kinases, four showed substantially improved activity (up to 350%) with designed peptides. Kinetic analyses revealed >2-fold reductions in the Michaelis constant (Km), indicating improved enzyme–substrate affinity. Designed peptides for MET and ROS1 exhibited reciprocal selectivity, with 4-fold and 11-fold preferences for their intended targets, respectively. This study demonstrates AI-driven structure-guided protein design as an effective approach for developing potent and selective kinase substrates, facilitating assay development for drug discovery and functional investigation of the kinome.
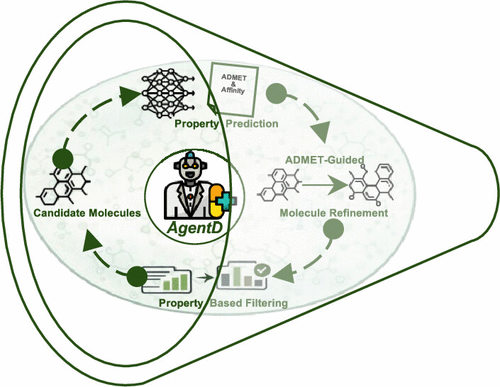
Large Language Model Agent for Modular Task Execution in Drug Discovery
Janghoon Ock - ,
Radheesh Sharma Meda - ,
Srivathsan Badrinarayanan - ,
Neha S. Aluru - ,
Achuth Chandrasekhar - , and
Amir Barati Farimani *
This publication is Open Access under the license indicated. Learn More
We present a modular framework powered by large language models (LLMs) that automates and streamlines key tasks across the early stage computational drug discovery pipeline. By combining LLM reasoning with domain-specific tools, the framework performs biomedical data retrieval, literature-grounded question answering via retrieval-augmented generation, molecular generation, multiproperty prediction, property-aware molecular refinement, and 3D protein–ligand structure generation. The agent autonomously retrieves relevant biomolecular information, including FASTA sequences, SMILES representations, and literature, and answers mechanistic questions with improved contextual accuracy compared to standard LLMs. It then generates chemically diverse seed molecules and predicted 75 properties, including ADMET-related and general physicochemical descriptors, which guids iterative molecular refinement. Across two refinement rounds, the number of molecules with QED >0.6 increased from 34 to 55. The number of molecules satisfying empirical drug-likeness filters also rose; for example, compliance with the Ghose filter increased from 32 to 55 within a pool of 100 molecules. The framework also employed Boltz-2 to generate 3D protein–ligand complexes and provide rapid binding affinity estimates for candidate compounds. These results demonstrate that the approach effectively supports molecular screening, prioritization, and structure evaluation. Its modular design enables flexible integration of evolving tools and models, providing a scalable foundation for AI-assisted therapeutic discovery.

KPGT-Fluor: A Graph Transformer Framework for Accurate Property Prediction of Fluorescent Dyes under Different Solvent Environment
Jintian Lyu - ,
Jiamin Zhong - ,
Nan Zhou - ,
Dadong Shen *- ,
Jiangcheng Xu - ,
Shaolong Lin - ,
Li Qin - ,
Zhao Chen *- , and
Kui Du *
Data-driven machine learning (ML) technologies have become increasingly prevalent in the prediction of the optical properties of fluorescent dyes, especially across diverse solvent environments─a key requirement for the rational design of small solvatochromic systems. Here, we introduce KPGT-Fluor, a novel adaptation of the Knowledge-guided Pretraining of Graph Transformer (KPGT) framework, designed to model solvent-dependent photophysical behavior. Through the integration of solvent molecular descriptors, KPGT-Fluor effectively captures solvent environmental effects that influence optical properties. KPGT-Fluor exhibits strong predictive performance, achieving mean absolute error (MAE) of 10.55 and 12.09 nm for absorption wavelengths (λabs) and emission wavelengths (λem), respectively. For the logarithm of the extinction coefficient (ε) and quantum yield (Φ), the MAE values are 0.104 and 0.081, demonstrating a high accuracy. Compared with the existing models, a comprehensive evaluation across the four key property prediction tasks shows that KPGT-Fluor exhibits a more balanced and competitive overall performance. To further demonstrate the effectiveness of the proposed framework, an external test set containing representative main ring structures was selected. Furthermore, two novel D–π–A molecules were synthesized, and their optical properties in different solvents were experimentally compared with KPGT-Fluor predictions. These results highlight KPGT-Fluor as a powerful tool for predicting and discovering solvatochromic materials.

Traj2Relax: A Trajectory-Supervised Method for Robust Structure Relaxation
Zhiyuan Liu - and
Quan Qian *
Structure relaxation plays a crucial role in atomic simulation and materials modeling, yet traditional first-principles approaches remain computationally expensive and, therefore, difficult to scale to high-throughput applications. In this work, we propose Traj2Relax, a trajectory-supervised structure relaxation framework based on conditional velocity field modeling. Instead of relying on explicit energy or force evaluations, Traj2Relax learns a time-dependent velocity field from geometric differences between successive configurations along real relaxation trajectories, enabling physically consistent structural convergence across a wide range of perturbation magnitudes. A time-scheduled noise mechanism is introduced during training to improve stability under highly distorted inputs, while deterministic integration during inference produces smooth, interpretable relaxation trajectories. Experimental results show that Traj2Relax achieves competitive accuracy under near-equilibrium conditions and demonstrates clear advantages under moderate to strong perturbations, where energy-driven and distribution-based relaxation methods tend to degrade. On representative inorganic crystal systems, Traj2Relax attains a root-mean-square deviation of 0.26 Å and a space-group consistency of 82.3% under equilibrium settings and maintains a root-mean-square deviation of 0.38 Å with a recovery rate of 5.8% under strong perturbations. The framework further supports deterministic, batch-parallel relaxation, yielding an order-of-magnitude improvement in inference throughput compared with iterative energy-minimization-based approaches. Overall, Traj2Relax provides an efficient and physically grounded alternative for learning-driven structure relaxation, particularly suited for high-throughput screening scenarios involving nonequilibrium or highly perturbed structures.
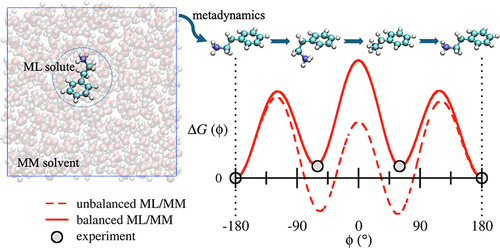
Prediction of Charged Small Molecule Conformations in Solution Using a Balanced ML/MM Potential
Christopher D. Williams - ,
Neil A. Burton - , and
Richard A. Bryce *
Reliably evaluating the most stable conformations of charged small organic molecules in solution poses a major challenge to computational chemistry, due to limitations in the accuracy or efficiency of conventional methodologies. In this paper, we present a hybrid machine learning/molecular mechanics (ML/MM) potential, with dynamic, conformationally dependent charges on the atoms in the ML region, that can be used to address this important challenge in drug design. By way of a case study, metadynamics-enhanced molecular dynamics simulations were used to compare the performance of several intermolecular potentials in evaluating the solution phase conformational free energy differences of pharmaceutically relevant ligands based on the protonated 2-phenylethylamine scaffold. A straightforward approach to an ML/MM potential, in which the solute’s intramolecular interactions are substituted by an ML model and making no other modifications to the force field, yields results inferior to a conventional fixed-charge MM potential, due to an imbalance created in the intra- and intermolecular interactions. To remedy this shortcoming, we present a new ML/MM potential that combines an accurate, trained-for-purpose ML model (PairFEQ-Net) of the charged ligand, with the polarizable SWM3 MM model of water. Crucially, by employing an empirical parameter to scale gas phase charges, the ML model predicts dynamic, solvent-polarized charges that embed the ligand in the solvent in a more balanced way. This ML/MM potential results in mean absolute errors in the prediction of conformational free energies of just 0.5 kcal mol–1, hence furnishing a possible route to the chemically accurate prediction of the shapes of charged organic molecules in aqueous solution.
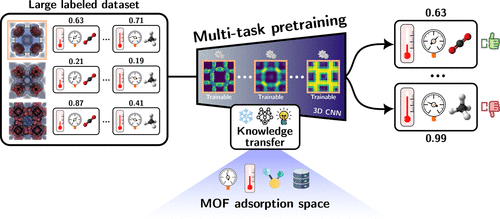
RetNeXt: A Pretrained Model for Transfer Learning Across the MOF Adsorption Space
Antonios P. Sarikas - ,
Konstantinos Gkagkas - , and
George E. Froudakis *
This publication is Open Access under the license indicated. Learn More
Because of their ultrahigh porosity and tunable chemistry, metal–organic frameworks (MOFs) have emerged as leading candidates for gas adsorption applications. Nevertheless, their combinatorial nature induces a vast chemical space, challenging traditional exploration methods. In recent years, machine learning (ML) predictive models have enabled large-scale screening, but they are typically developed for a single adsorption property. This entails that for a new property one must train a model from scratch, a process that requires large amounts of labeled data that are not always available. In our previous work, we demonstrated that combining the potential energy surface─a 3D energy image of the material─with a convolutional neural network improves sample efficiency compared to conventional ML approaches. Here we extend this framework by introducing multitask and transfer learning to foster generalization across gases and conditions, even in data-scarce scenarios. To this end, we developed RetNeXt, a multitask pretrained model on 3.2 million publicly available adsorption-related data, which can be readily adapted to new domains and adsorption tasks. RetNeXt outperforms conventional single-task transfer approaches and achieves up to a 100-fold increase in sample efficiency compared to training from scratch. As such, it can serve as a foundation for future advances in the data-driven adsorption modeling of MOFs.
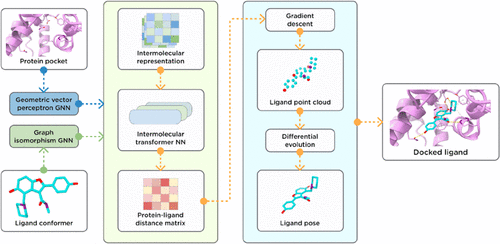
ArtiDock: Accurate Machine Learning Approach to Protein–Ligand Docking Optimized for High-Throughput Virtual Screening
Taras Voitsitskyi - ,
Ihor Koleiev - ,
Roman Stratiichuk - ,
Oleksandr Kot - ,
Roman Kyrylenko - ,
Illia Savchenko - ,
Vladyslav Husak - ,
Semen Yesylevskyy *- ,
Sergii Starosyla - , and
Alan Nafiiev
This publication is Open Access under the license indicated. Learn More
Classical protein–ligand docking has been a cornerstone technique in computational drug discovery for decades but has reached an accuracy and performance plateau. Recently introduced Machine Learning (ML)-based docking methods offer a promising paradigm shift, but their practical adoption is hampered by accuracy-to-speed trade-offs, inadequate benchmarking standards, and questionable chemical validity of predicted poses. In this study, we introduce ArtiDock─an ML-based docking technique optimized for high-throughput virtual screening applications. To evaluate ArtiDock, we developed a dedicated performance and accuracy benchmark for pocket-specific rigid protein–ligand docking, which mimics realistic industrial drug discovery scenarios and is based on the novel PLINDER data set. We demonstrate that ArtiDock is 29–38% more accurate in comparison to leading open-source and commercial classical docking techniques such as AutoDock, Vina, and Glide, while providing a low computational cost. ArtiDock notably excels in challenging docking scenarios involving unbound protein structures and binding sites containing ions and structured water molecules. Additionally, we demonstrated competitive accuracy of our approach at considerably higher throughput compared to a wide range of AI docking and AI cofolding methods using the PoseX benchmark. Our results show that ArtiDock could be considered as a method of choice in high-throughput virtual screening scenarios.
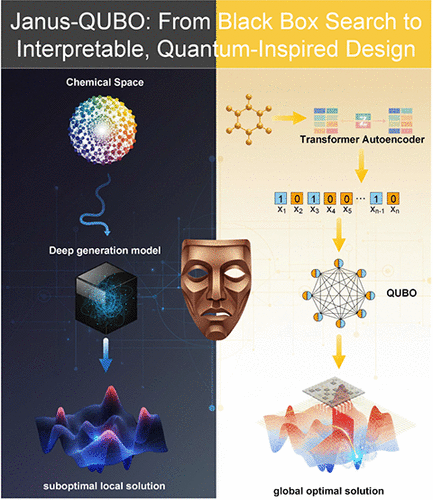
Janus-QUBO: A Duality-Aware Framework for Navigating Chemical Space with a Tunable Quantum-Inspired Landscape
Dinghao Liu - ,
Wenyu Zhu - ,
Yuanpeng Fu - ,
Xinyi Wang - ,
Yuchen Zhou - ,
Mengzhen Guo - , and
Jun Liao *
Discovering novel molecules within the vast chemical space is a central scientific challenge, increasingly delegated to deep generative models. However, the prevailing “black box” paradigm, built on continuous latent spaces, faces a fundamental mismatch between smooth optimization landscapes and inherently discrete molecular structures, often limiting global exploration. To overcome these limitations, we introduce Janus, a framework that recasts molecular design as a transparent, physics-inspired combinatorial optimization problem. At its core, Janus employs a Transformer-based autoencoder with a regularized binary bottleneck to map molecules into a compact discrete latent space. This representation enables the reformulation of molecular generation and optimization as a Quadratic Unconstrained Binary Optimization (QUBO) problem. This approach unlocks synergistic capabilities. For molecular generation, Janus leverages classical and quantum annealers to efficiently traverse the structured energy landscape, enabling the global discovery of diverse chemical scaffolds. Crucially, for molecular optimization, it moves beyond blind search by utilizing quantifiable feature interactions as machine-discovered SAR rules. This allows for rational, interpretable optimization─selectively modifying latent bits to enhance properties. Benchmarking against state-of-the-art methods reveals that this approach achieves superior multiobjective performance while preserving scaffold integrity, avoiding the structural fragmentation common in heuristic baselines. We validate the feasibility of the workflow on a quantum annealer and demonstrate its efficacy in drug-like property optimization. By unifying powerful combinatorial exploration with deep model interpretability, Janus establishes a robust framework for rational, quantum-assisted molecular design.
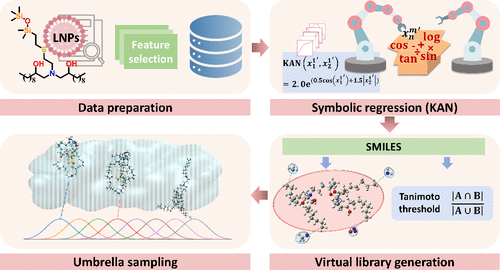
Accelerating Siloxane-Based Ionizable Lipid Design for LNPs with Data-Efficient Kolmogorov–Arnold Networks
Yujing Zhao - ,
Juntao Wang - ,
Yuxin Song - ,
Qilei Liu *- , and
Jiaqi Lin *
Ionizable lipids are fundamental to the efficacy of lipid nanoparticles (LNPs) in pivotal areas including mRNA vaccines. Their development, however, is hindered by intricate structure–property relationships and limited experimental data. To address these challenges, this study proposed a small-data-driven framework that pioneered the use of Kolmogorov–Arnold networks (KANs)─a symbolic regression-based machine learning (ML) approach─to accelerate the discovery of novel siloxane-based ionizable lipids. Using only 36 training samples, the resulting KAN model demonstrated high predictive accuracy for mRNA delivery efficiency (Qcv2 = 0.710), outperforming conventional ML models by an average absolute improvement of 0.627 in cross-validation and yielding explicit mathematical formulas. Combined with virtual screening and umbrella sampling simulations, the framework identified three candidate lipids with superior predicted performance. Molecular dynamics simulations validated that the optimal candidate achieved stronger binding affinity to the endosomal membrane, as evidenced by a 187% reduction (from −1.048 to −3.011 kcal/mol) in the binding free energy minimum compared to the best experimental control. This result aligns with the delivery efficiency predicted by the KAN model. Overall, the proposed framework establishes a data-efficient paradigm for ML-guided ionizable lipid design, bridging symbolic regression with molecular dynamics validation for next-generation LNP therapeutics.
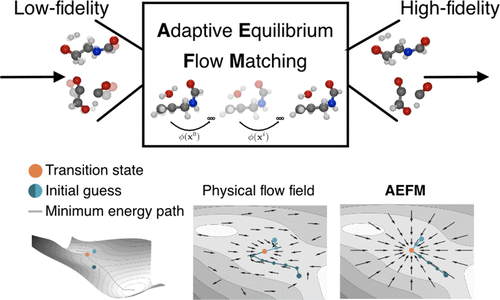
Adaptive Transition-State Refinement with Learned Equilibrium Flows
Samir Darouich - ,
Vinh Tong - ,
Tanja Bien - ,
Johannes Kästner *- , and
Mathias Niepert *
This publication is Open Access under the license indicated. Learn More
Identifying transition states (TSs), the high-energy configurations that molecules pass through during chemical reactions, is essential for understanding and designing chemical processes. However, accurately and efficiently identifying these states remains one of the most challenging problems in computational chemistry. In this work, we introduce a new generative AI approach that improves the quality of initial guesses for TS structures. Our method can be combined with a variety of existing techniques, including both machine-learning models and fast, approximate quantum methods, to refine their predictions and bring them closer to chemically accurate results. Applied to TS guesses from a state-of-the-art machine-learning model, our approach reduces the median structural error to 0.077 Å and lowers the median absolute error in reaction barrier heights to 0.40 kcal mol–1. When starting from a widely used tight-binding approximation, it increases the success rate of locating valid TSs by 41% and speeds up high-level quantum optimization by a factor of 3. By making TS searches more accurate, robust, and efficient, this method could accelerate reaction mechanism discovery and support the development of new materials, catalysts, and pharmaceuticals.
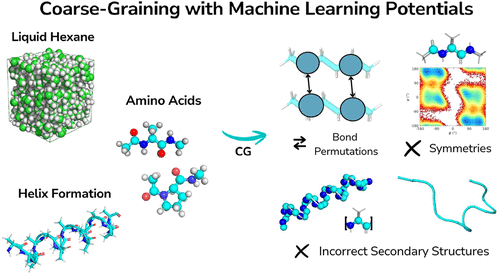
Mapping Still Matters: Coarse-Graining with Machine Learning Potentials
Franz Görlich - and
Julija Zavadlav *
This publication is Open Access under the license indicated. Learn More
Coarse-grained (CG) modeling enables molecular simulations to reach time and length scales inaccessible to fully atomistic methods. For classical CG models, the choice of mapping, that is, how atoms are grouped into CG sites, is a major determinant of accuracy and transferability. At the same time, the emergence of machine learning potentials (MLPs) offers new opportunities to build CG models that can in principle learn the true potential of the mean force for any mapping. In this work, we systematically investigate how the choice of mapping influences the representations learned by equivariant MLPs by studying liquid hexane, amino acids, and polyalanine. We find that when the length scales of bonded and nonbonded interactions overlap, unphysical bond permutations can occur. We also demonstrate that correctly encoding species and maintaining stereochemistry are crucial, as neglecting either introduces unphysical symmetries. Our findings provide practical guidance for selecting CG mappings compatible with modern architectures and guide the development of transferable CG models.
Reactive Neural Network Potential Developed for Asphalt Aging Systems Through Active Learning and Enhanced Sampling
Zhengwu Long - and
Lingyun You *
The atomic-scale mechanisms of asphalt oxidative aging remain poorly understood due to the chemical complexity of asphalt and limitations of conventional methods. Herein, we develop a reactive neural network potential (NNP) for asphalt-oxygen systems via active learning combined with enhanced sampling (well-tempered metadynamics). The NNP achieves quantum-mechanical accuracy while enabling large-scale molecular dynamics simulations. Coupled with multimodal experimental characterization, we uncover a sequential “dehydrogenation-oxidation-crosslinking” reaction network during aging, initiated by thiophene sulfur oxidation and followed by hydrogen abstraction, aromatization, and carbonyl formation. Temperature modulates the reaction landscape, shifting the preference from carbonylation-aromatization at low temperature to hydroxylation-aromatization at high temperature. We identify six parallel pathways with sulfoxide and carbonyl channels being dominant. Free energy analysis reveals that aging proceeds via successive polarization of C–H, O–H, C–O, and S–O bonds with energy barriers significantly lower than C–C cleavage. This work establishes a machine learning-accelerated computational framework for asphalt aging and provides guidance for designing durable pavement materials.
Chemical Information

Toward More Trustworthy QSAR: A Systematic Discussion on Data Set Partitioning
Shangyu Li - and
Peizhe Sun *
With the surge in QSAR model development, concerns about evaluation rigor, particularly regarding the influence of data splitting, have grown. Using five data sets of various sizes, we systematically assessed the effects of random splits (RS), similarity-based splits (SS), and random-seed variability on model generalizability under two scenarios: limited data for chemical screening and standard modeling with ample data. Both the choice of data set partitioning method and the selection of random seeds can substantially affect internal test performance, which may not reliably reflect true predictive capability. Although SS can improve internal test performance in many settings, these gains do not necessarily translate into stronger external generalizability. Moreover, under low sampling ratios, SS may perform worse than RS on both internal and external tests. This challenges the implicit assumption that rational splits optimized for internal performance universally improve model performance. Notably, variability across random seeds was high on internal tests in the smallest data set (R2: 0.453–0.783), whereas on the fixed external data set R2 varied less (0.633–0.672), regardless of applicability domain (AD) filtering. This undermined cross-study comparability and underscored the risk of overly optimistic conclusions. Our findings highlighted that test-set construction must be aligned with real-world application scenarios. Researchers should avoid relying on single or cherry-picked random seeds or unsuitable rational partitioning. Transparent, application-aligned partitioning protocols and AD methods should be employed to emphasize true external generalizability over potentially inflated internal metrics.
Computational Chemistry
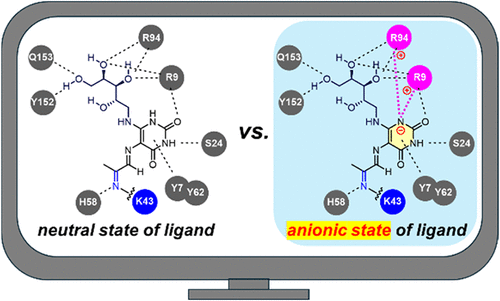
Elucidating Ligand Charge Effects in MR1 Cell-Surface Translocation Using Molecular Simulations
Toshiki Fujii - ,
Mitsugu Araki - ,
Shigeyuki Matsumoto - ,
Biao Ma - ,
Takao Otsuka - ,
Gert-Jan Bekker - ,
Narutoshi Kamiya - ,
Hiroaki Ohno - ,
Shinsuke Inuki *- , and
Yasushi Okuno *
The major histocompatibility complex class I-related protein 1 (MR1) is an antigen-presenting protein that binds its ligand in the endoplasmic reticulum and presents the resulting complex on the cell surface to regulate mucosal-associated invariant T (MAIT) cell function. The MAIT cells play an important role in infection defense and tissue repair and are involved in various pathological conditions. Therefore, MR1 and its ligands, which act as MAIT cell activation triggers, have garnered increasing attention. Notably, MR1 levels on the cell surface vary greatly depending on its ligands. However, the ligand recognition mechanism of MR1, as well as the structure–activity relationship of its ligands, remains poorly understood. In this study, we conducted computational analyses on the interactions between MR1 and its ligands and determined the stability of the MR1–ligand complex structures to clarify the chemical properties of ligands involved in the regulation of MR1 levels on the cell surface. Our findings provide evidence that covalent and noncovalent ligands bind to MR1 in an anionic state rather than in a previously assumed neutral state, thereby stabilizing the resulting MR1–ligand complex. Furthermore, neutralization of the positive charge derived from Arg9 upon binding of anionic ligands was identified as a key factor that contributes to the enhancement of its levels on the cell surface. Our results lay a strong foundation for further studies delving into the molecular mechanisms of ligand recognition and cell-surface localization of MR1 and will facilitate the design of ligands that activate MAIT cells.
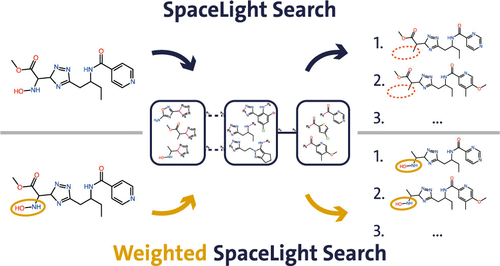
Guiding Similarity Search in Chemical Fragment Spaces with Weighted Fingerprints
Justin Lübbers - ,
Malte Schokolowski - ,
Uta Lessel - ,
Alexander Weber - , and
Matthias Rarey *
This publication is Open Access under the license indicated. Learn More
The introduction of chemical fragment spaces as a way to model large chemical spaces led to readily available compound libraries several orders of magnitude larger than seen before. The possibility of efficient similarity search based on molecular fingerprint comparison in such chemical fragment spaces was introduced by the SpaceLight algorithm for the first time. In this work, we introduce weighted SpaceLight, an enhancement that allows the algorithm to focus the search on important areas of a query molecule, increasing the local similarity while increasing variability in other areas, ultimately providing more structural control over the results. Due to the size of chemical fragment spaces, such customization methodologies become crucial to avoid millions of hits which have to be postfiltered. We demonstrate how weighted SpaceLight produces more molecules that preserve selected substructures during similarity search and how it can be adapted for different search scenarios. Combining global fingerprint similarity with a focus on specific substructures bridges the gap between existing search methods like SpaceLight and SpaceMACS and offers a new level of control for chemical space exploration in drug discovery.

xTB-Based High-Throughput Screening of TADF Emitters: 747-Molecule Benchmark
Jean-Pierre Tchapet Njafa *- ,
Elvira Vanelle Kameni Tcheuffa - ,
Aissatou Maghame Foumkpou - , and
Serge Guy Nana Engo
We validate semiempirical sTDA-xTB and sTD-DFT-xTB methods for high-throughput screening of thermally activated delayed fluorescence (TADF) emitters using 747 experimentally characterized molecules─the largest such benchmark to date. Our framework achieves >99% computational cost reduction versus TD-DFT while maintaining strong internal consistency (Pearson r ≈ 0.82) and reasonable agreement with 312 experimental singlet–triplet gaps (MAE ≈ 0.17 eV). Large-scale analysis statistically validates key design principles: D-A-D architectures outperform other motifs, and optimal torsional angles of 50°–90° maximize TADF efficiency, while PCA confirms a low-dimensional property space. This work establishes xTB methods as cost-effective tools for accelerating TADF discovery.
Computational Biochemistry
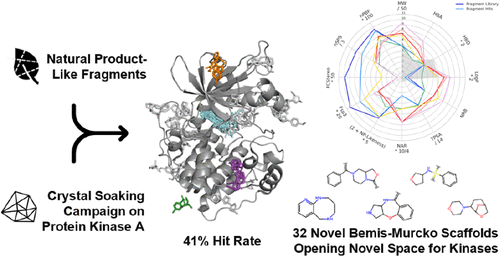
Natural Product-like Fragments Unlock Novel Chemotypes for a Kinase Target─Exploring Options beyond the Flatland
Anna Santura - ,
Janis Müller - ,
Madita Wolter - ,
Ina-Charlotte Tutzschky - ,
Moritz Ruf - ,
Alexander Metz - ,
Anna Sandner - ,
Stefan Merkl - ,
Gerhard Klebe - ,
Serghei Glinca *- , and
Paul Czodrowski *
This publication is Open Access under the license indicated. Learn More
In this study, we utilized a high-performance soaking system of protein kinase A (PKA) to perform a crystallographic screening of a natural product-like fragment library. We resolved 36 fragment-bound structures, corresponding to a hit rate of 41%. Nine fragments bound within the ATP site, nine peripherally, and 18 interacted with both the ATP and peripheral sites. One fragment binds to the same site as the approved allosteric kinase inhibitor asciminib, while another induces an unexpected conformational change. Systematic database mining revealed that both the fragments and their natural product parents have not been previously associated with PKA or kinase activity. A scaffold/chemotype analysis further underscored their novelty. Cheminformatics analyses confirmed that these fragments occupy a distinct chemical space, enriched in saturation, spatial complexity and molecular three-dimensional character compared to kinase binders from reference data sets. These properties have previously been linked to increased selectivity, reduced CYP450 inhibition, and higher overall clinical success rates.
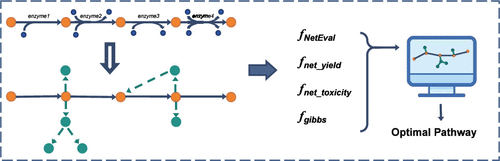
A Novel Metabolic Pathway Design Method Based on Evolutionary Algorithms and Metabolic Network Evaluation
Xin Zhao - ,
Xueying Sun - ,
Tao Zhang *- ,
Shuxin Cui - ,
Yahui Cao - ,
Bingzhi Li - ,
Heng Song - , and
Shuo Zheng
Metabolic pathway design is a fundamental aspect of metabolic engineering, playing a crucial role in the microbial synthesis of high-value compounds. While metabolic engineers recognize the prevalence of branching reactions─side reactions that divert metabolic flux toward nontarget compounds─current automated pathway design tools often focus primarily on linear pathway optimization. This focus may lead to incomplete efficiency assessments and suboptimal pathway selection due to unaccounted metabolic complexity. To address this gap, we introduce a novel metabolic pathway design method, EA-MNE (Evolutionary Algorithm-based Metabolic Network Evaluation). Within the EA-MNE method, we propose a new approach for expanding linear pathways into metabolic networks and two new evaluation criteria: (1) the number of effective branching reactions, which assesses the extent of branching impacts, and (2) the network theoretical yield, which precisely quantifies yield losses caused by branching reactions. Additionally, we integrate four key criteria─the number of effective branching reactions, network theoretical yield, network toxicity, and Gibbs free energy─for metabolic pathway design. This integrated approach provides a systematic solution for addressing branching reaction challenges, significantly improving both the accuracy of pathway evaluation and the synthetic efficiency of microbial systems.

Mechanism of Polyester Hydrolysis by Marine Bacterium PE-H Enzyme: an Atomistic and Thermodynamic Characterization
Samah Nassir - ,
Pedro Paiva - ,
Rui P. P. Neves - ,
Pedro A. Fernandes - ,
Achraf El Allali *- , and
Maria J. Ramos *
This publication is Open Access under the license indicated. Learn More
Polyethylene terephthalate (PET) is a widely used plastic due to its durability and adaptability; however, its resistance to natural degradation has led to severe accumulation in the environment. Recently, a PET-degrading marine bacterium, Pseudomonas aestusnigri, was identified and proposed for possible use in sustainable plastic recycling, particularly the PE-H enzyme, which hydrolyses PET with MHET as the main hydrolysis product. In this work, we investigate the reaction mechanism of PE-H through umbrella sampling hybrid quantum mechanics/molecular mechanics molecular dynamics simulations at the PBE/AMBER level. Our results show a two-stage reaction pathway: acylation and deacylation, both of which proceed stepwise via tetrahedral intermediate formation. We identified deacylation as the rate-limiting step with a free energy barrier of approximately 10.6 kcal·mol–1, which is relatively lower than the barrier of other PET hydrolyses. Our analysis suggests that structural features promoting oxyanion hole formation or enhancing substrate accommodation contribute to lower free energy barrier and promote catalysis. We highlight the role of the S171, H249 and D217 triad responsible for the catalysis of proton transfer and nucleophilic attack reactions, and of F98 and M172 responsible for the formation of the oxyanion hole contributing to the stabilization of tetrahedral intermediates formed along the path. These findings provide mechanistic insights into PE-H catalysis and suggest structural factors that could be extended to other enzymes, providing a basis for future studies to understand enzymatic plastic degradation.
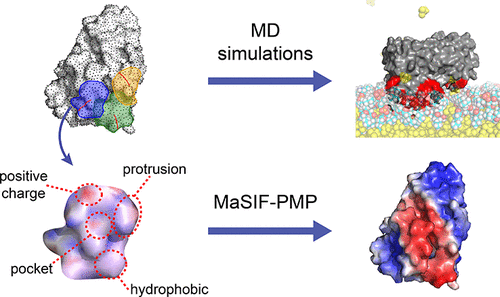
Decoding Protein–Membrane Binding Interfaces from Surface-Fingerprint-Based Geometric Deep Learning and Molecular Dynamics Simulations
ByungUk Park - and
Reid C. Van Lehn *
This publication is Open Access under the license indicated. Learn More
Predicting protein–membrane interactions is a formidable challenge due to the subtle physicochemical features that distinguish membrane-binding regions of a protein surface as well as the scarcity of experimentally resolved membrane-bound protein conformations. Here, we present MaSIF-PMP, a geometric deep learning model that leverages molecular surface fingerprints to predict interfacial binding sites (IBSs) of peripheral membrane proteins (PMPs). MaSIF-PMP integrates geometric and chemical surface features to produce spatially resolved IBS predictions. Compared to existing models, MaSIF-PMP achieves superior performance for IBS classification, while feature ablation studies and transfer learning analyses reveal distinct determinants governing protein–membrane versus protein–protein interactions. We further show that molecular dynamics (MD) simulations can validate model predictions, refine IBS labels, and capture composition-dependent membrane binding patterns. These results establish MaSIF-PMP as an effective framework for IBS prediction and highlight the potential of incorporating conformational dynamics from MD to improve both the model accuracy and biological interpretability.
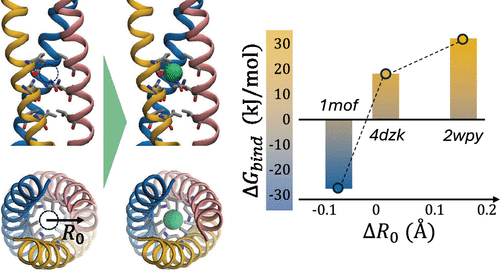
Chloride Binding in Trimeric Coiled Coils: Free Energy and Structural Determinants from Molecular Simulations
Riccardo Nifosì *- and
Luca Bellucci
This publication is Open Access under the license indicated. Learn More
Coiled coils, owing to their simple yet versatile architecture, serve as valuable model systems for both experimental and computational studies in protein science. Whereas the sequence–structure relationships that govern their oligomeric state and stability have been thoroughly investigated, important gaps remain, most notably regarding the role of central chloride ions coordinated by asparagine triads observed in several trimeric coiled-coil (TCC) crystal structures. To investigate the thermodynamics of chloride binding at this site, we performed extensive molecular simulations using metadynamics and alchemical free-energy calculations, both enhanced with replica exchange, to determine the chloride binding free energy (ΔGbind) in three TCCs of similar length but different stability (PDB IDs: 2wpy, 4dzk, 1mof). Despite the nearly identical local coordination environment, the computed ΔGbind values strongly depend on the overall protein structure, with variations in superhelical radius R0 upon ion removal systematically accompanying the observed binding thermodynamics. In particular, both the metastable TCC 2wpy─a variant of the GCN4 leucine-zipper domain previously shown to be unstable in the absence of chloride─and the synthetic design 4dzk exhibit highly unfavorable binding, suggesting that current biomolecular force fields may not fully capture either the stabilizing role of chloride or the conformational ensemble of the unbound state. By contrast, the calculated ΔGbind in 1mof, a fragment of the MoMuLV retroviral transmembrane protein, is favorable and is associated with the presence of an additional C-terminal leash domain that modulates the binding-site environment. These results identify TCCs as critical benchmarks for improving the description of anion–protein interactions and the balance between bound and unbound states in future force-field developments.

XRepDDA: An Interpretable Drug–Disease Association Prediction Framework Leveraging Pretrained Chemical Language Models
Chenyi Zhang - ,
Yun Zuo *- ,
Qiao Ning - ,
Sisi Yuan - ,
Zhaohong Deng - ,
Hongwei Yin - , and
Anjing Zhao
Drug repositioning aims to identify new indications for existing drugs, offering a cost-effective and time-efficient strategy for therapeutic development. Its core challenge lies in accurately predicting potential drug–disease associations (DDAs). However, existing computational approaches often suffer from inadequate drug representation, insufficient modeling of disease semantics, and imbalanced data distributions, which collectively limit predictive accuracy and generalization ability. To address these challenges, we propose an innovative framework, termed XRepDDA, that integrates multimodal feature representation with deep metric learning to improve DDA prediction accuracy and robustness. For drug representation, the SMI-TED pretrained chemical language model encodes SMILES sequences into chemically informative molecular embeddings. For disease representation, a hierarchical semantic graph based on the MeSH ontology is constructed together with a semantic-enhanced graph embedding strategy to capture hierarchical and semantic relationships among diseases. To mitigate class imbalance, we applied the AllKNN adaptive undersampling strategy. The prediction module is built upon an improved ModernNCA architecture, which learns a discriminative embedding space through deep metric learning. Experiments on multiple public benchmark data sets demonstrate that XRepDDA consistently outperforms diverse baseline models, including traditional machine learning, tree-based ensemble, and deep learning methods, achieving AUC and AUPR values of up to 0.9990 and 0.9991, respectively. Furthermore, molecular docking experiments on top-ranked candidate drugs for Alzheimer’s disease and stomach neoplasms provide in silico validation of predictive reliability. To enhance interpretability, a multilevel explainability framework is established, combining SHAP-based global feature attribution with attention mechanisms and molecular perturbation analyses to identify key features and pharmacophores at the local level. These results support the chemical interpretability and the biological plausibility of the predictions.

Molecular Dynamics-Enhanced Sampling Reveals Electrofusion Mechanisms and Pathways
Fei Guo *- ,
Xin Song - ,
Xinyu Peng - ,
Junjie Zhu - , and
Yu Zhang *
While electroporation mechanisms in biomembranes are well-established, the molecular basis of electrofusion remains unclear due to sampling limitations and artificial dismissal of prestalk membrane leakage. By integrating molecular dynamics simulations with enhanced sampling, employing coarse-grained (extended sampling for conformational exploration, requiring rationalized polarizable water settings) and atomistic force fields (high spatial resolution to capture details, necessitating overcoming prevalent sampling artifacts), we elucidate the field-strength-dependent free energy landscape of electrofusion (focusing on stalk formation) and noncanonical fusion pathways. A critical electric field threshold (Ec), validated by water dipole orientation, mass density, and transmembrane potential profiles, governs distinct regimes: for E < Ec, discontinuous aqueous defects emerge on noncontact monolayers, causing sluggish membrane deformation and attenuation of stalk formation energy under increasing field. For E > Ec, prestalk single bilayer leakage (resembling peptide-induced π-shaped pores, hence named) redistributes local fields, triggering cooperative rupture on opposing leaflets to form 2π-shaped fusion pores and inducing a precipitous drop in stalk formation energy with heightened field sensitivity. This threshold mechanism may unify kinetic disparities in synaptic transmission/viral fusion, partly attributable to bias variations imposed by fusion proteins. In summary, our work advances understanding of electrofusion mechanisms and pathways.

How Minor Sequence Changes Enable Mechanistic Diversity in MFS Transporters? An Atomic-Level Rationale for Symport Emergence in NarU
Tanner J. Dean - ,
Jiangyan Feng - , and
Diwakar Shukla *
Closely related membrane transporters can diverge sharply in their modes of transport despite minimal sequence differences, underscoring how minor structural features can alter the transport function. This divergence is exemplified in nitrate and nitrite transport across bacterial membranes, which supports anaerobic respiration and involves the major facilitator superfamily (MFS) transporters NarK and NarU. NarK operates as a nitrate/nitrite antiporter, whereas NarU’s mechanism remains unresolved, with evidence suggesting potential symport activity. Using extensive adaptive molecular dynamics simulations and Markov State Modeling, we mapped NarU’s conformational free-energy landscape and assessed how its behavior contrasts with mechanistic principles established for NarK. NarU follows a similar gating pathway but displays pronounced asymmetry favoring the outward-facing state and stabilizes an apo-occluded intermediate inaccessible to antiporters. This state arises from rotation of an arginine gating pair and a hinged glycine substitution that enhances gate flexibility. These sequence-dependent adaptations alter gating energetics and reprogram the scaffold to permit coupled cotransport. Our results show that the presence of a few strategic residue substitutions in the binding pocket and translocation pathway could alter the transport mechanism of transporters with high sequence and structural similarity.
Pharmaceutical Modeling
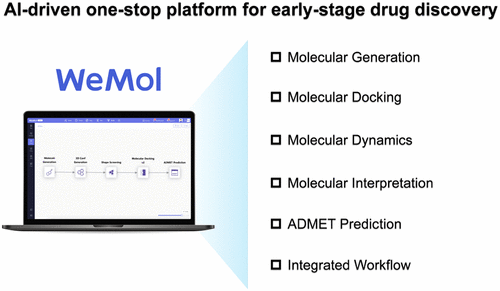
WeMol: A Cloud-Based and Zero-Code Platform for AI-Driven Molecular Design and Simulation
Haihan Liu - ,
Xin Yan - ,
Hao Fang *- ,
Hu Ge *- , and
Xuben Hou *
Artificial intelligence (AI) has demonstrated remarkable potential in reshaping modern drug discovery, yet its widespread adoption is hindered by fragmented tools, high technical barriers, and the lack of user-friendly interfaces. Here, we present WeMol, an AI-driven one-stop molecular computing platform designed to streamline early-stage drug discovery. WeMol integrates a series of modules, covering molecular similarity search, structure-based and AI-enhanced docking, ADMET prediction, molecular generation, and molecular dynamics simulations. The platform features a zero-code, cloud-based interface that enables researchers without programming expertise to construct and execute comprehensive computational workflows. By integrating advanced AI algorithms with practical applications, WeMol lowers the entry barrier for nonexperts and provides a versatile, accessible, and reproducible solution to accelerate early drug design and discovery.
Bioinformatics
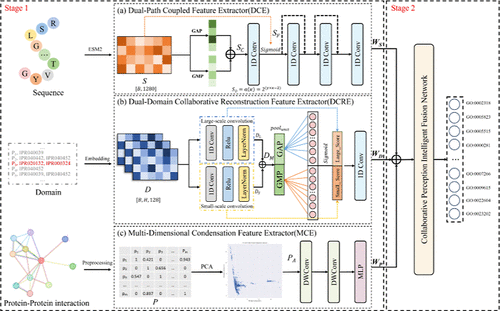
ME-PFP: An Ensemble Learning Approach Fusing Multi-Source Features for Protein Function Prediction
Haoxing Luo - ,
Yue Hu - ,
Chaolin Song - ,
Xinhui Li - ,
Yuyin Ma *- ,
Yurong Qian *- , and
Lei Deng
Proteins, as essential components of living organisms, play a critical role in both drug discovery and disease mechanism research. Multiple empirical studies have shown that there is a significant correlation between protein function and drug targets with therapeutic potential. Therefore, how to accurately and efficiently predict protein function is an urgent issue that needs to be addressed. Existing research faces challenges such as insufficient utilization of protein data and low heterogeneous fusion performance. In this paper, we propose ME-PFP, a novel ensemble learning framework that integrates sequence representations from a protein language model, domain, and protein–protein interaction data to improve protein function prediction. To effectively capture and utilize heterogeneous features, we design three specialized attention-based feature extractors tailored to each data modality. These features are then fused through a dynamic weighting strategy to enable complementary information exchange between different modalities, thereby improving protein function prediction performance. Extensive experiments on benchmark data sets show that ME-PFP significantly outperforms sequence-based and multisource fusion models. Notably, it achieved an average improvement of 13.23% on the human data set and 11.11% on the yeast data set. The experimental results show that this study not only improves the accuracy of protein function prediction, but also promotes progress in the field of computational biology.
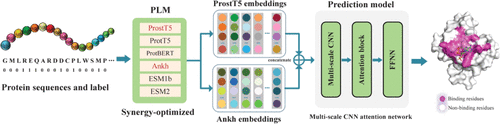
SOPE-MsL: Synergy-Optimized Protein Language Model Embeddings with Multiscale Learning for Interpretable Protein–Small-Molecule Binding-Site Prediction
Zhen Feng - ,
Gen Li - ,
Xiaoya Guan - ,
Hui Yu - ,
Xiaobo Zhou *- , and
Ke Li *
Protein–small-molecule interactions are fundamental to cellular regulation and represent critical targets for therapeutic intervention. Accurate identification of binding residues is essential for elucidating molecular recognition mechanisms and guiding the rational drug design. Experimental approaches, however, are often costly, time-consuming, and limited in scalability, while existing computational methods that rely on handcrafted features or single Protein Language Model (PLM) embeddings fail to capture comprehensive residue-level representations and overlook the potential synergistic effects among diverse PLMs. Here, we present SOPE-MsL, a synergy-optimized approach that integrates PLM embedding fusion with multiscale learning for binding-site prediction. Through a systematic evaluation of representative state-of-the-art PLMs from the ProtTrans, ESM, and Ankh families, we identified ProstT5 and Ankh as the most effective embedding pair. The fused embeddings are then processed by a network that combines multiscale convolutional operations with attention mechanisms, enabling the concurrent modeling of intricate local patterns and long-range dependencies. To address the pronounced class imbalance between binding and nonbinding residues, a weighted focal loss is employed. Beyond predictive performance, t-SNE and SHAP analyses further confirm the advantages of synergistic embedding fusion over single-model representations, providing residue-level interpretability. Extensive experiments across multiple benchmark data sets demonstrate that SOPE-MsL achieves competitive performance and provides a robust and interpretable tool for structure-aware sequence analysis and the identification of protein–small-molecule interaction sites.
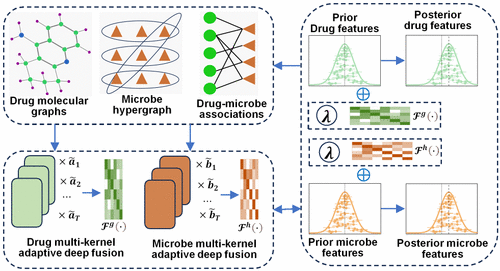
Variational Bayesian Multi-Kernel Adaptive Deep Fusion for Microbe-Related Drug Prediction
Yingjun Ma - ,
MingXu Luo - ,
Liyu Yan - , and
Yuanyuan Ma *
Exploring potential microbe-drug associations (MDAs) not only facilitates drug discovery and clinical treatment but also contributes to a deeper understanding of microbial mechanisms. However, most MDA discoveries rely on biological experiments, which are time-consuming and costly. Therefore, developing an effective computational model to predict novel MDAs is of great importance. In this study, we propose a Variational Bayesian Multi-Kernel Adaptive Deep Fusion (VBMKADF) model for MDA prediction. We first integrate multiomics data to construct drug molecular graphs and a microbe hypergraph. Then, we perform multilayer graph convolution and hypergraph convolution to extract multilevel similarities of drugs and microbes, respectively. An attention mechanism is subsequently introduced to adaptively fuse these multilevel similarities, which are then incorporated into the Bayesian logistic matrix factorization framework to guide the generation of latent variable distributions. Additionally, we develop a variational Expectation-Maximization algorithm for adaptive inference of model hyperparameters and latent variables, which also guides the training of the deep learning model. Experimental results on two benchmark data sets across three scenarios show that, compared to other state-of-the-art methods, VBMKADF achieves higher AUPR, AUC, and F1 scores in both balanced and highly imbalanced settings. Moreover, case studies further confirm that VBMKADF can serve as an effective tool for MDA prediction.
Retractions
Retraction of “The Use of DeepQSAR Models for The Discovery of Peptides with Enhanced Antimicrobial and Antibiofilm Potential”
Jiaying You - ,
Hazem Mslati - ,
Evan F. Haney - ,
Noushin Akhoundsadegh - ,
Robert E.W. Hancock - , and
Artem Cherkasov *

This publication is free to access through this site. Learn More
Mastheads
Issue Editorial Masthead
This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More