ASAP (As Soon As Publishable)
Description:

April 11, 2026
Accelerating Excited-State Calculations of Large Systems with Restricted Boltzmann Machines
Xiang-Yang Liu *- ,
Sheng-Rui Wang - ,
Dong-Yi Xiao - ,
Wei-Hai Fang - , and
Ganglong Cui *
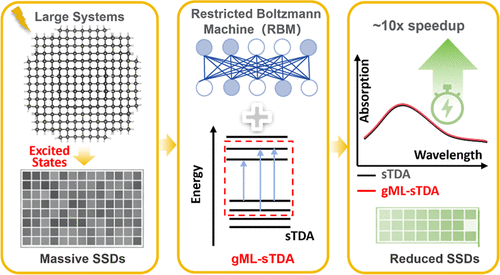
Accurate excited-state electronic-structure calculations for large systems remain challenging due to the prohibitive cost of constructing and screening the configuration space in conventional linear-response approaches. Here, we present a generative machine-learning-accelerated simplified Tamm–Dancoff approximation (gML-sTDA) method that enables efficient excited-state calculations for large systems. The key idea is to use restricted Boltzmann machines (RBM) as generative models to learn the distribution of important singly excited Slater determinants (SSDs) from previously calculated excited-state solutions and to propose new relevant SSDs in an iterative way to avoid exhaustively evaluating matrix elements over the huge configuration space. Benchmarks on finite single-walled carbon nanotube models up to 1114 atoms (16,424 basis functions) show that gML-sTDA reproduces sTDA reference excitation energies with mean absolute errors of ∼0.007–0.011 eV and yields essentially identical absorption spectra while providing substantial speedups. In particular, relative to the tight-threshold sTDA reference protocol, the overall acceleration ratio approaches ∼40× for the largest nanotube and remains close to ∼9× compared with the default-threshold sTDA-cut workflow. Additional applications to a silicon quantum dot, a large black-phosphorus supercell, and a rubrene cluster demonstrate consistent performance, with speedup factors of 7.4, 4.5, and 9.8, respectively, compared to those of the sTDA-cut. These results establish gML-sTDA as a practical and scalable route to excited-state electronic structure calculations in large-scale systems and provide an efficient starting point for applications requiring repeated excited-state evaluations.
April 10, 2026
Phonon-Focusing and Rattler-Mode Interference in Thermal Conductivity Transitions of the Breathing Metal–Organic Framework MIL-53
Masoumeh Mahmoudi Gahrouei - ,
Luping Han - ,
Oreoluwa Adesina - ,
Alathea Davies - ,
Quincy Reynolds - ,
Agnieszka Truszkowska - ,
P. Alex Greaney *- , and
Laura de Sousa Oliveira *
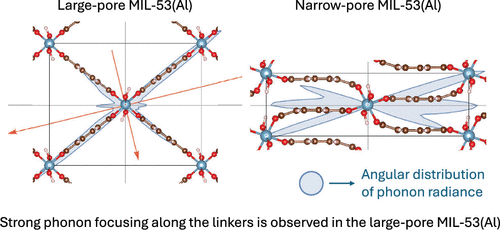
The metal–organic framework (MOF) MIL-53(Al) has a framework geometry with an unconstrained wine-rack mode that enables it to concertina between large-pore (lp) and narrow-pore (np) structures either under pressure or with the uptake of adsorbates. This article presents the results of equilibrium classical molecular dynamics simulations that show that, in these breathing MOFs, the thermal conductivity─an important property for gas sorption in high-surface-area materials─also changes between open and closed conformations. The observed conductivity change differs significantly from that of a network of resistors undergoing the same geometric change. The dispersion relations and phonon group velocities in both the lp and np configurations of MIL-53(Al) are computed using self-consistent-charge density functional-based tight binding (SCC-DFTB). These exhibit rattler-mode behavior and phonon-focusing effects. To provide a mechanistic understanding of these features, a reduced order model of the MOF architecture is presented that captures the lattice dynamics and the band avoidance of acoustic and rattler phonon bands. Moreover, it is shown that the observed phonon focusing is caused by band bending from the rattler modes.

Linear-Scaling and Memory-Efficient Implementation of van-der-Waals Interaction (DFT-D3) for Large Systems
Han-Zhi Luo - ,
Cheng Shang *- , and
Zhi-Pan Liu *
The van der Waals (vdW) interaction is ubiquitous in materials and is long-range by nature. To facilitate vdW-included atomic simulations in large systems with tens of thousands of atoms, we developed LASP-D3, a Compute Unified Device Architecture (CUDA) implementation of the DFT-D3 method on graphics processing unit (GPU) devices, which realizes fast vdW corrections compatible with state-of-the-art machine-learning potential calculations. Our implementation achieves a linear-scaling time complexity, O(N), for large periodic systems, being up to 2 orders of magnitude faster than all current versions for systems above 100,000 atoms, and significantly reduces GPU memory consumption compared to existing PyTorch-based GPU implementations. By combining LASP-D3 with the generalized global neural network potential developed by us, we show that the leading solid electrolyte LiTaCl6 can achieve high conductivity, where the vdW interaction plays a key role in governing Li-ion diffusion and the simulated conductivity reproduces experimental measurements.
April 8, 2026

Study of Arbitrarily Low Shear Rate Rheology Using Dissipative Particle Dynamics
Francesco De Roma - ,
Luca Maffioli - ,
Edward R. Smith *- , and
Antonio Buffo *

This publication is Open Access under the license indicated. Learn More
The use of dissipative particle dynamics (DPD) simulation to study the rheology of fluids under shear has always been of great interest to the research community. Despite being a powerful tool, a limitation of DPD is the need to use high shear rates to obtain viscosity results with a sufficiently high signal-to-noise ratio (SNR). This often leads to simulations with unrealistically large deformations that do not reflect typical stress conditions on the fluid. In this work, the transient time correlation function (TTCF) technique is used for a simple Newtonian DPD fluid to achieve high SNR results even at arbitrarily low shear rates. The applicability of the TTCF on DPD systems is assessed, and the modifications required by the nature of the DPD force field are discussed. The results showed that the standard error (SE) of viscosity values obtained with TTCF is consistently lower than that of the classic averaging procedure across all tested shear rates. Moreover, the SE resulted in a proportionality to the shear rate, leading to a constant SNR that does not decrease at lower shear rates. Additionally, the effect of trajectory mapping on DPD is studied, and a TTCF approach that does not require mappings is consolidated. Remarkably, the absence of mappings has not reduced the precision of the method compared with the more common mapped approach.
Scalable Distributed Memory Implementation of the Quasi-Adiabatic Propagator Path Integral
Roman Ovcharenko *- ,
Xiangyu Xu - , and
Benjamin P. Fingerhut *
This publication is Open Access under the license indicated. Learn More
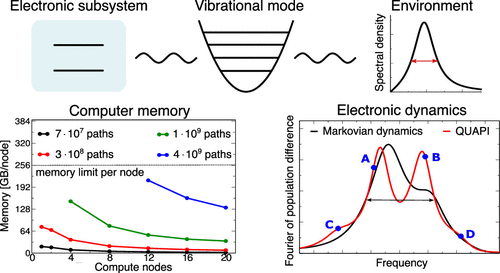
The accurate simulation of dissipative quantum dynamics subject to a non-Markovian environment poses persistent numerical challenges, in particular, for structured environments, where sharp mode resonances induce long-time system bath correlations. We present a scalable distributed memory implementation of the mask-assisted coarse graining of influence coefficients (MACGIC)-quasi-adiabatic propagator path integral (-QUAPI) method that exploits the memory resources of multiple compute nodes and mitigates the memory bottleneck of the method via a new premerging algorithm and a hash-based look-up (hMACGIC-QUAPI) while preserving numerical accuracy. The source code of the hMACGIC-QUAPI implementation is available in a publicly accessible Git repository under GPL license. The developed distributed memory implementation spreads the paths over the computing nodes by means of the MPI protocol, and efficient high level path management is achieved via the hash map-based implementation with constant access time. The efficiency of the implementation is demonstrated in large-scale dissipative quantum dynamics simulations that account for the coupling to a structured non-Markovian environment containing a sharp resonance, a setup for which convergence properties are investigated in depth. Broad applicability and the nonperturbative nature of the simulation method is illustrated via the tuning of the mode resonance frequency of the structured environment with respect to the characteristic system frequency. The simulations reveal a splitting of resonances due to a strong system–environment interaction and the emergence of sidebands due to multiexcitations of the bosonic mode that are not accounted for in perturbative approaches. The simulations demonstrate the versatility of the new hMACGIC-QUAPI method in the presence of strong non-Markovian system bath correlations.
April 7, 2026
Second-Order Perturbative Treatment of Spin–Orbit Coupling and Ground-State Electron Correlation
Yanzhao Lu - ,
Zhifan Wang - , and
Fan Wang *
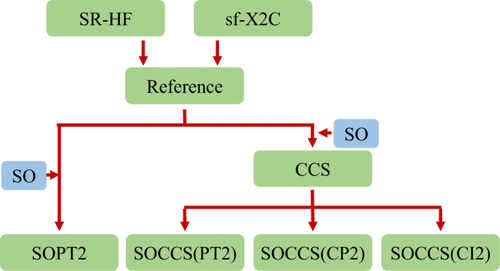
Relativistic effects, particularly spin–orbit coupling (SOC), are essential for accurately describing heavy-element systems, but their efficient treatment within correlated wave function frameworks remains a significant challenge. In this work, we develop four second-order approaches that simultaneously treat electron correlation and SOC within a coupled-cluster-based perturbative framework for closed-shell systems. SOC is introduced as a zeroth-order operator, and correlation effects are included through second-order perturbative expansions starting from a scalar-relativistic Hartree–Fock (SR-HF) reference. The methods consist of one SR-HF-based standard second-order perturbation theory with spin–orbit treated at zeroth order (SOPT2) and three variants derived from spin–orbit coupled cluster singles (SOCCS), all of which reduce to second-order Mo̷ller-Plesset perturbation theory (MP2) in the absence of SOC. Benchmark results for closed-shell sixth- and seventh-row atoms, ions, and halides show that the SOCCS-based schemes significantly improve upon SOPT2, with the CI-like variant achieving the highest accuracy. Accurate 2Π SOC splittings are also obtained using the equation-of-motion coupled-cluster theory at the singles and doubles level (EOM-CCSD) with perturbatively generated cluster operators. Overall, these approaches offer an efficient and reliable framework for incorporating SOC into correlated electronic structure calculations for systems with strong relativistic effects.

Quantum Seniority-Based Subspace Expansion: Linear Combinations of Short-Circuit Unitary Transformations for the Electronic Structure Problem
Smik Patel - ,
Praveen Jayakumar - ,
Rick Huang - ,
Tao Zeng - , and
Artur F. Izmaylov *
Quantum SENiority-based Subspace Expansion (Q-SENSE) is a hybrid quantum-classical algorithm that interpolates between the Variational Quantum Eigensolver (VQE) and Configuration Interaction (CI) methods. It constructs Hamiltonian matrix elements on a quantum device and solves the resulting eigenvalue problem classically. Unlike other expansion-based methods, such as Quantum Subspace Expansion (QSE), Quantum Krylov Algorithms, and the Non-Orthogonal Quantum Eigensolver, Q-SENSE introduces seniority operators as artificial symmetries to construct orthogonal basis states. This seniority-symmetry-based approach reduces one of the primary limitations of VQE on near-term quantum hardware─circuit depth─at the cost of measuring additional matrix elements. The artificial symmetries also reduce the number of Hamiltonian terms that must be measured, as only a small fraction of the terms couple basis states in different seniority subspaces. With all these merits, Q-SENSE offers a scalable and resource-efficient route to quantum advantage on near-term quantum devices and in the early fault-tolerant regime.
StochasticGW-GPU: Rapid Quasi-Particle Energies for Molecules beyond 10,000 Atoms
Phillip S. Thomas - ,
Minh Nguyen - ,
Dimitri Bazile - ,
Tucker Allen - ,
Barry Y. Li - ,
Wenfei Li - ,
Mauro Del Ben - ,
Jack Deslippe - , and
Daniel Neuhauser *
StochasticGW is a code for computing accurate quasi-particle (QP) energies of molecules and material systems in the GW approximation. StochasticGW utilizes the stochastic Resolution of the Identity (sROI) technique to enable a massively parallel implementation with computational costs that scale semilinearly with system size, allowing the method to access systems with tens of thousands of electrons. We introduce a new implementation, StochasticGW–GPU, for which the main bottleneck steps have been ported to GPUs and give substantial performance improvements over previous versions of the code. We showcase the new code by computing band gaps of hydrogenated silicon clusters (SixHy) containing up to 10,001 atoms and 35,144 electrons, and we obtain individual QP energies with a statistical precision of better than ±0.03 eV with times-to-solution of less than 1 h.
How Well Can AI and Physics-Based Simulations Predict the Probability a Cryptic Pocket Is Open?
Si Zhang - ,
Justin J. Miller - , and
Gregory R. Bowman *
Identifying and understanding cryptic pockets remains a compelling goal in drug discovery, as they can offer new avenues for targeting proteins that are otherwise challenging to modulate. While artificial intelligence (AI) methods for structure prediction have been revolutionary, they have not learned enough physics to characterize the rest of a protein’s conformational ensemble, such as structures with cryptic pockets. This limitation has motivated the development of new AI-based methods for characterizing ensembles, or properties thereof, such as AlphaFlow, BioEmu, PocketMiner, and CryptoBank. Here, we benchmark how well these models and physics-based molecular dynamics (MD) simulations can recapitulate the thermodynamics of known cryptic pockets in Ebola VP35 and TEM β-lactamase. These two pockets were chosen because the probability that they are open and the impact of point mutations have been well characterized experimentally. Multiple methods are remarkably successful at predicting whether a mutation will increase or decrease the probability of cryptic pocket opening. However, none can reliably predict the absolute probability of pocket opening. MD is very close for the two wild-type proteins, but all the methods struggle for pockets with small probabilities of opening experimentally (e.g., less than 1%). BioEmu and PocketMiner capture some trends between variants with experimental populations over 1% but have systematic errors and poorer performance for rare pockets (e.g., < 1% open experimentally). These results highlight the promise of AI- and simulation-based strategies for cryptic pocket characterization while pointing to the need for further improvements to achieve robust predictors.
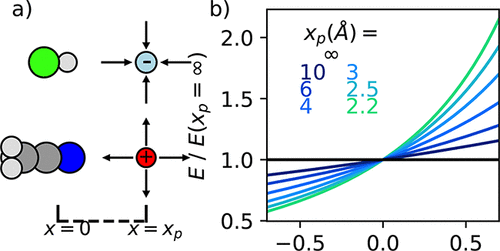
The Interplay of Pauli Repulsion, Electrostatics, and Field Inhomogeneity for Blueshifting and Redshifting Vibrational Probe Molecules
R. Allen LaCour *- ,
Ruoqi Zhao - , and
Teresa Head-Gordon *
Many molecules’ vibrational frequencies are sensitive to intermolecular electric fields, enabling them to probe the field in complex molecular environments. However, it is often unclear whether the probe is responding to the local electric field or other types of intermolecular interactions, inhibiting interpretation of the frequency and effectiveness as probes. This is especially true for molecules whose vibrational frequencies blueshift instead of the more typical redshift in hydrogen bonding configurations. Here, we computationally investigate the causes of redshifting versus blueshifting over a range of vibrational reporters. First, we apply adiabatic energy decomposition analysis to a paradigmatic set of probes, finding that redshifting only occurs when electrostatic interactions are strong enough to overcome the dominant and large blueshifting contribution of Pauli repulsion. Furthermore, we demonstrate that field inhomogeneity can further shift the frequency of many probes substantially to either reinforce or counteract the shift expected from a homogeneous field. We find that redshifting is reinforced by electric field inhomogeneity, otherwise field inhomogeneity further weakens the electrostatic contribution relative to Pauli repulsion, leading to blueshifting. Further calculations indicate that the probe’s response to field inhomogeneity can be understood by considering the mass of the atoms involved in the stretching mode and sign of the electric field. In explaining the interplay of different intermolecular interactions and field inhomogeneity for many probes, our results should enable the use and interpretation of spectroscopic probes and their connection to electric fields in more complex systems.
April 6, 2026
On the Feasibility of Exact Unitary Transformations for Many-Body Hamiltonians
Praveen Jayakumar - ,
Tao Zeng - , and
Artur F. Izmaylov *
Exact unitary transformations play a central role in the analysis and simulation of many-body quantum systems, yet the conditions under which they can be carried out exactly and efficiently remain incompletely understood. We show that exact transformations arise whenever the adjoint action of a unitary’s generator defines a linear map within a finite-dimensional operator space. In this regime, there exists a finite-degree polynomial that annihilates the adjoint map, rendering the Baker–Campbell–Hausdorff (BCH) expansion finite. We identify the role of Lie algebras and their modules in producing finite BCH expansions in all known cases. This perspective brings together previously disparate examples of exact transformations under a single unifying principle and clarifies how algebraic relations between generators and transformed operators determine the polynomial degree of the transformation. We illustrate this framework for previously known cases of efficient unitary transformations including unitary coupled-cluster and Pauli product generators. Using this framework, we propose a new class of Fermionic generators that can be used for efficient transformations. The result establishes sufficient algebraic conditions for when exact unitary transformations are possible and provides new strategies for reducing their computational cost in quantum simulation and constructing feasible unitary transformations.
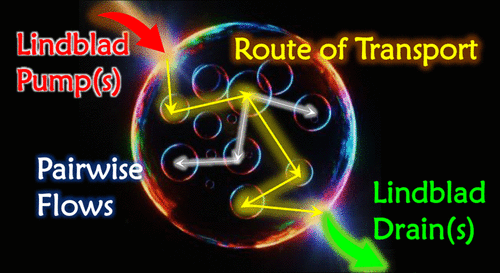
Routes of Transport in the Path Integral Lindblad Dynamics through State-to-State Analysis
Devansh Sharma - and
Amartya Bose *
Analyzing routes of transport for open quantum systems with nonequilibrium initial conditions, such as exciton transport aggregates, is extremely challenging. The state-to-state approach [A. Bose, and P. L. Walters, J. Chem. Theory Comput. 2023, 19, 15, 4828–4836] has proven to be a useful method for understanding transport mechanisms in quantum systems interacting with dissipative thermal baths. However, real systems are often exposed to processes that may lead to either increase or decrease of the number of excitations in the aggregate. Such pumping or draining processes can also affect the routes of transport. We extend the state-to-state analysis to account for approximate Lindbladian descriptions of generic dissipative, pumping and decohering processes acting on a system, which is exchanging energy with a thermal bath. The exchange of energy between the system and the environment is incorporated in a numerically exact manner. This Lindblad state-to-state analysis framework is able to unravel the internal transport pathways along with the effect of the external empirical processes and how the thermal solvents modulate these transport routes. Using this new state-to-state formalism, we demonstrate different mechanistic aspects, including the establishment of steady-state excitonic currents in molecular aggregates under the simultaneous influence of pumps and drains whose dynamics is simulated using the path integral Lindblad dynamics [A. Bose, J. Phys. Chem. Lett. 2024, 15, 12, 3363–3368]. It is especially lucrative that in the absence of such processes, the current method reduces to the standard state-to-state approach. We believe that this Lindblad state-to-state method promises to be a unique tool for understanding the dynamics of open quantum systems subject to a host of additional processes with unprecedented granularity, enabling unique questions to be asked about these systems of great complexity.
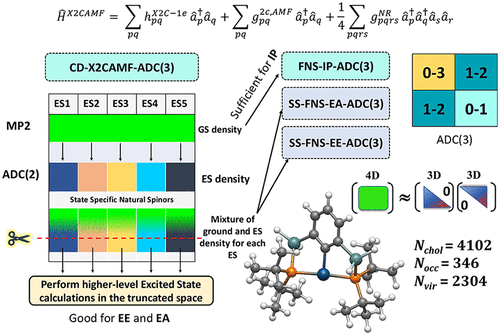
A Low Cost Relativistic Algebraic Diagrammatic Construction Method Based on Cholesky Decomposition and Frozen Natural Spinors for Electronic Ionization, Attachment and Excitation Energy Problem
Sudipta Chakraborty - ,
Kamal Majee - , and
Achintya Kumar Dutta *
We present an efficient relativistic implementation of algebraic diagrammatic construction (ADC) theory up to the third order for the treatment of electronic ionization potentials (IP), electron affinities (EA), and excitation energies (EE) in heavy-element systems using an exact two-component atomic mean-field (X2CAMF) Hamiltonian. The approach combines Cholesky decomposition (CD) of two-electron integrals with frozen natural spinors (FNS) to significantly reduce the computational cost without compromising accuracy. To improve the description of excited states, we have implemented a state-specific frozen natural spinor (SS-FNS) framework and applied it to both electron affinity and excitation energy calculations. In addition to the standard relativistic ADC(3) method, we investigate a semiempirically scaled variant in which the third-order contribution to the ADC secular matrix is multiplied by a scaling factor (x), denoted as FNS/SS-FNS-[ADC(2)+x(3)]. This [ADC(2)+x(3)] approach shows systematic improvements over conventional ADC(3) in a variety of cases. Substantial computational savings are achieved through the use of FNS and SS-FNS schemes when compared to canonical calculations, resulting in significant speedups for ionization, attachment, and excitation energy computations. The current implementation accurately reproduces the canonical four-component ADC(3) results while significantly reducing computational cost. The efficiency and robustness of the method are demonstrated through applications to medium and large-sized molecular systems, including systems with 70 atoms and over 2600 basis functions.
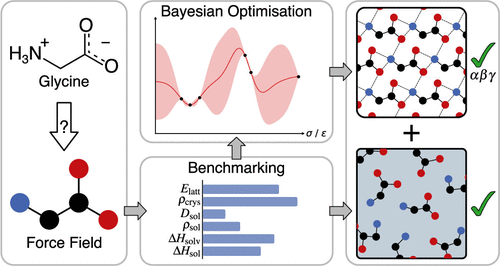
Assessment and Optimization of Force Fields for Glycine Polymorphism and Solution Properties
James W. Meadows - ,
Sharon J. Cooper - ,
Mark A. Miller - , and
Mark R. Wilson *
This publication is Open Access under the license indicated. Learn More
Molecular dynamics studies of glycine crystal growth require a force field that accurately reproduces both solution- and crystal-phase properties for all three ambient-pressure polymorphs. However, many studies use force fields validated only for α-glycine, which often yield poor crystal properties. Here, we extensively evaluate 18 force field variants (10 OPLS and 8 GAFF) and recalibrate the parameters of the best-performing models using multiobjective Bayesian optimization. Crystal lattice energies and densities are computed for α-, β-, and γ-glycine, and the mechanical stability of these polymorphs is tested over a range of temperatures. Solution densities and concentration-dependent self-diffusion coefficients are calculated in conjunction with three water models. Using alchemical hydration free energy calculations, we also obtain solvation and solution enthalpies. Optimization of the nonbonded parameters in OPLS force field variants improves the prediction of crystal properties for all polymorphs simultaneously. We present a force field that correctly reproduces the relative polymorph stability, remains mechanically stable beyond ambient temperatures, and gives excellent agreement with experimental lattice energies, crystal densities, and solution enthalpies. This optimized model is expected to provide accurate insight into the mechanisms controlling glycine polymorphism in complex environments. Moreover, the optimization framework developed here provides a general approach for improving force fields of other molecular crystals.
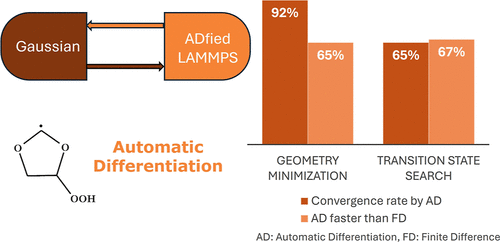
Automatic Differentiation for Enhanced Potential Energy Surface Navigation: Improved Minimum and Transition State Search in Molecular Dynamics
Indu Sekhar Roy - ,
Krep Lukas - ,
Enia Léon Mudimu - ,
Markus Towara - , and
Kai Leonhard *
Reaction models are essential for understanding chemical reactions, but modeling them is a time-demanding process. Automated reaction space exploration techniques, such as ChemTraYzer-TAD, can simplify this process. However, finding transition states (TS) remains a hurdle. TS geometries are crucial for calculating reaction rate constants. Quantum mechanical methods are computationally expensive for TS geometry searches, while reactive molecular mechanics, like ReaxFF, offer faster calculations. Accurate TS searches require second derivatives of energy. State-of-the-art ReaxFF implementations can provide these derivatives only through finite differentiation (FD), which introduces noise. Automatic differentiation (AD) can provide more accurate second derivatives. Hence, this work integrates AD into the classical molecular dynamics code LAMMPS for calculations of second derivatives, presenting ADfied LAMMPS. By interfacing ADfied LAMMPS with the Gaussian computational chemistry suite, LMP-Gau is developed, enabling efficient geometry optimization, frequency calculations, and reaction path following for any force field. LMP-Gau demonstrates improved energy minimization for stable molecules in comparison to standard LAMMPS methods. It is also used successfully to find transition states in 1,3-dioxolane oxidation, demonstrating improved convergence with AD compared to FD.
April 5, 2026

Speeding Up Hartree–Fock in JuliaChem with Density Fitting
John J. Hayes - and
Mark S. Gordon *
This publication is Open Access under the license indicated. Learn More
In this work, the density fitting (DF) approximation is added to the restricted Hartree–Fock (RHF) implementation in the JuliaChem computational chemistry code. Utilizing a DF algorithm that uses symmetry and integral screening, a significant reduction in time to compute the Fock matrix is achieved. The symmetry and screening DF-RHF techniques were adapted to be performed on graphics processing units (GPUs), which are well suited to perform the matrix multiplications that comprise the bulk of the Fock build time in DF-RHF. The JuliaChem DF-RHF GPU algorithm employs a novel approach that automatically switches between two DF-RHF algorithms depending on the number of basis functions in the calculation. The JuliaChem GPU DF-RHF implementation demonstrates up to 2× speedup for Fock build times compared to the existing best-in-class GPU DF-RHF implementation by operating directly on screened intermediate matrices. Due to the high portability of the Julia language code, the JuliaChem CPU and GPU DF-RHF implementations could be benchmarked on a variety of CPU and GPU architectures from multiple hardware vendors.
April 3, 2026

Decomposition of Molecular Charge and Spin Transfer Global Indexes into Atomic Group Contributions
Carlo Gatti *- ,
Yann Danten - , and
Christine Frayret
This publication is Open Access under the license indicated. Learn More
We recently introduced a model for decomposing the global charge transfer (CT) excitation indexes proposed by Le Bahers, Adamo, and Ciofini (Le Bahers, T. J. Chem. Theory Comput. 2011, 7, 2498–2506) into contributions from molecular subdomains (Gatti, C. J. Phys. Chem. A 2022, 126, 6314–6328), together with a software tool, DOCTRINE (atomic group Decomposition Of the Charge TRansfer INdExes), which implements this approach. DOCTRINE has been successfully applied to several excited states (ESs) of a push–pull compound in different solvent environments. In this work, we extend our previous model to spin-polarized systems by introducing, in addition to the global CT excitation indexes, their analogous electron spin transfer (ST) indexes. These can also be decomposed into chemically significant contributions from molecular subdomains. This extension provides a set of related CT and ST descriptors, enabling a visual and quantitative differentiation of the behavior of electronic charge and spin transfers. The updated DOCTRINE_SPIN version of the software now includes computation of ST indexes and their associated descriptors, broadening the applicability of the method to spin-resolved electronic excitations. Our CT and ST decomposition model is applicable to any partitioning of real space, whether fuzzy or disjoint and exhaustive. However, we apply it in terms of chemically relevant molecular subdomains based on the Atoms in Molecules (AIM) Bader’s basins, taking advantage of associating intra- and inter-subdomain contributions with rigorously defined quantum objects that retain clear chemical meaning. The model allows for a quantitative evaluation of subdomain contributions to the CT, the ST, and their excitation lengths, and to the charge- and spin-transfer dipole moments. Although these global indexes can be derived either from electron and spin density increments or from their depletions upon excitation, the subdomain contributions obtained from the two distributions generally differ. This distinction helps to determine whether a given property’s contribution from a subdomain is dominated by one of the distributions or whether both play a significant role. As an initial application of our spin-polarized model extension, we selected a π-conjugated (acceptor–donor–acceptor) compound (TMTQ), composed of a central 1,6-methano[10]annulene (M10A) and 5-dicyanomethyl-thiophene (DT) peripheries in an exo geometry. TMTQ exhibits a singlet–triplet energy gap of only 4.9 kcal/mol, with the singlet state being more stable than the triplet. This small energy gap arises from the different weights of nearly degenerate mesomeric structures with distinct electron delocalization patterns. The electronic charge (and spin) transfers occurring upon excitation of the singlet and triplet ground states (GS) (S0 and T1) to their first five excited states (S1–S5 and T2–T6) are characterized and compared, highlighting their distinct features, the role of ST on CT when both transfers are possible, and the resulting effects on electron and spin delocalizations.
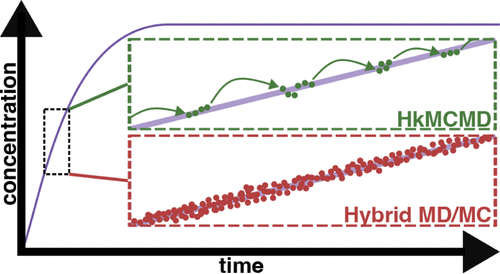
Escaping Vibrational Purgatory: Hybrid kMC/MD Algorithms for Atomistic Simulations of Slow Reaction Chemistry
Dylan M. Gilley - ,
Vignesh Sathyaseelan - , and
Brett M. Savoie *
Atomistic simulations provide essential mechanistic insights into chemical processes, yet many important phenomena in chemistry and materials science occur on time scales that are inaccessible to molecular dynamics. Existing computational approaches force a choice between atomic resolution on relatively short time scales or phenomenological descriptions of long-time behavior. Compounding this difficulty, state-of-the-art hybrid methods inadequately address common phenomena such as spatial heterogeneity and disparate reaction kinetics landscapes. Here, this gap is addressed with the introduction of the Hybrid kinetic Monte Carlo/Molecular Dynamics (HkMCMD) algorithm, which decouples reactive event selection from vibrational dynamics to enable the use of kMC for time evolution. The algorithm incorporates three key components: (1) kMC-based timekeeping that advances time according to reactive events rather than atomic vibrations; (2) dynamic reaction rate scaling that detects and escapes pseudosteady states in which fast reactions dominate; and (3) spatially resolved diffusion calculations that capture heterogeneous transport with a voxel-based analysis. Validation on model systems demonstrates accurate dynamics across nanosecond to second time scales, computational savings of up to 4 orders of magnitude for systems with disparate reaction rates, and a quantitatively accurate treatment of diffusion-limited kinetics. This approach enables atomic-scale investigation of previously inaccessible slow chemical processes while retaining full configurational detail between reactive events.
April 1, 2026
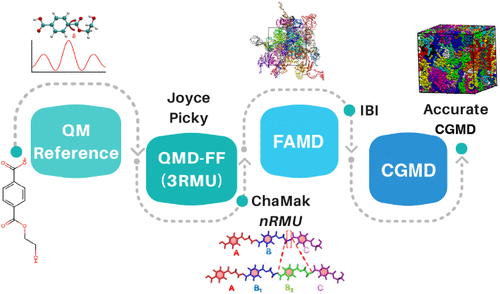
From Quantum Mechanics to Coarse-Grained Models: Bridging the Gap toward Polymer Rational Design
Abderrahmane Semmeq - ,
Andoni Ugartemendia - ,
Alessandro Mossa - ,
Serena Coiai - ,
Giorgia Brancolini *- , and
Giacomo Prampolini *
This publication is Open Access under the license indicated. Learn More
In the rational design of novel polymers, the role of simulation methods based on classical physics is often hindered by the limited accuracy and transferability of the available models, at both the full-atomistic (FA) and coarse-grained (CG) level. Here, we introduce a first-principles-based, fully modular computational protocol for the generation of accurate and consistent FA and CG force fields, tailored on a specific material, and requiring as the sole input the chemical formula of one repeating monomeric unit of the target polymer. The proposed workflow is aimed to connect, across multiple scales, Quantum Mechanical (QM) calculations, FA and CG quantum-mechanically derived force fields (QMD-FFs), and Molecular Dynamics (MD), integrating them into a single, consistent, and reproducible framework. The protocol is tested on poly(ethylene terephthalate) (PET), a well-known polymeric material, widely used in the packaging industry. MD simulations carried out with our FA and CG QMD-FFs are found to significantly outperform standard general-purpose models in predicting key properties such as density, glass transition temperature, as well as the intra- and supra-molecular structure. Such improvement is traced back to the accuracy of the parent QM description by controlling the adherence of the lower level models to the reference set, monitoring this flow of information at each step of the applied procedure. The performances obtained for PET confirm the reliability of a general and tunable approach, which supports systematic refinement and hence stands as a promising tool for in silico design of novel polymers. Subjected to further automation, the procedure could also be integrated into computational machine-learning-based high-throughput schemes, paving the way toward an efficient data-driven polymer discovery.
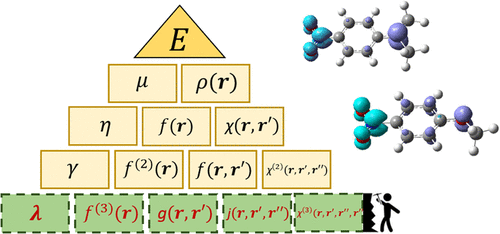
Extending Conceptual DFT to Fourth Order: From Quartic Curvature to Third-Order Fukui Response
Guillaume Hoffmann *- ,
Olivier Aroule - ,
Rémi Grincourt - ,
Henry Chermette - , and
Christophe Morell
We extend the framework of conceptual density functional theory (DFT) to include fourth-order energy derivatives. We present the series of quantum reactivity descriptors at this order for the canonical and the grand canonical ensembles. After introducing a direct computational methodology, we investigate and describe the quartic curvature λ and the third-order Fukui function f(3)(r). These higher-order descriptors capture nonlinear aspects of electronic reactivity that go beyond the conventional concepts of chemical potential, hardness, or Fukui functions. The global descriptor λ, obtained via finite-difference approximations of frontier orbital energies, quantifies the curvature of the chemical hardness and offers insight into the electronic (in)stability of molecular systems under charge perturbations. The local descriptor f(3)(r), derived as the third-order response of the electron density, reveals spatially resolved regions of charge-transfer sensitivity. We apply these descriptors to a series of push–pull organic molecules and open-shell systems, demonstrating their ability to highlight differences in internal charge transfer character and electronic delocalization. The results support the use of fourth-order conceptual DFT tools as chemically meaningful indicators of reactivity beyond the harmonic regime.
March 31, 2026
Limitations of Cluster-Trained MLIPs for Liquid Density and Diffusivity
Viktor Svahn - ,
Ioan-Bogdan Magdău - ,
Samuel P. Niblett - ,
Gábor Csányi - ,
Kersti Hermansson - , and
Jolla Kullgren *
This publication is Open Access under the license indicated. Learn More
Machine-learned interatomic potentials (MLIPs) based on quantum-mechanical data are often used as a means to combine the performance of classical force-fields with the accuracy of electronic structure methods. In this work, MLIPs based on the MACE architecture were trained, starting from two publicly available data sets: one based on periodic structures, and the other based on molecular cluster data. Two rather challenging liquid properties are in focus, density and diffusivity, here for the battery-relevant ethylene carbonate and ethyl methyl carbonate solvents and mixtures thereof. The focus of our study is the uncertainties in the generated MLIP models themselves (calculated for committees of models with different regression seeds and different training set sizes) and how these uncertainties reflect on the MD-simulated target properties. The second focus point is whether these uncertainties are small enough to allow the comparison and assessment of different density functional theory (DFT) functionals; here, only a small number of them are compared, but the workflow opens up for a more extensive assessment of many DFT functionals. We find that all our MACE-MLIPs, both cluster-trained ones and the periodic-structure-trained ones, produce stable 1 ns NPT trajectories, regardless of training set size and cluster composition, but the MACE-MLIPs trained on cluster data (labeled with the hybrid ωB97X-D3 functional) are found to be sensitive to both the random training seed and the data selection, resulting in large uncertainties on the simulated diffusivity and density values.

An Efficient PCM Scheme for ESA Oscillator Strengths within the Unrelaxed TD-DFT Approximation
Jakub Širůček - ,
Boris Le Guennic - ,
Denis Jacquemin *- ,
Benedetta Mennucci - , and
Lorenzo Cupellini *
We propose a TD-DFT protocol for computing unrelaxed excited-state absorption (ESA) oscillator strengths in solution. Our model is formulated within the popular PCM framework and includes both linear-response and state-specific solvent effects through the cLR2 scheme. This protocol can be applied in two regimes: fast and slow. The former corresponds to situations where the time scale of the entire photophysical process is too short to allow relaxation of the nuclear degrees of freedom of the solvent, whereas the latter allows such relaxation. For selected illustrative examples of S1–Sn ESA transitions in organic dyes, we compare solvated and gas phase transition energies, oscillator strengths, and transition dipole moments. This analysis reveals that solvent-induced shifts in oscillator strengths are predominantly driven by the variations of the transition dipole moment. The magnitude of the solvent effect is strongly system- and state-dependent. For transitions for which S0-geometry states could be unambiguously assigned to their S1-geometry counterparts, we found that the two solvation regimes can lead to significantly different effects on the ESA transition properties. This observation is further confirmed by comparing the two solvation regimes, as well as gas phase results, with experimental ESA spectra extracted from transient spectroscopy measurements. Our scheme exhibits clear improvement over the in vacuo outcomes and correctly reproduces the main regions with intense ESA, although an unambiguous choice between the two regimes remains challenging.
March 30, 2026

Nuclear Spin–Spin Coupling Constants from Auxiliary Density Functional Theory
Bernardo Zuniga-Gutierrez *- ,
Luis G. Cota - ,
Jesús N. Pedroza-Montero - ,
Mark R. Pederson - , and
Andreas M. Köster
The working equations for the calculation of indirect spin–spin coupling constants (NSSCCs) within the framework of auxiliary density functional theory (ADFT) are presented. The individual contributions to the NSSCC, namely the Fermi-contact, spin-dipole, paramagnetic spin–orbit and diamagnetic spin–orbit terms, are calculated as analytic second-order ADFT energy derivatives. For the calculation of the perturbed density matrix elements, auxiliary density perturbation theory (ADPT) is used. To improve the computational performance, analytic kernel implementations for the local density approximation and the generalized gradient approximation are employed. To validate our new ADPT NSSCC implementation we compare ADFT calculated NSSCC values with results from other theoretical methods and experiments for a series of small molecules. This validation shows that ADPT NSSCCs are of similar quality as their Kohn–Sham counterparts and compare favorable with post Hartree–Fock results. To demonstrate the computational efficiency of our new implementation and its parallel scaling, benchmark calculations of ADPT NSSCCs in amylose chains are presented. These results show that NSSCCs of nanosystems with more than 1000 atoms can be routinely calculated with the here presented approach employing moderate-sized parallel architectures.

Accelerated Combinatorial Drug Design for Human Immunodeficiency Virus Resistance through Seeded Multisite λ-Dynamics
Paige E. Bowling - ,
Jonah Z. Vilseck - , and
Charles L. Brooks III*
The seemingly endless emergence of drug-resistant mutations poses a significant challenge in developing effective therapeutics, particularly for rapidly evolving pathogens like human immunodeficiency virus (HIV). Understanding how small molecular changes can confer resilience against diverse protein mutants is crucial for next-generation drug design. In the work presented here, we apply multisite λ-dynamics (MSλD) to navigate a chemical space of unprecedented size of over 12,000 protein–ligand combinations of HIV-1 Reverse Transcriptase (HIV-RT) and non-nucleoside inhibitors. We simultaneously explore the indole and indolizine inhibitor scaffolds against the wild-type protein, as well as several key resistance-conferring mutations at residues 181 and 188 within the active site. Our simulations show that smaller substituents, such as hydrogen, fluorine, or chlorine atoms, are preferred at all three sites on both drug scaffolds. We find that the binding affinity rankings of the strongest inhibitors remain highly conserved across the entire mutational panel, supporting the identification of truly resilient universal binders. Furthermore, the indolizine scaffold predicts a more favorable binding landscape across the mutants compared to the indole scaffold, suggesting a higher intrinsic resilience. We also map the limits of therapeutic efficacy, which show the Y188I mutation as a resistance hotspot that markedly reduces binding affinity for all tested compounds. To efficiently sample this vast alchemical and mutational space, we introduce a bias seeding method that leverages solvent-phase free energy simulations, calculations already necessary for estimating the relative free energies. This work establishes a powerful and efficient framework for understanding drug resistance landscapes and guiding the design of robust antiviral therapies capable of overcoming mutation-driven escape in systems like HIV-RT.

AmberTorchPB: A Unified Framework for Poisson–Boltzmann-Based Reaction Field Energy Calculation via Tensor Computation
Yongxian Wu - ,
Qiankang Wang - ,
Robin Jiang - , and
Ray Luo *
This publication is Open Access under the license indicated. Learn More
Electrostatic interactions are pivotal to understanding biomolecular structure and function, with the Poisson–Boltzmann (PB) equation serving as a cornerstone for modeling these phenomena in ionic solutions; however, the application of PB solvers to large-scale macromolecular assemblies is currently impeded by significant computational bottlenecks and a fragmented software ecosystem rooted in legacy architectures, which collectively struggle to exploit the capabilities of modern high-performance computing (HPC). While traditional methods grapple with scalability and hardware adaptation, tensor abstraction utilized in contemporary deep learning has emerged as a transformative paradigm for efficiently managing hardware heterogeneity, memory optimization, and mixed-precision arithmetic. Capitalizing on this advancement, we introduce AmberTorchPB, a unified, extensible, and accelerator-aware framework built upon LibTorch designed to modernize biomolecular electrostatics. By abstracting low-level data management, AmberTorchPB enables a single algorithmic implementation to seamlessly support diverse sparse matrix layouts, numerical precisions, and computing devices. We demonstrate the framework’s versatility by implementing and benchmarking a suite of iterative solvers, thereby providing a robust C++ backend that facilitates rapid prototyping, rigorous benchmarking, and the deployment of high-fidelity electrostatic simulations on heterogeneous architectures.
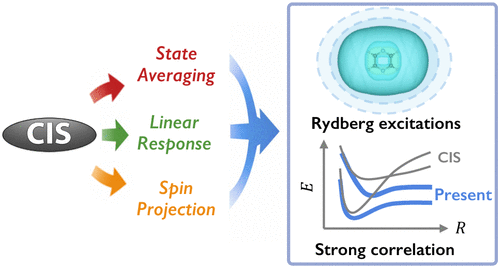
Leveraging Configuration Interaction Singles for Qualitative Descriptions of Ground and Excited States: State-Averaging, Linear-Response, and Spin-Projection
Takashi Tsuchimochi *- and
Benjamin Mokhtar
This publication is Open Access under the license indicated. Learn More
While configuration interaction singles (CIS) provides a computationally efficient description of excited states, it systematically overestimates excitation energies and performs poorly for strongly correlated systems, partly due to the lack of orbital relaxation and the strong ground-state bias of Hartree–Fock orbitals. To address these limitations, we present a unified variational framework that extends CIS by incorporating orbital optimization in state-specific and state-averaged forms (SSCIS and SACIS), linear-response orbital relaxation via a double-CIS scheme (DCIS), and spin-symmetry breaking and restoration (ECIS). In spin-projected state-averaged formulations, standard multistate parametrizations are no longer valid because the projection operator breaks the unitary invariance of orbital rotations and induces nonorthogonal couplings among states. By formulating a rigorous state-averaged objective in the projected subspace, we derive analytic electronic gradients and Hessians and enable robust optimization using a trust-region augmented Hessian algorithm. Benchmark calculations show that spin projection alone significantly exacerbates the CIS overestimation in weakly correlated systems, whereas combining spin projection with state averaging or double-CIS corrections substantially reduces errors, particularly for Rydberg excitations. We further demonstrate that state averaging and spin projection provide complementary and essential benefits in strongly correlated regimes, as illustrated by the bond dissociation of hydrogen fluoride and nitrogen.

Energy-Screened Many-Body Expansion for Protein–Ligand Interactions: Examining Convergence for Metalloenzymes Through Seven–Body Interactions
Paige E. Bowling - ,
Dustin R. Broderick - , and
John M. Herbert *
This publication is Open Access under the license indicated. Learn More
Fragment-based quantum chemistry is a powerful strategy for calculating protein–ligand interaction energies using quantum chemistry methods. Rigorous convergence often requires hundreds of atoms in the protein binding-site model, especially if that model is constructed using distance-based criteria to select amino acid residues, while three- and four-body calculations exhibit instability related to combinatorial proliferation in the number of subsystem calculations. Here, we report an energy-based screening protocol for the many-body expansion applied to protein–ligand interactions, implemented in the open-source Fragme∩t code. Using a combination of aggressive screening based on semiempirical quantum chemistry, with an improved graph-theoretical algorithm to eliminate unimportant subsystems, we are able to perform n-body calculations up to n = 7 using density functional theory in triple-ζ basis sets. Distance cutoffs further reduce the cost without compromising accuracy. Rapid and stable convergence of the many-body expansion is obtained by n = 4, for a pair of metalloenzymes in which a divalent ion coordinates directly to the ligand. As compared to previous results that relied solely on distance cutoffs, oscillations in the n-body corrections are reduced or eliminated, although residual errors remain in one case. This work demonstrates that benchmark-quality protein–ligand interaction energies can be systematically converged using a method with excellent parallel efficiency and scalability.
March 27, 2026
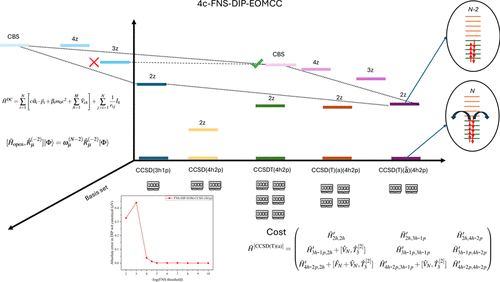
Reduced-Cost Four-Component Relativistic Double Ionization Potential Equation-of-Motion Coupled-Cluster Approaches with 4-Hole–2-Particle Excitations and Three-Body Clusters
Tamoghna Mukhopadhyay - ,
Madhubani Mukherjee - ,
Karthik Gururangan - ,
Piotr Piecuch *- , and
Achintya Kumar Dutta
This publication is Open Access under the license indicated. Learn More
The double ionization potential (DIP) equation-of-motion (EOM) coupled-cluster (CC) method with 4-hole–2-particle (4h-2p) excitations on top of the CC with singles, doubles, and triples calculation, abbreviated as DIP-EOMCCSDT(4h-2p), along with its perturbative DIP-EOMCCSD(T)(a)(4h-2p) approximation, are extended to a relativistic four-component (4c) framework. In addition, we introduce and test a new computationally practical DIP-EOMCC approach, which we call DIP-EOMCCSD(T)(ã)(4h-2p), that approximates the treatment of 4h-2p correlations within the DIP-EOMCCSD(T)(a)(4h-2p) method and reduces the N8 scaling characterizing DIP-EOMCCSDT(4h-2p) and DIP-EOMCCSD(T)(a)(4h-2p) to N7 with the system size N. Further improvements in computational efficiency are obtained using the frozen natural spinor (FNS) approximation to reduce the numbers of unoccupied spinors entering the correlated steps of the DIP-EOMCC calculations according to a well-defined occupation-number-based threshold. The resulting 4c-FNS-DIP-EOMCC approaches are used to compute DIPs for the series of inert gas atoms from argon to radon as well as the vertical DIPs in Cl2, Br2, HBr, and HI, which have been experimentally examined in the past. We demonstrate that, when using complete basis set extrapolations and FNS truncation threshold of 10–4.5, the 4c-FNS-DIP-EOMCCSD(T)(ã)(4h-2p) calculations are capable of predicting DIPs in agreement with experimental data, improving upon their nonrelativistic and spin-free scalar-relativistic counterparts, particularly when examining DIPs characterized by stronger spin–orbit coupling effects.

Correction to “Excited-State Absorption: Reference Oscillator Strengths, Wave Function, and TDDFT Benchmarks”
Jakub Širůček - ,
Boris Le Guennic *- ,
Yann Damour - ,
Pierre-François Loos *- , and
Denis Jacquemin *

This publication is free to access through this site. Learn More
March 26, 2026

Machine Learning-Enhanced Orbital-Free Density Functional Theory
Min Chen - ,
Michele Pavanello - ,
Wenhui Mi - ,
Manabu Ihara - , and
Sergei Manzhos *
Orbital-free density functional theory (OF-DFT) is the ultimate large-scale ab initio method, allowing calculations with 106 atoms and beyond to be done relatively routinely with relatively modest computational resources. The key bottleneck to its wider adoption in applications is the accuracy of kinetic energy functionals (KEF). An important restriction is also the availability and accuracy of pseudopotentials (PP) that can be used with OF-DFT. Machine learning (ML) has recently emerged as a viable approach to construct KEFs and OF-DFT-suited PPs, expanding the domains of applicability of OF-DFT, as well as to predict electron density. We review works to date on ML-based construction of KEFs, PPs, and related works on ML of electron density and discuss the use of various ML methods (from neural networks to kernel regressions to symbolic regressions), the data aspect of the problem, connections to other applications, and perspectives of ML-based OF-DFT going forward.
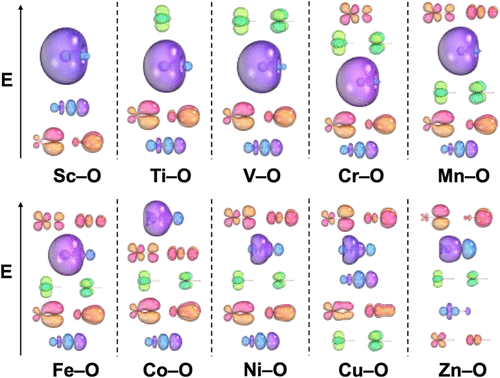
The Electronic Structure and Properties of First-Row Transition Metal Oxides
João G. F. Romeu *- ,
Nickolas A. Joyner - , and
David A. Dixon
The chemical and electronic properties of all diatomic 3d transition metal oxides are investigated to further understand their bonding and growing involvement in atmospheric science. High-level CCSD(T) and spin–orbit icMRCI+Q calculations were used to predict the potential energy curves (PECs) for the ground state and the low-lying states for TiO (3Δ), VO (4Σ–), CrO (5Π), MnO (6Σ+), FeO (5Δ), CoO (4Δ), CuO (2Π), and ZnO (1Σ+). The inclusion of spin–orbit effects is critical for the determination of the ground states in FeO and CoO. Vibrational frequencies of each transition metal oxide are also calculated with CCSD(T) and icMRCI+Q. For the vibrational frequency calculations, the performance of using Hartree–Fock and PW91 reference orbitals was evaluated. The use of PW91 reference orbitals was found to provide better agreement with literature values for the vibrational frequencies and aids in property prediction of highly multireference transition metal oxides. The calculated vibrational frequencies are found to be in reasonable agreement with prior experimental and computational results. Bond dissociation energies (BDEs) of these systems were calculated at the Feller–Peterson–Dixon (FPD) level and are 158.5 kcal/mol (ScO), 158.2 kcal/mol (TiO), 150.1 kcal/mol (VO), 106.2 kcal/mol (CrO), 83.6 kcal/mol (MnO), 97.0 kcal/mol (FeO), 91.4 kcal/mol (CoO), 90.8 kcal/mol (NiO), 65.9 kcal/mol (CuO), and 35.6 kcal/mol (ZnO).

ORCA Meets Python─The ORCA Python Interface OPI
Tim Tetenberg - ,
Hagen Neugebauer *- ,
Christoph Plett - ,
Nakul Santhosh - ,
Markus Bursch *- , and
Christoph Riplinger *
This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
The ORCA program suite is one of the most widely used quantum chemistry software packages. It features a wide range of electronic structure methods and algorithms for the prediction of molecular chemical properties, reactivity, and spectroscopy. In this paper, we present a fully featured ORCA Python Interface termed OPI to drastically increase the accessibility of ORCA’s method portfolio and enable efficient automation of quantum chemical workflows. OPI is an open-source Python library that provides straightforward low-level access to ORCA’s input, execution, and output with a few lines of Python code. In the following, we introduce OPI version 2.0 and its key features, also outlining its general architecture. In addition, we demonstrate its application through several diverse examples of quantum chemical workflows. These examples include a system-dependent optimal-tuning procedure for range-separated hybrid functionals, generation of training data for machine learning purposes, orbital localization and visualization for chemical education, and calculations with density functional ensembles. Finally, we outline the current status of OPI and future plans for its development. OPI is compatible with ORCA ≥ 6.1.1 and Python ≥ 3.11. The project, its code, and its including documentation are available at https://github.com/faccts/opi. OPI is also available through PyPI (https://pypi.org/project/orca-pi).
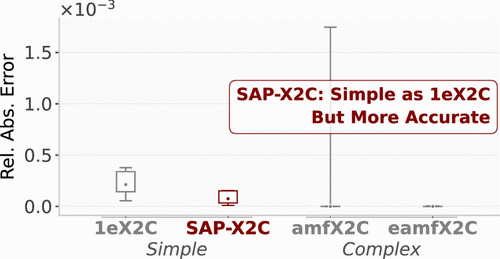
SAP-X2C: Optimally-Simple Two-Component Relativistic Hamiltonian with Size-Intensive Picture Change
Kshitijkumar A. Surjuse - and
Edward F. Valeev *
This publication is Open Access under the license indicated. Learn More
We present a simple relativistic exact 2-component (X2C) Hamiltonian that models two-electron picture-change effects using Lehtola’s superposition of atomic potentials (SAP) [S. Lehtola, J. Chem. Theory Comput. 15, 1593–1604 (2019)]. The SAP-X2C approach retains the low cost and technical simplicity of the popular 1-electron X2C (1eX2C) predecessor but is significantly more accurate and has a well-defined thermodynamic limit, making it applicable to extended systems (such as large molecules and periodic crystals). The assessment of the SAP-X2C-based Hartree–Fock total and spinor energies, spin–orbit splittings, equilibrium bond distances, and harmonic vibrational frequencies suggests that SAP-X2C is similar to the more complex atomic mean-field (AMF) X2C counterparts in its ability to approximate the 4-component Dirac–Hartree–Fock reference.
March 25, 2026
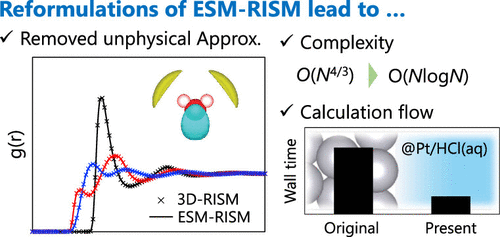
An Efficient Implementation of the ESM-RISM Method for Simulating Electrode/Electrolyte Interfaces
Satoshi Hagiwara *- and
Minoru Otani *
We present an efficient implementation of an effective screening medium method combined with a reference interaction site model (ESM-RISM). The ESM-RISM theory is a straightforward extension of the three-dimensional RISM (3D-RISM) theory, which solves the Laue-represented RISM equation to describe atomistic-scale phenomena at the electrified electrode/electrolyte interface. However, the original ESM-RISM approach employed an approximate solution susceptibility, which leads to deviations of the ESM-RISM results from the 3D-RISM level and requires higher computational costs than 3D-RISM. In this study, we analytically addressed these limitations and applied the present implementation to a water molecule in bulk solution and a Pt(111)/HCl aqueous solution interface. The results for a hydrated water molecule show that the current implementation cures the different approximation levels between ESM-RISM and 3D-RISM. In addition, the analytical reformulation enables us to reduce the computational complexity of evaluating the Laue-represented RISM equation from O(N4/3) to O(NlogN). Subsequently, we re-examined the computational flow of ESM-RISM and found that the present scheme reasonably reduces the wall time required to reach convergence in the overall ESM-RISM calculation. The results of the application to the Pt(111)/HCl aqueous solution interface demonstrated that the current implementation enabled us to capture the properties of the electrified interface.
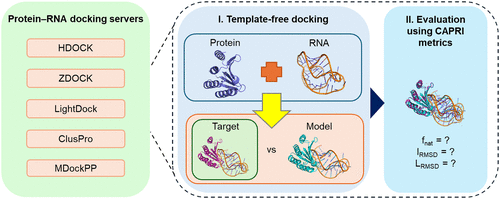
Evaluation of protein–RNA Docking Web Servers for Template-Free Docking and Comparison with the AlphaFold Server
Nicole Hui Hsuen Chong - ,
Eunice Zi Ting Tan - ,
Clara Hui En Tan - ,
Harishiga Ilangovan - ,
Yu Jing Lim - ,
Aarushi Srivastava - , and
Yaw Sing Tan *
Protein–RNA docking is a valuable tool for predicting the structures of protein–RNA complexes, which allow us to understand the structural basis for gene expression and regulation, thus facilitating drug development. Despite the development of several protein–RNA docking programs, the field remains relatively underdeveloped compared to protein–protein docking, and a systematic comparison of these programs in terms of accuracy and efficiency is still lacking. Recent advances in deep learning-based structure prediction, such as AlphaFold 3, offer a promising alternative for modeling protein–RNA complexes. Here, we have compiled a consolidated benchmark data set of 235 protein–RNA complexes (freely available at https://github.com/tanys-group/protein-rna-docking-benchmark), which were curated from PDB structures deposited up to July 2024, to assess the performance of five template-free docking web servers and the AlphaFold Server. Among the docking web servers, HDOCK performed the best, achieving success rates of 31.1% and 44.7% within the top 1 and top 5 predictions, respectively, as assessed by CAPRI (Critical Assessment of PRedicted Interactions) metrics. Although AlphaFold 3 outperformed all the docking web servers with an overall success rate of 87.0% in its top 5 predictions, it failed in nine cases where docking approaches succeeded and showed a markedly lower success rate of 40% for protein–RNA complexes outside its training set, comparable to that of HDOCK (35%). Our study provides valuable insights into the strengths and limitations of current protein–RNA docking servers and AlphaFold 3, offering practical guidance for selecting the appropriate tool for protein–RNA complex structure prediction. These results also suggest that hybrid approaches combining physics-based and machine learning methods hold significant promise for achieving higher prediction accuracy.
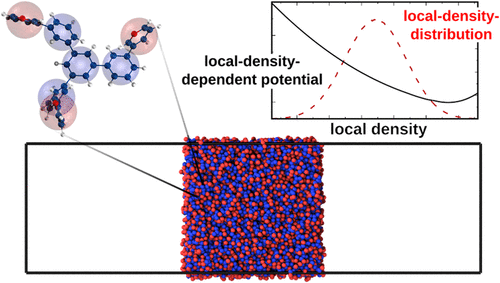
Accurate Coarse-Graining of Conjugated Organic Molecules in Melts and Thin Films Using Density-Dependent Potentials
Sayan Dutta - ,
Maria C. Lesniewski - ,
Muhammad Nawaz Qaisrani - ,
W. G. Noid - ,
Denis Andrienko - , and
Arash Nikoubashman *
Conjugated organic molecules play a central role in a wide range of optoelectronic devices, including organic light-emitting diodes, organic field-effect transistors, and organic solar cells. A major bottleneck in the computational design of these materials is the discrepancy between simulation and experimental time and length scales. Coarse-graining (CG) offers a promising solution to bridge this gap by reducing redundant degrees of freedom and smoothing the potential energy landscape, thereby significantly accelerating molecular dynamics simulations. However, standard CG models are typically parametrized from homogeneous bulk simulations and assume density-independent effective interactions. As a consequence, they often fail to replicate inhomogeneous systems, such as (free-standing) thin films, due to an incorrect representation of interfacial properties. In this work, we develop a CG parametrization strategy that incorporates local-density-dependent potentials to capture material heterogeneities. We evaluate the methodology by simulating free-standing films and comparing interfacial orientational order parameters between all-atom and CG simulations. The resulting CG models accurately reproduce bulk densities and radial distribution functions as well as molecular orientations at the thin film interface. This work paves the way for reliable, computation-driven predictions of atomically resolved interfacial ordering in organic molecular systems.
March 24, 2026
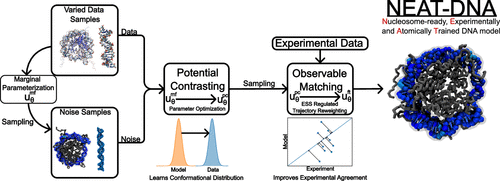
NEAT-DNA: A Chemically Accurate, Sequence-Dependent Coarse-Grained Model for Large-Scale DNA Simulations
Ivan Riveros - and
Bin Zhang *
DNA’s physical properties play a central role in genome organization and regulation, but simulating its behavior across biologically relevant scales remains a major computational challenge. Coarse grained DNA models have enabled faster simulations, yet they often sacrifice chemical accuracy or produce unphysical conformations, limiting their utility for studying genome structure. A key difficulty has been constructing a model that is both efficient enough for large-scale simulations and faithful to the molecular mechanics of DNA. Here, we introduce NEAT-DNA, a new coarse-grained DNA model that resolves longstanding limitations in physical realism and parameter optimization. By combining a physically principled energy formulation with a unified training framework that integrates data from both atomistic simulations and experiments, NEAT-DNA accurately reproduces sequence-dependent structure and flexibility while remaining computationally efficient. This approach marks a significant advance over previous models, which either lacked sequence specificity or introduced distortions inconsistent with experimental observations. NEAT-DNA bridges this gap, offering a high-fidelity yet tractable representation of DNA suitable for exploring chromatin folding. More broadly, it provides a foundation for large-scale simulations that couple molecular detail with gene-level chromatin organization, opening new avenues for predictive modeling in structural genomics.

High-Precision Solvation Free Energy Calculation via Multi-Input Linear Correction in 3D-RISM Theory
Yutaka Maruyama *- and
Norio Yoshida
This study introduces the multi-input linear correction (MILC) method to enhance the accuracy of solvation free energy (SFE) calculations with the three-dimensional reference interaction site model (3D-RISM) theory. While the 3D-RISM theory offers significant computational efficiency compared with molecular simulation-based methods, conventional energy functionals─such as the Singer–Chandler or Gaussian fluctuation formulas─often suffer from systematic overestimation of SFEs. In general, partial molar volume (PMV) corrections are employed to account for errors related to hydrophobic and cavity formation energies. To further improve the prediction accuracy, the MILC method employs a physically motivated data-driven approach that incorporates not only the standard SFE components but also physical quantities obtained when the solute atomic charges are set to zero. Accordingly, the method requires two independent 3D-RISM calculations per solute. The robustness and transferability of the MILC method were rigorously validated using a nested cross-validation protocol on 628 molecules in the FreeSolv database (excluding carboxylic acids). By averaging over 10 conformers per molecule, the method achieved a mean absolute deviation (MAD) of 0.38 kcal/mol relative to the benchmark Bennett acceptance ratio (BAR) method. Furthermore, we demonstrate that the MILC method outperforms conventional volume-based corrections and provides predictive performance superior or comparable to complex machine learning models while maintaining high physical interpretability. This level of accuracy and statistical reliability demonstrates that the 3D-RISM theory with the MILC method is a robust and efficient tool for high-throughput applications in drug discovery and material design.
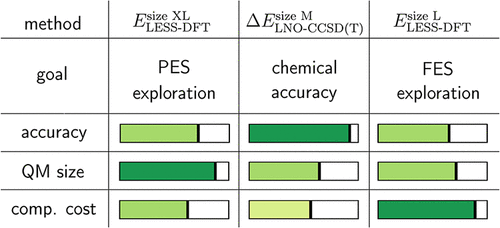
Systematically Improvable and Locality Accelerated Enzymatic Reactivity Modeling: Toward Chemical Accuracy at Affordable Cost
Dénes Berta - ,
József Csóka - ,
Gyula Samu - , and
Péter R. Nagy *
This publication is Open Access under the license indicated. Learn More
Quantum mechanics/molecular mechanics (QM/MM) is the cornerstone of computational enzymology. Herein, we address an outstanding challenge in QM/MM, namely, simultaneous access to accurate QM methodology and a large QM subsystem at an affordable computational cost. First, local natural orbital (LNO)-based CCSD(T) is employed for chemically accurate energetics and as a reference for choosing density functional theory (DFT) models. Next, reliable hybrid DFT methods are selected, with large QM subsystem selections suitable also for reaction barriers. Then, quantum embedding, especially accelerated via our recent local embedded subsystem (LESS) approach, is used to reduce the cost of DFT calculations to a few core hours, even with large QM sizes up to ca. 400 QM atoms. By combining these advanced methods, we propose a Locality Accelerated (by LESS and LNO) and Systematically Improvable (LASI) scheme for QM/MM simulations. It benefits from the strengths of a converged QM size in its DFT component, affordability for many configurations via quantum embedding, and, if needed, CCSD(T) accuracy for energetics. The protocol is validated through the study of challenging, representative, and clinically relevant enzyme-catalyzed phosphate hydrolysis. Based on these results, we establish generally applicable guidelines to set up the components of the LASI protocol. The flexibility and affordability of LASI, both in large-scale QM and QM/MM contexts, make it broadly applicable for the predictive computational description of enzyme reactivity and beyond.
March 23, 2026
QC Lab: A Python Package for Quantum–Classical Dynamics
Alex Krotz - ,
Antonio J. Garzón-Ramírez - ,
Ethan Byrd - ,
Ken Miyazaki - , and
Roel Tempelaar *
QC Lab is an open-source Python package for quantum–classical (QC) dynamics simulations aimed to promote the development of QC algorithms, and their application to a wide variety of relevant model problems. It follows a modular design that facilitates cross-compatibility between algorithms and models. By decomposing algorithms and models into a series of tasks and ingredients that can be substituted and reused, it minimizes development efforts and code redundancy. In this Paper, we introduce the first stable version of QC Lab, and describe its design philosophy.
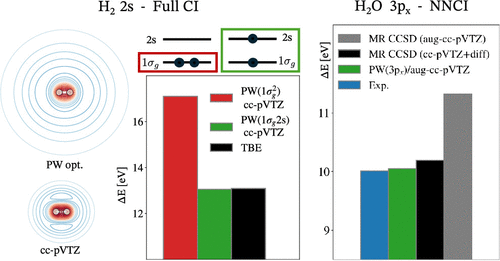
Orbital Optimization and Neural-Network-Assisted Configuration Interaction Calculations of Rydberg States
Gianluca Levi *- ,
Max Kroesbergen - ,
Louis Thirion - ,
Yorick L. A. Schmerwitz - ,
Elvar Ö. Jónsson - ,
Pavlo Bilous - ,
Philipp Hansmann *- , and
Hannes Jónsson *
Rydberg excited states of molecules pose a challenge for electronic structure calculations because of their highly diffuse electron distribution. Even large and elaborate atomic basis sets tend to underrepresent the long-range tail, overly confining the Rydberg state. An approach is presented here where the molecular orbitals are variationally optimized for the excited state using a plane wave basis set in a Hartree–Fock calculation, followed by a configuration interaction calculation. The use of excited state optimized orbitals greatly enhances the convergence of the many-body calculation, as illustrated by a full configuration interaction calculation of the 2s Rydberg state of H2. A neural-network-based selective configuration interaction approach is then applied to calculations of 3s and 3p states of H2O and NH3. The obtained values of excitation energy are in close agreement with experimental measurements as well as previous many-body calculations where sufficiently diffuse atomic basis sets were used. Calculations using atomic basis sets lacking extra diffuse functions, such as aug-cc-pVTZ, give significantly higher estimates due to confinement of the Rydberg states.
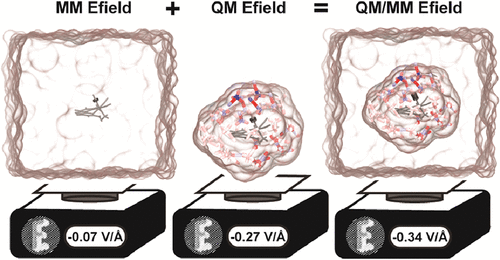
pyEF: A Python Framework for QM and QM/MM Atom-Wise Electric Field Analysis
Melissa T. Manetsch - ,
David W. Kastner - ,
Yuriy Román-Leshkov - , and
Heather J. Kulik *
This publication is Open Access under the license indicated. Learn More
We introduce pyEF, a software package for computing molecular electric fields, electrostatic interaction energies, and electrostatic potentials from quantum mechanical (QM) atom-centered multipole expansions with atom-wise decomposable contributions. We demonstrate the computational efficiency and accuracy of this QM-derived electric field evaluation tool through several tests. To assess the influence of the underlying QM method and charge partitioning scheme on these electrostatic quantities, we analyze over 250 configurations of an acetone solute molecule in five solvents of variable polarity. We find that electric field calculations are highly sensitive to the choice of charge partitioning method. Even among real-space charge schemes, acetone Stark tuning rates differ by up to a factor of 2. Benchmarking computed solvent dipole moments against experimental bulk values, we conclude that the CM5, ADCH, and Hirshfeld-I charge schemes most reliably capture solvent electrostatics and therefore provide a more faithful foundation for computing electric fields. When constructed from these real-space charges, electric fields are nearly insensitive to basis set size and monotonically increase in magnitude with higher Fock exchange. We also demonstrate efficient convergence of QM electrostatics when more distant molecules are represented solely by MM point charges, reducing computational overhead. Leveraging these findings, we demonstrate the use of pyEF to deduce environmental effects on a transition metal complex from a Ga4L612– nanocage and quantify the dominant role of organic linkers in orchestrating electrostatic preorganization.
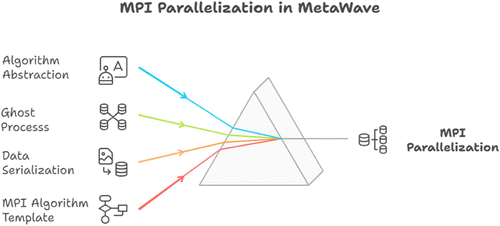
Unified MPI Parallelization of Wave Function Methods: iCIPT2 as a Showcase
Qingpeng Wang - ,
Ning Zhang *- , and
Wenjian Liu *
The integration of quantum chemical methods with high-performance computing is indispensable for handling large systems with modest accuracy or even small systems but with high accuracy. Continuing with the unified implementation of nonrelativistic and relativistic wave function methods within the MetaWave platform (J. Phys. Chem. A 2025, 129, 5170), we present here a unified MPI parallelization of the methods by abstracting every computational step of a method as a dynamically scheduled loop via ghost process, followed by a global reduction of local results from each node. The algorithmic abstraction enables the use of a single MPI template in various steps of different methods. Taking iCIPT2 [J. Chem. Theory Comput. 2021, 17, 949] as a showcase, the parallel efficiencies achieve 94% and 89% on 16 nodes (1024 cores) for the perturbation and whole calculations, respectively. Further combined with an improved algorithm for the matrix-vector product in the matrix diagonalization and an orbital-configuration-based semistochastic estimator for the perturbation correction, this renders large active space calculations possible, so as to obtain benchmarks for the automerization of cyclobutadiene, ground-state energy of benzene, and potential energy profile of ozone. It is also shown that the error of iCIPT2 follows a power law with respect to the number of configuration state functions.
March 21, 2026

Comprehensive Comparison of Molecular Fragmentation Schemes for Proteins
Katharina Rüther - ,
Ken Bunge - ,
Lasse M. Hilmer - ,
Janine Hellmers - , and
Carolin König *
This publication is Open Access under the license indicated. Learn More
Conventional quantum chemical (QC) methods exhibit a steep computational scaling with respect to the number of atoms in the investigated system. Hence, working with larger systems like peptides or even proteins becomes computationally unfeasible with traditional QC methods. One way to overcome this challenge is through molecular fragmentation methods. Many different flavours of molecular fragmentation schemes based on different partitionings have been suggested in the literature, but have hardly been compared numerically. Our group has recently reported a common formalism for molecular fragmentation schemes, which enables a consistent benchmark of different approaches. Here, we assess the performance of the molecular fractionation with hydrogen caps (MFHC), the pair–pair approximation to the generalized many-body expansion (pp–GMBE), the molecules-in-molecules (MIM) approach, and the kernel energy method (KEM) within this general framework. Our benchmark includes single- and multilevel schemes as well as an electrostatic embedding of the fragments in point charges of the whole system. The energies and computational demand of a chosen set of proteins are evaluated with the different methods within the framework. This enables a rare numerical comparison between the different schemes. Of the compared methods, our implementation of pp–GMBE yields the best agreement with supermolecular QC reference calculations, while MFHC with additional pair couplings offers a good cost–accuracy ratio.
March 20, 2026

An Algorithm for Atom-Centered Lossy Compression of the Atomic Orbital Basis in Density Functional Theory Calculations
Anthony O. Lara - ,
Justin J. Talbot - ,
Zhe Wang - , and
Martin Head-Gordon *
Large atomic-orbital (AO) basis sets of at least triple and preferably quadruple-ζ (QZ) size are required to adequately converge Kohn–Sham density functional theory (DFT) calculations toward the complete basis set limit. However, incrementing the cardinal number by one nearly doubles the AO basis dimension, and the computational cost scales as the cube of the AO dimension, so this is very computationally demanding. In this work, we develop and test a threshold-based natural atomic orbital (NAO) scheme in which ϵ-NAOs are obtained as eigenfunctions of atomic blocks of the density matrix in a one-center orthogonalized representation. This enables compression of the AO basis that is optimal for a given threshold, 10–ϵ, by discarding NAOs with occupation numbers below that threshold. Extensive pilot test calculations using the Hartree–Fock functional and taking the converged density matrix as input suggest that a threshold of 10–5 can yield a compression factor (ratio of AO to compressed ϵ-NAO dimension) between 2.5 and 4.5 for the QZ pc-3 basis. The errors in relative energies are typically less than 0.1 kcal/mol when the compressed basis is used instead of the uncompressed basis. Between 10 and 100 times smaller errors (i.e., usually less than 0.01 kcal/mol) can be obtained with a threshold 10–7, while the compression factor is typically between 2 and 2.5.

Nuclear Quantum Effects on the Equation of State of Water: Insights from the Potential Energy Landscape Formalism
Ali Eltareb *- ,
Gustavo E. Lopez *- , and
Nicolas Giovambattista *
This publication is Open Access under the license indicated. Learn More
We apply the potential energy landscape (PEL) formalism for quantum liquids, together with path-integral (PI) computer simulations, to derive the equation of state (EOS) for both equilibrium and supercooled water over a wide range of temperatures and pressures. The PEL-EOS for water, which includes nuclear quantum effects (NQE), is in very good agreement with the PI computer simulations, particularly in the proximity of water’s liquid–liquid critical point (LLCP). Relative to the classical case, including NQE shifts the overall phase diagram of water toward lower temperatures and slightly lower pressures. In particular, the LLCP temperature and pressure are shifted by ΔTc ≈ 18 K and ΔPc ≈ 49 MPa, with a minor change in the LLCP density, Δρc ≈ 0.002 g/cm3. These values of (ΔPc, ΔTc, Δρc) represent, approximately, a maximum shift for the location of the LLCP for H2O due to isotope substitution (H2O → D2O → T2O). Additionally, NQE also affect the shape of the density and LL spinodal lines in the P-T plane. The PEL of (q-TIP4P/F) water is Gaussian, allowing for the evaluation of the configurational entropy SIS(T, V) and Kauzmann temperature, TK(V). NQE reduce the TK(V) of water by 5–20 K depending on the density, consistent with the observed increase in water diffusion coefficient D at low temperatures upon the inclusion of quantum fluctuations. Notably, the Adam–Gibbs relationship, which relates D and SIS, holds remarkably well at all densities studied. From the perspective of the PEL formalism, NQE primarily modify the curvature of water’s PEL basins while the corresponding IS remain unchanged, isomorphic to the IS of classical water. The PEL-based approach employed in this work is versatile and physically intuitive, suitable for calculating the free energy and EOS of quantum liquids beyond water.
March 19, 2026
Complete Active Space Self-Consistent Field with GPU-Accelerated Density Fitting
Ruiyan Wang - ,
Yuanheng Wang - ,
Lixin Lu - ,
Diptarka Hait - , and
Todd J. Martínez *
The complete active space self-consistent field (CASSCF) method is essential for describing complex photochemical processes, but its application in ab initio molecular dynamics is often limited by the computational cost associated with four-center two-electron repulsion integrals (ERIs). We implement the atomic orbital (AO)-based GPU-accelerated density fitting (DF) approximation for CASSCF within the TeraChem software package. Validation on salicylaldimine demonstrates that the DF approximation introduces negligible errors in relative energies, yielding excitation energies accurate to within 10 microHartrees of the integral-direct reference. The DF-CASSCF implementation achieves significant computational speedups, accelerating total energy and gradient calculations by more than an order of magnitude for small- to medium-sized systems with large AO basis sets. We demonstrate the practical utility of this approach through ab initio multiple spawning dynamics simulations of excited-state intramolecular proton transfer (ESIPT). The DF-CASSCF trajectories reproduce the photodynamics of the reference simulations while reducing the total wall time (for a single GPU) by a factor of 3–30, depending on the choice of the basis set. This work significantly lowers the barrier for high-throughput, high-accuracy multireference simulations on modern GPU architectures.
Induced Dipole Calculation with E(3)-Equivariant Neural Networks and Multipole Field Perturbation
Shiyue Yang - and
Jing Huang *
Polarizable force fields based on induced dipoles are widely implemented in molecular dynamics simulations of biological systems to explicitly capture electric induction effects. The iterative computation of induced dipoles suffers from convergence issue in large systems. We describe the implementation of an E(3)-equivariant neural network for predicting induced dipoles in polar solvent systems to avoid the numerical iterations. The neural network is combined with a physics-informed loss function to enable the use of artificially perturbed training data sets. The architecture is validated on water systems, benchmarked across varying densities, system sizes and ice polymorphs, and further integrated into molecular dynamics simulations. We demonstrate that perturbation-based data augmentation substantially enhances model transferability across diverse chemical environments, while physics-informed loss alone offers limited gains in generalization.

PNcsp+: A Periodic Number-Based Crystal Structure Prediction Method Enhanced by Machine Learning
Cem Oran - ,
Riccarda Caputo - ,
Pierre Villars - , and
Adem Tekin *
This publication is Open Access under the license indicated. Learn More
Crystal structure prediction (CSP) is central to materials discovery, yet its efficiency and interpretability remain limited by the vast configurational space and reliance on costly local optimizations. Although template-based and machine-learning (ML) approaches have improved exploration, many approaches still require large data sets, complex similarity metrics, or opaque generative pipelines. In this work, we introduce PNcsp+, an enhanced and chemically interpretable CSP framework that uses the Mendeleev Periodic Number (PN) as a transparent descriptor of elemental similarity. PNcsp+ expands the original implementation through a larger prototype library, an improved data management strategy, and ML-assisted prototype scoring by combining cutting-edge neural network models such as MACE, M3GNet, and ALIGNN-FF. Despite its simplicity, PNcsp+ reaches state-of-the-art performance. In evaluations on the CSPBench data set─a curated set of 180 benchmark crystal structures for assessing CSP methods─our approach surpasses alternative methods by achieving 86.1% space group accuracy and 85.0% structure matching accuracy within the Top-5 predictions, all without structure relaxations. Moreover, our case study on several hybrid systems, including ammonium and methylammonium cations, demonstrated that molecular components emerge autonomously in the predicted lattices, guided solely by PN-derived similarity relationships. Overall, PNcsp+ shows that fundamental periodic trends, combined with targeted ML-based evaluation, offer an efficient, scalable, and interpretable CSP framework, enabling accelerated discovery across both inorganic and hybrid chemical spaces.
Gaussian Charge-Based Electrostatic Embedding Scheme for Solid-State Excited-State Modeling
Alexandre Huguet - ,
Ilaria Ciofini *- , and
Frédéric Labat *
Electrostatic embedding schemes represent an affordable and accurate alternative to more costly approaches to model the excited-state properties of crystalline materials. They are commonly based on point-charge (PC) formalisms, which may lead to numerical instabilities and unphysical electron density (de)localization. In this work, we introduce and validate a Gaussian charge-based (GC) electrostatic embedding scheme for solid-state excited-state calculations based on analytical Ewald lattice summations. The implementation is first validated by systematically comparing electrostatic potentials obtained using PC and GC formalisms at the atomic sites of a broad and representative data set of 530 crystalline structures, covering all space-group types and crystallographic settings. Excellent agreement between PC and GC electrostatic potentials is obtained when the Gaussian width parameter is appropriately chosen. In particular, from the overlap of two GC distributions, we propose a criterion based on the minimal interatomic distance to define an upper bound of the Gaussian width parameter, which still maintains reasonable agreement with the PC-based Ewald potentials for embedded excited-state calculations. The GC embedding scheme is then applied to the modeling of excited-state properties of crystalline imidazole using embedded monomer and hydrogen-bonded dimer models in vacuum, dielectric media, and crystalline environments. The results demonstrate that, although both PC and GC embeddings yield excitation energies in very good agreement with GW-BSE and optimally tuned range-separated hybrid calculations, GC avoids the excessive electron density contraction observed with PC. Overall, the proposed GC-based electrostatic embedding scheme therefore offers a robust and physically sound alternative to PC models for excited-state calculations in solids and constitutes a promising framework for both future methodological developments and practical applications in embedded excited-state calculations.
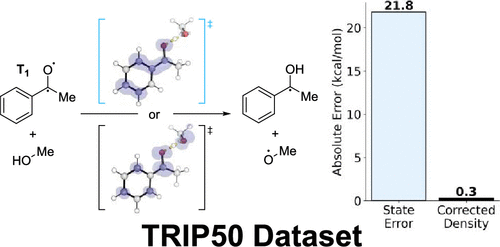
Fundamental Study of Density Functional Theory Applied to Triplet State Reactivity: Introduction of the TRIP50 Data Set
William B. Hughes - ,
Mihai V. Popescu *- , and
Robert S. Paton *
This publication is Open Access under the license indicated. Learn More
The recent development of organic visible-light active photosensitizers has enabled the development of many novel triplet transformations, the mechanistic studies of which often rely on computation due to the short lifetime of the excited state intermediates. However, in contrast to studies of ground state reactivity, there has been little discussion of the best practices when using density functional theory to model triplet state reactions. Here, we report the first benchmark of density functionals on triplet reaction mechanisms. Barrier heights and thermodynamic values were computed for a set of 50 organic reactions using 45 functionals, with reference values obtained using high-level DLPNO–CCSD(T) calculations extrapolated to the complete basis set limit. In the course of this study, we observed a common tendency for triplet SCF calculations to converge non-Aufbau solutions, resulting in catastrophic predictions in both thermochemistry and activation energy barriers and leading to errors as high as 26.4 kcal/mol. Modifications to the initial SCF guess are proposed as a solution to such errors, enabling accurate comparison of functional performance. Range-separated hybrid functionals were found to consistently outperform their non-range-separated versions, while rungs below hybrid meta-GGA produce high errors compared to reference values. We recommend the best-performing single hybrid functionals ωM06, ωB97M, M06–2X, and M05–2X for their balance of high accuracy and computational efficiency.

ReVesicle: Curation and Equilibration of Lipid Vesicles for Mesoscale Simulations
Matteo Castelli - ,
Lorenzo Casalino *- , and
Rommie E. Amaro *
This publication is Open Access under the license indicated. Learn More
Molecular dynamics simulations provide essential atomistic insights into the organization and dynamics of complex biological membranes. However, the equilibration of large-scale, curved lipid assemblies at the all-atom level remains a significant challenge. Standard construction approaches, such as wrapping planar bilayers onto spherical meshes, frequently reverberate into structural instabilities, including membrane holes, infiltrated water, lipid flipping, and nonequilibrated densities, which hinder stable production simulations. Here, we present ReVesicle, an iterative equilibration protocol designed to restore and stabilize quasi-spherical lipid vesicles with complex compositions and large dimensions. The proposed protocol combines selective identification and removal of infiltrated water molecules and flipped lipids with short nonequilibrium MD cycles and anisotropic pressure equilibration. These steps are organized into a modular, iterative sequence that progressively recovers bilayer continuity while preserving the vesicle geometry and enabling global density relaxation. Local vacuum-induced stress generated during nonequilibrium phases promotes lipid tail melting and hole curation, while anisotropic equilibration allows relaxation of box dimensions and system density. To demonstrate the robustness of ReVesicle, we applied the protocol to six biologically realistic vesicle systems: synaptic vesicles, plasma membranes, late endosomes, exosomes, mitochondria-derived vesicles, and the HIV-1 lipid envelope. These systems span diameters from 40 to 105 nm and reach total sizes of up to ∼150 million atoms with heterogeneous and asymmetric lipid compositions. Across all cases, ReVesicle consistently converges to continuous, tightly packed bilayers. Structural and biophysical analyses, including vesicle diameter, sphericity, area per lipid, and lipid acyl-chain order parameters, indicate preservation of quasi-spherical geometry and structural integrity. Overall, ReVesicle provides a reproducible framework for equilibrating large, heterogeneous lipid vesicles suitable for downstream all-atom simulations of complex biological environments.
March 18, 2026
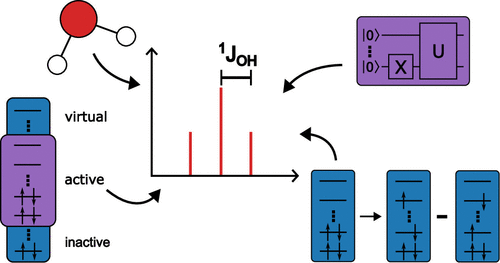
Orbital-Optimized Unitary Coupled Cluster for Indirect Nuclear Spin–Spin Coupling Constants within a Quantum Linear Response Framework
Juliane H. Fuglsbjerg *- ,
Peter Reinholdt - ,
Erik Kjellgren - ,
Phillip W. K. Jensen - ,
Sonia Coriani - ,
Jacob Kongsted - , and
Stephan P. A. Sauer
We present a quantum linear response (qLR) approach within an active space framework for computing indirect nuclear spin–spin coupling constants, a key ingredient in NMR spectra predictions. The method employs the unitary coupled cluster (UCC) ansatz and its orbital-optimized variant (ooUCC), both suitable for quantum computing implementations, to evaluate spin–spin coupling constants via qLR. Test calculations on five small molecules are compared with CASCI, CASSCF, CCSD, and CC3 results. qLR with UCC/ooUCC yields spin–spin coupling constants comparable to those of classical methods. We further examine the role of orbital optimization and find that ooUCC markedly affects the computed couplings; the orbital-optimized results show better agreement with CCSD and CC3. These findings indicate that orbital response is important for accurate NMR coupling predictions within quantum-computing-friendly correlated methods.

Unitary Coupled-Cluster Based Self-Consistent Electron Propagator Theory for Electron-Detached and Electron-Attached States: A Quadratic Unitary Coupled-Cluster Singles and Doubles Method and Benchmark Calculations
Yu Zhang - and
Junzi Liu *
A unitary coupled-cluster (UCC)-based self-consistent electron propagator theory (EPT) is proposed for the description of electron-detached and electron-attached states. Two practical schemes, termed IP/EA-UCC3 and IP/EA-qUCCSD, are developed and implemented within the UCC singles and doubles (UCCSD) framework using the perturbative and commutator-based truncation strategies for the similarity-transformed Hamiltonian H̅. The numerical performance of these UCC-based EPT methods is evaluated primarily using full configuration interaction (FCI) reference data and compared with established approaches, including IP/EA-ADC(3), IP/EA-ADC(4) and IP/EA-EOM-CCSD. Benchmark calculations demonstrate that IP-qUCCSD achieves the highest overall accuracy among Hermitian ionized-state methods for one-hole (1h)-dominated IPs of closed-shell systems, with a mean absolute deviation (MAD) of 0.19 eV and standard deviation (SD) of 0.13 eV. Remarkably, despite the absence of triple-excitation contributions, IP-qUCCSD outperforms the higher-order ADC(4) method. For one-particle (1p)-dominated EA calculations, all tested methods exhibit comparable accuracy.

Li–P–S Electrolyte Materials as a Benchmark for Machine-Learned Interatomic Potentials
Natascia L. Fragapane - and
Volker L. Deringer *
This publication is Open Access under the license indicated. Learn More
With the growing availability of machine-learned interatomic potential (MLIP) models for materials simulations, there is an increasing demand for robust, automated, and chemically informed benchmarking methodologies. In response, we here introduce LiPS-25, a curated benchmark data set for a canonical series of solid-state electrolyte materials from the Li2S–P2S5 pseudobinary compositional line, including crystalline and amorphous configurations. Together with the data set, we present a suite of performance tests that range from conventional numerical error metrics to physically motivated evaluation tasks. With a focus on graph-based MLIP architectures, we then show examples of using this data set to conduct numerical experiments, systematically assessing (i) the effect of hyperparameters on task-level performance and (ii) the fine-tuning behavior of selected pretrained (“foundational”) MLIP models. Beyond the Li–P–S solid-state electrolytes, we expect that such benchmarks and accompanying code can be readily adapted to other material systems.
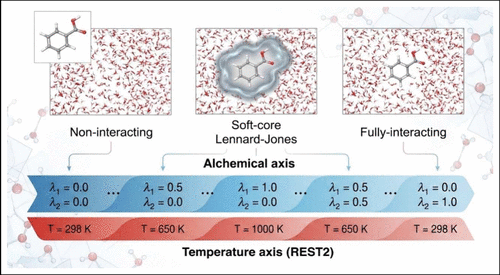
Accurate Hydration Free Energy Calculations for Diverse Organic Molecules With a Machine Learning Force Field
Xiaowei Xie - ,
John L. Weber - ,
Mats Svensson - ,
Ryne C. Johnston - ,
Edward D. Harder - , and
Leif D. Jacobson *
Free energy perturbation (FEP) calculations using classical force fields remain the dominant approach for large-scale, computational drug discovery efforts, but the accuracy is fundamentally limited by simplified forms that cannot quantitatively reproduce ab initio methods without significant fine-tuning. Machine Learning force fields (MLFFs) offer a promising avenue to retain quantum mechanical accuracy with significantly reduced computational cost compared with ab initio molecular dynamics (AIMD) simulations. Thus far, direct applications of ML force fields to FEP calculations lack systematic protocols and extensive benchmarking. In this work, we take a step in this direction by presenting a general and robust workflow for solvation (hydration) free energy (HFE) calculations which is independent of the details of the particular MLFF architecture used. Combining a broadly trained ML force field, Organic_MPNICE, with sufficient statistical and conformational sampling empowered by the solute-tempering technique, affords sub-kcal/mol average errors in HFE predictions relative to experimental estimates. This approach outperforms state-of-the-art classical force fields and DFT-based implicit solvation models on a diverse set of 59 organic molecules and provides a route to ab initio-quality HFE predictions, advancing the use of ML force fields in thermodynamic property prediction.

One-Body Properties and Their Perturbative Accuracy with Aufbau Suppressed Coupled Cluster Theory
Conor Bready - ,
Harrison Tuckman - , and
Eric Neuscamman *
We derived and implemented the calculation of the one-body reduced density matrix for Aufbau suppressed coupled cluster theory, from which excited state natural orbitals and one-body properties, like atomic populations and dipole moments, are obtained. We utilized the natural orbitals to refine the ASCC solution for simple valence and Rydberg systems, exploring the process of repeatedly solving the ASCC equations in successive natural orbital bases to achieve independence from the starting molecular orbitals. For dipole moments in small molecules where high-level comparison data is available, we find that the accuracy of ASCC essentially matches that of linear response and equation-of-motion coupled cluster as long as care is taken to preserve the response’s perturbative completeness.

Accurate Core-Level Ionization Energies from an Affordable Second-Order Approach
Dávid Mester *- and
Mihály Kállay
This publication is Open Access under the license indicated. Learn More
An approach is proposed for the accurate calculation of core-level ionization potentials (IPs) using second-order methods. The assessed theoretical frameworks are based on the iterative second-order algebraic-diagrammatic construction [ADC(2)] and configuration interaction singles with perturbative second-order correction [CIS(D)] methods. Here, our efficient implementations of IP-ADC(2) and IP-CIS(D) [Mester, D.; Kállay, M. J. Chem. Theory Comput. 2023, 19, 3982–3995] are combined with the core–valence separation (CVS) approximation, thereby enabling ionization from the core region. The approaches exhibit highly favorable scaling behavior: the computational cost is practically cubic, with only a mild prefactor that depends on the number of active core orbitals. Furthermore, the resulting wave function-based methods are combined with spin-scaling techniques and successfully extended to double-hybrid (DH) functionals without any necessary modifications in the implementation of the second-order corrections. The performance of the proposed methods was thoroughly assessed in benchmark calculations. Our results demonstrate that the iterative treatment of double excitations is essential, underscoring the necessity of the more advanced DH ansatz. Moreover, the SOS0-PBE0-2/CVS-IP-ADC(2) approach is highly competitive with more expensive higher-level coupled-cluster methods. Finally, the second-order correction introduces only a negligible overhead in the overall computational time─only about 1 min for a 61-atom azafullerene molecule using triple-ζ basis sets─thereby enabling accurate calculations for extended molecular systems.
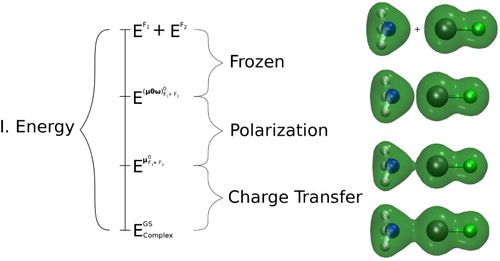
Constrained Multipole Moment Density Functional Theory for the Frozen Contribution in Non-Covalent Complexes
Javier Carmona-Espíndola *- and
José L. Gázquez
The constrained dipole moment density functional theory allows one to control the magnitude and the components of the molecular dipole moment. In this work, we present a methodology which can control the dipole, quadrupole, and octupole moments in a variational and nonempirical way. This development allows us to estimate the individual (dipole, quadrupole, and octupole) and the combined (dipole–quadrupole, dipole–octupole, quadrupole–octupole, and dipole–quadrupole–octupole) multipole contributions in the formation of the ground state of the complex, taking as reference the frozen state of the complex. These contributions allow us to introduce an approximation to the variational frozen state and the corresponding frozen contribution. To test the reliability of the theoretical development, we study four sets of noncovalent complexes from the literature with a total of 24 systems. The individual and the combined multipole contributions results reveal the nature of the interaction between fragments according to these multipole moments, and the rather fast convergence of the multipole expansion, which, according to the results obtained, indicates that by including just the dipole, quadrupole, and octupole moments, one can describe the frozen state reasonably well.
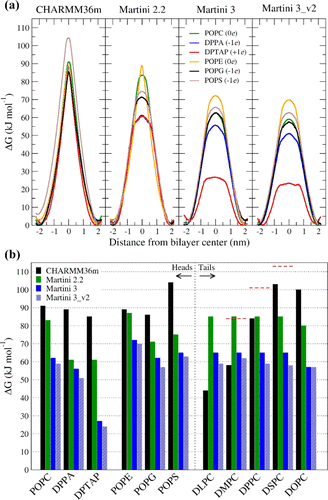
Correction to “Martini 3 Limitations in Phospholipid Flip-Flop”
Ondřej Kroutil - ,
Ladislav Bartoš - ,
Ivo Kabelka - , and
Robert Vácha *
This publication is Open Access under the license indicated. Learn More
March 17, 2026
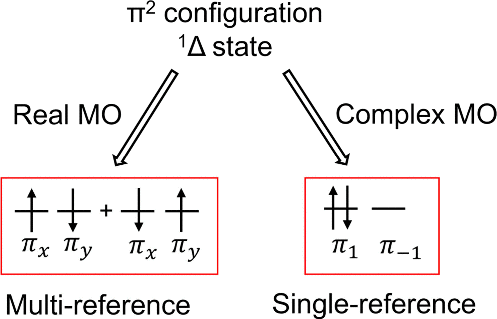
Single-Reference Methods Based on Complex Orbital in Electronic Structure Calculations for High-Symmetry Systems
Shun Li - ,
Zhifan Wang - ,
Zhihua Luo - , and
Fan Wang *
In nonrelativistic and scalar-relativistic electronic structure calculations, molecular orbitals (MOs) are usually chosen as real functions and multideterminant wave functions are required to describe multireference (MR) states. However, certain specific MR states in atoms, linear molecules, and nonlinear molecules possessing real two-dimensional irreducible representations can be represented by a single determinant when complex MOs are employed. For atoms and linear molecules, MOs that are eigenfunctions of the angular momentum operator are used, and the resulting single-determinants states are labeled by the angular momentum quantum numbers. Within density functional theory (DFT), an angular-momentum symmetry-broken method, analogous to the spin-symmetry broken method, is also developed for selected MR states in atoms and linear molecules. The performance of MP2, CCSD, CCSD(T), and DFT using complex MOs is assessed for low-spin states of some p- and d-block atoms, diatomic and nonlinear molecules, and for high-spin states of selected transition-metal diatomic molecules. CCSD(T) with complex MOs generally yields highly accurate results when applicable, while DFT provides reasonable accuracy with appropriately chosen exchange-correlation functionals.

Bias and Its Control in Stochastic Approaches to Electronic-Structure Theory
Pavel Savchenko - ,
Sayak Adhikari - ,
Efrat Hadad - ,
Eran Rabani *- , and
Roi Baer *
This publication is Open Access under the license indicated. Learn More
Stochastic formulations of electronic-structure theory often reduce computational cost by replacing exact contractions with statistical estimates obtained from random samples, a procedure that inherently introduces random fluctuations and systematic bias. The fluctuations decay as M–1/2 with the number of samples M, whereas the bias generated in nonlinear or self-consistent settings decays as M–1 and can remain significant for moderate M. To control this bias we employ the jackknife-2 estimator, which reduces its leading term to O(M−2) with only modest extra cost. We examine bias formation and removal in three settings: (i) stochastic treatments of the Markovian master equation using bundled dissipators, (ii) stochastic Kohn–Sham density functional theory for warm dense hydrogen, and (iii) stochastic evaluation of the Hubbard-model partition function. The first two settings have been presented in earlier works; accordingly, we review them only briefly and focus primarily on the issue of bias control. The Hubbard-model application is entirely new. For this case, we present two approaches: a direct estimator, which has large variance but no bias, and a “midway transition probability” (ΣMTP) estimator, which has smaller variance but introduces bias. Applying the jackknife-2 procedure to the ΣMTP estimator controls this bias and yields a substantially lower total error than the direct estimator. Across all cases, jackknife bias removal markedly improves the accuracy and reliability of stochastic electronic-structure calculations without increasing the computational cost.
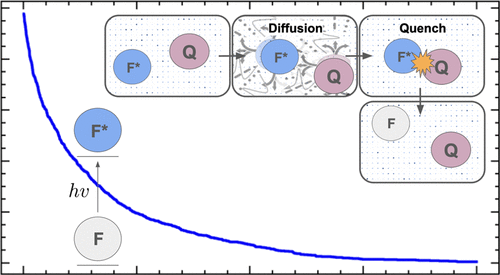
Reaction-Diffusion Dynamics Simulations of Bimolecular Quenching in Solution
Simon A. Liedtke - ,
Martin Trulsson - , and
Petter Persson *
This publication is Open Access under the license indicated. Learn More
A computational method to simulate bimolecular quenching reactions using coarse-grained reaction-diffusion dynamics is presented and applied to the quenching of molecular photosensitizers in solution. The simulations describe photoinduced reactions involving explicit excited states of light-harvesting species together with intrinsic deactivation, as well as collision quenching from separate quencher species. The simulation methodology is applied to time-resolved quenching of light-harvesting Fe(III) complexes in electron-donating solvents as a prototype system for reaction-diffusion dynamics of experimental interest over a wide range of quencher concentrations. The results show clear signatures for the transition from classical diffusion-limited Stern–Volmer dynamics to close-contact quencher-photosensitizer interactions at high quencher concentrations, and the simulations are used to elucidate physically realistic photosensitizer-quenching collision interaction parameters for photoinduced dynamics beyond the classical Stern–Volmer model. The simulation method provides the means to directly model and analyze system kinetics and dynamics beyond standard theoretical equations, opening up significant opportunities to simulate a broad range of reactions in solutions.
March 16, 2026
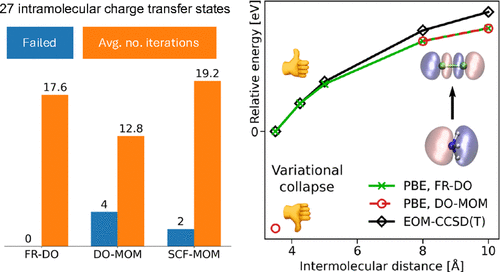
Freeze-and-Release Direct Optimization Method for Variational Calculations of Excited Electronic States
Yorick L. A. Schmerwitz *- ,
Elli Selenius - , and
Gianluca Levi *
This publication is Open Access under the license indicated. Learn More
Variational optimization of orbitals in time-independent density functional calculations of excited electronic states presents a significant challenge, as excited states typically correspond to saddle points on the electronic energy landscape. The optimization can be particularly difficult if the excitation involves significant rearrangement of the electron density, as for charge-transfer excitations. A simple strategy for variational orbital optimization of excited states is presented. The approach involves minimizing the energy while freezing the orbitals directly involved in the excitation, followed by a fully unconstrained saddle-point optimization. Both steps of this freeze-and-release strategy are carried out using direct optimization algorithms with the same computational scaling as ground-state calculations. The performance of the method is extensively assessed in calculations of intramolecular and intermolecular charge-transfer excited states of organic molecules and molecular dimers using a generalized gradient approximation functional. It is found that the freeze-and-release direct optimization approach can avoid variational collapse to spurious, charge-delocalized solutions for cases where conventional algorithms based on the maximum overlap method fail. For intermolecular charge transfer, the orbital-optimized calculations are found to provide the correct dependency of the energy on the donor–acceptor separation without requiring long-range exact exchange, something common time-dependent density functional theory approaches fail to achieve.
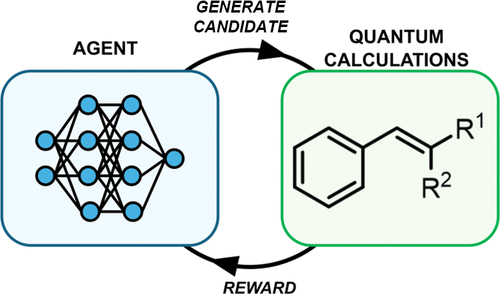
Quantum Chemistry-Driven Molecular Inverse Design of Stable Isomers with Data-Free Reinforcement Learning
Francesco Calcagno *- ,
Luca Serfilippi - ,
Giorgio Franceschelli - ,
Marco Garavelli - ,
Mirco Musolesi - , and
Ivan Rivalta *
This publication is Open Access under the license indicated. Learn More
The inverse design (ID) of molecules remains one of the greatest challenges in chemistry. Machine learning and artificial intelligence (AI) methods are increasingly employed to generate candidate molecules with tailored properties but mostly rely on pretraining over large data sets, which introduces bias. Here, we present a data-free generative AI model called PROTEUS that integrates reinforcement learning with on-the-fly quantum mechanical calculations to enable the de novo design of molecules from first-principles. The AI tool uses a custom syntax and hierarchical learning architecture to navigate the chemical space without prior knowledge, optimizing the desired chemical property. We demonstrate the efficiency of our software by solving complex molecular design tasks related to the maximization of isomerization energy gaps for styrene derivatives. By solving ID problems for which the exact solutions are known, PROTEUS proved to be robust and flexible enough to perform a broad exploration of different chemical spaces while successfully exploiting chemical rewards. This framework opens new avenues for quantum chemistry-driven unbiased molecular design, offering a flexible and scalable strategy to address design challenges in chemistry.

Toward Reaction Vessel Mimicry: Machine Learning-Assisted Automated Exploration of Alkene Polymerization and Its Transferability
Sagar Ghorai - ,
Ruben Staub - ,
Yu Harabuchi - ,
Takashi Nakano - ,
Alexandre Varnek - , and
Satoshi Maeda *
Automated reaction path network exploration and product identification through kinetic analysis are essential for mimicking real reaction vessels. A common practice involves using inexpensive semiempirical methods for initial exploration, followed by energy refinement using more accurate density functional theory (DFT) methods. However, semiempirical methods often yield less accurate reaction kinetics, making them unsuitable for efficient exploration and reliable product prediction. Here, we demonstrate the advantages of iterative training of a delta-learning neural network potential (ΔNNP) for automated reaction path exploration. Using ethylene polymerization catalyzed by the [ZrCp2CH3]+ catalyst as a model system, we achieve DFT-level accuracy by learning the energy difference between DFT and semiempirical methods. Training the ΔNNP on reaction path networks involving one and two ethylene molecules with the catalyst successfully captures all key elementary steps─initiation, propagation, and termination─which can then be extended to study the polymerization of up to six ethylene molecules. Furthermore, a minimally trained ethylene polymerization model provides a robust foundation for propylene polymerization. We also explore the influence of a cocatalyst on the polymerization elementary step network through additional iterative training. Beyond polymerization, this framework can incorporate other ZrCp2-mediated chemistry, such as metallacycle formation, with minimal additional training─yielding several new metallacycle structures. Overall, this iterative training framework is particularly effective for reactions involving repeated analogous elementary steps, such as polymer growth. The approach enables the model to handle increasingly complex reactions, representing an important step toward realistic mimicking of reaction vessels.
Uncertainty-Driven Deep-Ensemble Temporal Convolutional Networks for Predicting Chemical Reaction Dynamics
Zhengzheng Dang - ,
Lei Cheng - ,
Zhichen Tang - ,
Zeyu Zhang - ,
Yu Tian - ,
Jixin Wu - ,
Yide Chang - ,
Chi Chen - , and
Yanming Wang *
Chemical reaction dynamics, critical to advancing technologies in energy, environment, and materials, can in principle be captured via reactive molecular dynamics (RMD). Time-series machine learning algorithms enable prediction of species evolution information hidden in these simulations, but long-horizon autoregressive accuracy often degrades due to error accumulation, and generating sufficiently large and diverse RMD data sets across operating conditions is computationally expensive. Here, we propose DEAL-TCN (deep-ensemble active learning with temporal convolutional networks), an active-learning framework that effectively disentangles the high-dimensional complexity of reaction dynamics spanning species, time, and operating conditions. It adopts the query-by-committee strategy to select informative conditions and leverages one-dimensional temporal convolutions to capture interspecies and long timescale couplings, enabling efficient modeling and long-term prediction of species evolution. In a prototypical case study of Mo–O–S precursors, DEAL-TCN robustly identifies and accurately predicts the concentration evolution of all involved chemical species across orthogonally designed test sets spanning a broad parameter range (1100–1500 K, 2–6 atm, and a feed ratio of 1/25–1/15). Given only the first 50 ps of each trajectory as input, the model attains a mean prediction error of 4.8% at the picosecond level while maintaining a mean error of 18.2% over the subsequent 0.45 ns, representing improvements of ∼29% and 24% over baseline LSTM and Transformer architectures, respectively. Meanwhile, DEAL-TCN outperforms random sampling in 98.8% of active-learning iterations with the same labeling budget. These results underscore DEAL-TCN’s potential as a scalable and generalizable approach for mechanistic discovery, reaction design, and optimization.
Bias-Deletion Metadynamics Revealing Volume–Rotation Coupling Mechanisms in Metal–Organic Frameworks
Konstantin Stracke - and
Jack D. Evans *
Enhanced sampling methods in molecular dynamics simulations often struggle to handle interdependent modes in complex systems. This leads to nonergodic simulations characterized by hysteresis, dependence on initial configurations, and the need for intricate collective variables. We introduce bias-deletion metadynamics, a technique that conditionally deletes biases to enable repeated exploration of transition pathways and systematic refinement of the sampled configurational space. The underlying free-energy surface is accurately recovered by rescaling the resulting probability distribution by the number of repetitions. We benchmark bias-deletion metadynamics on alanine dipeptide, demonstrating its ability to handle recurrent rotational motion where conventional metadynamics can fail. We further show its power by resolving the distinct phases of CAU-13 using only a volume bias, a known challenge for other methods. Finally, by applying this approach to a series of isoreticular metal–organic frameworks (MIL-53(Al), NU-2002, MIL-cub, and NU-2000), we uncover the intricate coupling between linker rotation and framework volume, revealing how linker dimensionality dictates volume-specific rotational preferences.
March 15, 2026
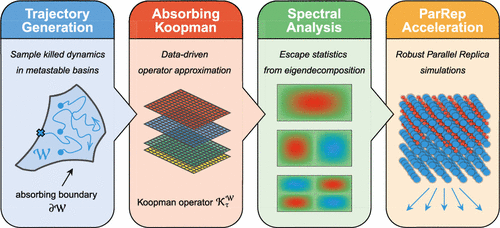
Data-Driven Characterization and Acceleration of Metastable Dynamics Using Koopman Operators
Julien Luzzatto - ,
Feliks Nüske - ,
Nicolas G. Hadjiconstantinou - , and
Danny Perez *
Many physical and biological systems evolve through metastable dynamics characterized by long intervals during which the trajectory remains confined to a small region of the configuration space punctuated by rare but rapid transitions between such regions. Accurately quantifying both the local relaxation and the first-escape behavior from each metastable set is central to many applications including enabling the simulation of long-time dynamics. In this work, we extend well-established data-driven methods for estimating Koopman operators to the setting of quasi-stationary distributions (QSDs) by enforcing absorbing boundary conditions on metastable states. We show that this absorbing Koopman formulation reliably recovers the spectral properties governing relaxation and escape using only short-trajectory data. Finally, we show how these spectral estimates naturally couple with a general parallel-in-time simulation scheme, enabling rigorous and substantial extensions of the time scales accessible to direct simulation of complex metastable systems.
March 12, 2026
Energy Landscape Analysis of Membrane Proteins Using NMR-Based Hybrid Restraint Potentials
Diksha Dewan - ,
Yifei Wang - ,
Alfonso De Simone - , and
David J. Wales *
This publication is Open Access under the license indicated. Learn More
Most biomolecular simulations depend on the quality of empirical force fields, and the use of hybrid restraint potentials has emerged as a promising approach. In this contribution, we extend the application of hybrid potentials to membrane proteins by developing optimized restraints derived from experimentally determined NMR data. NMR chemical shift, chemical shift anisotropy, dipolar coupling, and NOE distance information are combined with appropriately weighted empirical force fields to study two transmembrane systems, namely sarcolipin and phospholamban. To remedy the problems of rare events and broken ergodicity, the energy landscape framework, including basin-hopping global optimization and discrete path sampling, is employed for exploring the underlying energy landscapes. Much of the appeal of the hybrid potential approach is the ability to study membrane proteins in the absence of conventional explicit or implicit solvent and lipid molecules, thereby simplifying the sampling of complex biomolecular conformational spaces. Our results suggest that the hybridization of NMR constraints as penalty energies with empirical force fields improves global optimization and energy landscape analysis by excluding experimentally incompatible structures.
February 25, 2026

Local Vibrational Mode Analysis of Phonon Dispersion Relations in Crystals
Mateusz Mojsak - ,
Filippo Bodo - ,
Alessandro Erba - ,
Adam A. L. Michalchuk *- , and
Elfi Kraka *
This publication is Open Access under the license indicated. Learn More
We present a general framework for performing local vibrational mode analysis of vibrations in crystalline materials at arbitrary wavevectors throughout the Brillouin zone. The approach enables phonon dispersion relations to be interpreted in terms of chemically meaningful interatomic interactions and structural motifs, providing direct insight into the microscopic origins of the phonon behavior in periodic systems. We demonstrate the methodology for representative one-, two-, and three-dimensional materials including polymeric chains, graphene, and prototypical rock-salt and perovskite crystals. Across these systems, the analysis reveals how specific bonding patterns and structural features govern phonon dispersion relations. This framework provides a quantitative tool for the chemically intuitive analysis of phonon spectra and offers a pathway toward the rational design of phonon-dependent properties in crystalline materials.
February 17, 2026
Volume Dependence of Contact Angle in Nanodroplets: Interface Layering Contributions beyond the Standard Line Tension
Qinlin Wan - ,
Xingjun Hu *- ,
Yanghui Zhang - ,
Li Xin - ,
Firoz Alam - ,
Yingai Jin - ,
Zewei Wang - ,
Hongyang Liang - ,
Lihua Liu - ,
Jinglong Zhang - ,
Yufei Luo - ,
Peng Guo - ,
Tianming Yu - ,
Yang Xiao - ,
Jingyu Wang *- ,
Hongda Shi - ,
Shen Chen - , and
Daqian Wang
The volume dependence of the contact angle (VDCA) of a droplet at the nanoscale is primarily ascribed to the standard line tension. Researchers have identified additional volume-dependent sources that arise from the liquid layering structure within the three-phase contact zone (TPZ). These sources introduce systematic errors in the analysis of wettability and the measurement of standard line tension in nanodroplets. Therefore, this study examines the effect of the liquid layering structure on the VDCA and contrasts it with the standard line tension. The mechanism is elucidated through a thermodynamic theoretical analysis of the liquid at the adsorbed layer in cylindrical droplets: the liquid layering structure influences the contact angle by altering the liquid density distribution and the corresponding Gibbs adsorption. The validity of the theoretical analysis is confirmed through molecular dynamics simulations of parallel liquid films and cylindrical droplets. This study analyzes the effects of sources associated with liquid layering, including normal solid capillary force and Laplace pressure. The findings demonstrate that the influence of the liquid layering structure on the VDCA is first order in relation to the contact curvature, comparable to the contribution from the standard line. Furthermore, this study examines how the layering liquid structure influences the wettability of nanodroplets, offering a new thermodynamic perspective for understanding the origins of line tension in experimental contexts.
February 13, 2026
Toward Doubly Local Double Hybrid Functionals Using Neural-Network Local Mixing Functions
Nóra Kovács - ,
Szymon Śmiga - ,
Martin Kaupp *- , and
Artur Wodyński *
This publication is Open Access under the license indicated. Learn More
The first optimization and evaluation of “doubly local double hybrid” (DLDH) functionals is reported. DLDHs provide a position-dependent mixing not only of exact and semi-local exchange but at the same time also of second-order perturbational (PT2) and semilocal correlation. Both admixtures are governed by local mixing functions (LMFs) in coordinate space. The LMFs have been trained as neural networks, where the correlation LMF is either trained independently or fixed during training to the square of the exchange LMF (DL2DH variant). Training and evaluation has been done for different combinations of W4–17 atomization-energy and BH76 barrier test sets with the Slim16 or Slim20 subset of the large GMTKN55 benchmark suite. Comparison is also made to local double hybrids exhibiting constant PT2 admixtures and to local hybrids, trained in the same way, and an application to the argon-benzene dissociation curve is provided. As training-set choices are currently limited by the availability of only an inefficient implementation of the PT2 energy density, this work serves as an initial evaluation of feasibility before implementing more efficient versions. Graphical comparisons of differently trained LMFs for both exchange and correlation provide appreciable insights into the function of DLDHs. Interestingly, we find that the best training runs make the addition of empirical dispersion corrections obsolete for optimum performance for either the Slim16 or Slim20 subsets. This can be traced to very large values of the correlation LMFs in spatial regions where noncovalent interactions originate. This observation indicates possible advantages of position-dependent PT2 admixtures in DLDHs, while the position-dependent exchange admixture offers the potential for subsequent inclusion of strong-correlation effects.