

About the Cover:
Editorials
Introduction to Markov State Modeling of Conformational Dynamics
Gregory R. Bowman *- ,
Cecilia Clementi *- , and
Xuhui Huang *

This publication is free to access through this site. Learn More
Reviews
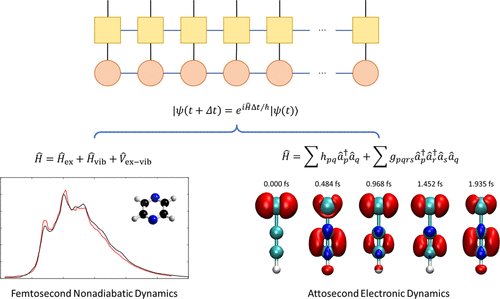
The Time-Dependent Density Matrix Renormalization Group Method for Nonadiabatic Dynamics and Electronic Dynamics
Xiaoyu Xie *- ,
Yihe Xu - ,
Ulrich Schollwöck - , and
Haibo Ma *
Recent advances in experimental techniques have enabled precise characterization of fundamental nonadiabatic and even electronic dynamics in molecules, on ultrafast time scales reaching femtosecond to attosecond resolution. However, accurate theoretical simulation of these ultrafast chemical dynamics processes in large systems remains challenging, largely due to the overwhelming number of degrees of freedom (DoFs) and the pronounced many-body correlations. In recent years, by leveraging efficient decomposition schemes for high-dimensional wave function and operator tensors, the time-dependent density matrix renormalization group (TD-DMRG) has emerged as a powerful and accurate quantum dynamics method for simulating nonadiabatic and electron dynamics in large chemical systems, in conjunction with realistic electron/exciton-vibration/phonon models or ab initio quantum chemistry many-electron Hamiltonians. This review outlines the fundamentals of TD-DMRG for chemical dynamics, covering matrix product state/operator (MPS/MPO) frameworks and algorithms from ground-state calculations to time evolution. We discuss thermal/environmental effects and compare TD-DMRG with other tensor network methods such as multiconfiguration time-dependent Hartree (MCTDH) and multilayer MCTDH (ML-MCTDH). Demonstrated applications include simulations of pyrazine absorption, singlet fission in rubrene crystal, and charge migration in chloroacetylene cation. These show TD-DMRG’s capability for modeling complex ultrafast processes from femtoseconds to attoseconds with controlled accuracy.
Dynamics
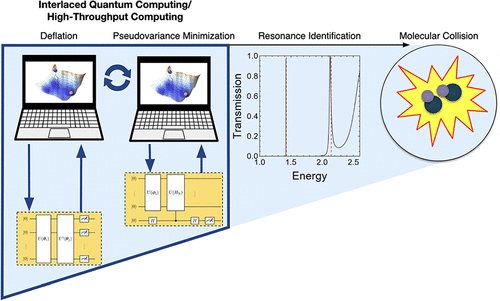
Molecular Resonance Identification in Complex Absorbing Potentials via Integrated Quantum Computing and High-Throughput Computing
Jingcheng Dai - ,
Atharva Vidwans - ,
Eric H. Wan - ,
Alexander X. Miller - , and
Micheline B. Soley *

This publication is Open Access under the license indicated. Learn More
Recent advancements in quantum algorithms have reached a state where we can consider how to capitalize on quantum and classical computational resources to accelerate molecular resonance state identification. Here, we identify molecular resonances with a method that combines quantum computing with classical high-throughput computing (HTC). This algorithm, which we term qDRIVE (the quantum deflation resonance identification variational eigensolver), exploits the complex absorbing potential formalism to distill the problem of molecular resonance identification into a network of hybrid quantum-classical variational quantum eigensolver tasks and harnesses HTC resources to execute these interconnected but independent tasks both asynchronously and in parallel, a strategy that minimizes wall time to completion. We show qDRIVE successfully identifies resonance energies and wave functions in simulated quantum processors with current and planned specifications, which bodes well for qDRIVE’s ultimate application in disciplines ranging from photocatalysis to quantum control and places a spotlight on the potential offered by integrated heterogeneous quantum computing/HTC approaches in computational chemistry.
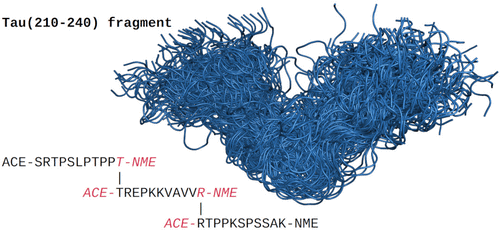
Evaluating the Impact of Phosphorylation on the Dynamics of the Tau Protein Proline-Rich Region
Johannes Stöckelmaier - ,
Giovanni Polato - ,
Jozef Hritz - , and
Chris Oostenbrink *
This publication is Open Access under the license indicated. Learn More
The proline-rich region of the tubulin-associated unit (TAU) protein is of substantial interest in understanding neurodegenerative diseases due to its interaction with bridging integrator 1 (BIN1). The associated gene BIN1 is substantially associated with the development of Alzheimer’s disease. Previous studies have underlined the importance of the conformation of the proline-rich region of TAU and the effect of its phosphorylation. In this study, we investigate the change in compactness between a four times phosphorylated TAU fragment (210–240) compared to the unphosphorylated (non-P) form using computational means. We apply our Ensemble Reconstruction from Fragments (ERF) approach to create two unbiased conformational ensembles from which a reweighted ensemble is derived that agrees with observables from nuclear magnetic resonance experiments. The resulting shift of the radius of gyration indicates a preference for relatively compact conformations for the non-P form, while the restraints derived from the experimental data do not substantially influence the compactness of the phosphorylated peptide.
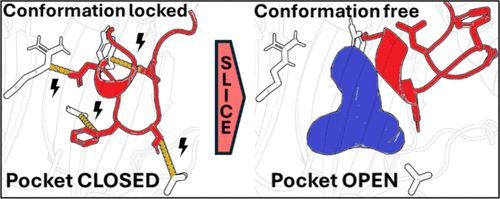
Exploring the Structural Basis of Cryptic Pocket Formation Driven by Extensive Protein Conformational Changes in Drug Targets
Martijn P. Bemelmans - ,
Alberto Borsatto - ,
Simone Marsili - ,
Francesco L. Gervasio *- , and
Vineet Pande *
This publication is Open Access under the license indicated. Learn More
Allosteric pockets that typically only emerge in the presence of a binder, known as cryptic pockets, can provide an avenue for drug discovery in challenging pharmaceutical targets. However, protein conformations exposing cryptic pockets are generally short-lived and can require significant structural rearrangements that complicate their discovery in experiment and simulation. Here, we investigate the structural basis of cryptic pocket formation in drug targets characterized by extensive dynamics using simulation-based methods. We find that functional protein segments can be anchored by local intramolecular contacts and that disrupting these interactions drives undirected large conformational changes to form cryptic pockets in PRMT5, PRMT6, SMARCA2, Abl1, and PI3Kα. Perturbing the contact networks with benzene probes, elevated temperature, or scaled protein–water interactions could not facilitate these structural dynamics here, indicating that complex mechanisms involving high-energy barriers are necessary to form ligandable cryptic pockets. Based on these limitations, a new computational approach was developed to guide conformational sampling by local interactions surrounding functional protein segments, termed “SLICE” (sampling by local interaction-guided conformational exploration). Across multiple pharmaceutically relevant proteins, our simulations aid in understanding and rapidly exploring the large-scale structural plasticity governed by the local protein environment around functional segments that can be leveraged for drug discovery.

MaxwellLink: A Unified Framework for Self-Consistent Light–Matter Simulations
Xinwei Ji - ,
Andres Felipe Bocanegra Vargas - ,
Gang Meng - , and
Tao E. Li *
A major challenge in light–matter simulations is bridging the disparate time and length scales of electrodynamics and molecular dynamics. Current computational approaches often rely on heuristic approximations of either the electromagnetic (EM) or the material component, hindering the exploration of complex light–matter systems. Herein, MaxwellLink─a modular, open-source Python framework─is developed for the massively parallel, self-consistent propagation of classical EM fields interacting with a large heterogeneous molecular ensemble. The package utilizes a robust TCP/UNIX socket interface to couple EM solvers with a wide range of molecular drivers. In this initial release, MaxwellLink supports EM solvers spanning from single-mode cavities to full-feature three-dimensional finite-difference time-domain (FDTD) engines and molecules described by multilevel open quantum systems, force-field and first-principles molecular dynamics, and nonadiabatic real-time Ehrenfest dynamics. With the socket-based architecture, users can seamlessly switch between levels of theory of either the EM solver or molecules without modifying the counterpart. Moreover, the EM engine and molecular drivers scale independently across multiple high-performance computing (HPC) nodes, facilitating large-scale simulations previously inaccessible to existing numerical schemes. The versatility and accuracy of this code are further demonstrated through applications including superradiance, radiative energy transfer, vibrational strong coupling in Bragg resonators, and plasmonic heating of molecular gases. By providing a unified, extensible engine, MaxwellLink potentially offers a powerful platform for exploring emerging phenomena across the research fronts of spectroscopy, quantum optics, plasmonics, and polaritonics.
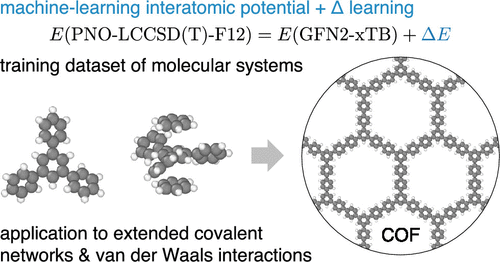
Machine-Learning Interatomic Potentials Achieving CCSD(T) Accuracy for Systems with Extended Covalent Networks and van der Waals Interactions
Yuji Ikeda *- ,
Axel Forslund - ,
Pranav Kumar - ,
Yongliang Ou - ,
Jong Hyun Jung - ,
Andreas Köhn - , and
Blazej Grabowski
This publication is Open Access under the license indicated. Learn More
Machine-learning interatomic potentials (MLIPs) enable large-scale atomistic simulations at moderate computational cost while retaining ab initio accuracy. In recent years, MLIPs trained on coupled-cluster data─particularly CCSD(T), which includes single, double, and perturbative triple excitations─have emerged as a promising route to achieve chemical accuracy (1 kcal/mol) beyond the limits of density functional theory (DFT) and to incorporate nonempirical van der Waals (vdW) interactions. Most existing approaches are, however, still not straightforwardly applicable for systems with extended covalent networks such as covalent organic frameworks (COFs) due to the limited availability of CCSD(T) under periodic boundary conditions. Here we present a methodology to train MLIPs with CCSD(T) accuracy for systems with extended covalent networks. The approach is based on the Δ-learning method with a dispersion-corrected tight-binding baseline and an MLIP trained on the differences of the target CCSD(T) energies from the baseline. This Δ-learning strategy enables training on compact molecular fragments while preserving transferability toward the periodic systems. Dispersion interactions are accounted for by including vdW-bound multimers in the training set, and the combination with a vdW-aware tight-binding baseline allows the formally local MLIP to attain CCSD(T)-level accuracy even for systems dominated by long-range vdW forces. The resulting potential yields root-mean-square energy errors below 0.4 meV/atom on both training and test sets and reproduces electronic total atomization energies, bond lengths, harmonic vibrational frequencies, and intermolecular interaction energies for benchmark molecular systems. We apply the method to a prototypical quasi-two-dimensional covalent organic framework (COF) composed of carbon and hydrogen. The COF structure, interlayer binding energies, and hydrogen absorption are analyzed at CCSD(T) accuracy. Overall, the developed methodology opens a practical route to large-scale atomistic simulations for systems with extended covalent networks and vdW interactions with chemical accuracy.
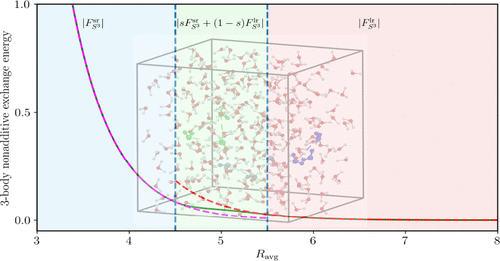
How Long-Range Are Three-Body “Exchange” Interactions in Liquid Water?
Ommair Ishaque *- ,
John W. Melkumov - , and
Krzysztof Szalewicz
Three-body interactions in water play a crucial role in accurately modeling its structural and thermodynamic properties. These interactions consist of a polarization term that decays as an inverse power of the intermolecular separations Rab and a term that is usually assumed to describe exchange interactions and decay exponentially. Due to the complexity of fitting the latter term at large Rab, it is often damped or truncated beyond a certain distance, also because the computational cost of including three-body effects in molecular simulations scales as N3 with the number of molecules, compared to the N2 scaling of two-body interactions. Here, investigations of the impact of long-range three-body exchange interactions on the results of such simulations have been performed by systematically extending the average Rab of trimers included. It is demonstrated that these long-range effects are important for accurately describing the density of liquid water, ρ(T), as a function of temperature, but are essentially negligible for several other properties of water. The effects of three-body damping onset on ρ(T) are larger than they would have been with an exponential decay; however, it is shown here that the decay is dominated by exponential components only at fairly small Rab, while for large Rab, the nonpolarization three-body effects decay as 1/Rabn. These findings are rationalized by calculations with the symmetry-adapted perturbation theory. Another reason for the importance of three-body effects is their N3 scaling. Clearly, long-range three-body exchange interactions should be included in high-accuracy water models. It is shown that the reason these interactions have such large effects on ρ(T) is their extreme anisotropy affecting the structure of liquid water. Our work also sheds light on discrepancies between the theory and experiment for ρ(T).

Quantum Dynamics Description of Reactions with Seven Atoms: Application to the OH + CHD3
Zhaojun Zhang *- ,
Fabien Gatti *- , and
Dong H. Zhang *
High-dimensional quantum dynamics theoretical studies of polyatomic molecules are extremely challenging due to the exponential scaling of the required basis set size with system dimensionality. Here, we report a novel C3v model for treating CZ3-type molecules, which can be widely applied to polyatomic reactions involving methane-like species. By substantially reducing the angular momentum coupling basis functions, this model effectively decreases both the number of basis functions and the computational time for quantum dynamics simulations. The results for OH + CHD3 reactions demonstrate the reliability and applicability of the method. They also confirm the applicability of the Polanyi rules to this reaction. This study extends the boundaries of quantum dynamics investigations on elementary reactions and provides critical insights for developing precise quantum and classical theories capable of treating reactions with even more atoms.
Statistical Mechanics
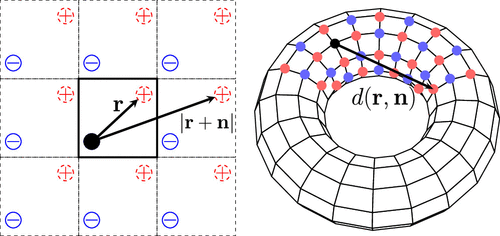
The Madelung Problem of Finite Crystals
Yihao Zhao - ,
Yang He - , and
Zhonghan Hu *
The Coulomb potential at an interior ion in a finite crystal of size p is given by a linear superposition of contributions from displacement vectors r = (x, y, z) to its neighbors. This additive structure underlies universal relationships among Madelung constants and applies to both standard periodic boundary conditions and alternative Clifford supercells. Each pairwise contribution decomposes into three physically distinct components: a periodic bulk term, a quadratic boundary term, and a finite-size correction, whose leading order term is [24r4−40(x4+y4+z4)]/[93(2p+1)2] for cubic crystals with unit lattice constant. Combining this decomposition with linear superposition yields a rapidly convergent direct-summation scheme, accurate even at p = 1 (33 unit cells), enabling hands-on calculations of Madelung constants for a wide range of ionic crystals.
Quantum Electronic Structure
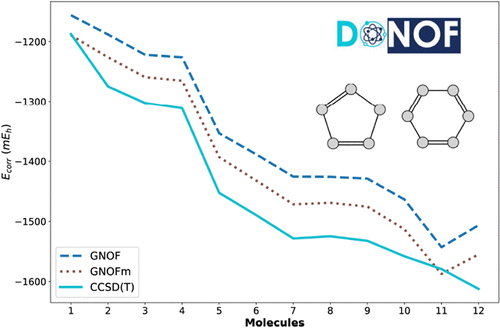
5- and 6-Membered Rings: A Natural Orbital Functional Study
Ion Mitxelena *- ,
Juan Felipe Huan Lew-Yee *- , and
Mario Piris *
This publication is Open Access under the license indicated. Learn More
The Global Natural Orbital Functional (GNOF) provides a straightforward approach to capture most electron correlation effects without needing perturbative corrections or limited active spaces selection. In this work, we evaluate both the original GNOF and its modified variant, GNOFm, on a set of twelve 5- and 6-membered molecular rings, systems characterized primarily by dynamic correlation. This reference set is vital as it comprises essential substructures of more complex molecules. We report complete-basis-set limit correlation energies for GNOF, GNOFm, and the benchmark CCSD(T) method. Across the Dunning basis sets, both functionals deliver a balanced and accurate description of the molecular set, with GNOFm showing small but systematic improvements while preserving the overall robustness of the original formulation. These results confirm the reliability of the GNOF family and its ability to capture dynamic correlation effects.
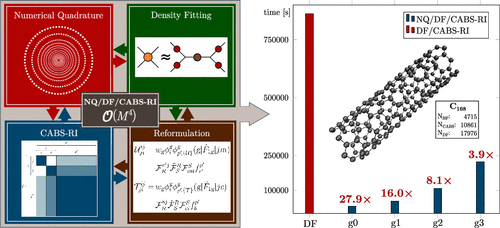
Formulation of an Efficient 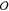 (M4)-Scaling Explicitly Correlated MP2-F12 Correction by Combining Numerical Quadrature with Density Fitting and CABS-RI
(M4)-Scaling Explicitly Correlated MP2-F12 Correction by Combining Numerical Quadrature with Density Fitting and CABS-RI
Lars Urban - ,
Henryk Laqua - ,
Travis H. Thompson - , and
Christian Ochsenfeld *
This publication is Open Access under the license indicated. Learn More
We present a novel approach that combines numerical quadrature with density fitting and CABS-RI for the evaluation of exchange-type intermediates in RI-MP2-F12 theory, rigorously reducing the formal and practical scaling of the total correction from O(M5) to O(M4). Our new hybrid NQ/DF/CABS-RI ansatz is based directly on our previously developed NQ/CABS-RI method for the efficient evaluation of 6c3e integrals [Urban, L.; Laqua, H; Thompson, T. H.; Ochsenfeld, C. J. Chem. Theory Comput. 2024, 20, 3706–3718] and extends this approach to the optimized computation of products of 4c2e integrals. In this framework, the main exchange-type intermediates V, X, and B are reformulated, resulting in more compact expressions, increased shared computations, and fewer CABS-RI insertions. We introduce efficient algorithms that cover all exchange-type contributions, including advantageous batching of integrals. Benchmarks show that NQ/DF/CABS-RI achieves mean errors below 0.01 kcal/mol for noncovalent interaction and isomerization energies already with small to modest grid sizes, while the numerical precision can be adjusted to balance computational cost. Empirical scaling was determined using linear glycine chains, demonstrating the expected O(M4) behavior for the rate-determining steps, with the remaining exchange-type expressions scaling nearly linearly. Compared with an idealized DF/CABS-RI implementation, our approach achieves speedups of roughly one order of magnitude for the most expensive steps with virtually no loss of numerical accuracy. Systems with strongly delocalized electronic structures benefit particularly. For a nanotube with 168 carbon atoms, the computational time for the most demanding expressions is reduced from 9.97 to 1.25 days, bringing the cost much closer to that of conventional DF-MP2. At present, NQ/DF/CABS-RI achieves efficient O(M4) scaling, and further cost reductions are anticipated through the introduction of integral screening based on Cholesky orbitals, which will be explored in future work.
Local Pair Natural Orbital-Based Coupled-Cluster Theory through Full Quadruples (DLPNO–CCSDTQ)
Andy Jiang - ,
Devin A. Matthews - ,
David Poole - ,
Connor G. Briggs - ,
Justin M. Turney - ,
C. David Sherrill - , and
Henry F. Schaefer III *
This publication is Open Access under the license indicated. Learn More
In this work, we implement a local pair natural orbital-based coupled-cluster method through the full treatment of quadruple excitations (CCSDTQ). The domain-based local pair natural orbital (DLPNO) approach, which has successfully been applied to lower levels of coupled-cluster theory, is utilized in our algorithm, and thus our algorithm is called DLPNO-CCSDTQ. For simplicity in the working equations and in the implementation, we t1-dress the two-electron integrals as well as Fock matrix elements. Our method can recover CCSDTQ-CCSDT and CCSDTQ-CCSDT(Q) energy differences on the order of 0.01–0.05 kcal mol–1, even at a loose quadruples natural orbital (QNO) occupation number cutoff of 3.33 × 10–6. To highlight the capabilities of our code and its potential future applications, we showcase computations that would be intractable with canonical CCSDTQ, such as the benzene dimer, (H2O)17, and adamantane. With sufficient computing resources, computations up to 15 heavy atoms (40 atoms overall) may be feasible for fully bonded 3D systems.
Ionization Potentials at Mean-Field Computational Cost: The Extended Koopmans’ Framework for pCCD
Seyedehdelaram Jahani - ,
Katharina Boguslawski *- , and
Paweł Tecmer *
We introduce a mean-field-like computational model for calculating ionization potentials (IPs) based on the pair Coupled Cluster Doubles (pCCD) wave function. Specifically, our model combines the extended Koopmans’ theorem (EKT) with the advantages of a variationally orbital-optimized (oo)-pCCD ansatz. The computational cost of the EKT(pCCD) method is negligible (O(N3)) as the response 1- and 2-particle reduced density matrices used to construct the generalized Fock matrix are readily available after an oo-pCCD calculation. We benchmarked our new computational model for IPs of atoms, small molecules, and a set of organic acceptor molecules against experimental and theoretical reference data. The EKT(pCCD) model significantly improves upon the modified Koopmans’ approach [J. Chem. Phys. 162, 184110 (2025)], and the obtained IPs are comparable to those of computationally more expensive IP-EOM-pCCD-based models, approaching CCSD(T) reference values (with a mean error of 0.05 eV). Most importantly, the EKT(pCCD) approach is almost independent of the basis set size, and reliable IPs are already obtained with small basis sets.
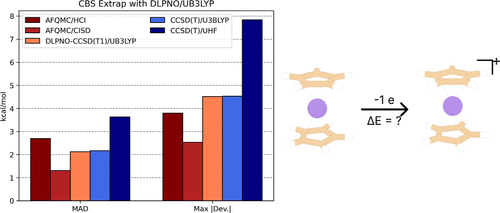
Evaluating Multiconfigurational Trials for Accurate Phaseless Auxiliary-Field Quantum Monte Carlo on 3d Transition Metal Complexes
Hung T. Vuong - ,
Ankit Mahajan - ,
John L. Weber - ,
James Shee *- ,
David R. Reichman *- , and
Richard A. Friesner *
In this study, we evaluate multiconfigurational trial wave function protocols for phaseless auxiliary field quantum Monte Carlo (ph-AFQMC) on transition metal containing systems. First, we benchmark vertical ionization potentials for 22 3d transition metal complexes against published high-accuracy ph-AFQMC values in a double-ζ basis set. We then compute the vertical ionization potential for a set of six metallocenes using our best-performing protocol, alongside ph-AFQMC using a configuration interaction singles and doubles (CISD) trial state. We also analyze the performance of canonical coupled-cluster theory with singles, doubles and perturbative triples (CCSD(T)), as well as its local approximation using domain-based local pair natural orbitals (DLPNO–CCSD(T1)) using different reference orbitals. To reach the complete-basis-set (CBS) limit, we examine several extrapolation schemes and report CBS-limit ph-AFQMC and CCSD(T) values alongside experimental results. We find that ph-AFQMC with the best-performing trial in a triple-ζ basis, followed by CBS correction from DLPNO–CCSD(T1) with unrestricted B3LYP reference orbitals, yields small deviations from experiment at modest cost. Using a CISD trial state in ph-AFQMC gives the closest agreement with experiment (errors <2 kcal/mol), albeit with lower scalability.
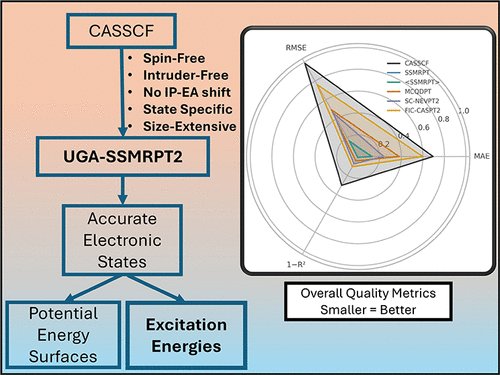
UGA-SSMRPT2 - A Multireference Perturbation Theory Predicting Accurate Electronic Excitation Energies in Diverse Molecular Systems
Shamik Chanda - ,
Pratyush Bhattacharjya - ,
Avijit Sen - , and
Sangita Sen *
UGA-SSMRPT2, the spin-free perturbative analogue of Mukerjee’s State-Specific Multireference Coupled Cluster Theory (MkMRCC), is known to be successful for size-extensive and intruder-free construction of dissociation curves. This work demonstrates that UGA-SSMRPT2 is also an accurate and computationally inexpensive framework for computing the excitation energies. The method achieves near-chemical accuracy for the vast majority of π → π*, n → π*, charge-transfer, valence-Rydberg, and Rydberg excited states commonly used for benchmarking electronic structure theories for excited states. Our results demonstrate that UGA-SSMRPT2 excitation energies lie within 0.20 eV of EOM-CCSD and/or well-established theoretical best estimates, often surpassing the popular MRPT2 approaches like NEVPT2, CASPT2, and MCQDPT while typically requiring smaller active spaces. Its state-specific formulation circumvents the well-known intruder-state problem and eliminates the need for empirical parameters, such as IPEA shifts in CASPT2. This work proposes UGA-SSMRPT2 as a robust and scalable approach for modeling challenging electronically excited states.
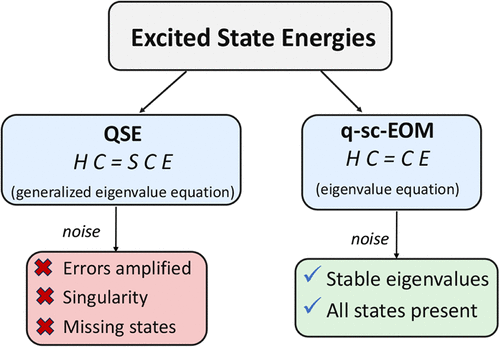
Generalized Eigenvalue Problem in Subspace-Based Excited-State Methods for Quantum Computers
Prince Frederick Kwao - ,
Srivathsan Poyyapakkam Sundar - ,
Brajesh Gupt - , and
Ayush Asthana *
This publication is Open Access under the license indicated. Learn More
Solving challenging problems in quantum chemistry is one of the most promising applications of quantum computers. Within the quantum algorithms proposed for problems in excited-state quantum chemistry, subspace-based quantum algorithms, including quantum subspace expansion (QSE), quantum equation of motion (qEOM), and quantum self-consistent equation-of-motion (q-sc-EOM), are promising for pre-fault-tolerant quantum devices. The working equation of QSE and qEOM requires solving a generalized eigenvalue equation with associated matrix elements measured on a quantum computer. Our careful analytical and numerical analysis of the standard and generalized eigenvalue problems, especially in the context of excited-state methods, shows that the errors in eigenvalues magnify drastically with an increase in the condition number of the overlap matrix when a generalized eigenvalue equation is solved in the presence of statistical sampling errors. This makes methods such as QSE unstable for errors that are unavoidable when using quantum computers. Further, at very high condition numbers of the overlap matrix, the QSE’s working equation could not be solved without any additional steps in the presence of sampling errors, as it becomes ill-conditioned. It was possible to use the thresholding technique in this case to solve the equation, but the solutions achieved had missing excited states, which may be a problem for future chemical studies. We also show that excited-state methods that have an eigenvalue equation as the working equation, such as q-sc-EOM, do not have the problems associated with the condition number and could be generally more stable to errors and therefore more suitable candidates for excited-state quantum chemistry calculations using quantum computers.

Interpolative Separable Density Fitting on Adaptive Real Space Grids
Hai Zhu - ,
Chia-Nan Yeh - ,
Miguel A. Morales - ,
Leslie Greengard - ,
Shidong Jiang *- , and
Jason Kaye *
We generalize the interpolative separable density fitting (ISDF) method, used for compressing the four-index electron repulsion integral (ERI) tensor, to incorporate adaptive real space grids for potentially highly localized single-particle basis functions. To do so, we employ a fast adaptive algorithm, the recently introduced dual-space multilevel kernel-splitting method, to solve the Poisson equation for the ISDF auxiliary basis functions. The adaptive grids are generated by using a high-order accurate, black-box procedure that satisfies a user-specified error tolerance. Our algorithm relies on the observation, which we prove, that an adaptive grid resolving the pair densities appearing in the ERI tensor can be straightforwardly constructed from one that resolves the single-particle basis functions, with the number of required grid points differing only by a constant factor. We find that the ISDF compression efficiency for the ERI tensor with highly localized basis sets is comparable to that for smoother basis sets compatible with uniform grids. To demonstrate the performance of our procedure, we consider several molecular systems with all-electron basis sets that are intractable using uniform grid-based methods. Our work establishes a pathway for scalable many-body electronic structure simulations with arbitrary smooth basis functions, making simulations of phenomena such as core-level excitations feasible on a large scale.
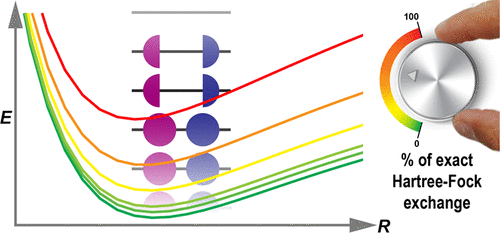
Origin of the Singlet Excited Electronic Energy Shifts in ΔSCF with Fractional Occupation Numbers and Hybrid Density Functionals
Momir Mališ *- and
Sandra Luber
Singlet excited electronic states can be directly constructed with the ΔSCF method using a single electronic density by applying fractional occupation numbers. While the advantages and disadvantages of such a DFT-based ΔSCF method were demonstrated in several studies, the spurious energy shift of the singlet electronic states when constructed using hybrid DFT functionals remains unexplained. Here, we explain in detail the origin of the effect of losing the idempotence properties of density matrices on the DFT energy terms with hybrid-based DFT functionals, as well as a procedure to eliminate the artificial shift from ΔSCF singlet excited state energies when obtained using the hybrid-based DFT functionals.

Solving the Scaled Schrödinger Equation with SAC–CI and Electrostatic Force Formalism. Ground, Excited, and Ionized States of the Benzene Molecule
Hiroshi Nakatsuji *
An exact theory for solving the scaled Schrödinger equation (SSE) has been extended to molecular ground, excited, and ionized states that satisfy the Hellmann–Feynman electrostatic force (ESF) theorem, a necessary condition for the exact wave function. This is realized by applying the free complete-element (FC) or SSE theory to the symmetry-adapted-cluster (SAC) configuration-interaction (CI) wave functions that are designed to satisfy the ESF theorem. The resultant theory, called the FC or SSE(SAC–CI, ESF) theory, is not only highly accurate but also predictive, using both energetic and force theoretic concepts. Here, this theory is applied to a benzene molecule, a 42-electron molecule, the largest system yet treated by our exact theory. Notably, the theory resolved complex features of higher singlet and triplet excited states of a benzene molecule, which had previously eluded conventional methods.
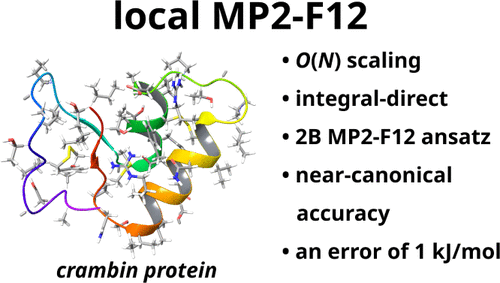
A Linear-Scaling Integral-Direct Explicitly Correlated Second-Order Møller–Plesset Approach
Mihály Kállay *- ,
Péter R. Nagy - ,
Bence Ladóczki - , and
Dávid Mester
This publication is Open Access under the license indicated. Learn More
We present an integral-direct, iteration-free, linear-scaling, local explicitly correlated second-order Møller–Plesset (MP2-F12) approach, extending our previous local MP2 method [J. Chem. Theory Comput. 2016, 12, 4897]. The correlation contributions for individual electron pairs are computed within domains defined by the corresponding localized orbitals, while the correlation energies of spatially distant electron pairs are determined via multipole expansions. All the various types of integrals are computed and transformed directly, thereby precluding the need for integral storage and yielding asymptotically constant memory as well as negligible disk I/O demand. Another competitive advantage is the implementation of the 2B MP2-F12 ansatz, the most complete one so far with local approximations, enabling excellent basis set convergence even with double-ζ basis sets. Our validation studies indicate that the approach recovers at least 99.9% of the canonical MP2-F12 correlation energy and yields reaction energies with a mean error of less than 1 kJ/mol. With respect to the complete basis set limit of MP2, the error of our local approach is just slightly larger than that of canonical MP2-F12. With the new local MP2-F12 approach, we were able to compute the correlation energy for a small protein containing 644 atoms, which is the largest system ever considered in an explicitly correlated calculation.

Solvation Lies Within: Simulating Condensed-Phase Properties from Local Electronic Structures
Kasper F. Schaltz - ,
Jonas Greiner - ,
Filippo Lipparini - , and
Janus J. Eriksen *
In transitions between different environmental settings, a molecular system inevitably undergoes a range of detectable changes, and the ability to accurately simulate such responses, e.g., in the form of shifts to molecular energies, remains an important challenge across physical chemistry. Based on an exact decomposition of total energies from Kohn–Sham density functional theory in a basis of spatially localized molecular orbitals, the present work outlines a robust protocol for sampling the effect of solvation within homogeneous condensed phases by focusing on perturbations to local electronic structures only. We report chemically intuitive results for binding energies of water, ethanol, and acetonitrile that all display fast convergence with respect to the bulk size. Results stay largely invariant with respect to the choice of basis set while reflecting differences in density functional approximations, and our protocol thus allows for a physically sound and efficient estimation of general effects related to bulk solvation.
Reaction Mechanisms
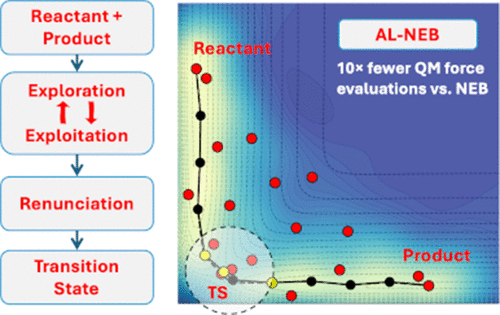
An Active Learning Algorithm for Identifying Transition States on a Potential Energy Surface
Sandra Liz Simon - ,
Nitin Kaistha *- , and
Vishal Agarwal *
Mapping reaction pathways on complex potential energy surfaces (PESs) and locating transition states (TSs) is often used for understanding chemical reaction mechanism(s). The nudged elastic band (NEB) method is widely used for this purpose, but it becomes computationally expensive for large systems due to the repeated evaluation of energies and forces. We present an active learning algorithm coupled with the nudged elastic band, AL-NEB, for efficient convergence to the TS. AL-NEB constructs a surrogate PES and actively selects training points in two phases: (a) Exploration-Exploitation and (b) Renunciation. Strategies have been introduced for making the algorithm efficient and stable. We show the efficacy of the algorithm on several 2D analytical potentials, HCN isomerization, keto-enol tautomerization, and high-dimensional heptamer island diffusion (up to 525 degrees of freedom). In all cases, AL-NEB locates the “exact” TS on the chosen model chemistry with an order-of-magnitude fewer force evaluations than the standard NEB, demonstrating its scalability and efficiency.

RxnNet: An AI Framework for Reaction Mechanism Discovery─A Case Study of Carbocations
Shani Zev - ,
Michal Roth - ,
Jishnu Narayanan S J - , and
Dan T. Major *
Understanding complex chemical reaction cascades remains a major challenge in chemistry. Scalable investigation of their thermodynamic and kinetic properties requires the use of automated reaction prediction tools, which is a rapidly growing area in the study of chemical reactivity. However, the systematic exploration of intricate reaction networks involving highly reactive intermediates continues to pose significant difficulties. Here, we introduce RxnNet, a novel artificial intelligence-assisted platform for the automated prediction of chemical reaction mechanisms. RxnNet integrates heuristic rules with domain-specific chemical knowledge including stereochemistry, regiochemistry, conformational preferences, and isotope labeling, to construct mechanistically informed reaction networks. These networks are represented as graphs and are coupled with on-the-fly quantum chemical evaluations to identify all feasible intermediates and transition states. In this work, we apply RxnNet to carbocation chemistry, a notoriously complex and computationally demanding type of reaction. We demonstrate the method’s capabilities by analyzing three multistep reactions with known mechanisms, each of which poses significant challenges even for expert computational and synthetic chemists. RxnNet provides a robust approach for uncovering reaction mechanisms, which can accelerate the understanding and design of transformations in complex chemical systems.
Molecular Mechanics
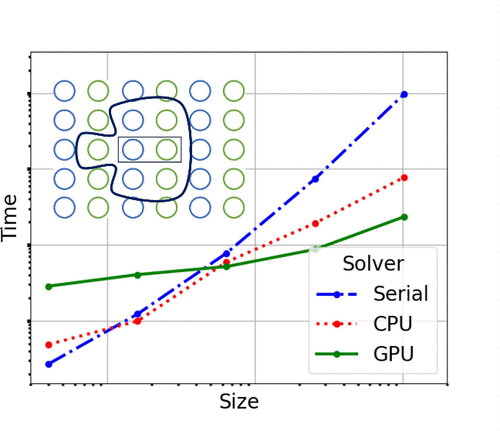
KinCat: Kinetic Monte Carlo Parallel Computations of Surface Kinetics in Heterogeneous Catalysis
Craig Daniels *- ,
Nathan V. Roberts - ,
Kyungjoo Kim - , and
Habib N. Najm
Kinetic Monte Carlo (KMC) simulations are broadly used to investigate chemical and materials systems where a balance between atomic detail and diffusion or reaction time scales is needed. Here we present KinCat, an open-source 2D KMC package designed for use in lattice-KMC studies of surface kinetics in heterogeneous catalytic systems. It is written in C++ and uses Kokkos to facilitate use on a variety of shared-memory CPU/GPU/accelerator systems. We demonstrate the performance scaling of KinCat on GPU and CPU architectures, using CO oxidation on RuO2 as a model system. KinCat efficiently manages large lattice KMC simulations using a parallel domain-decomposition algorithm.

A Hybrid Physics-Driven Neural Network Force Field for Liquid Electrolytes
Junmin Chen - ,
Qian Gao - ,
Yange Lin - ,
Miaofei Huang - ,
Zheng Cheng - ,
Wei Feng - ,
Jianxing Huang *- ,
Bo Wang *- , and
Kuang Yu *
Electrolyte design plays an important role in the development of lithium–ion batteries and sodium-ion batteries. Battery electrolytes feature a large design space composed of different solvents, additives, and salts, which is difficult to explore experimentally. High-fidelity molecular simulation can accurately predict the bulk properties of electrolytes by employing accurate potential energy surfaces, thus guiding the molecule and formula engineering. At present, the overly simplified classic force fields rely heavily on experimental data for fine-tuning, thus its predictive power on microscopic level is under question. In contrast, the newly emerged machine learning interatomic potential (MLIP) can accurately reproduce the ab initio data, demonstrating excellent fitting ability. However, it is still haunted by problems such as low transferability, insufficient stability in the prediction of bulk properties, and poor training cost scaling. Therefore, it cannot yet be used as a robust and universal tool for the exploration of electrolyte design space. In this work, we introduce a highly scalable and fully bottom-up force field construction strategy called PhyNEO-Electrolyte. It adopts a hybrid physics-driven and data-driven method that relies only on monomer and dimer EDA (energy decomposition analysis) data. With a careful separation of long/short-range and nonbonding/bonding interactions, we rigorously restore the long-range asymptotic behavior, which is critical in the description of electrolyte systems. Through this approach, we significantly improve the data efficiency of MLIP training, allowing us to achieve much larger chemical space coverage using much less data while retaining reliable quantitative prediction power in bulk phase calculations. PhyNEO-Electrolyte thus serves as an important tool for future electrolyte optimization.
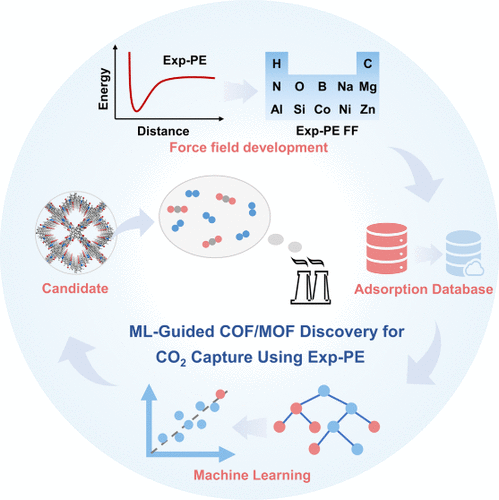
CO2 Capture from Flue Gas: A High-Fidelity Force Field and Machine Learning Framework for Adsorbent Discovery
Yunjie Lang - ,
Yuwei Pan - ,
Mengqian Xu - ,
Xin Wei - ,
Ran Duan - ,
Shaohuang Li - ,
Dong Zhai - ,
Lei Sun - ,
Weiqiao Deng - , and
Li Yang *
CO2 from flue gas is central to mitigating fossil-fuel-derived emissions, where adsorbent performance directly dictates process energy efficiency and process cost. Although machine learning (ML) has emerged as a powerful tool for accelerating adsorbent discovery, its predictive accuracy is fundamentally limited by the physical reliability of the underlying training data, a manifestation of the “garbage in, garbage out” (GIGO) problem. Most existing CO2 adsorption databases rely on Lennard-Jones (LJ) force fields, whose deficiencies in describing CO2–CO2 and CO2–framework interactions, particularly at high pressures, introduce systematic bias into the ML models. To address this, we developed a physically accurate van der Waals force field based on an Exp-PE potential and constructed a high-fidelity CO2 adsorption database. Building on this data set, we introduce quadrupole-responsive descriptors that explicitly capture the anisotropic electrostatics of CO2, leading to improved ML predictive accuracy. This framework identifies high-performing COF/MOF adsorbents, including COF-50 (ΔNCO2 = 13.58 mol/kg) and COF-364 (ΔNCO2 = 12.43 mol/kg), whose working capacities exceed those of current reported porous materials.
Spectroscopy and Excited States
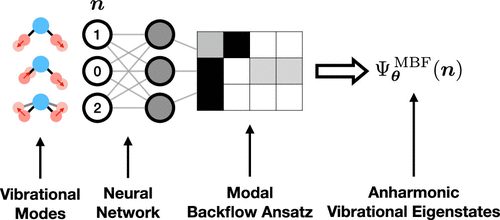
Modal Backflow Neural Quantum States for Anharmonic Vibrational Calculations
Lexin Ding *- and
Markus Reiher *
This publication is Open Access under the license indicated. Learn More
Neural quantum states (NQS) are a promising ansatz for solving many-body quantum problems due to their inherent expressiveness. Yet this expressiveness can only be harnessed efficiently for treating identical particles if the suitable physical knowledge is hardwired into the neural network itself. For electronic structure, NQS based on backflow determinants have been shown to be a powerful ansatz for capturing strong correlation. By contrast, the analogue for bosons, backflow permanents, is unpractical due to the steep cost of computing the matrix permanent and due to the lack of particle conservation in common bosonic problems. To circumvent these obstacles, we introduce a modal backflow (MBF) NQS design and demonstrate its efficacy by solving the anharmonic vibrational problem. To accommodate the demand of high accuracy in spectroscopic calculations, we implement a selected-configuration scheme for evaluating physical observables and gradients, replacing the standard stochastic approach based on Monte Carlo sampling. A vibrational self-consistent field calculation is conveniently carried out within the MBF network, which serves as a pretraining step to accelerate and stabilize the optimization. In applications to both artificial and ab initio Hamiltonians, we find that the MBF network is capable of delivering spectroscopically accurate zero-point energies and vibrational transitions in all anharmonic regimes.
Condensed Matter, Interfaces, and Materials

Large-Scale Calculation of Vibrational Sum Frequency Generation Spectra of Aqueous Interfaces
Patrik Musil - ,
Ondřej Kroutil - ,
Simone Pezzotti - ,
Marie-Pierre Gaigeot *- , and
Milan Předota *
We present a software to calculate phase-resolved resonant vibrational sum-frequency generation (vSFG) susceptibility χ(2)(ω) of water and hydroxyls at planar interfaces, e.g., air/water or solid/liquid or (bio)membrane/liquid interfaces of aqueous solutions. The released code (i) reads several formats of molecular trajectories, both from ab initio (AIMD) and classical MD (CMD), (ii) calculates instantaneous surfaces to allow flexible interfaces, (iii) is written in FORTRAN, parallelized by OpenMP and optimized for memory usage, (iv) allows processing of systems up of tens of thousand atoms and for unlimited simulation time, and (v) includes many tunable processing parameters. The code and its documentation are available via GitHub. Flexible models of water and surface hydroxyl (if evaluated) (CMD or AIMD) must be used. The derivatives of the polarizability tensors and dipole moments with the change of O–H distance must be calculated externally by ab initio methods and provided as input data. We present the impact of various parameters of the MD simulations (simulation length, nonbonded interaction cutoff, size of the system, and thermostat relaxation time) as well as of the processing code (filter relaxation, cutoff of cross-terms) and provide representative results for air/water, charged quartz (101)/aqueous solution, and neutral α-alumina (0001)/aqueous solution interfaces. Further extensions are planned to distinguish signals from specific O–H or C–H bonds of interfacial molecules.
Polymers and Biopolymers
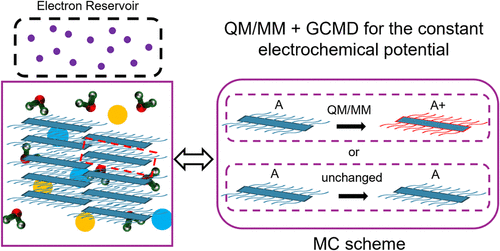
Simulation of Charge Distribution and Microstructure in Semicrystalline Polymeric Ionic-Electronic Conductors Using Classical Simulation at Constant Electrochemical Potential
Zixuan Wei *- ,
Hesam Makki - ,
Paola Carbone - , and
Alessandro Troisi
This publication is Open Access under the license indicated. Learn More
Understanding how charge distributions on aggregated chains change with microstructure under constant electrochemical potential is crucial for elucidating the behavior of polymeric organic mixed ionic–electronic conductors (OMEICs), yet it remains difficult to study. To address this challenge, we introduce a methodology to perform classical atomistic simulations of doped semiconductors at a constant electrochemical potential. The method allows individual polymer chains to be oxidized and reduced, taking into account their individual redox potentials and the externally tunable electrochemical potential. The implementation follows a grand-canonical molecular dynamics (GC-MD) scheme, with the local modulation of the redox potential being described by a QM/MM Hamiltonian. Applied to a semicrystalline polymer with ordered layered and lamellar structures, the method reproduces the experimentally observed minimal structural changes over the electrochemical potentials and charging levels considered. Near the redox potential, charging levels fluctuate more strongly, and variations in the interlamellar angle (defined by the normal of adjacent lamellae) are most pronounced. Moreover, analysis of the local environment reveals no detectable correlation between a chain’s redox reaction and the charge distribution of neighboring chains, except at the most negative potentials, where redox events occur preferentially in more positively charged surroundings. Lastly, examination of individual chains shows minimal chain–chain charge correlation, and the single-chain conformation remains closely linked to its redox behavior. Overall, this work provides a robust framework for investigating charge distributions in dynamically doped systems and offers new conceptual routes for studying polymer structural responses under constant electrochemical potentials.
Biomolecular Systems
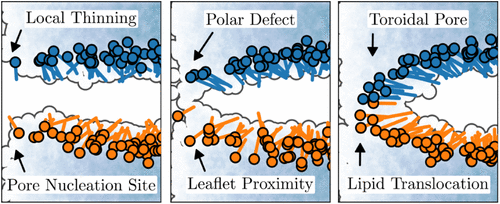
Membrane Pore Formation Unveiled by ∞RETIS Path Sampling: From Thinning to Flip-Flop
Daniel Tianhou Zhang *- ,
Lukas Baldauf - ,
Grzegorz Lazarski - ,
Titus S. van Erp - , and
Wataru Shinoda *
This publication is Open Access under the license indicated. Learn More
Pore formation in lipid bilayers plays a vital role in membrane fusion, transport, and signaling. Yet, its detailed mechanism remains elusive due to the limitations of conventional simulation methods. To overcome this, we apply a newly developed path sampling technique, the asynchronous and infinite swap version of Replica Exchange Transition Interface Sampling (∞RETIS), to study pore formation in a dimyristoylphosphatidylcholine (DMPC) bilayer modeled with the CHARMM36m force field. Our results reveal a sequence of tightly coupled events: pore nucleation sites are determined by early-stage thinning, and the progress into a metastable pore requires a combination of polar defects and close proximity between lipids across opposite leaflets. Using Inf-init, an initiation protocol based on ∞RETIS, rare trajectories can be generated starting directly from equilibrium simulations. Inf-init and ∞RETIS simulations reveal that lipid flip-flop occurs exclusively via local membrane thinning, and pore closure often results in asymmetric lipid distributions.
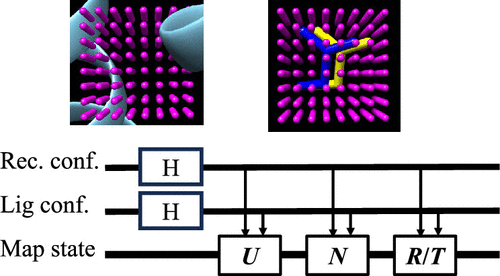
Quantum Inner Product Scoring with Grid-Based Maps for Structure-Based Virtual Screening
Pei-Kun Yang *
Structure-based virtual screening is fundamentally constrained by the combinatorial growth of configurational spaces arising from receptor conformations, ligand identities, conformations, and spatial degrees of freedom. We reformulate protein–ligand interaction energy calculation as a linear-algebraic problem defined on shared Cartesian grids. Within this framework, electrostatic and van der Waals interaction energies are expressed as inner products between receptor potential maps and ligand charge and atom-type occupancy vectors. Ligand translations and rotations are represented as unitary operations acting on independent spatial registers, enabling systematic reuse of grid information across large pose ensembles within a unified computational formulation while explicitly evaluating interaction energies for each receptor–ligand configuration via inner products. We implement inner-product estimation using the Hadamard test and validate the formulation through systematic comparisons with classical atom-based and map-based energy evaluations. Across multiple receptor–ligand systems, we demonstrate that the proposed representation preserves energetic ordering in the low-energy regime relevant to structure-based virtual screening, while remaining robust under finite-sampling conditions. By exposing the tensorized structure underlying interaction-energy evaluation, this work establishes a representation-level formulation for map-based virtual screening compatible with both classical and quantum computational paradigms.
Structure Prediction

HighRelax: Physics-Based Refinement of Deep Learning Protein Predictions with Noncanonical Amino Acids
Sen Cao - ,
Chengyun Zhang - ,
Ning Zhu - ,
Chongyang Li - ,
Qingyi Mao - ,
Zhigang Cao - ,
Yutong Ge - ,
Yaling Wu - ,
Juan Guo - ,
Qiang Cao - ,
Jingjing Guo - ,
Zhiguo Wang *- , and
Hongliang Duan *
Noncanonical amino acids (NCAAs) have emerged as essential building blocks in protein engineering and peptide drug development owing to their advantages in enhancing metabolic stability, membrane permeability, and resistance to proteolytic degradation. Accurate construction of 3D protein structures containing NCAAs is crucial for elucidating their functions, understanding molecular interactions, and enabling rational design. However, integrating NCAAs into state-of-the-art protein structure prediction frameworks─such as AlphaFold3─often results in chirality violations, steric clashes, and local geometric distortions. These issues likely reflect limited parametrization of nonstandard residues within current models. To address these challenges, we expanded the AMBER force field covering 139 NCAAs, and we developed an enhanced Amber-relax protocol named HighRelax. Unlike conventional workflows that are restricted to linear peptides composed of canonical amino acids, HighRelax is compatible with complex systems containing NCAAs and cyclic peptides and can be seamlessly integrated with structures generated by state-of-the-art models such as AlphaFold3. Our results demonstrate that HighRelax effectively reduces steric clashes, restores residue chirality, and improves overall structural quality. This method provides a general postprocessing strategy for refining NCAA-containing structures, facilitating their applications in molecular simulation, peptide drug design, and protein engineering.
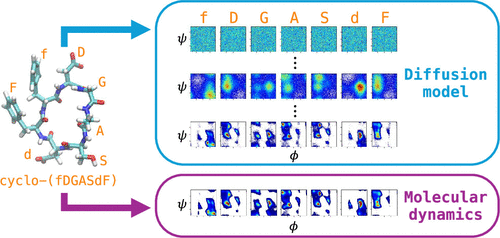
Fast Generation of Simulation-Quality Structural Ensembles of Mixed-Chirality Cyclic Peptides via Diffusion Models
Nomindari Bayaraa - ,
Maxim Secor - ,
Marc L. Descoteaux - , and
Yu-Shan Lin *
Cyclic peptides are an emerging therapeutic modality, with recent computational efforts focusing on the design of cyclic peptides that predominantly adopt a single conformation. However, many cyclic peptides adopt multiple conformations in solution, existing as structural ensembles. This conformational flexibility is often integral to their function: chameleonic switching between alternative states can enhance membrane permeability, and specific conformations may be required for molecular recognition and binding. Consequently, the ability to predict their structural ensembles is crucial for advancing the de novo design of cyclic peptide therapeutics. Here, we introduce diffusion models to efficiently and accurately predict structural ensembles of mixed-chirality cyclic peptides. The models are trained directly on molecular dynamics (MD) simulation data; in particular, each frame of the simulation becomes a single training instance in which a structure is represented as sine and cosine values of backbone dihedral angles. The trained diffusion model can not only generate MD-quality structures of cyclic peptides, but also the generated structures follow the Boltzmann distribution sampled in the MD simulation, enabling a deeper understanding of the physicochemical basis of cyclic peptide properties and allowing efficient computational design of cyclic peptides targeting biologically relevant systems.
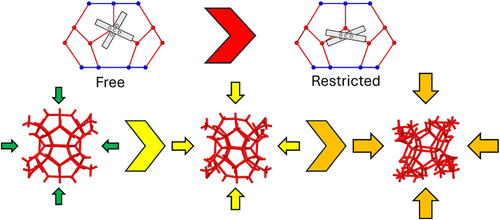
Atomistic Modeling of Methane and Carbon Dioxide Structure I Gas Hydrates under Pressure: Guest Effects and Properties
Samuel Mathews - ,
Xiaodan Zhu - ,
André Guerra - ,
Phillip Servio - , and
Alejandro Rey *
Gas hydrates are potential candidates in future energy sources while simultaneously providing structures with extensive applications in carbon capture and storage, gas transport, and important separation processes. Prior research in the field considers the dynamics of the water molecule backbone, in particular. We investigated the pressure-enthalpy landscape and mechanical stability envelope of sI methane and carbon dioxide hydrates simulated using DFT. We investigated the effect of revPBE + DFT-D2 and SCAN + rVV10 and their treatment of the exchange-correlation interactions. We examined the zero-pressure material properties, finding that revPBE comparatively underbinds the interactions, causing more flexible structures with large equilibrium volumes. Under pressure, the carbon dioxide molecule was found to align itself parallel to the hexagonal faces of the large cage despite the functional used. Additionally, the property differences are caused by the ability of the carbon dioxide molecule to rotate and disperse the changes in the energy landscape in ways that methane molecules cannot. This computational methodology describes the elastic stability of gas hydrates, marginal stability, and critical differences across important molecular interactions, confirming experimentally observed restrictions in guest molecule rotations and novel pressure behaviors under hydrostatic loads.
Additions and Corrections
Correction to “From PECs to Spectrum and From Spectrum to PECs: A Morse Protocol for Diatomic X-ray Absorption”
Minrui Wei - ,
Lu Zhang - ,
Junxiang Zuo *- ,
Guangjun Tian - , and
Weijie Hua *
This publication is free to access through this site. Learn More
Mastheads
Issue Editorial Masthead
This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More