



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
The Effect of Exogenous Acid Identity on Iron Tetraphenylporphyrin-Catalyzed CO2 ReductionClick to copy article linkArticle link copied!
- Kaeden TeindlKaeden TeindlDepartment of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, CanadaMore by Kaeden Teindl
- Eva M. Nichols*Eva M. Nichols*Email: [email protected]Department of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, CanadaMore by Eva M. Nichols
Abstract
Iron tetraphenylporphyrin (FeTPP) is a privileged electrocatalyst for the 2e–/2H+ reduction of CO2 to CO. FeTPP-catalyzed CO2 reduction typically employs phenol as an exogenous acid to promote the rate-limiting proton-coupled electron transfer. Beyond the observation that catalytic rates increase with decreasing pKa, the effects of acid identity on reaction kinetics are largely unexplored. Herein, we report rates of FeTPP-catalyzed CO2 reduction with structurally diverse O–H, N–H, and C–H acids. While many of these acids follow the expected Brønsted relationship, there are several notable exceptions: the fluorinated alcohols hexafluoroisopropanol (log(kcat) = 4.54) and 2,2,2-trifluoroethanol (log(kcat) = 3.55)─and the N–H acid imidazole (log(kcat) = 4.41)─display catalytic rates that are several times greater than rates obtained with similarly acidic phenols. Amides with pKas < 19 (in dimethyl sulfoxide) display similar activity as comparably acidic O–H acids, while rates obtained with less acidic amides are ∼2 orders of magnitude slower than O–H donors of similar pKa. Each C–H acid affords poor activity. An Eyring analysis suggests that acids enforcing less ordered transition states afford superior kinetics. This study reveals that acid pKa is only one relevant parameter for altering catalytic rates, and judicious selection of the acid is crucial for enhancing catalytic rates.
This publication is licensed for personal use by The American Chemical Society.
1. Introduction
Figure 1
Figure 1. Comparison of strategies to improve catalytic activity, where the catalyst is synthetically modified to include a secondary sphere proton relay (left) or the identity of the exogenous acid is altered (right).
2. Results and Discussion
2.1. Evaluating the Activity of Structurally Diverse Exogenous Acids by Cyclic Voltammetry
Figure 2
Figure 2. A) Structure of FeTPP and each of the structurally diverse exogenous acids. Values in parentheses indicate pKa values reported in DMSO. The cyclic voltammograms obtained with each (B) O–H, (C) N–H, and (D) C–H exogenous acid (CV conditions: 0.5 mM catalyst in dry DMF, 0.1 M TBAPF6, 50 mM acid, and 0.23 M CO2; scan rate = 0.1 V s–1). E) The kcat values of each exogenous acid as a function of pKa; kcat values are averaged over at least four different acid concentrations. Black line and symbols correspond to a Brønsted plot constructed from a series of phenols of tunable acidity reported in ref (21). Colors and symbols correspond to the exogenous acids displayed in panel A.
2.2. Controlled Potential Electrolysis to Evaluate Selectivity and Stability for Each Exogenous Acid Family
2.3. Effects of Structurally Diverse Exogenous Acids on CO2 Reduction Kinetics
| exogenous acid | catalytic rate law (kobs =) | log (kcat) |
|---|---|---|
| 4-trifluoromethyl phenola | kcat[CO2][acid] | 4.74 |
| 3-fluorophenola | 4.38 | |
| phenola | 3.84 | |
| 4-methoxy phenola | 3.58 | |
| 1-naphtholb | kcat[CO2][acid] | 4.04 ± 0.03 |
| 2-naphtholb | kcat[CO2][acid] | 3.70 ± 0.06 |
| 2,6-di-tert-butyl phenolb | kcat[CO2][acid]1/2 | 2.28 ± 0.03 |
| [NEt4][HCO3]b | kcat[CO2][acid] | 3.38 ± 0.08 |
| HFIPb | kcat[CO2][acid] | 4.54 ± 0.08 |
| TFEb | kcat[CO2][acid] | 3.55 ± 0.08 |
| pentafluorophenyl acetamideb | kcat[CO2][acid] | 4.40 ± 0.04 |
| 3,5-bis(trifluoromethyl)phenyl acetamideb | kcat[CO2][acid] | 3.39 ± 0.04 |
| 4-(trifluoromethyl)phenyl acetamideb | kcat[CO2][acid]1/2 | 1.98 ± 0.03 |
| acetanilideb | kcat[CO2][acid]1/2 | 1.79 ± 0.04 |
| benzanilideb,c |  | 1.63 ± 0.02 |
| imidazoleb | kcat[CO2][acid] | 4.41 ± 0.05 |
| fluoreneb,c |  | 1.65 ± 0.02 |
| acetylacetoneb | kcat[CO2][acid]1/2 | 2.52 ± 0.03 |
| no acidc,d |  | 1.64 ± 0.03 |
Kinetic parameters for each exogenous phenol used to construct the Brønsted relationship are obtained from ref (21).
kcat values are averages of at least four different acid concentrations.
The proposed second-order dependence on CO2 is based on the rate law reported in ref (58), in which FeTPP CO2 reduction is performed under aprotic conditions; we presume this reaction order holds under conditions wherein a zeroth-order dependence on acid is observed.
kcat value measured for the case of “no acid” is an average of three separate experiments, rather than different acid concentrations. All reported uncertainties represent one standard deviation.
Figure 3
Figure 3. Eyring plots for HFIP, phenol, and 3,5-bis(trifluoromethyl)phenyl acetamide. Colors and symbols correspond to the acids shown on the left. Kinetics were obtained from variable temperature CV experiments (T = 313, 298, 273, and 263 K) in dry DMF with 0.5 mM catalyst, 0.1 M TBAPF6, 50 mM acid, and 1 atm CO2; scan rate = 0.1 V s–1. Each data point is the average of two separate replicates.
| exogenous acid | ΔS‡ (e.u.) | ΔH‡ (kcal mol–1) |
|---|---|---|
| HFIP | –18 ± 8 | 6 ± 2 |
| phenol | –35 ± 3 | 2 ± 1 |
| 3,5-bis(trifluoromethyl) phenyl acetamide | –31 ± 4 | 4 ± 1 |
Uncertainties for each activation parameter are reported to two standard deviations.
2.4. Implications for Electronic Scaling Relationships
Figure 4
Figure 4. Electronic scaling plot (black symbols) showing the correlation between log(kcat) and E1/2(FeI/0) for the Fe complexes of meso-tetra(4-trifluoromethoxyphenyl) porphyrin, tetraphenyl porphyrin, and meso-tetra(4-methoxyphenyl) porphyrin (left to right). Colored symbols indicate the rate constants obtained for FeTPP with HFIP (red star) and 3,5-bis(trifluoromethyl)phenyl acetamide (blue triangle). log(kcat) values are averaged over several acid concentrations. The yellow region (top left) indicates region of fast kinetics and mild reduction potentials, while the purple region indicates the opposite (lower right). Kinetic parameters were obtained in dry DMF with 0.5 mM catalyst, 0.1 M TBAPF6, 50 mM acid, and 1 atm CO2; scan rate = 0.1 V s–1
3. Conclusions
4. Experimental Section
4.1. General Methods for Electrochemistry
4.2. Controlled Potential Electrolysis Experiments
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.5c05122.
pKa determination, electrochemical data, gas chromatography, and Eyring analysis (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Eva M. Nichols - Department of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada;
 https://orcid.org/0000-0002-3718-7273;
https://orcid.org/0000-0002-3718-7273;
- Kaeden Teindl - Department of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada;https://orcid.org/0009-0005-3367-6845
K.T. and E.M.N. conceived the project and wrote the manuscript. K.T. performed all experimental work and data analysis.
Acknowledgments
This work was supported by the University of British Columbia, the Natural Sciences and Engineering Research Council of Canada (RGPIN-2021-03691 and DGECR-2021-00427), and the Research Corporation for Science Advancement (27752). K.T. acknowledges support from a British Columbia Graduate Fellowship, as well as UBC chemistry for a Head’s departmental scholarship. E.M.N. gratefully acknowledges NSERC for support as a Canada Research Chair and CIFAR for support as an Azrieli Global Scholar.
References
This article references 76 other publications.
- 1Appel, A. M.; Bercaw, J. E.; Bocarsly, A. B.; Dobbek, H.; DuBois, D. L.; Dupuis, M.; Ferry, J. G.; Fujita, E.; Hille, R.; Kenis, P. J. A.; Kerfeld, C. A.; Morris, R. H.; Peden, C. H. F.; Portis, A. R.; Ragsdale, S. W.; Rauchfuss, T. B.; Reek, J. N. H.; Seefeldt, L. C.; Thauer, R. K.; Waldrop, G. L. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621– 6658, DOI: 10.1021/cr300463yGoogle ScholarThere is no corresponding record for this reference.
- 2Bushuyev, O. S.; De Luna, P.; Dinh, C. T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S. O.; Sargent, E. H. What Should We Make with CO2 and How Can We Make It?. Joule 2018, 2, 825– 832, DOI: 10.1016/j.joule.2017.09.003Google ScholarThere is no corresponding record for this reference.
- 3Morris, A. J.; Meyer, G. J.; Fujita, E. Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels. Acc. Chem. Res. 2009, 42, 1983– 1994, DOI: 10.1021/ar9001679Google ScholarThere is no corresponding record for this reference.
- 4Benson, E. E.; Kubiak, C. P.; Sathrum, A. J.; Smieja, J. M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89– 99, DOI: 10.1039/B804323JGoogle ScholarThere is no corresponding record for this reference.
- 5Beley, M.; Collin, J. P.; Ruppert, R.; Sauvage, J. P. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: study of the factors affecting the efficiency and the selectivity of the process. J. Am. Chem. Soc. 1986, 108, 7461– 7467, DOI: 10.1021/ja00284a003Google ScholarThere is no corresponding record for this reference.
- 6Beley, M.; Collin, J.-P.; Ruppert, R.; Sauvage, J.-P. Nickel(II)-cyclam: an extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc., Chem. Commun. 1984, 1315– 1316, DOI: 10.1039/c39840001315Google ScholarThere is no corresponding record for this reference.
- 7Fisher, B. J.; Eisenberg, R. Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt. J. Am. Chem. Soc. 1980, 102, 7361– 7363, DOI: 10.1021/ja00544a035Google ScholarThere is no corresponding record for this reference.
- 8Sampson, M. D.; Nguyen, A. D.; Grice, K. A.; Moore, C. E.; Rheingold, A. L.; Kubiak, C. P. Manganese Catalysts with Bulky Bipyridine Ligands for the Electrocatalytic Reduction of Carbon Dioxide: Eliminating Dimerization and Altering Catalysis. J. Am. Chem. Soc. 2014, 136, 5460– 5471, DOI: 10.1021/ja501252fGoogle ScholarThere is no corresponding record for this reference.
- 9Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem., Int. Ed. 2011, 50, 9903– 9906, DOI: 10.1002/anie.201103616Google ScholarThere is no corresponding record for this reference.
- 10Ishida, H.; Tanaka, K.; Tanaka, T. Electrochemical CO2 reduction catalyzed by ruthenium complexes [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)Cl]+. Effect of pH on the formation of CO and HCOO. Organometallics 1987, 6, 181– 186, DOI: 10.1021/om00144a033Google ScholarThere is no corresponding record for this reference.
- 11Bolinger, C. M.; Story, N.; Sullivan, B. P.; Meyer, T. J. Electrocatalytic reduction of carbon dioxide by 2,2’-bipyridine complexes of rhodium and iridium. Inorg. Chem. 1988, 27, 4582– 4587, DOI: 10.1021/ic00298a016Google ScholarThere is no corresponding record for this reference.
- 12Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy= 2, 2′-bipyridine). J. Chem. Soc., Chem. Commun. 1984, 328– 330, DOI: 10.1039/C39840000328Google ScholarThere is no corresponding record for this reference.
- 13Zee, D. Z.; Nippe, M.; King, A. E.; Chang, C. J.; Long, J. R. Tuning Second Coordination Sphere Interactions in Polypyridyl–Iron Complexes to Achieve Selective Electrocatalytic Reduction of Carbon Dioxide to Carbon Monoxide. Inorg. Chem. 2020, 59, 5206– 5217, DOI: 10.1021/acs.inorgchem.0c00455Google ScholarThere is no corresponding record for this reference.
- 14Fernández, S.; Franco, F.; Casadevall, C.; Martin-Diaconescu, V.; Luis, J. M.; Lloret-Fillol, J. A Unified Electro- and Photocatalytic CO2 to CO Reduction Mechanism with Aminopyridine Cobalt Complexes. J. Am. Chem. Soc. 2020, 142, 120– 133, DOI: 10.1021/jacs.9b06633Google ScholarThere is no corresponding record for this reference.
- 15Kang, P.; Meyer, T. J.; Brookhart, M. Selective electrocatalytic reduction of carbon dioxide to formate by a water-soluble iridium pincer catalyst. Chem. Sci. 2013, 4, 3497– 3502, DOI: 10.1039/c3sc51339dGoogle ScholarThere is no corresponding record for this reference.
- 16DuBois, D. L.; Miedaner, A.; Haltiwanger, R. C. Electrochemical reduction of carbon dioxide catalyzed by [Pd (triphosphine)(solvent)](BF4)2 complexes: synthetic and mechanistic studies. J. Am. Chem. Soc. 1991, 113, 8753– 8764, DOI: 10.1021/ja00023a023Google ScholarThere is no corresponding record for this reference.
- 17Slater, S.; Wagenknecht, J. H. Electrochemical reduction of carbon dioxide catalyzed by Rh(diphos)2Cl. J. Am. Chem. Soc. 1984, 106, 5367– 5368, DOI: 10.1021/ja00330a064Google ScholarThere is no corresponding record for this reference.
- 18Dubois, D. L. Development of Transition Metal Phosphine Complexes as Electrocatalysts for CO2 and CO Reduction. Comments Inorg. Chem. 1997, 19, 307– 325, DOI: 10.1080/02603599708032743Google ScholarThere is no corresponding record for this reference.
- 19Teindl, K.; Cai, A. Y.; Graham, D.; Branch, K. L.; Farhat, R.; Sahu, S.; Hallas, A. M.; Nichols, E. M. Electrochemical formation of methane, ethylene, and ethane with a bimetallic nickel complex: Is CO2 the source of carbon?. Dalton Trans. 2025, DOI: 10.1039/d5dt01205hGoogle ScholarThere is no corresponding record for this reference.
- 20Nichols, E. M.; Derrick, J. S.; Nistanaki, S. K.; Smith, P. T.; Chang, C. J. Positional effects of second-sphere amide pendants on electrochemical CO2 reduction catalyzed by iron porphyrins. Chem. Sci. 2018, 9, 2952– 2960, DOI: 10.1039/C7SC04682KGoogle ScholarThere is no corresponding record for this reference.
- 21Teindl, K.; Patrick, B. O.; Nichols, E. M. Linear Free Energy Relationships and Transition State Analysis of CO2 Reduction Catalysts Bearing Second Coordination Spheres with Tunable Acidity. J. Am. Chem. Soc. 2023, 145, 17176– 17186, DOI: 10.1021/jacs.3c03919Google ScholarThere is no corresponding record for this reference.
- 22Branch, K. L.; Johnson, E. R.; Nichols, E. M. Porphyrin Aggregation under Homogeneous Conditions Inhibits Electrocatalysis: A Case Study on CO2 Reduction. ACS Cent. Sci. 2024, 10, 1251– 1261, DOI: 10.1021/acscentsci.4c00121Google ScholarThere is no corresponding record for this reference.
- 23Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90– 94, DOI: 10.1126/science.1224581Google ScholarThere is no corresponding record for this reference.
- 24Costentin, C.; Passard, G.; Robert, M.; Savéant, J.-M. Pendant Acid–Base Groups in Molecular Catalysts: H-Bond Promoters or Proton Relays? Mechanisms of the Conversion of CO2 to CO by Electrogenerated Iron(0)Porphyrins Bearing Prepositioned Phenol Functionalities. J. Am. Chem. Soc. 2014, 136, 11821– 11829, DOI: 10.1021/ja506193vGoogle ScholarThere is no corresponding record for this reference.
- 25Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. Dissection of Electronic Substituent Effects in Multielectron–Multistep Molecular Catalysis. Electrochemical CO2-to-CO Conversion Catalyzed by Iron Porphyrins. J. Phys. Chem. C 2016, 120, 28951– 28960, DOI: 10.1021/acs.jpcc.6b09947Google ScholarThere is no corresponding record for this reference.
- 26Sinha, S.; Warren, J. J. Unexpected Solvent Effect in Electrocatalytic CO2 to CO Conversion Revealed Using Asymmetric Metalloporphyrins. Inorg. Chem. 2018, 57, 12650– 12656, DOI: 10.1021/acs.inorgchem.8b01814Google ScholarThere is no corresponding record for this reference.
- 27Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639– 16644, DOI: 10.1021/jacs.6b07014Google ScholarThere is no corresponding record for this reference.
- 28Costentin, C.; Drouet, S.; Passard, G.; Robert, M.; Savéant, J.-M. Proton-Coupled Electron Transfer Cleavage of Heavy-Atom Bonds in Electrocatalytic Processes. Cleavage of a C–O Bond in the Catalyzed Electrochemical Reduction of CO2. J. Am. Chem. Soc. 2013, 135, 9023– 9031, DOI: 10.1021/ja4030148Google ScholarThere is no corresponding record for this reference.
- 29Nguyen, B. X.; Sonea, A.; Warren, J. J. Further Understanding the Roles of Solvent, Brønsted Acids, and Hydrogen Bonding in Iron Porphyrin-Mediated Carbon Dioxide Reduction. Inorg. Chem. 2023, 62, 17602– 17611, DOI: 10.1021/acs.inorgchem.3c01855Google ScholarThere is no corresponding record for this reference.
- 30Gotico, P.; Boitrel, B.; Guillot, R.; Sircoglou, M.; Quaranta, A.; Halime, Z.; Leibl, W.; Aukauloo, A. Second-Sphere Biomimetic Multipoint Hydrogen-Bonding Patterns to Boost CO2 Reduction of Iron Porphyrins. Angew. Chem., Int. Ed. 2019, 58, 4504– 4509, DOI: 10.1002/anie.201814339Google ScholarThere is no corresponding record for this reference.
- 31Sonea, A.; Branch, K. L.; Warren, J. J. The Pattern of Hydroxyphenyl-Substitution Influences CO2 Reduction More Strongly than the Number of Hydroxyphenyl Groups in Iron-Porphyrin Electrocatalysts. ACS Catal. 2023, 13, 3902– 3912, DOI: 10.1021/acscatal.2c06275Google ScholarThere is no corresponding record for this reference.
- 32Amanullah, S.; Gotico, P.; Sircoglou, M.; Leibl, W.; Llansola-Portoles, M. J.; Tibiletti, T.; Quaranta, A.; Halime, Z.; Aukauloo, A. Second Coordination Sphere Effect Shifts CO2 to CO Reduction by Iron Porphyrin from Fe(0) to Fe(I). Angew. Chem., Int. Ed. 2024, 136, e202314439 DOI: 10.1002/anie.202314439Google ScholarThere is no corresponding record for this reference.
- 33Margarit, C. G.; Schnedermann, C.; Asimow, N. G.; Nocera, D. G. Carbon Dioxide Reduction by Iron Hangman Porphyrins. Organometallics 2019, 38, 1219– 1223, DOI: 10.1021/acs.organomet.8b00334Google ScholarThere is no corresponding record for this reference.
- 34Margarit, C. G.; Asimow, N. G.; Gonzalez, M. I.; Nocera, D. G. Double Hangman Iron Porphyrin and the Effect of Electrostatic Nonbonding Interactions on Carbon Dioxide Reduction. J. Phys. Chem. Lett. 2020, 11, 1890– 1895, DOI: 10.1021/acs.jpclett.9b03897Google ScholarThere is no corresponding record for this reference.
- 35Sen, P.; Mondal, B.; Saha, D.; Rana, A.; Dey, A. Role of 2nd sphere H-bonding residues in tuning the kinetics of CO2 reduction to CO by iron porphyrin complexes. Dalton Trans. 2019, 48, 5965– 5977, DOI: 10.1039/C8DT03850CGoogle ScholarThere is no corresponding record for this reference.
- 36Teindl, K.; Jacobs, D.; Panetier, J. A.; Nichols, E. M. Highly Acidic Second Coordination Spheres Promote In Situ Formation of Iron Phlorins Exhibiting Fast and Selective CO2 Reduction. J. Am. Chem. Soc. 2025, DOI: 10.26434/chemrxiv-2025-5zzvbGoogle ScholarThere is no corresponding record for this reference.
- 37Derrick, J. S.; Loipersberger, M.; Nistanaki, S. K.; Rothweiler, A. V.; Head-Gordon, M.; Nichols, E. M.; Chang, C. J. Templating Bicarbonate in the Second Coordination Sphere Enhances Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins. J. Am. Chem. Soc. 2022, 144, 11656– 11663, DOI: 10.1021/jacs.2c02972Google ScholarThere is no corresponding record for this reference.
- 38Guo, K.; Li, X.; Lei, H.; Guo, H.; Jin, X.; Zhang, X.-P.; Zhang, W.; Apfel, U.-P.; Cao, R. Role-Specialized Division of Labor in CO2 Reduction with Doubly-Functionalized Iron Porphyrin Atropisomers. Angew. Chem., Int. Ed. 2022, 61, e202209602 DOI: 10.1002/anie.202209602Google ScholarThere is no corresponding record for this reference.
- 39Martin, D. J.; Mayer, J. M. Oriented Electrostatic Effects on O2 and CO2 Reduction by a Polycationic Iron Porphyrin. J. Am. Chem. Soc. 2021, 143, 11423– 11434, DOI: 10.1021/jacs.1c03132Google ScholarThere is no corresponding record for this reference.
- 40Nichols, E. M.; Chang, C. J. Urea-Based Multipoint Hydrogen-Bond Donor Additive Promotes Electrochemical CO2 Reduction Catalyzed by Nickel Cyclam. Organometallics 2019, 38, 1213– 1218, DOI: 10.1021/acs.organomet.8b00308Google ScholarThere is no corresponding record for this reference.
- 41Cypcar, A. D.; Bui, K. M.; Yang, J. Y. Suppressing H2 Evolution with Sterically Encumbered Proton Sources to Improve the Faradaic Efficiency for CO2 Reduction to Formate. ACS Catal. 2024, 14, 14517– 14523, DOI: 10.1021/acscatal.4c03809Google ScholarThere is no corresponding record for this reference.
- 42Madsen, M. R.; Rønne, M. H.; Heuschen, M.; Golo, D.; Ahlquist, M. S. G.; Skrydstrup, T.; Pedersen, S. U.; Daasbjerg, K. Promoting Selective Generation of Formic Acid from CO2 Using Mn(bpy)(CO)3Br as Electrocatalyst and Triethylamine/Isopropanol as Additives. J. Am. Chem. Soc. 2021, 143, 20491– 20500, DOI: 10.1021/jacs.1c10805Google ScholarThere is no corresponding record for this reference.
- 43Margarit, C. G.; Asimow, N. G.; Costentin, C.; Nocera, D. G. Tertiary Amine-Assisted Electroreduction of Carbon Dioxide to Formate Catalyzed by Iron Tetraphenylporphyrin. ACS Energy Lett. 2020, 5, 72– 78, DOI: 10.1021/acsenergylett.9b02093Google ScholarThere is no corresponding record for this reference.
- 44Martin, D. J.; Mercado, B. Q.; Mayer, J. M. Combining scaling relationships overcomes rate versus overpotential trade-offs in O2 molecular electrocatalysis. Sci. Adv. 2020, 6, eaaz3318 DOI: 10.1126/sciadv.aaz3318Google ScholarThere is no corresponding record for this reference.
- 45Cook, E. N.; Courter, I. M.; Dickie, D. A.; Machan, C. W. Controlling product selectivity during dioxygen reduction with Mn complexes using pendent proton donor relays and added base. Chem. Sci. 2024, 15, 4478– 4488, DOI: 10.1039/D3SC02611FGoogle ScholarThere is no corresponding record for this reference.
- 46Martin, D. J.; Wise, C. F.; Pegis, M. L.; Mayer, J. M. Developing Scaling Relationships for Molecular Electrocatalysis through Studies of Fe-Porphyrin-Catalyzed O2 Reduction. Acc. Chem. Res. 2020, 53, 1056– 1065, DOI: 10.1021/acs.accounts.0c00044Google ScholarThere is no corresponding record for this reference.
- 47Lazouski, N.; Steinberg, K. J.; Gala, M. L.; Krishnamurthy, D.; Viswanathan, V.; Manthiram, K. Proton Donors Induce a Differential Transport Effect for Selectivity toward Ammonia in Lithium-Mediated Nitrogen Reduction. ACS Cat 2022, 12, 5197– 5208, DOI: 10.1021/acscatal.2c00389Google ScholarThere is no corresponding record for this reference.
- 48Maran, F.; Celadon, D.; Severin, M. G.; Vianello, E. Electrochemical determination of the pKa of weak acids in N, N-dimethylformamide. J. Am. Chem. Soc. 1991, 113, 9320– 9329, DOI: 10.1021/ja00024a041Google ScholarThere is no corresponding record for this reference.
- 49Bordwell, F. G.; Cheng, J. Substituent effects on the stabilities of phenoxyl radicals and the acidities of phenoxyl radical cations. J. Am. Chem. Soc. 1991, 113, 1736– 1743, DOI: 10.1021/ja00005a042Google ScholarThere is no corresponding record for this reference.
- 50Tshepelevitsh, S.; Trummal, A.; Haav, K.; Martin, K.; Leito, I. Hydrogen-Bond Donicity in DMSO and Gas Phase and Its Dependence on Brønsted Acidity. J. Phys. Chem. A 2017, 121, 357– 369, DOI: 10.1021/acs.jpca.6b11115Google ScholarThere is no corresponding record for this reference.
- 51Bordwell, F. G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456– 463, DOI: 10.1021/ar00156a004Google ScholarThere is no corresponding record for this reference.
- 52Teindl, K.; Reid, J. P.; Nichols, E. M. Promoting selective electrochemical CO2 reduction under unconventionally acidic conditions through secondary coordination sphere positioning. Chem. Sci. 2025, 16, 19226– 19234, DOI: 10.1039/D5SC04700EGoogle ScholarThere is no corresponding record for this reference.
- 53Kütt, A.; Tshepelevitsh, S.; Saame, J.; Lõkov, M.; Kaljurand, I.; Selberg, S.; Leito, I. Strengths of Acids in Acetonitrile. Eur. J. Org. Chem. 2021, 2021, 1407– 1419, DOI: 10.1002/ejoc.202001649Google ScholarThere is no corresponding record for this reference.
- 54Bordwell, F. G.; Ji, G. Z. Effects of structural changes on acidities and homolytic bond dissociation energies of the hydrogen-nitrogen bonds in amidines, carboxamides, and thiocarboxamides. J. Am. Chem. Soc. 1991, 113, 8398– 8401, DOI: 10.1021/ja00022a029Google ScholarThere is no corresponding record for this reference.
- 55Matthews, W. S.; Bares, J. E.; Bartmess, J. E.; Bordwell, F. G.; Cornforth, F. J.; Drucker, G. E.; Margolin, Z.; McCallum, R. J.; McCollum, G. J.; Vanier, N. R. Equilibrium acidities of carbon acids. VI. Establishment of an absolute scale of acidities in dimethyl sulfoxide solution. J. Am. Chem. Soc. 1975, 97, 7006– 7014, DOI: 10.1021/ja00857a010Google ScholarThere is no corresponding record for this reference.
- 56Olmstead, W. N.; Bordwell, F. G. Ion-pair association constants in dimethyl sulfoxide. J. Org. Chem. 1980, 45, 3299– 3305, DOI: 10.1021/jo01304a033Google ScholarThere is no corresponding record for this reference.
- 57Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide by Iron(0) Porphyrins: Synergystic Effect of Weak Brönsted Acids. J. Am. Chem. Soc. 1996, 118, 1769– 1776, DOI: 10.1021/ja9534462Google ScholarThere is no corresponding record for this reference.
- 58Hammouche, M.; Lexa, D.; Savéant, J. M.; Momenteau, M. Catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. J. Electroanal. Chem. Interfac. Electrochem. 1988, 249, 347– 351, DOI: 10.1016/0022-0728(88)80372-3Google ScholarThere is no corresponding record for this reference.
- 59Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide by Iron(0) Porphyrins. Synergistic Effect of Lewis Acid Cations. J. Phys. Chem. 1996, 100, 19981– 19985, DOI: 10.1021/jp9618486Google ScholarThere is no corresponding record for this reference.
- 60Dixon, D. A.; Dobbs, K. D.; Valentini, J. J. Amide-water and amide-amide hydrogen bond strengths. J. Phys. Chem. 1994, 98, 13435– 13439, DOI: 10.1021/j100102a001Google ScholarThere is no corresponding record for this reference.
- 61Vallejo Narváez, W. E.; Jiménez, E. I.; Romero-Montalvo, E.; Sauza-de la Vega, A.; Quiroz-García, B.; Hernández-Rodríguez, M.; Rocha-Rinza, T. Acidity and basicity interplay in amide and imide self-association. Chem. Sci. 2018, 9, 4402– 4413, DOI: 10.1039/C8SC01020JGoogle ScholarThere is no corresponding record for this reference.
- 62Kraft, A.; Osterod, F. Self-association of branched and dendritic aromatic amides. J. Chem. Soc., Perkin Trans. 1998, 1, 1019– 1026, DOI: 10.1039/a800100fGoogle ScholarThere is no corresponding record for this reference.
- 63Sassone, D.; Bocchini, S.; Fontana, M.; Salvini, C.; Cicero, G.; Re Fiorentin, M.; Risplendi, F.; Latini, G.; Amin Farkhondehfal, M.; Pirri, F.; Zeng, J. Imidazole-imidazolate pair as organo-electrocatalyst for CO2 reduction on ZIF-8 material. Appl. Energy 2022, 324, 119743, DOI: 10.1016/j.apenergy.2022.119743Google ScholarThere is no corresponding record for this reference.
- 64Izadyar, M.; Rezaeian, M.; Victorov, A. Theoretical study on the absorption of carbon dioxide by DBU-based ionic liquids. Phys. Chem. Chem. Phys. 2020, 22, 20050– 20060, DOI: 10.1039/D0CP03612AGoogle ScholarThere is no corresponding record for this reference.
- 65Zhu, X.; Song, M.; Xu, Y. DBU-Based Protic Ionic Liquids for CO2 Capture. ACS Sustainable Chem. Eng. 2017, 5, 8192– 8198, DOI: 10.1021/acssuschemeng.7b01839Google ScholarThere is no corresponding record for this reference.
- 66Shu, H.; Xu, Y. Tuning the strength of cation coordination interactions of dual functional ionic liquids for improving CO2 capture performance. Int. J. Greenhouse Gas Control 2020, 94, 102934, DOI: 10.1016/j.ijggc.2019.102934Google ScholarThere is no corresponding record for this reference.
- 67Darcy, J. W.; Koronkiewicz, B.; Parada, G. A.; Mayer, J. M. A Continuum of Proton-Coupled Electron Transfer Reactivity. Acc. Chem. Res. 2018, 51, 2391– 2399, DOI: 10.1021/acs.accounts.8b00319Google ScholarThere is no corresponding record for this reference.
- 68Morris, W. D.; Mayer, J. M. Separating proton and electron transfer effects in three-component concerted proton-coupled electron transfer reactions. J. Am. Chem. Soc. 2017, 139, 10312– 10319, DOI: 10.1021/jacs.7b03562Google ScholarThere is no corresponding record for this reference.
- 69Rhile, I. J.; Markle, T. F.; Nagao, H.; DiPasquale, A. G.; Lam, O. P.; Lockwood, M. A.; Rotter, K.; Mayer, J. M. Concerted proton–electron transfer in the oxidation of hydrogen-bonded phenols. J. Am. Chem. Soc. 2006, 128, 6075– 6088, DOI: 10.1021/ja054167+Google ScholarThere is no corresponding record for this reference.
- 70Markle, T. F.; Darcy, J. W.; Mayer, J. M. A new strategy to efficiently cleave and form C–H bonds using proton-coupled electron transfer. Sci. Adv. 2018, 4, eaat5776 DOI: 10.1126/sciadv.aat5776Google ScholarThere is no corresponding record for this reference.
- 71Abraham, M. H.; Grellier, P. L.; Prior, D. V.; Duce, P. P.; Morris, J. J.; Taylor, P. J. Hydrogen bonding. Part 7. A scale of solute hydrogen-bond acidity based on log K values for complexation in tetrachloromethane. J. Chem. Soc., Perkin Trans. 1989, 2, 699– 711, DOI: 10.1039/p29890000699Google ScholarThere is no corresponding record for this reference.
- 72Rovira, C.; Kunc, K.; Hutter, J.; Ballone, P.; Parrinello, M. Equilibrium Geometries and Electronic Structure of Iron–Porphyrin Complexes: A Density Functional Study. J. Phys. Chem. A 1997, 101, 8914– 8925, DOI: 10.1021/jp9722115Google ScholarThere is no corresponding record for this reference.
- 73Rovira, C.; Kunc, K.; Hutter, J.; Ballone, P.; Parrinello, M. A comparative study of O2, CO, and NO binding to iron–porphyrin. Int. J. Quantum Chem. 1998, 69, 31– 35, DOI: 10.1002/(SICI)1097-461X(1998)69:1<31::AID-QUA5>3.0.CO;2-YGoogle ScholarThere is no corresponding record for this reference.
- 74Abraham, M. H.; Grellier, P. L.; Prior, D. V.; Morris, J. J.; Taylor, P. J. Hydrogen bonding. Part 10. A scale of solute hydrogen-bond basicity using log K values for complexation in tetrachloromethane. J. Chem. Soc., Perkin Trans. 1990, 2, 521– 529, DOI: 10.1039/p29900000521Google ScholarThere is no corresponding record for this reference.
- 75Anslyn, E. V.; Dougherty, D. A. Modern Physical Organic Chemistry. 1 ed. University Science Books 2006, 1104, DOI: 10.1002/anie.200585348Google ScholarThere is no corresponding record for this reference.
- 76Teindl, K.; Nichols, E. M. The Effect of Exogenous Acid Identity on Iron Tetraphenylporphyrin-Catalyzed CO2 Reduction. ChemRxiv 2025, DOI: 10.26434/chemrxiv-2025-n2h1qGoogle ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract

Figure 1

Figure 1. Comparison of strategies to improve catalytic activity, where the catalyst is synthetically modified to include a secondary sphere proton relay (left) or the identity of the exogenous acid is altered (right).
Figure 2

Figure 2. A) Structure of FeTPP and each of the structurally diverse exogenous acids. Values in parentheses indicate pKa values reported in DMSO. The cyclic voltammograms obtained with each (B) O–H, (C) N–H, and (D) C–H exogenous acid (CV conditions: 0.5 mM catalyst in dry DMF, 0.1 M TBAPF6, 50 mM acid, and 0.23 M CO2; scan rate = 0.1 V s–1). E) The kcat values of each exogenous acid as a function of pKa; kcat values are averaged over at least four different acid concentrations. Black line and symbols correspond to a Brønsted plot constructed from a series of phenols of tunable acidity reported in ref (21). Colors and symbols correspond to the exogenous acids displayed in panel A.
Figure 3

Figure 3. Eyring plots for HFIP, phenol, and 3,5-bis(trifluoromethyl)phenyl acetamide. Colors and symbols correspond to the acids shown on the left. Kinetics were obtained from variable temperature CV experiments (T = 313, 298, 273, and 263 K) in dry DMF with 0.5 mM catalyst, 0.1 M TBAPF6, 50 mM acid, and 1 atm CO2; scan rate = 0.1 V s–1. Each data point is the average of two separate replicates.
Figure 4
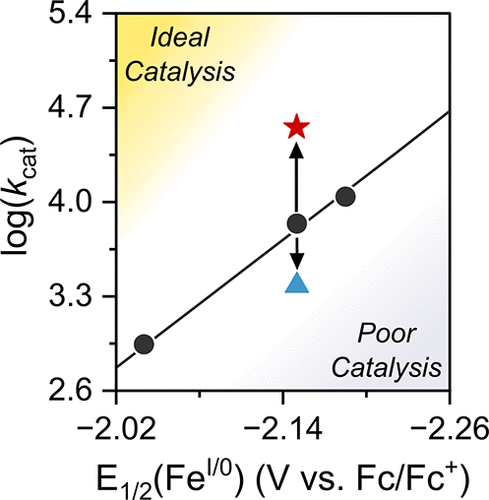
Figure 4. Electronic scaling plot (black symbols) showing the correlation between log(kcat) and E1/2(FeI/0) for the Fe complexes of meso-tetra(4-trifluoromethoxyphenyl) porphyrin, tetraphenyl porphyrin, and meso-tetra(4-methoxyphenyl) porphyrin (left to right). Colored symbols indicate the rate constants obtained for FeTPP with HFIP (red star) and 3,5-bis(trifluoromethyl)phenyl acetamide (blue triangle). log(kcat) values are averaged over several acid concentrations. The yellow region (top left) indicates region of fast kinetics and mild reduction potentials, while the purple region indicates the opposite (lower right). Kinetic parameters were obtained in dry DMF with 0.5 mM catalyst, 0.1 M TBAPF6, 50 mM acid, and 1 atm CO2; scan rate = 0.1 V s–1
References
This article references 76 other publications.
- 1Appel, A. M.; Bercaw, J. E.; Bocarsly, A. B.; Dobbek, H.; DuBois, D. L.; Dupuis, M.; Ferry, J. G.; Fujita, E.; Hille, R.; Kenis, P. J. A.; Kerfeld, C. A.; Morris, R. H.; Peden, C. H. F.; Portis, A. R.; Ragsdale, S. W.; Rauchfuss, T. B.; Reek, J. N. H.; Seefeldt, L. C.; Thauer, R. K.; Waldrop, G. L. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621– 6658, DOI: 10.1021/cr300463yThere is no corresponding record for this reference.
- 2Bushuyev, O. S.; De Luna, P.; Dinh, C. T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S. O.; Sargent, E. H. What Should We Make with CO2 and How Can We Make It?. Joule 2018, 2, 825– 832, DOI: 10.1016/j.joule.2017.09.003There is no corresponding record for this reference.
- 3Morris, A. J.; Meyer, G. J.; Fujita, E. Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels. Acc. Chem. Res. 2009, 42, 1983– 1994, DOI: 10.1021/ar9001679There is no corresponding record for this reference.
- 4Benson, E. E.; Kubiak, C. P.; Sathrum, A. J.; Smieja, J. M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89– 99, DOI: 10.1039/B804323JThere is no corresponding record for this reference.
- 5Beley, M.; Collin, J. P.; Ruppert, R.; Sauvage, J. P. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: study of the factors affecting the efficiency and the selectivity of the process. J. Am. Chem. Soc. 1986, 108, 7461– 7467, DOI: 10.1021/ja00284a003There is no corresponding record for this reference.
- 6Beley, M.; Collin, J.-P.; Ruppert, R.; Sauvage, J.-P. Nickel(II)-cyclam: an extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc., Chem. Commun. 1984, 1315– 1316, DOI: 10.1039/c39840001315There is no corresponding record for this reference.
- 7Fisher, B. J.; Eisenberg, R. Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt. J. Am. Chem. Soc. 1980, 102, 7361– 7363, DOI: 10.1021/ja00544a035There is no corresponding record for this reference.
- 8Sampson, M. D.; Nguyen, A. D.; Grice, K. A.; Moore, C. E.; Rheingold, A. L.; Kubiak, C. P. Manganese Catalysts with Bulky Bipyridine Ligands for the Electrocatalytic Reduction of Carbon Dioxide: Eliminating Dimerization and Altering Catalysis. J. Am. Chem. Soc. 2014, 136, 5460– 5471, DOI: 10.1021/ja501252fThere is no corresponding record for this reference.
- 9Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem., Int. Ed. 2011, 50, 9903– 9906, DOI: 10.1002/anie.201103616There is no corresponding record for this reference.
- 10Ishida, H.; Tanaka, K.; Tanaka, T. Electrochemical CO2 reduction catalyzed by ruthenium complexes [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)Cl]+. Effect of pH on the formation of CO and HCOO. Organometallics 1987, 6, 181– 186, DOI: 10.1021/om00144a033There is no corresponding record for this reference.
- 11Bolinger, C. M.; Story, N.; Sullivan, B. P.; Meyer, T. J. Electrocatalytic reduction of carbon dioxide by 2,2’-bipyridine complexes of rhodium and iridium. Inorg. Chem. 1988, 27, 4582– 4587, DOI: 10.1021/ic00298a016There is no corresponding record for this reference.
- 12Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy= 2, 2′-bipyridine). J. Chem. Soc., Chem. Commun. 1984, 328– 330, DOI: 10.1039/C39840000328There is no corresponding record for this reference.
- 13Zee, D. Z.; Nippe, M.; King, A. E.; Chang, C. J.; Long, J. R. Tuning Second Coordination Sphere Interactions in Polypyridyl–Iron Complexes to Achieve Selective Electrocatalytic Reduction of Carbon Dioxide to Carbon Monoxide. Inorg. Chem. 2020, 59, 5206– 5217, DOI: 10.1021/acs.inorgchem.0c00455There is no corresponding record for this reference.
- 14Fernández, S.; Franco, F.; Casadevall, C.; Martin-Diaconescu, V.; Luis, J. M.; Lloret-Fillol, J. A Unified Electro- and Photocatalytic CO2 to CO Reduction Mechanism with Aminopyridine Cobalt Complexes. J. Am. Chem. Soc. 2020, 142, 120– 133, DOI: 10.1021/jacs.9b06633There is no corresponding record for this reference.
- 15Kang, P.; Meyer, T. J.; Brookhart, M. Selective electrocatalytic reduction of carbon dioxide to formate by a water-soluble iridium pincer catalyst. Chem. Sci. 2013, 4, 3497– 3502, DOI: 10.1039/c3sc51339dThere is no corresponding record for this reference.
- 16DuBois, D. L.; Miedaner, A.; Haltiwanger, R. C. Electrochemical reduction of carbon dioxide catalyzed by [Pd (triphosphine)(solvent)](BF4)2 complexes: synthetic and mechanistic studies. J. Am. Chem. Soc. 1991, 113, 8753– 8764, DOI: 10.1021/ja00023a023There is no corresponding record for this reference.
- 17Slater, S.; Wagenknecht, J. H. Electrochemical reduction of carbon dioxide catalyzed by Rh(diphos)2Cl. J. Am. Chem. Soc. 1984, 106, 5367– 5368, DOI: 10.1021/ja00330a064There is no corresponding record for this reference.
- 18Dubois, D. L. Development of Transition Metal Phosphine Complexes as Electrocatalysts for CO2 and CO Reduction. Comments Inorg. Chem. 1997, 19, 307– 325, DOI: 10.1080/02603599708032743There is no corresponding record for this reference.
- 19Teindl, K.; Cai, A. Y.; Graham, D.; Branch, K. L.; Farhat, R.; Sahu, S.; Hallas, A. M.; Nichols, E. M. Electrochemical formation of methane, ethylene, and ethane with a bimetallic nickel complex: Is CO2 the source of carbon?. Dalton Trans. 2025, DOI: 10.1039/d5dt01205hThere is no corresponding record for this reference.
- 20Nichols, E. M.; Derrick, J. S.; Nistanaki, S. K.; Smith, P. T.; Chang, C. J. Positional effects of second-sphere amide pendants on electrochemical CO2 reduction catalyzed by iron porphyrins. Chem. Sci. 2018, 9, 2952– 2960, DOI: 10.1039/C7SC04682KThere is no corresponding record for this reference.
- 21Teindl, K.; Patrick, B. O.; Nichols, E. M. Linear Free Energy Relationships and Transition State Analysis of CO2 Reduction Catalysts Bearing Second Coordination Spheres with Tunable Acidity. J. Am. Chem. Soc. 2023, 145, 17176– 17186, DOI: 10.1021/jacs.3c03919There is no corresponding record for this reference.
- 22Branch, K. L.; Johnson, E. R.; Nichols, E. M. Porphyrin Aggregation under Homogeneous Conditions Inhibits Electrocatalysis: A Case Study on CO2 Reduction. ACS Cent. Sci. 2024, 10, 1251– 1261, DOI: 10.1021/acscentsci.4c00121There is no corresponding record for this reference.
- 23Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90– 94, DOI: 10.1126/science.1224581There is no corresponding record for this reference.
- 24Costentin, C.; Passard, G.; Robert, M.; Savéant, J.-M. Pendant Acid–Base Groups in Molecular Catalysts: H-Bond Promoters or Proton Relays? Mechanisms of the Conversion of CO2 to CO by Electrogenerated Iron(0)Porphyrins Bearing Prepositioned Phenol Functionalities. J. Am. Chem. Soc. 2014, 136, 11821– 11829, DOI: 10.1021/ja506193vThere is no corresponding record for this reference.
- 25Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. Dissection of Electronic Substituent Effects in Multielectron–Multistep Molecular Catalysis. Electrochemical CO2-to-CO Conversion Catalyzed by Iron Porphyrins. J. Phys. Chem. C 2016, 120, 28951– 28960, DOI: 10.1021/acs.jpcc.6b09947There is no corresponding record for this reference.
- 26Sinha, S.; Warren, J. J. Unexpected Solvent Effect in Electrocatalytic CO2 to CO Conversion Revealed Using Asymmetric Metalloporphyrins. Inorg. Chem. 2018, 57, 12650– 12656, DOI: 10.1021/acs.inorgchem.8b01814There is no corresponding record for this reference.
- 27Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639– 16644, DOI: 10.1021/jacs.6b07014There is no corresponding record for this reference.
- 28Costentin, C.; Drouet, S.; Passard, G.; Robert, M.; Savéant, J.-M. Proton-Coupled Electron Transfer Cleavage of Heavy-Atom Bonds in Electrocatalytic Processes. Cleavage of a C–O Bond in the Catalyzed Electrochemical Reduction of CO2. J. Am. Chem. Soc. 2013, 135, 9023– 9031, DOI: 10.1021/ja4030148There is no corresponding record for this reference.
- 29Nguyen, B. X.; Sonea, A.; Warren, J. J. Further Understanding the Roles of Solvent, Brønsted Acids, and Hydrogen Bonding in Iron Porphyrin-Mediated Carbon Dioxide Reduction. Inorg. Chem. 2023, 62, 17602– 17611, DOI: 10.1021/acs.inorgchem.3c01855There is no corresponding record for this reference.
- 30Gotico, P.; Boitrel, B.; Guillot, R.; Sircoglou, M.; Quaranta, A.; Halime, Z.; Leibl, W.; Aukauloo, A. Second-Sphere Biomimetic Multipoint Hydrogen-Bonding Patterns to Boost CO2 Reduction of Iron Porphyrins. Angew. Chem., Int. Ed. 2019, 58, 4504– 4509, DOI: 10.1002/anie.201814339There is no corresponding record for this reference.
- 31Sonea, A.; Branch, K. L.; Warren, J. J. The Pattern of Hydroxyphenyl-Substitution Influences CO2 Reduction More Strongly than the Number of Hydroxyphenyl Groups in Iron-Porphyrin Electrocatalysts. ACS Catal. 2023, 13, 3902– 3912, DOI: 10.1021/acscatal.2c06275There is no corresponding record for this reference.
- 32Amanullah, S.; Gotico, P.; Sircoglou, M.; Leibl, W.; Llansola-Portoles, M. J.; Tibiletti, T.; Quaranta, A.; Halime, Z.; Aukauloo, A. Second Coordination Sphere Effect Shifts CO2 to CO Reduction by Iron Porphyrin from Fe(0) to Fe(I). Angew. Chem., Int. Ed. 2024, 136, e202314439 DOI: 10.1002/anie.202314439There is no corresponding record for this reference.
- 33Margarit, C. G.; Schnedermann, C.; Asimow, N. G.; Nocera, D. G. Carbon Dioxide Reduction by Iron Hangman Porphyrins. Organometallics 2019, 38, 1219– 1223, DOI: 10.1021/acs.organomet.8b00334There is no corresponding record for this reference.
- 34Margarit, C. G.; Asimow, N. G.; Gonzalez, M. I.; Nocera, D. G. Double Hangman Iron Porphyrin and the Effect of Electrostatic Nonbonding Interactions on Carbon Dioxide Reduction. J. Phys. Chem. Lett. 2020, 11, 1890– 1895, DOI: 10.1021/acs.jpclett.9b03897There is no corresponding record for this reference.
- 35Sen, P.; Mondal, B.; Saha, D.; Rana, A.; Dey, A. Role of 2nd sphere H-bonding residues in tuning the kinetics of CO2 reduction to CO by iron porphyrin complexes. Dalton Trans. 2019, 48, 5965– 5977, DOI: 10.1039/C8DT03850CThere is no corresponding record for this reference.
- 36Teindl, K.; Jacobs, D.; Panetier, J. A.; Nichols, E. M. Highly Acidic Second Coordination Spheres Promote In Situ Formation of Iron Phlorins Exhibiting Fast and Selective CO2 Reduction. J. Am. Chem. Soc. 2025, DOI: 10.26434/chemrxiv-2025-5zzvbThere is no corresponding record for this reference.
- 37Derrick, J. S.; Loipersberger, M.; Nistanaki, S. K.; Rothweiler, A. V.; Head-Gordon, M.; Nichols, E. M.; Chang, C. J. Templating Bicarbonate in the Second Coordination Sphere Enhances Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins. J. Am. Chem. Soc. 2022, 144, 11656– 11663, DOI: 10.1021/jacs.2c02972There is no corresponding record for this reference.
- 38Guo, K.; Li, X.; Lei, H.; Guo, H.; Jin, X.; Zhang, X.-P.; Zhang, W.; Apfel, U.-P.; Cao, R. Role-Specialized Division of Labor in CO2 Reduction with Doubly-Functionalized Iron Porphyrin Atropisomers. Angew. Chem., Int. Ed. 2022, 61, e202209602 DOI: 10.1002/anie.202209602There is no corresponding record for this reference.
- 39Martin, D. J.; Mayer, J. M. Oriented Electrostatic Effects on O2 and CO2 Reduction by a Polycationic Iron Porphyrin. J. Am. Chem. Soc. 2021, 143, 11423– 11434, DOI: 10.1021/jacs.1c03132There is no corresponding record for this reference.
- 40Nichols, E. M.; Chang, C. J. Urea-Based Multipoint Hydrogen-Bond Donor Additive Promotes Electrochemical CO2 Reduction Catalyzed by Nickel Cyclam. Organometallics 2019, 38, 1213– 1218, DOI: 10.1021/acs.organomet.8b00308There is no corresponding record for this reference.
- 41Cypcar, A. D.; Bui, K. M.; Yang, J. Y. Suppressing H2 Evolution with Sterically Encumbered Proton Sources to Improve the Faradaic Efficiency for CO2 Reduction to Formate. ACS Catal. 2024, 14, 14517– 14523, DOI: 10.1021/acscatal.4c03809There is no corresponding record for this reference.
- 42Madsen, M. R.; Rønne, M. H.; Heuschen, M.; Golo, D.; Ahlquist, M. S. G.; Skrydstrup, T.; Pedersen, S. U.; Daasbjerg, K. Promoting Selective Generation of Formic Acid from CO2 Using Mn(bpy)(CO)3Br as Electrocatalyst and Triethylamine/Isopropanol as Additives. J. Am. Chem. Soc. 2021, 143, 20491– 20500, DOI: 10.1021/jacs.1c10805There is no corresponding record for this reference.
- 43Margarit, C. G.; Asimow, N. G.; Costentin, C.; Nocera, D. G. Tertiary Amine-Assisted Electroreduction of Carbon Dioxide to Formate Catalyzed by Iron Tetraphenylporphyrin. ACS Energy Lett. 2020, 5, 72– 78, DOI: 10.1021/acsenergylett.9b02093There is no corresponding record for this reference.
- 44Martin, D. J.; Mercado, B. Q.; Mayer, J. M. Combining scaling relationships overcomes rate versus overpotential trade-offs in O2 molecular electrocatalysis. Sci. Adv. 2020, 6, eaaz3318 DOI: 10.1126/sciadv.aaz3318There is no corresponding record for this reference.
- 45Cook, E. N.; Courter, I. M.; Dickie, D. A.; Machan, C. W. Controlling product selectivity during dioxygen reduction with Mn complexes using pendent proton donor relays and added base. Chem. Sci. 2024, 15, 4478– 4488, DOI: 10.1039/D3SC02611FThere is no corresponding record for this reference.
- 46Martin, D. J.; Wise, C. F.; Pegis, M. L.; Mayer, J. M. Developing Scaling Relationships for Molecular Electrocatalysis through Studies of Fe-Porphyrin-Catalyzed O2 Reduction. Acc. Chem. Res. 2020, 53, 1056– 1065, DOI: 10.1021/acs.accounts.0c00044There is no corresponding record for this reference.
- 47Lazouski, N.; Steinberg, K. J.; Gala, M. L.; Krishnamurthy, D.; Viswanathan, V.; Manthiram, K. Proton Donors Induce a Differential Transport Effect for Selectivity toward Ammonia in Lithium-Mediated Nitrogen Reduction. ACS Cat 2022, 12, 5197– 5208, DOI: 10.1021/acscatal.2c00389There is no corresponding record for this reference.
- 48Maran, F.; Celadon, D.; Severin, M. G.; Vianello, E. Electrochemical determination of the pKa of weak acids in N, N-dimethylformamide. J. Am. Chem. Soc. 1991, 113, 9320– 9329, DOI: 10.1021/ja00024a041There is no corresponding record for this reference.
- 49Bordwell, F. G.; Cheng, J. Substituent effects on the stabilities of phenoxyl radicals and the acidities of phenoxyl radical cations. J. Am. Chem. Soc. 1991, 113, 1736– 1743, DOI: 10.1021/ja00005a042There is no corresponding record for this reference.
- 50Tshepelevitsh, S.; Trummal, A.; Haav, K.; Martin, K.; Leito, I. Hydrogen-Bond Donicity in DMSO and Gas Phase and Its Dependence on Brønsted Acidity. J. Phys. Chem. A 2017, 121, 357– 369, DOI: 10.1021/acs.jpca.6b11115There is no corresponding record for this reference.
- 51Bordwell, F. G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456– 463, DOI: 10.1021/ar00156a004There is no corresponding record for this reference.
- 52Teindl, K.; Reid, J. P.; Nichols, E. M. Promoting selective electrochemical CO2 reduction under unconventionally acidic conditions through secondary coordination sphere positioning. Chem. Sci. 2025, 16, 19226– 19234, DOI: 10.1039/D5SC04700EThere is no corresponding record for this reference.
- 53Kütt, A.; Tshepelevitsh, S.; Saame, J.; Lõkov, M.; Kaljurand, I.; Selberg, S.; Leito, I. Strengths of Acids in Acetonitrile. Eur. J. Org. Chem. 2021, 2021, 1407– 1419, DOI: 10.1002/ejoc.202001649There is no corresponding record for this reference.
- 54Bordwell, F. G.; Ji, G. Z. Effects of structural changes on acidities and homolytic bond dissociation energies of the hydrogen-nitrogen bonds in amidines, carboxamides, and thiocarboxamides. J. Am. Chem. Soc. 1991, 113, 8398– 8401, DOI: 10.1021/ja00022a029There is no corresponding record for this reference.
- 55Matthews, W. S.; Bares, J. E.; Bartmess, J. E.; Bordwell, F. G.; Cornforth, F. J.; Drucker, G. E.; Margolin, Z.; McCallum, R. J.; McCollum, G. J.; Vanier, N. R. Equilibrium acidities of carbon acids. VI. Establishment of an absolute scale of acidities in dimethyl sulfoxide solution. J. Am. Chem. Soc. 1975, 97, 7006– 7014, DOI: 10.1021/ja00857a010There is no corresponding record for this reference.
- 56Olmstead, W. N.; Bordwell, F. G. Ion-pair association constants in dimethyl sulfoxide. J. Org. Chem. 1980, 45, 3299– 3305, DOI: 10.1021/jo01304a033There is no corresponding record for this reference.
- 57Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide by Iron(0) Porphyrins: Synergystic Effect of Weak Brönsted Acids. J. Am. Chem. Soc. 1996, 118, 1769– 1776, DOI: 10.1021/ja9534462There is no corresponding record for this reference.
- 58Hammouche, M.; Lexa, D.; Savéant, J. M.; Momenteau, M. Catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. J. Electroanal. Chem. Interfac. Electrochem. 1988, 249, 347– 351, DOI: 10.1016/0022-0728(88)80372-3There is no corresponding record for this reference.
- 59Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide by Iron(0) Porphyrins. Synergistic Effect of Lewis Acid Cations. J. Phys. Chem. 1996, 100, 19981– 19985, DOI: 10.1021/jp9618486There is no corresponding record for this reference.
- 60Dixon, D. A.; Dobbs, K. D.; Valentini, J. J. Amide-water and amide-amide hydrogen bond strengths. J. Phys. Chem. 1994, 98, 13435– 13439, DOI: 10.1021/j100102a001There is no corresponding record for this reference.
- 61Vallejo Narváez, W. E.; Jiménez, E. I.; Romero-Montalvo, E.; Sauza-de la Vega, A.; Quiroz-García, B.; Hernández-Rodríguez, M.; Rocha-Rinza, T. Acidity and basicity interplay in amide and imide self-association. Chem. Sci. 2018, 9, 4402– 4413, DOI: 10.1039/C8SC01020JThere is no corresponding record for this reference.
- 62Kraft, A.; Osterod, F. Self-association of branched and dendritic aromatic amides. J. Chem. Soc., Perkin Trans. 1998, 1, 1019– 1026, DOI: 10.1039/a800100fThere is no corresponding record for this reference.
- 63Sassone, D.; Bocchini, S.; Fontana, M.; Salvini, C.; Cicero, G.; Re Fiorentin, M.; Risplendi, F.; Latini, G.; Amin Farkhondehfal, M.; Pirri, F.; Zeng, J. Imidazole-imidazolate pair as organo-electrocatalyst for CO2 reduction on ZIF-8 material. Appl. Energy 2022, 324, 119743, DOI: 10.1016/j.apenergy.2022.119743There is no corresponding record for this reference.
- 64Izadyar, M.; Rezaeian, M.; Victorov, A. Theoretical study on the absorption of carbon dioxide by DBU-based ionic liquids. Phys. Chem. Chem. Phys. 2020, 22, 20050– 20060, DOI: 10.1039/D0CP03612AThere is no corresponding record for this reference.
- 65Zhu, X.; Song, M.; Xu, Y. DBU-Based Protic Ionic Liquids for CO2 Capture. ACS Sustainable Chem. Eng. 2017, 5, 8192– 8198, DOI: 10.1021/acssuschemeng.7b01839There is no corresponding record for this reference.
- 66Shu, H.; Xu, Y. Tuning the strength of cation coordination interactions of dual functional ionic liquids for improving CO2 capture performance. Int. J. Greenhouse Gas Control 2020, 94, 102934, DOI: 10.1016/j.ijggc.2019.102934There is no corresponding record for this reference.
- 67Darcy, J. W.; Koronkiewicz, B.; Parada, G. A.; Mayer, J. M. A Continuum of Proton-Coupled Electron Transfer Reactivity. Acc. Chem. Res. 2018, 51, 2391– 2399, DOI: 10.1021/acs.accounts.8b00319There is no corresponding record for this reference.
- 68Morris, W. D.; Mayer, J. M. Separating proton and electron transfer effects in three-component concerted proton-coupled electron transfer reactions. J. Am. Chem. Soc. 2017, 139, 10312– 10319, DOI: 10.1021/jacs.7b03562There is no corresponding record for this reference.
- 69Rhile, I. J.; Markle, T. F.; Nagao, H.; DiPasquale, A. G.; Lam, O. P.; Lockwood, M. A.; Rotter, K.; Mayer, J. M. Concerted proton–electron transfer in the oxidation of hydrogen-bonded phenols. J. Am. Chem. Soc. 2006, 128, 6075– 6088, DOI: 10.1021/ja054167+There is no corresponding record for this reference.
- 70Markle, T. F.; Darcy, J. W.; Mayer, J. M. A new strategy to efficiently cleave and form C–H bonds using proton-coupled electron transfer. Sci. Adv. 2018, 4, eaat5776 DOI: 10.1126/sciadv.aat5776There is no corresponding record for this reference.
- 71Abraham, M. H.; Grellier, P. L.; Prior, D. V.; Duce, P. P.; Morris, J. J.; Taylor, P. J. Hydrogen bonding. Part 7. A scale of solute hydrogen-bond acidity based on log K values for complexation in tetrachloromethane. J. Chem. Soc., Perkin Trans. 1989, 2, 699– 711, DOI: 10.1039/p29890000699There is no corresponding record for this reference.
- 72Rovira, C.; Kunc, K.; Hutter, J.; Ballone, P.; Parrinello, M. Equilibrium Geometries and Electronic Structure of Iron–Porphyrin Complexes: A Density Functional Study. J. Phys. Chem. A 1997, 101, 8914– 8925, DOI: 10.1021/jp9722115There is no corresponding record for this reference.
- 73Rovira, C.; Kunc, K.; Hutter, J.; Ballone, P.; Parrinello, M. A comparative study of O2, CO, and NO binding to iron–porphyrin. Int. J. Quantum Chem. 1998, 69, 31– 35, DOI: 10.1002/(SICI)1097-461X(1998)69:1<31::AID-QUA5>3.0.CO;2-YThere is no corresponding record for this reference.
- 74Abraham, M. H.; Grellier, P. L.; Prior, D. V.; Morris, J. J.; Taylor, P. J. Hydrogen bonding. Part 10. A scale of solute hydrogen-bond basicity using log K values for complexation in tetrachloromethane. J. Chem. Soc., Perkin Trans. 1990, 2, 521– 529, DOI: 10.1039/p29900000521There is no corresponding record for this reference.
- 75Anslyn, E. V.; Dougherty, D. A. Modern Physical Organic Chemistry. 1 ed. University Science Books 2006, 1104, DOI: 10.1002/anie.200585348There is no corresponding record for this reference.
- 76Teindl, K.; Nichols, E. M. The Effect of Exogenous Acid Identity on Iron Tetraphenylporphyrin-Catalyzed CO2 Reduction. ChemRxiv 2025, DOI: 10.26434/chemrxiv-2025-n2h1qThere is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.5c05122.
pKa determination, electrochemical data, gas chromatography, and Eyring analysis (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.