



This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Structural, Thermodynamic, and Spectroscopic Characterization of Diphosgene and TriphosgeneClick to copy article linkArticle link copied!
- Sven RingelbandSven RingelbandDepartment of Chemistry, Philipps University Marburg, Marburg 35043, GermanyMore by Sven Ringelband
- Stewart F. ParkerStewart F. ParkerISIS Pulsed Neutron and Muon Facility, STFC Rutherford Appleton Laboratory, Chilton OX11 0QX, U.K.More by Stewart F. Parker
- Frank Tambornino*Frank Tambornino*Email: [email protected]Department of Chemistry, Philipps University Marburg, Marburg 35043, GermanyMore by Frank Tambornino
Inorganic Chemistry
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
Phosgene (COCl2) is an important industrial reagent but its gaseous state and limited availability limit laboratory use. Diphosgene and triphosgene are safer surrogates, yet their solid-state structures and vibrational properties remain poorly documented. Here we report a comprehensive investigation of both compounds combining crystallography, calorimetry, spectroscopy, and quantum chemical calculations. A new polymorph of diphosgene (β-diphosgene) was discovered. Differential scanning calorimetry revealed rare cold crystallization behavior, i.e., crystallization of a supercooled melt only during subsequent heating. Solid-state DFT calculations reproduced lattice parameters and clarified the thermodynamic balance between the polymorphs. Infrared, Raman, and inelastic neutron scattering spectra of diphosgene and triphosgene were measured and fully assigned with the aid of periodic DFT calculations, providing the first complete solid-state vibrational characterization of these compounds.
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Introduction
Scheme 1
| properties | phosgene | diphosgene | triphosgene |
|---|---|---|---|
| phase at 300 K | colorless gas | colorless liquid | colorless solid |
| molecular formula | COCl2 | C2O2Cl4 | C3O3Cl6 |
| bp. [°C] | 7.56 | 128 | 203–206 |
| mp. [°C] | –127.8 | –57 | 79–83 |
| density [g cm–3] | 1.38 (20 °C) | 1.65 (14 °C) | 1.629 (80 °C) |
| vapor pressure [bar] | 1.09 (10 °C) | 0.74–0.77 (20 °C) | 0.12 (25 °C) |
| LC50 [mg m3]a | 7.2 | 13.9 | 41.5 |
| kobs [s–1]b | 1.7×10–2 | 9.1×10–4 | 1.0×10–4 |
Experimental Section
X-Ray Diffraction
Growth of Single Crystals of Triphosgene
Growth of Single Crystals of β-Diphosgene
Hirshfeld Surface Analysis
Differential Scanning Calorimetry
Vibrational Spectroscopy
Quantum Chemical Calculations
Results and Discussion
Crystal Structure of β-Diphosgene
Figure 1
Figure 1. Asymmetric units of the crystal structures of α-diphosgene (a, measured at 150 K) (37) and β-diphosgene (b, measured at 100 K) with ellipsoids drawn at 60% probability level. Hirshfeld surfaces of α-diphosgene (c) and both independent molecules of β-diphosgene (d). Atoms are drawn with arbitrary radii. Short contacts are marked with dashed lines.
Comparison of the Crystal Structures of α- and β-Diphosgene
| α-diphosgene | β-diphosgene | ||
|---|---|---|---|
| 150 K | 100 K | 200 K | |
| a [Å] | 5.5578(5) | 11.6703(3) | 11.7485(5) |
| b [Å] | 14.2895(12) | 7.3870(2) | 7.4109(2) |
| c [Å] | 8.6246(7) | 15.3222(4) | 15.4942(6) |
| β [°] | 102.443(2) | 94.574(2) | 94.581(3) |
| V [Å3] | 668.86(10) | 1316.70(6) | 1344.72(9) |
| T [K] | 150 | 100 | 200 |
| Z | 4 | 8 | 8 |
Figure 2
Figure 2. Arrangement of molecule “pairs” in the crystal structure of β-diphosgene. The asymmetric unit is shown in the circle with the two crystallographically independent molecules with different patterns.
Redetermination of the Crystal Structure of Triphosgene
Figure 3
Figure 3. Molecular structure of one independent triphosgene molecule in the solid state measured at 100 K. Atoms are drawn with 60% displacement ellipsoids.
Molecular Structure Comparison Both of α- and β-Diphosgene, and Triphosgene
| α-diphosgenec (37) | β-diphosgene (1) | β-diphosgene (2) | triphosgene | phosgene (40) | dimethyl carbonateb, (41) | methyl chloroformate (42) | trifluoromethyl chloroformate (43) | |
|---|---|---|---|---|---|---|---|---|
| C═O | 1.1802(12) | 1.172(5) | 1.167(5) | 1.188(5) | 1.184(2) | 1.219(2) | 1.195(2) | 1.164(4) |
| (CO)–O | 1.365(1) | 1.356(5) | 1.362(5) | 1.361(6)a | – | 1.337(2) | 1.309(2) | 1.367(4) |
| CX3–O | 1.419(1) | 1.418(5) | 1.407(5) | 1.414(5)a | – | 1.456(2) | 1.462(2) | 1.386(3) |
| (CO)–Cl | 1.729(1) | 1.732(4) | 1.735(4) | – | 1.725(2)a | – | 1.7502(13) | 1.716(3) |
| C–X | 1.7627(9)a | 1.759(4)a | 1.759(4)a | 1.762(5)a | – | – | – | 1.310(4) |
| ∠O═C–O | 127.86(9) | 127.8(4) | 128.1(4) | 128.7(4)a | – | 125.58(11) | 128.78(11) | 126.9(3) |
| ∠X–C═O | 124.80(7) | 125.2(3) | 124.8(3) | – | 123.9(2) | – | 122.47(10) | 125.3(2) |
| ∠X–C–O | 107.34(6) | 107.0(3) | 107.0(3) | – | – | – | 108.75(9) | 107.8(2) |
Mean values.
Neutron powder data at 82 K.
SCXRD at 150 K.
Parameters in [Å] and [°].
Differential Scanning Calorimetry of Diphosgene
Figure 4
Figure 4. Low-temperature differential scanning calorimetry data of diphosgene. Three consecutive measurements were performed. The curves for solidification and melting temperature coincide. For details of the fit see Supporting Information.
Quantum Chemical Calculations
Figure 5
Figure 5. Difference in entropy (─) and Gibbs free energy (---) between β- and α-diphosgene at different temperatures. Level of theory: DFT-PBE0-D3(BJ-ABC)/def2-TZVP.
Vibrational Spectroscopy of Diphosgene and Triphosgene
Figure 6
Figure 6. Vibrational spectra of diphosgene: (a) infrared (liquid at room temperature), (b) Raman (liquid at room temperature), (c) Raman (solid, 193 K) and (d) INS (solid at 15 K).
Figure 7
Figure 7. Vibrational spectra of triphosgene in the solid state: (a) infrared (room temperature), (b) Raman (liquid at 350 K), (c) Raman (solid at room temperature) and (d) INS (15 K).
Diphosgene
Triphosgene
Assignment of the Spectra of α-Diphosgene and Triphosgene
Figure 8
Figure 8. Comparison of measured ((a) and (c)) and calculated ((b) and (d)) INS spectra of α-diphosgene and triphosgene.
| α-diphosgene | triphosgene | ||||||
|---|---|---|---|---|---|---|---|
| infrareda | Raman | INS | assignment | infrared | Raman | INS | assignment |
| 69 m, 74 w | |||||||
| 84 s | 42 m | Libration | |||||
| 91 m, 95 m | 56 s,vbr | Libration | |||||
| 74 s | CCl3 rock | ||||||
| 85 m | 89 s | CCl3 rock | |||||
| 117 m/126 m | 101 s | 103 s | CCl3 rock | ||||
| 143 m/147 m | O(2)C(2) + C(2)O(3) ip torsion | ||||||
| 141 m | 144 s | CCl3 rock | 161 w | 162 s | CCl3 rock | ||
| 177 m | 175 s | O(1)C(2)O(3) bend | |||||
| 240 m | 238 s | Cl(1)–C(1)–O(1) ip bend | 241 m | 242 s | CCl3 asym bend | ||
| 249 m | 246 s | CCl3 asym bend | 256 sh | 255 s | CCl3 asym bend | ||
| 254 s | CCl3 asym bend | 268 m | CCl3 asym bend | ||||
| 326 w | 326 m | C(1)–O(1)–C(2) + C(2)–O(2)–C(3) oop bend | |||||
| 337 m | 338 s | C(1)–O(1)–C(2) ip bend | 355 w | 357 m | C(1)–O(1)–C(2) + C(2)–O(2)–C(3) ip bend | ||
| 363 w | 361 m | CCl3 oop sym bend | |||||
| 390 s | O(1) oop bend | 384 m | 383 m | Skeletal deformation | |||
| 402 sh | 402 m | Skeletal deformation | |||||
| 401 vs | 401 s | CCl3 sym bend | 415 vs | 413 s | CCl3 ip sym bend | ||
| 495 s | 498 vs | 494 s | Cl(1)–C(1) = O(2) bend | ||||
| 584 s | 589 w | 589 vs | CCl3 sym stretch | 530 m | CCl3 oop sym stretch | ||
| 676 s | 678 s | 684 m | CCl3 ip sym stretch | ||||
| 665 m | 671 s | C(1) oop bend | |||||
| 759 s | 764 w | 770 w | CCl3 asym stretch | 753 m | CCl3 asym stretch | ||
| 807 vs | 817 w | 821 m | CCl3 asym stretch | 806 vs,br | CCl3 asym stretch | ||
| 823 w | 827 m | CCl3 asym stretch | |||||
| 891 vw | 891 w | CCl3 asym stretch | |||||
| 908 s/925 sh | 915 w | 917 w,br | O(1)–C(1) stretch | 914 vs | C(1)–O(1) + O(3)–C(3) out-of-phase stretch | ||
| 957 w/972 w | 963 w | C(1)–O(1) + O(3)–C(3) ip stretch | |||||
| 965 vs | 965 w | Cl(1)–C(1) stretch | |||||
| 1042 vs,br | 1069 w | C(1)–O(1) stretch | 1109 w | 1103 vw | O(1)–C(2) + C(2)–O(3) ip stretch | ||
| 1171 vs,br | O(1)–C(2) + C(2)–O(3) oop stretch | ||||||
| 1800 vs | 1803 w | C(1) = O(2) stretch | 1818 s,br | 1819 w | C(2)═ O(2) stretch | ||
w = weak, m = medium, s = strong, v = very, br = broad, sh = shoulder atom numbering: diphosgene Cl(1)–C(1)(=O(2))–O(1)–C(2)Cl3 and triphosgene Cl3C(1)–O(1)–C(2)(=O(2))–O(3)–CCl3.
| syma stretch | asym stretch | sym bend | asym bend | rock | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cm–1 | IR | R | INS | cm–1 | IR | R | INS | cm–1 | IR | R | INS | cm–1 | IR | R | INS | cm–1 | IR | R | INS | |
| HCCl3 (52−54) | 670 | s | s | w | 756 | vs | w | w | 368 | vw | s | w | 258/268 | w | s | w | ||||
| hexachloroethane (55) | 432 | no | vs | n/a | 780 | vs | no | n/a | 288 | no | vs | n/a | 276 | w | no | n/a | 167 | m | no | n/a |
| trichloroacetic acid (56−59) | 459 | m | vs | n/a | 830/704 | vs | m | n/a | 283 | n/a | s | n/a | 280 | n/a | m | n/a | 209/218 | n/a | s | n/a |
| diphosgene | 589 | s | w | vs | 770/821 | s | w | m | 401 | n/a | vs | s | 246/254 | n/a | m | s | 117–144 | n/a | m | s |
| triphosgene | 530/684 | s | s | m | 753–891 | s | w | m | 415 | n/a | vs | s | 242–268 | n/a | m | s | 74–62 | n/a | m | s |
| mean | 561 | 788 | 351 | 262 | 156 | |||||||||||||||
| standard deviation | 106 | 58 | 56 | 14 | 50 | |||||||||||||||
sym = symmetric; asym = asymmetric; IR, R and INS are the infrared, Raman and INS intensities; no. = not observed; n/a = not available (outside of spectral range of the instrument or not measured).
Conclusion
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.5c05882.
Supporting Information crystallographic data, additional crystal structure pictures, Hirshfeld surface analysis, details on quantum chemical calculations and vibrational spectroscopy (PDF) cif file of β-diphosgene at 200 K (CIF) cif file of β-diphosgene at 100 K (CIF) cif file of triphosgene at 100 K (CIF) (PDF)
Deposition Numbers 2516310–2516312 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via the joint Cambridge Crystallographic Data Centre (CCDC) and Fachinformationszentrum Karlsruhe Access Structures service.
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Frank Tambornino - Department of Chemistry, Philipps University Marburg, Marburg 35043, Germany;
 https://orcid.org/0000-0003-3538-6049;
https://orcid.org/0000-0003-3538-6049;
- Stewart F. Parker - ISIS Pulsed Neutron and Muon Facility, STFC Rutherford Appleton Laboratory, Chilton OX11 0QX, U.K.;https://orcid.org/0000-0002-3228-2570
Acknowledgments
F.T. thanks the Deutsche Forschungsgemeinschaft for funding (grant No. TA 1357/5-1). The STFC Rutherford Appleton Laboratory is thanked for access to neutron beam facilities via RB2400072 (TOSCA). (60)
References
This article references 60 other publications.
- 1Cotarca, L.; Lange, C.; Meurer, K.; Pauluhn, J. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp 1– 30.Google ScholarThere is no corresponding record for this reference.
- 2Su, W.; Zhong, W.; Bian, G.; Shi, X.; Zhang, J. RECENT ADVANCES IN THE CHEMISTRY OF TRICHLOROMETHYL CHLOROFORMATE AND Bis-(TRICHLOROMETHYL) CARBONATE. Org. Prep. Proced. Int. 2004, 36, 499– 547, DOI: 10.1080/00304940409355972Google ScholarThere is no corresponding record for this reference.
- 3Cotarca, L.; Delogu, P.; Nardelli, A.; Šunjić, V. Bis(Trichloromethyl) Carbonate in Organic Synthesis. Synthesis 1996, 1996, 553– 576, DOI: 10.1055/s-1996-4273Google ScholarThere is no corresponding record for this reference.
- 4Ganiu, M. O.; Nepal, B.; Van Houten, J. P.; Kartika, R. A Decade Review of Triphosgene and Its Applications in Organic Reactions. Tetrahedron 2020, 76, 131553, DOI: 10.1016/j.tet.2020.131553Google ScholarThere is no corresponding record for this reference.
- 5Pasquato, L.; Modena, G.; Cotarca, L.; Delogu, P.; Mantovani, S. Conversion of Bis(Trichloromethyl) Carbonate to Phosgene and Reactivity of Triphosgene, Diphosgene, and Phosgene with Methanol1. J. Org. Chem. 2000, 65, 8224– 8228, DOI: 10.1021/jo000820uGoogle ScholarThere is no corresponding record for this reference.
- 6Hood, H. P.; Murdock, H. R. Superpalite. J. Phys. Chem. 1919, 23, 498– 512, DOI: 10.1021/j150196a004Google ScholarThere is no corresponding record for this reference.
- 7Eckert, H.; Auerweck, J. Solvent-Free and Safe Process for the Quantitative Production of Phosgene from Triphosgene by Deactivated Imino-Based Catalysts. Org. Process Res. Dev. 2010, 14, 1501– 1505, DOI: 10.1021/op100239nGoogle ScholarThere is no corresponding record for this reference.
- 8Cotarca, L.; Geller, T.; Répási, J. Bis(Trichloromethyl)Carbonate (BTC, Triphosgene): A Safer Alternative to Phosgene?. Org. Process Res. Dev. 2017, 21, 1439– 1446, DOI: 10.1021/acs.oprd.7b00220Google ScholarThere is no corresponding record for this reference.
- 9Hentschel, W. Ueber Gechlorte Ameisensäuremethyläther Und Verwandte Körper. J. Prakt. Chem. 1887, 36 (36), 305– 317, DOI: 10.1002/prac.18870360129Google ScholarThere is no corresponding record for this reference.
- 10Councler, C. Kohlensaures Methyl. Ber. Dtsch. Chem. Ges. 1880, 13 (13), 1697– 1699, DOI: 10.1002/cber.188001302116Google ScholarThere is no corresponding record for this reference.
- 11Hentschel, W. Ueber Gechlorte Ameisensäuremethyläther Und Verwandte Körper. J. Prakt. Chem. 1887, 36 (36), 99– 113, DOI: 10.1002/prac.18870360110Google ScholarThere is no corresponding record for this reference.
- 12Hentschel, W. Ueber Gechlorte Ameisensäuremethyläther Und Verwandte Körper. J. Prakt. Chem. 1887, 36 (36), 468– 480, DOI: 10.1002/prac.18870360139Google ScholarThere is no corresponding record for this reference.
- 13Hales, J. L.; Jones, J. I.; Kynaston, W. The Infrared Absorption Spectra of Some Chloroformates and Carbonates. The Structure of “Di- and Tri-Phosgene”. J. Chem. Soc. 1957, 0, 618– 625, DOI: 10.1039/JR9570000618Google ScholarThere is no corresponding record for this reference.
- 14Sørensen, A. M.; Rosenqvist, E.; Åse, K.; Omfeldt, M.; Lagerlund, I.; Ehrenberg, L. The Crystal Structure of Bistrichloromethylcarbonate, Triphosgene. Acta Chem. Scand. 1971, 25, 169– 174, DOI: 10.3891/acta.chem.scand.25-0169Google ScholarThere is no corresponding record for this reference.
- 15Pauluhn, J. Acute Nose-Only Inhalation Exposure of Rats to Di- and Triphosgene Relative to Phosgene. Inhalation Toxicol. 2011, 23, 65– 73, DOI: 10.3109/08958378.2010.542501Google ScholarThere is no corresponding record for this reference.
- 16Sheldrick, G. M. SHELXT – Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr., Sect. A:Found. Adv. 2015, 71, 3– 8, DOI: 10.1107/S2053273314026370Google ScholarThere is no corresponding record for this reference.
- 17Sheldrick, G. M. Crystal Structure Refinement withSHELXL. Acta. Crystallogr. C Struct. Chem. 2015, 71, 3– 8, DOI: 10.1107/S2053229614024218Google ScholarThere is no corresponding record for this reference.
- 18Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339– 341, DOI: 10.1107/S0021889808042726Google ScholarThere is no corresponding record for this reference.
- 19Brandenburg, K.; Putz, H. DIAMOND, Program for X-Ray Structure Analysis; Crystal Impact GbR: Bonn, Germany, 1999.Google ScholarThere is no corresponding record for this reference.
- 20Spackman, M. A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19– 32, DOI: 10.1039/B818330AGoogle ScholarThere is no corresponding record for this reference.
- 21Spackman, P. R.; Turner, M. J.; McKinnon, J. J.; Wolff, S. K.; Grimwood, D. J.; Jayatilaka, D.; Spackman, M. A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006– 1011, DOI: 10.1107/S1600576721002910Google ScholarThere is no corresponding record for this reference.
- 22Parker, S. F.; Fernandez-Alonso, F.; Ramirez-Cuesta, A. J.; Tomkinson, J.; Rudic, S.; Pinna, R. S.; Gorini, G.; Fernández Castañon, J. Recent and Future Developments on TOSCA at ISIS. J. Phys.: Conf. Ser. 2014, 554, 012003, DOI: 10.1088/1742-6596/554/1/012003Google ScholarThere is no corresponding record for this reference.
- 23Pinna, R. S.; Rudić, S.; Parker, S. F.; Armstrong, J.; Zanetti, M.; Škoro, G.; Waller, S. P.; Zacek, D.; Smith, C. A.; Capstick, M. J.; McPhail, D. J.; Pooley, D. E.; Howells, G. D.; Gorini, G.; Fernandez-Alonso, F. The Neutron Guide Upgrade of the TOSCA Spectrometer. Nuclear Inst. and Methods in Physics Research A 2018, 896, 68– 74, DOI: 10.1016/j.nima.2018.04.009Google ScholarThere is no corresponding record for this reference.
- 24https://www.isis.stfc.ac.uk/Pages/home.aspx (accessed 12 1, 2025).Google ScholarThere is no corresponding record for this reference.
- 25Arnold, O. Mantid─Data Analysis and Visualization Package for Neutron Scattering and μ SR Experiments. Nucl. Instrum. Methods Phys. Res., Sect. A 2014, 764, 156– 166, DOI: 10.1016/j.nima.2014.07.029Google ScholarThere is no corresponding record for this reference.
- 26Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C. M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; Kirtman, B. Quantum-mechanical Condensed Matter Simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360 DOI: 10.1002/wcms.1360Google ScholarThere is no corresponding record for this reference.
- 27Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865– 3868, DOI: 10.1103/PhysRevLett.77.3865Google ScholarThere is no corresponding record for this reference.
- 28Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0Model. J. Chem. Phys. 1999, 110, 6158– 6170, DOI: 10.1063/1.478522Google ScholarThere is no corresponding record for this reference.
- 29Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297, DOI: 10.1039/b508541aGoogle ScholarThere is no corresponding record for this reference.
- 30Monkhorst, H. J.; Pack, J. D. Special Points for Brillouin-zone Integrations. Phys. Rev. B: 1976, 13, 5188– 5192, DOI: 10.1103/PhysRevB.13.5188Google ScholarThere is no corresponding record for this reference.
- 31Pascale, F.; Zicovich-Wilson, C. M.; López Gejo, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The Calculation of the Vibrational Frequencies of Crystalline Compounds and Its Implementation in the CRYSTAL Code. J. Comput. Chem. 2004, 25, 888– 897, DOI: 10.1002/jcc.20019Google ScholarThere is no corresponding record for this reference.
- 32Clark, S. J.; Segall, M. D.; Pickard, C. J.; Hasnip, P. J.; Probert, M. I. J.; Refson, K.; Payne, M. C. First Principles Methods Using CASTEP. Z. Kristallogr. 2005, 220, 567– 570, DOI: 10.1524/zkri.220.5.567.65075Google ScholarThere is no corresponding record for this reference.
- 33Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005, DOI: 10.1103/PhysRevLett.102.073005Google ScholarThere is no corresponding record for this reference.
- 34Refson, K.; Tulip, P. R.; Clark, S. J. Variational Density-Functional Perturbation Theory for Dielectrics and Lattice Dynamics. Phys. Rev. B 2006, 73, 155114, DOI: 10.1103/PhysRevB.73.155114Google ScholarThere is no corresponding record for this reference.
- 35Gonze, X.; Charlier, J.-C.; Allan, D.; Teter, M. Interatomic Force Constants from First Principles: The Case of α-Quartz. Phys. Rev. B 1994, 50, 13035– 13038, DOI: 10.1103/PhysRevB.50.13035Google ScholarThere is no corresponding record for this reference.
- 36Dymkowski, K.; Parker, S. F.; Fernandez-Alonso, F.; Mukhopadhyay, S. AbINS: The Modern Software for INS Interpretation. Physica B: Condensed Matter 2018, 551, 443– 448, DOI: 10.1016/j.physb.2018.02.034Google ScholarThere is no corresponding record for this reference.
- 37Arce, V. B.; Della Védova, C. O.; Downs, A. J.; Parsons, S.; Romano, R. M. Trichloromethyl Chloroformate (“Diphosgene”), ClC(O)OCCl3: Structure and Conformational Properties in the Gaseous and Condensed Phases. J. Org. Chem. 2006, 71, 3423– 3428, DOI: 10.1021/jo052260aGoogle ScholarThere is no corresponding record for this reference.
- 38Spek, A. L. Structure Validation in Chemical Crystallography. Acta Crystallogr., Sect. D:Biol. Crystallogr. 2009, 65, 148– 155, DOI: 10.1107/S090744490804362XGoogle ScholarThere is no corresponding record for this reference.
- 39Bondi, A. Van Der Waals Volumes Radii. J. Phys. Chem. 1964, 68, 441– 451, DOI: 10.1021/j100785a001Google ScholarThere is no corresponding record for this reference.
- 40Ringelband, S.; Pfeiffer, J.; Fortes, A. D.; Howard, C. M.; Parker, S. F.; Karttunen, A. J.; Tambornino, F. Crystal Structures and Intermolecular Interactions in α- and β-phosgene. Angew. Chem., Int. Ed. 2025, 65, e17323 DOI: 10.1002/anie.202517323Google ScholarThere is no corresponding record for this reference.
- 41Whitfield, P. S. Low-Temperature Crystal Structures of the Solvent Dimethyl Carbonate. Powder Diffr. 2023, 38, 100– 111, DOI: 10.1017/S088571562300009XGoogle ScholarThere is no corresponding record for this reference.
- 42Ringelband, S.; Tambornino, F. Crystal Structure of Methyl Chloroformate. Acta Cryst. E 2025, 81, 987– 990, DOI: 10.1107/S2056989025008369Google ScholarThere is no corresponding record for this reference.
- 43Erben, M. F.; Della Védova, C. O.; Boese, R.; Willner, H.; Oberhammer, H. Trifluoromethyl Chloroformate, ClC(O)OCF3: Structure, Conformation, and Vibrational Analysis Studied by Experimental and Theoretical Methods. J. Phys. Chem. A 2004, 108, 699– 706, DOI: 10.1021/jp036966pGoogle ScholarThere is no corresponding record for this reference.
- 44Pyykkö, P.; Atsumi, M. Molecular Double-Bond Covalent Radii for Elements Li–E112. Chem.–Eur. J. 2009, 15, 12770– 12779, DOI: 10.1002/chem.200901472Google ScholarThere is no corresponding record for this reference.
- 45Bratsch, S. G. A Group Electronegativity Method with Pauling Units. J. Chem. Educ. 1985, 62, 101, DOI: 10.1021/ed062p101Google ScholarThere is no corresponding record for this reference.
- 46Pingping, Z.; Dezhu, M. Study on the Double Cold Crystallization Peaks of Poly(Ethylene Terephthalate) 3. The Influence of the Addition of Calcium Carbonate (CaCO3). Eur. Polym. J. 2000, 36, 2471– 2475, DOI: 10.1016/S0014-3057(00)00042-2Google ScholarThere is no corresponding record for this reference.
- 47Mitchell, P. C. H., Parker, S. F., Ramirez-Cuesta, A. J., Tomkinson, J., Eds.; Vibrational Spectroscopy with Neutrons: With Applications in Chemistry, Biology, Materials Science and Catalysis; Series on Neutron Techniques and Applications v; World Scientific: Singapore Hackensack, N.J, 2005; Vol. 3.Google ScholarThere is no corresponding record for this reference.
- 48Perrichon, A.; Bovo, C.; Parker, S. F.; Raspino, D.; Armstrong, J.; García Sakai, V. Overview of Planned Upgrade to the Secondary Spectrometer of TOSCA. Nuclear Inst. and Methods in Physics Research, A 2023, 1047, 167899, DOI: 10.1016/j.nima.2022.167899Google ScholarThere is no corresponding record for this reference.
- 49Bellamy, L. J. The Infrared Spectra of Complex Molecules, 3rd ed.; Chapman & Hall: Wiley: London: New York, 1975.Google ScholarThere is no corresponding record for this reference.
- 50Colthup, N. B.; Daly, L. H.; Wiberley, S. E. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: Boston, 1990.Google ScholarThere is no corresponding record for this reference.
- 51Lin-Vien, D.; Colthup, N. B.; Fateley, W. G.; Grasselli, J. G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Academic Press: Boston, 1991.Google ScholarThere is no corresponding record for this reference.
- 52Andrews, B.; Anderson, A.; Torrie, B. Raman and Infrared Spectra of Crystalline Chloroform. Chem. Phys. Lett. 1984, 104, 65– 70, DOI: 10.1016/0009-2614(84)85306-3Google ScholarThere is no corresponding record for this reference.
- 53Stanila, D.; Smith, W.; Anderson, A. Infrared Spectra of Chloroform at High Pressures. Spectrosc. Lett. 2002, 35, 703– 713, DOI: 10.1081/SL-120014941Google ScholarThere is no corresponding record for this reference.
- 54Nolasco, M. M.; Coimbra, M. M.; Parker, S. F.; Vaz, P. D.; Ribeiro-Claro, P. J. A. Structural Dynamics of Chloromethanes through Computational Spectroscopy: Combining INS and DFT. Molecules 2022, 27, 7661, DOI: 10.3390/molecules27217661Google ScholarThere is no corresponding record for this reference.
- 55Woost, B.; Bougeard, D. Vibrational Spectra and Phase Transitions of Crystalline Hexachloroethane. J. Chem. Phys. 1986, 84, 4810– 4817, DOI: 10.1063/1.449968Google ScholarThere is no corresponding record for this reference.
- 56Adams, J.; Kim, H. The Infrared Spectra of Anhydrous Trichloroacetic Acid and Oxalic Acid. Spectrochim. Acta, Part A 1973, 29, 675– 677, DOI: 10.1016/0584-8539(73)80097-2Google ScholarThere is no corresponding record for this reference.
- 57Trichloroacetic Acid Raman Spectrum. Available at: https://www.chemicalbook.com/SpectrumEN_76-03-9_Raman.htm (accessed 12 1, 2025).Google ScholarThere is no corresponding record for this reference.
- 58Trichloroacetic Acid Infrared Spectrum. Available at: https://www.chemicalbook.com/SpectrumEN_76-03-9_IR1.htm (accessed 12 1, 2025).Google ScholarThere is no corresponding record for this reference.
- 59Yadav, R.; Kumar, M.; Singh, R.; Singh, P.; Jaiswal, S.; Srivastav, G.; Prasad, R. Ab Initio Determination of Molecular Geometries and Vibrational Frequencies of CX3 COOH (X = H, F, Cl, Br). Spectrochim. Acta 2008, 71, 1565– 1570, DOI: 10.1016/j.saa.2008.06.016Google ScholarThere is no corresponding record for this reference.
- 60Tambornino, F.; Parker, S.; Ringelband, S. The Inelastic Neutron Scattering Spectra of Diphosgene and Triphosgene; STFC ISIS Neutron and Muon Source, 2024.Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inorganic Chemistry
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
Scheme 1
 Scheme 1. Representation of Diphosgene (Left) and Triphosgene (Right)
Scheme 1. Representation of Diphosgene (Left) and Triphosgene (Right)Figure 1
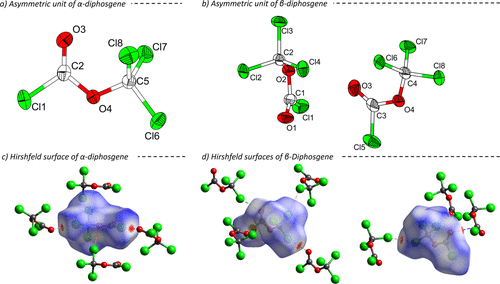
Figure 1. Asymmetric units of the crystal structures of α-diphosgene (a, measured at 150 K) (37) and β-diphosgene (b, measured at 100 K) with ellipsoids drawn at 60% probability level. Hirshfeld surfaces of α-diphosgene (c) and both independent molecules of β-diphosgene (d). Atoms are drawn with arbitrary radii. Short contacts are marked with dashed lines.
Figure 2
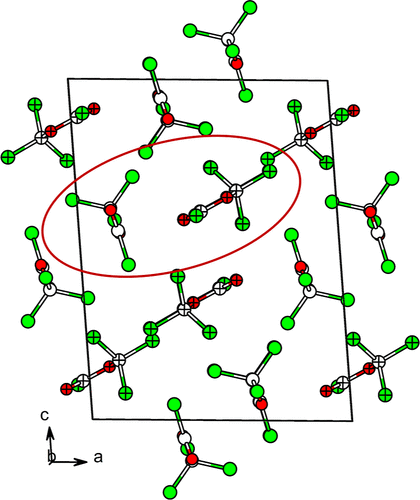
Figure 2. Arrangement of molecule “pairs” in the crystal structure of β-diphosgene. The asymmetric unit is shown in the circle with the two crystallographically independent molecules with different patterns.
Figure 3

Figure 3. Molecular structure of one independent triphosgene molecule in the solid state measured at 100 K. Atoms are drawn with 60% displacement ellipsoids.
Figure 4
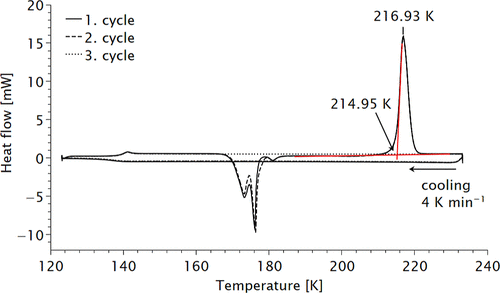
Figure 4. Low-temperature differential scanning calorimetry data of diphosgene. Three consecutive measurements were performed. The curves for solidification and melting temperature coincide. For details of the fit see Supporting Information.
Figure 5

Figure 5. Difference in entropy (─) and Gibbs free energy (---) between β- and α-diphosgene at different temperatures. Level of theory: DFT-PBE0-D3(BJ-ABC)/def2-TZVP.
Figure 6

Figure 6. Vibrational spectra of diphosgene: (a) infrared (liquid at room temperature), (b) Raman (liquid at room temperature), (c) Raman (solid, 193 K) and (d) INS (solid at 15 K).
Figure 7
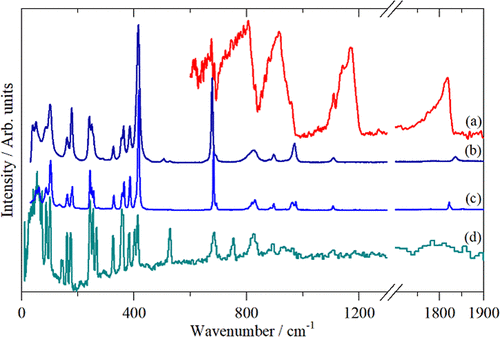
Figure 7. Vibrational spectra of triphosgene in the solid state: (a) infrared (room temperature), (b) Raman (liquid at 350 K), (c) Raman (solid at room temperature) and (d) INS (15 K).
Figure 8
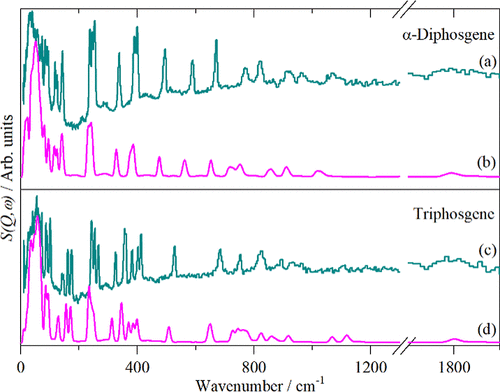
Figure 8. Comparison of measured ((a) and (c)) and calculated ((b) and (d)) INS spectra of α-diphosgene and triphosgene.
References
This article references 60 other publications.
- 1Cotarca, L.; Lange, C.; Meurer, K.; Pauluhn, J. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp 1– 30.There is no corresponding record for this reference.
- 2Su, W.; Zhong, W.; Bian, G.; Shi, X.; Zhang, J. RECENT ADVANCES IN THE CHEMISTRY OF TRICHLOROMETHYL CHLOROFORMATE AND Bis-(TRICHLOROMETHYL) CARBONATE. Org. Prep. Proced. Int. 2004, 36, 499– 547, DOI: 10.1080/00304940409355972There is no corresponding record for this reference.
- 3Cotarca, L.; Delogu, P.; Nardelli, A.; Šunjić, V. Bis(Trichloromethyl) Carbonate in Organic Synthesis. Synthesis 1996, 1996, 553– 576, DOI: 10.1055/s-1996-4273There is no corresponding record for this reference.
- 4Ganiu, M. O.; Nepal, B.; Van Houten, J. P.; Kartika, R. A Decade Review of Triphosgene and Its Applications in Organic Reactions. Tetrahedron 2020, 76, 131553, DOI: 10.1016/j.tet.2020.131553There is no corresponding record for this reference.
- 5Pasquato, L.; Modena, G.; Cotarca, L.; Delogu, P.; Mantovani, S. Conversion of Bis(Trichloromethyl) Carbonate to Phosgene and Reactivity of Triphosgene, Diphosgene, and Phosgene with Methanol1. J. Org. Chem. 2000, 65, 8224– 8228, DOI: 10.1021/jo000820uThere is no corresponding record for this reference.
- 6Hood, H. P.; Murdock, H. R. Superpalite. J. Phys. Chem. 1919, 23, 498– 512, DOI: 10.1021/j150196a004There is no corresponding record for this reference.
- 7Eckert, H.; Auerweck, J. Solvent-Free and Safe Process for the Quantitative Production of Phosgene from Triphosgene by Deactivated Imino-Based Catalysts. Org. Process Res. Dev. 2010, 14, 1501– 1505, DOI: 10.1021/op100239nThere is no corresponding record for this reference.
- 8Cotarca, L.; Geller, T.; Répási, J. Bis(Trichloromethyl)Carbonate (BTC, Triphosgene): A Safer Alternative to Phosgene?. Org. Process Res. Dev. 2017, 21, 1439– 1446, DOI: 10.1021/acs.oprd.7b00220There is no corresponding record for this reference.
- 9Hentschel, W. Ueber Gechlorte Ameisensäuremethyläther Und Verwandte Körper. J. Prakt. Chem. 1887, 36 (36), 305– 317, DOI: 10.1002/prac.18870360129There is no corresponding record for this reference.
- 10Councler, C. Kohlensaures Methyl. Ber. Dtsch. Chem. Ges. 1880, 13 (13), 1697– 1699, DOI: 10.1002/cber.188001302116There is no corresponding record for this reference.
- 11Hentschel, W. Ueber Gechlorte Ameisensäuremethyläther Und Verwandte Körper. J. Prakt. Chem. 1887, 36 (36), 99– 113, DOI: 10.1002/prac.18870360110There is no corresponding record for this reference.
- 12Hentschel, W. Ueber Gechlorte Ameisensäuremethyläther Und Verwandte Körper. J. Prakt. Chem. 1887, 36 (36), 468– 480, DOI: 10.1002/prac.18870360139There is no corresponding record for this reference.
- 13Hales, J. L.; Jones, J. I.; Kynaston, W. The Infrared Absorption Spectra of Some Chloroformates and Carbonates. The Structure of “Di- and Tri-Phosgene”. J. Chem. Soc. 1957, 0, 618– 625, DOI: 10.1039/JR9570000618There is no corresponding record for this reference.
- 14Sørensen, A. M.; Rosenqvist, E.; Åse, K.; Omfeldt, M.; Lagerlund, I.; Ehrenberg, L. The Crystal Structure of Bistrichloromethylcarbonate, Triphosgene. Acta Chem. Scand. 1971, 25, 169– 174, DOI: 10.3891/acta.chem.scand.25-0169There is no corresponding record for this reference.
- 15Pauluhn, J. Acute Nose-Only Inhalation Exposure of Rats to Di- and Triphosgene Relative to Phosgene. Inhalation Toxicol. 2011, 23, 65– 73, DOI: 10.3109/08958378.2010.542501There is no corresponding record for this reference.
- 16Sheldrick, G. M. SHELXT – Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr., Sect. A:Found. Adv. 2015, 71, 3– 8, DOI: 10.1107/S2053273314026370There is no corresponding record for this reference.
- 17Sheldrick, G. M. Crystal Structure Refinement withSHELXL. Acta. Crystallogr. C Struct. Chem. 2015, 71, 3– 8, DOI: 10.1107/S2053229614024218There is no corresponding record for this reference.
- 18Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339– 341, DOI: 10.1107/S0021889808042726There is no corresponding record for this reference.
- 19Brandenburg, K.; Putz, H. DIAMOND, Program for X-Ray Structure Analysis; Crystal Impact GbR: Bonn, Germany, 1999.There is no corresponding record for this reference.
- 20Spackman, M. A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19– 32, DOI: 10.1039/B818330AThere is no corresponding record for this reference.
- 21Spackman, P. R.; Turner, M. J.; McKinnon, J. J.; Wolff, S. K.; Grimwood, D. J.; Jayatilaka, D.; Spackman, M. A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006– 1011, DOI: 10.1107/S1600576721002910There is no corresponding record for this reference.
- 22Parker, S. F.; Fernandez-Alonso, F.; Ramirez-Cuesta, A. J.; Tomkinson, J.; Rudic, S.; Pinna, R. S.; Gorini, G.; Fernández Castañon, J. Recent and Future Developments on TOSCA at ISIS. J. Phys.: Conf. Ser. 2014, 554, 012003, DOI: 10.1088/1742-6596/554/1/012003There is no corresponding record for this reference.
- 23Pinna, R. S.; Rudić, S.; Parker, S. F.; Armstrong, J.; Zanetti, M.; Škoro, G.; Waller, S. P.; Zacek, D.; Smith, C. A.; Capstick, M. J.; McPhail, D. J.; Pooley, D. E.; Howells, G. D.; Gorini, G.; Fernandez-Alonso, F. The Neutron Guide Upgrade of the TOSCA Spectrometer. Nuclear Inst. and Methods in Physics Research A 2018, 896, 68– 74, DOI: 10.1016/j.nima.2018.04.009There is no corresponding record for this reference.
- 24https://www.isis.stfc.ac.uk/Pages/home.aspx (accessed 12 1, 2025).There is no corresponding record for this reference.
- 25Arnold, O. Mantid─Data Analysis and Visualization Package for Neutron Scattering and μ SR Experiments. Nucl. Instrum. Methods Phys. Res., Sect. A 2014, 764, 156– 166, DOI: 10.1016/j.nima.2014.07.029There is no corresponding record for this reference.
- 26Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C. M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; Kirtman, B. Quantum-mechanical Condensed Matter Simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360 DOI: 10.1002/wcms.1360There is no corresponding record for this reference.
- 27Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865– 3868, DOI: 10.1103/PhysRevLett.77.3865There is no corresponding record for this reference.
- 28Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0Model. J. Chem. Phys. 1999, 110, 6158– 6170, DOI: 10.1063/1.478522There is no corresponding record for this reference.
- 29Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297, DOI: 10.1039/b508541aThere is no corresponding record for this reference.
- 30Monkhorst, H. J.; Pack, J. D. Special Points for Brillouin-zone Integrations. Phys. Rev. B: 1976, 13, 5188– 5192, DOI: 10.1103/PhysRevB.13.5188There is no corresponding record for this reference.
- 31Pascale, F.; Zicovich-Wilson, C. M.; López Gejo, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The Calculation of the Vibrational Frequencies of Crystalline Compounds and Its Implementation in the CRYSTAL Code. J. Comput. Chem. 2004, 25, 888– 897, DOI: 10.1002/jcc.20019There is no corresponding record for this reference.
- 32Clark, S. J.; Segall, M. D.; Pickard, C. J.; Hasnip, P. J.; Probert, M. I. J.; Refson, K.; Payne, M. C. First Principles Methods Using CASTEP. Z. Kristallogr. 2005, 220, 567– 570, DOI: 10.1524/zkri.220.5.567.65075There is no corresponding record for this reference.
- 33Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005, DOI: 10.1103/PhysRevLett.102.073005There is no corresponding record for this reference.
- 34Refson, K.; Tulip, P. R.; Clark, S. J. Variational Density-Functional Perturbation Theory for Dielectrics and Lattice Dynamics. Phys. Rev. B 2006, 73, 155114, DOI: 10.1103/PhysRevB.73.155114There is no corresponding record for this reference.
- 35Gonze, X.; Charlier, J.-C.; Allan, D.; Teter, M. Interatomic Force Constants from First Principles: The Case of α-Quartz. Phys. Rev. B 1994, 50, 13035– 13038, DOI: 10.1103/PhysRevB.50.13035There is no corresponding record for this reference.
- 36Dymkowski, K.; Parker, S. F.; Fernandez-Alonso, F.; Mukhopadhyay, S. AbINS: The Modern Software for INS Interpretation. Physica B: Condensed Matter 2018, 551, 443– 448, DOI: 10.1016/j.physb.2018.02.034There is no corresponding record for this reference.
- 37Arce, V. B.; Della Védova, C. O.; Downs, A. J.; Parsons, S.; Romano, R. M. Trichloromethyl Chloroformate (“Diphosgene”), ClC(O)OCCl3: Structure and Conformational Properties in the Gaseous and Condensed Phases. J. Org. Chem. 2006, 71, 3423– 3428, DOI: 10.1021/jo052260aThere is no corresponding record for this reference.
- 38Spek, A. L. Structure Validation in Chemical Crystallography. Acta Crystallogr., Sect. D:Biol. Crystallogr. 2009, 65, 148– 155, DOI: 10.1107/S090744490804362XThere is no corresponding record for this reference.
- 39Bondi, A. Van Der Waals Volumes Radii. J. Phys. Chem. 1964, 68, 441– 451, DOI: 10.1021/j100785a001There is no corresponding record for this reference.
- 40Ringelband, S.; Pfeiffer, J.; Fortes, A. D.; Howard, C. M.; Parker, S. F.; Karttunen, A. J.; Tambornino, F. Crystal Structures and Intermolecular Interactions in α- and β-phosgene. Angew. Chem., Int. Ed. 2025, 65, e17323 DOI: 10.1002/anie.202517323There is no corresponding record for this reference.
- 41Whitfield, P. S. Low-Temperature Crystal Structures of the Solvent Dimethyl Carbonate. Powder Diffr. 2023, 38, 100– 111, DOI: 10.1017/S088571562300009XThere is no corresponding record for this reference.
- 42Ringelband, S.; Tambornino, F. Crystal Structure of Methyl Chloroformate. Acta Cryst. E 2025, 81, 987– 990, DOI: 10.1107/S2056989025008369There is no corresponding record for this reference.
- 43Erben, M. F.; Della Védova, C. O.; Boese, R.; Willner, H.; Oberhammer, H. Trifluoromethyl Chloroformate, ClC(O)OCF3: Structure, Conformation, and Vibrational Analysis Studied by Experimental and Theoretical Methods. J. Phys. Chem. A 2004, 108, 699– 706, DOI: 10.1021/jp036966pThere is no corresponding record for this reference.
- 44Pyykkö, P.; Atsumi, M. Molecular Double-Bond Covalent Radii for Elements Li–E112. Chem.–Eur. J. 2009, 15, 12770– 12779, DOI: 10.1002/chem.200901472There is no corresponding record for this reference.
- 45Bratsch, S. G. A Group Electronegativity Method with Pauling Units. J. Chem. Educ. 1985, 62, 101, DOI: 10.1021/ed062p101There is no corresponding record for this reference.
- 46Pingping, Z.; Dezhu, M. Study on the Double Cold Crystallization Peaks of Poly(Ethylene Terephthalate) 3. The Influence of the Addition of Calcium Carbonate (CaCO3). Eur. Polym. J. 2000, 36, 2471– 2475, DOI: 10.1016/S0014-3057(00)00042-2There is no corresponding record for this reference.
- 47Mitchell, P. C. H., Parker, S. F., Ramirez-Cuesta, A. J., Tomkinson, J., Eds.; Vibrational Spectroscopy with Neutrons: With Applications in Chemistry, Biology, Materials Science and Catalysis; Series on Neutron Techniques and Applications v; World Scientific: Singapore Hackensack, N.J, 2005; Vol. 3.There is no corresponding record for this reference.
- 48Perrichon, A.; Bovo, C.; Parker, S. F.; Raspino, D.; Armstrong, J.; García Sakai, V. Overview of Planned Upgrade to the Secondary Spectrometer of TOSCA. Nuclear Inst. and Methods in Physics Research, A 2023, 1047, 167899, DOI: 10.1016/j.nima.2022.167899There is no corresponding record for this reference.
- 49Bellamy, L. J. The Infrared Spectra of Complex Molecules, 3rd ed.; Chapman & Hall: Wiley: London: New York, 1975.There is no corresponding record for this reference.
- 50Colthup, N. B.; Daly, L. H.; Wiberley, S. E. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: Boston, 1990.There is no corresponding record for this reference.
- 51Lin-Vien, D.; Colthup, N. B.; Fateley, W. G.; Grasselli, J. G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Academic Press: Boston, 1991.There is no corresponding record for this reference.
- 52Andrews, B.; Anderson, A.; Torrie, B. Raman and Infrared Spectra of Crystalline Chloroform. Chem. Phys. Lett. 1984, 104, 65– 70, DOI: 10.1016/0009-2614(84)85306-3There is no corresponding record for this reference.
- 53Stanila, D.; Smith, W.; Anderson, A. Infrared Spectra of Chloroform at High Pressures. Spectrosc. Lett. 2002, 35, 703– 713, DOI: 10.1081/SL-120014941There is no corresponding record for this reference.
- 54Nolasco, M. M.; Coimbra, M. M.; Parker, S. F.; Vaz, P. D.; Ribeiro-Claro, P. J. A. Structural Dynamics of Chloromethanes through Computational Spectroscopy: Combining INS and DFT. Molecules 2022, 27, 7661, DOI: 10.3390/molecules27217661There is no corresponding record for this reference.
- 55Woost, B.; Bougeard, D. Vibrational Spectra and Phase Transitions of Crystalline Hexachloroethane. J. Chem. Phys. 1986, 84, 4810– 4817, DOI: 10.1063/1.449968There is no corresponding record for this reference.
- 56Adams, J.; Kim, H. The Infrared Spectra of Anhydrous Trichloroacetic Acid and Oxalic Acid. Spectrochim. Acta, Part A 1973, 29, 675– 677, DOI: 10.1016/0584-8539(73)80097-2There is no corresponding record for this reference.
- 57Trichloroacetic Acid Raman Spectrum. Available at: https://www.chemicalbook.com/SpectrumEN_76-03-9_Raman.htm (accessed 12 1, 2025).There is no corresponding record for this reference.
- 58Trichloroacetic Acid Infrared Spectrum. Available at: https://www.chemicalbook.com/SpectrumEN_76-03-9_IR1.htm (accessed 12 1, 2025).There is no corresponding record for this reference.
- 59Yadav, R.; Kumar, M.; Singh, R.; Singh, P.; Jaiswal, S.; Srivastav, G.; Prasad, R. Ab Initio Determination of Molecular Geometries and Vibrational Frequencies of CX3 COOH (X = H, F, Cl, Br). Spectrochim. Acta 2008, 71, 1565– 1570, DOI: 10.1016/j.saa.2008.06.016There is no corresponding record for this reference.
- 60Tambornino, F.; Parker, S.; Ringelband, S. The Inelastic Neutron Scattering Spectra of Diphosgene and Triphosgene; STFC ISIS Neutron and Muon Source, 2024.There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.5c05882.
Supporting Information crystallographic data, additional crystal structure pictures, Hirshfeld surface analysis, details on quantum chemical calculations and vibrational spectroscopy (PDF) cif file of β-diphosgene at 200 K (CIF) cif file of β-diphosgene at 100 K (CIF) cif file of triphosgene at 100 K (CIF) (PDF)
Deposition Numbers 2516310–2516312 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via the joint Cambridge Crystallographic Data Centre (CCDC) and Fachinformationszentrum Karlsruhe Access Structures service.
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.