



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
ORCA Meets Python─The ORCA Python Interface OPIClick to copy article linkArticle link copied!
- Tim Tetenberg
- Hagen Neugebauer*Hagen Neugebauer*E-mail: [email protected]FACCTs GmbH, Cologne 50677, GermanyMore by Hagen Neugebauer
- Christoph Plett
- Nakul Santhosh
- Markus Bursch*
- Christoph Riplinger*Christoph Riplinger*E-mail: [email protected]FACCTs GmbH, Cologne 50677, GermanyMore by Christoph Riplinger
Abstract
The ORCA program suite is one of the most widely used quantum chemistry software packages. It features a wide range of electronic structure methods and algorithms for the prediction of molecular chemical properties, reactivity, and spectroscopy. In this paper, we present a fully featured ORCA Python Interface termed OPI to drastically increase the accessibility of ORCA’s method portfolio and enable efficient automation of quantum chemical workflows. OPI is an open-source Python library that provides straightforward low-level access to ORCA’s input, execution, and output with a few lines of Python code. In the following, we introduce OPI version 2.0 and its key features, also outlining its general architecture. In addition, we demonstrate its application through several diverse examples of quantum chemical workflows. These examples include a system-dependent optimal-tuning procedure for range-separated hybrid functionals, generation of training data for machine learning purposes, orbital localization and visualization for chemical education, and calculations with density functional ensembles. Finally, we outline the current status of OPI and future plans for its development. OPI is compatible with ORCA ≥ 6.1.1 and Python ≥ 3.11. The project, its code, and its including documentation are available at https://github.com/faccts/opi. OPI is also available through PyPI (https://pypi.org/project/orca-pi).
This publication is licensed for personal use by The American Chemical Society.
1. Introduction
2. Project Philosophy
3. Architecture of OPI
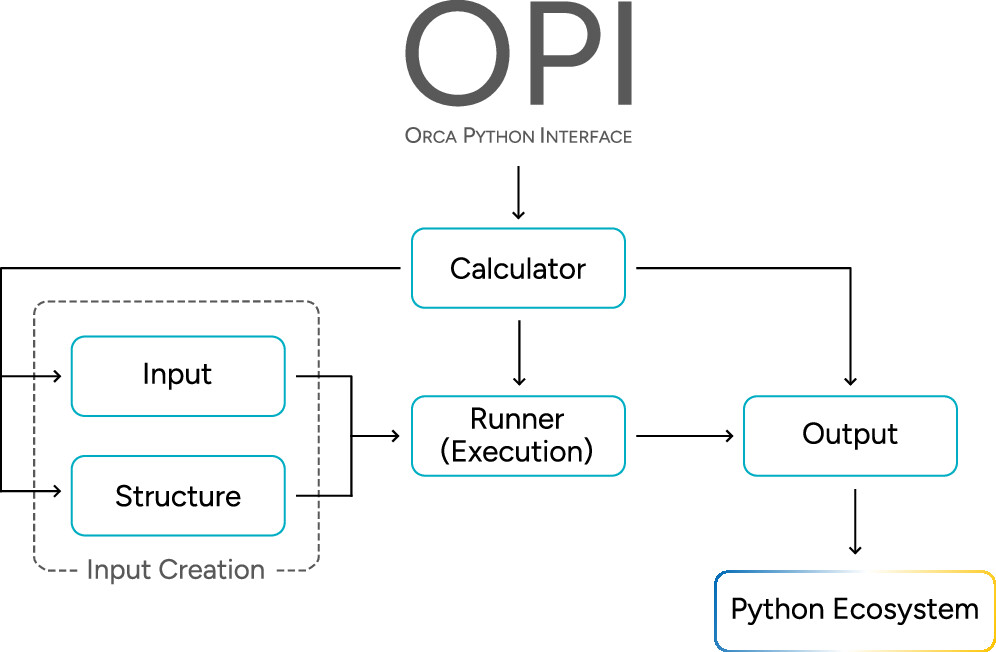
Figure 1
Figure 1. Schematic outline of OPI’s general structure and its five major classes: The Calculator class combines most of the functionality. The other major classes facilitate input creation (Structure and Input), job execution (Runner), and data extraction (Output). OPI benefits from Python’s ecosystem to visualize, analyze, or postprocess the data further and may be integrated into it at any point.
3.1. The Calculator Class
Figure 2
Figure 2. OPI example that shows the initialization of the Calculator class.
3.2. Input Generation: The Input and Structure Classes
3.2.1. The ORCA Input
Figure 3
Figure 3. Example of a simple ORCA input containing all major input elements: Simple input keywords (blue), key-value options (orange), block options (purple), and the coordinates block (black).
3.2.2. General Remarks on Input Generation with OPI
3.2.3. Input: Simple Input Keywords
Figure 4
Figure 4. OPI example of how to add simple keywords to the Calculator.
3.2.4. Input: Block Inputs
Figure 5
Figure 5. OPI example of how to add the maxiter options from ORCA’s %geom block to a job definition in OPI.
Figure 6
Figure 6. An example showing how to specify key-value options in OPI.
3.2.5. Input: Arbitrary Strings
Figure 7
Figure 7. A code example showcasing how to add not defined or arbitrary input to the ORCA input in OPI.
Figure 8
Figure 8. Excerpt of a minimal ORCA input file showing the three principal positions for input keywords defined in OPI: top: At the very top of the file; before_coords: Right before the coordinates block; bottom: At the very end of the file.
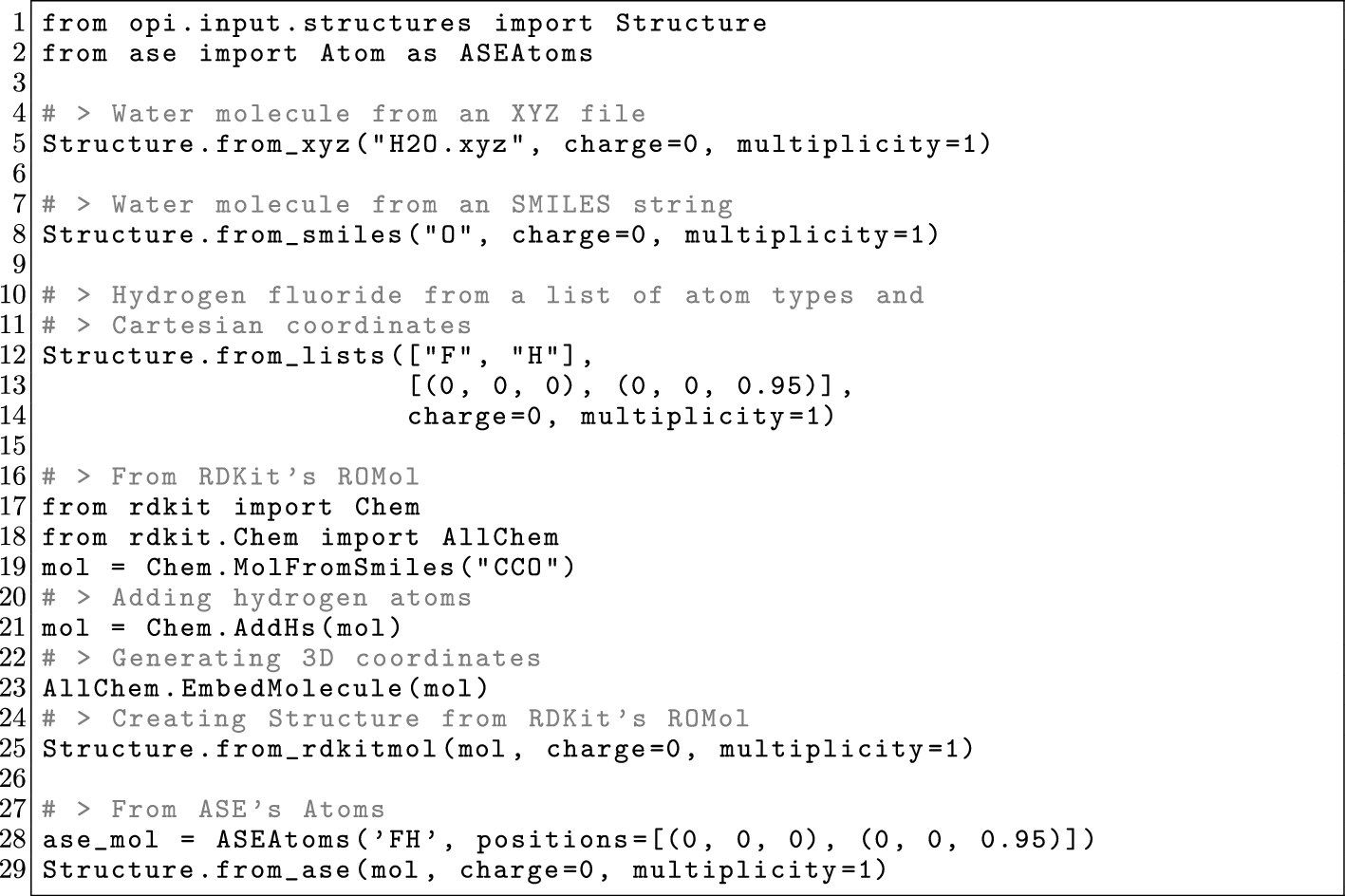
3.2.6. Structure: Structure Data
Figure 9
Figure 9. OPI example showcasing the different methods to create a Structure object from different sources.
4. Job Execution: The Runner Class
Figure 10
Figure 10. Example of an OPI configuration file specifying the location of ORCA and Open MPI. The file adheres to the TOML format.
Figure 11
Figure 11. OPI example showing how to create the ORCA input and execute the job after configuring the compute resources.
4.1. Output Handling: The Output Class
| 1. | A binary GBW file, which contains geometry, basis set, and wave function information. These data can be extracted into a JSON format using an ORCA utility. | ||||
| 2. | A structured property file, which contains computed quantities such as energies, thermochemical, and spectral data. This file can also be converted to JSON format. | ||||
| 3. | A free-form text-based output file collected from the STDOUT stream of the program. This file is intended for humans to read, but contains some information that is not present in the machine-readable files. | ||||
Figure 12
Figure 12. OPI example showing the creation of an Output from an existing Calculator object and parsing of the respective JSON files.
Figure 13
Figure 13. OPI example of how to access GBW and property file data from the Output object after parsing the files.
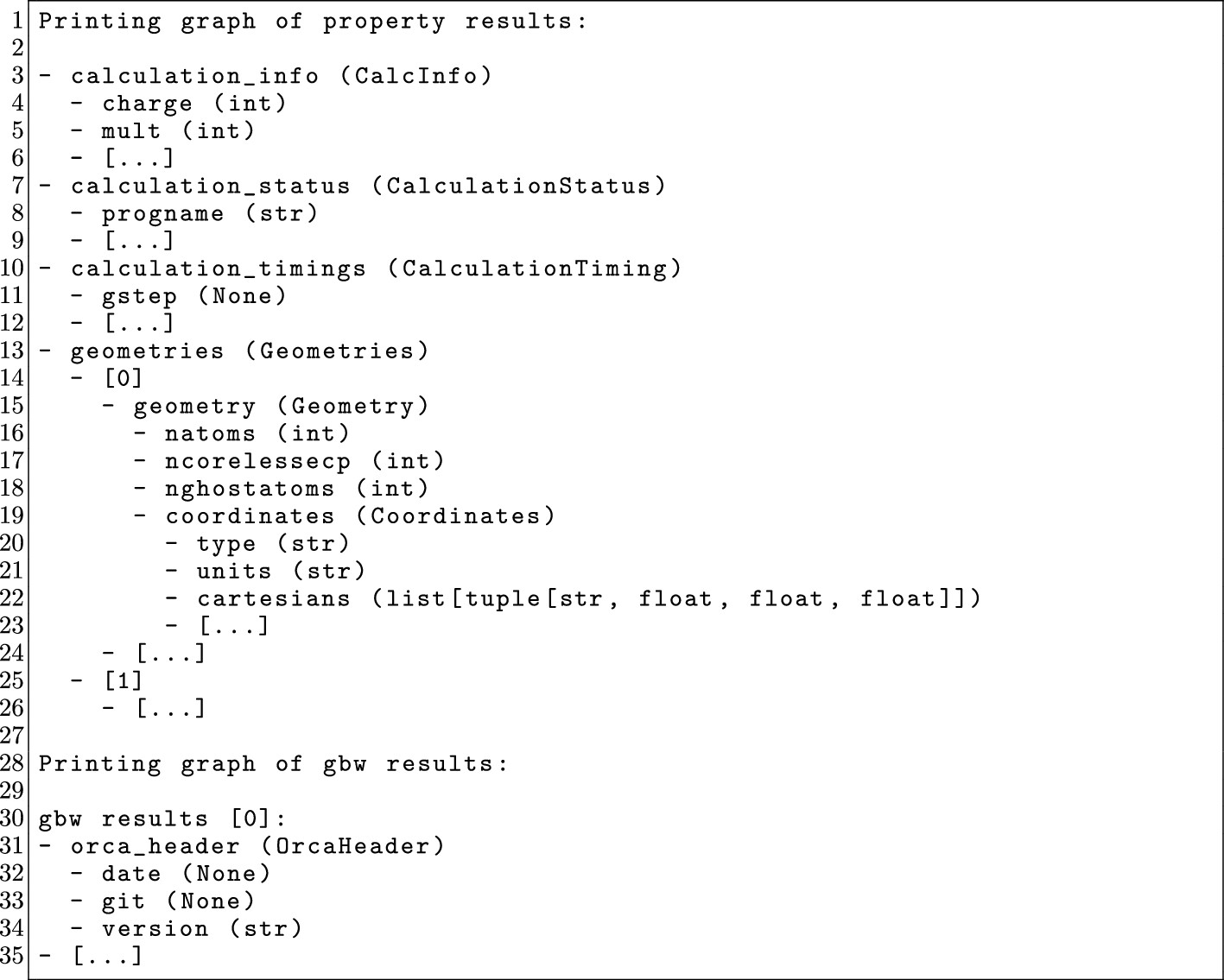
Figure 14
Figure 14. Shortened output example of the output.print_graph() method. Omitted parts are designated with [...].
4.1.1. Output: getter Methods, the Grepper Class, and Health-Checks
Figure 15
Figure 15. OPI has a series of getter methods to swiftly fetch properties from completed ORCA calculations. This example show the get_final_energy() method, which returns the final energy of the final structure.
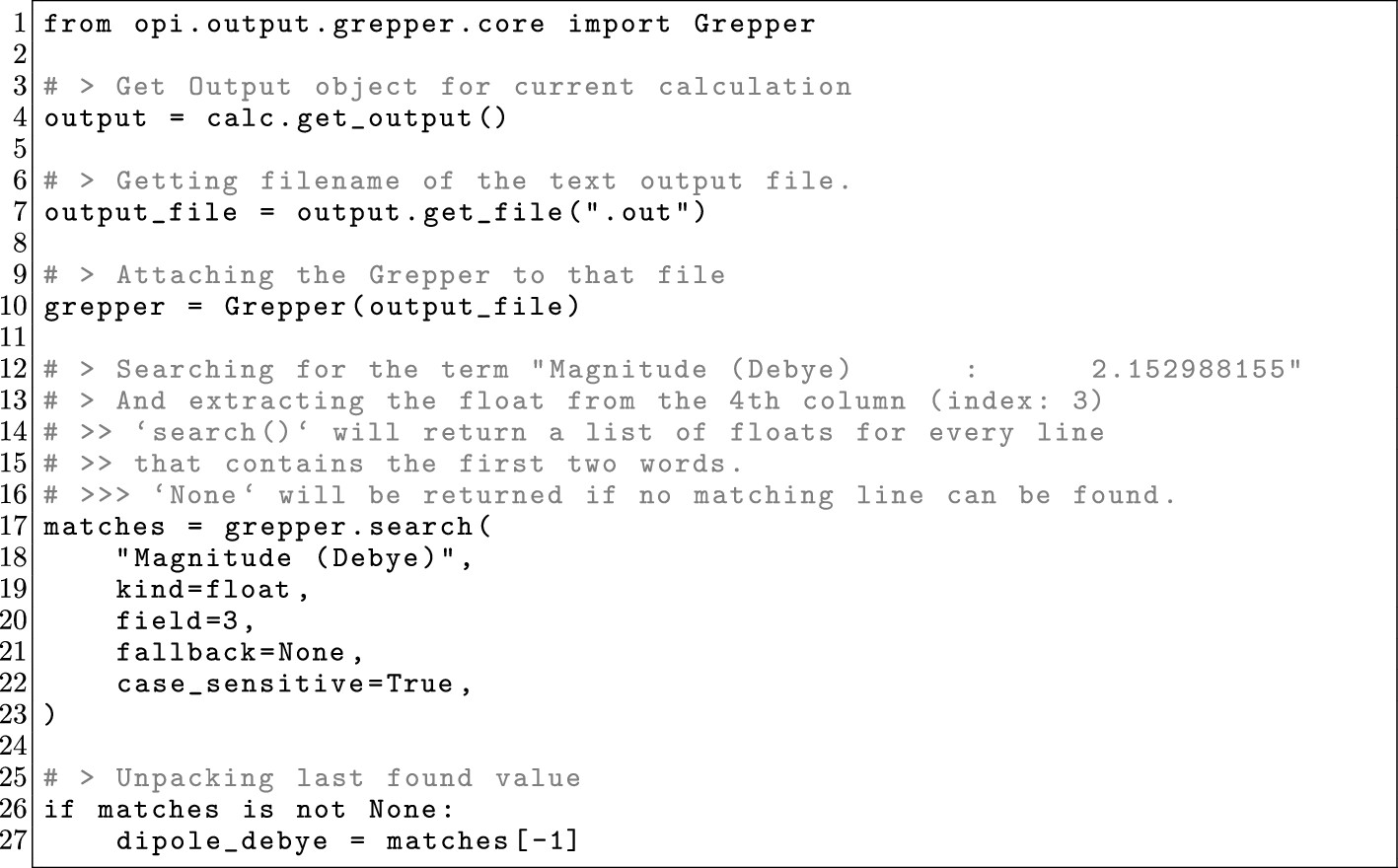
Figure 16
Figure 16. Minimal OPI example of the Grepper class, showcasing how to use the class to search for the line containing “Magnitude (Debye): 2.152988155” in the ORCA output of a completed calculation and extracting the value.
Figure 17
Figure 17. OPI example of predefined health-check routines.
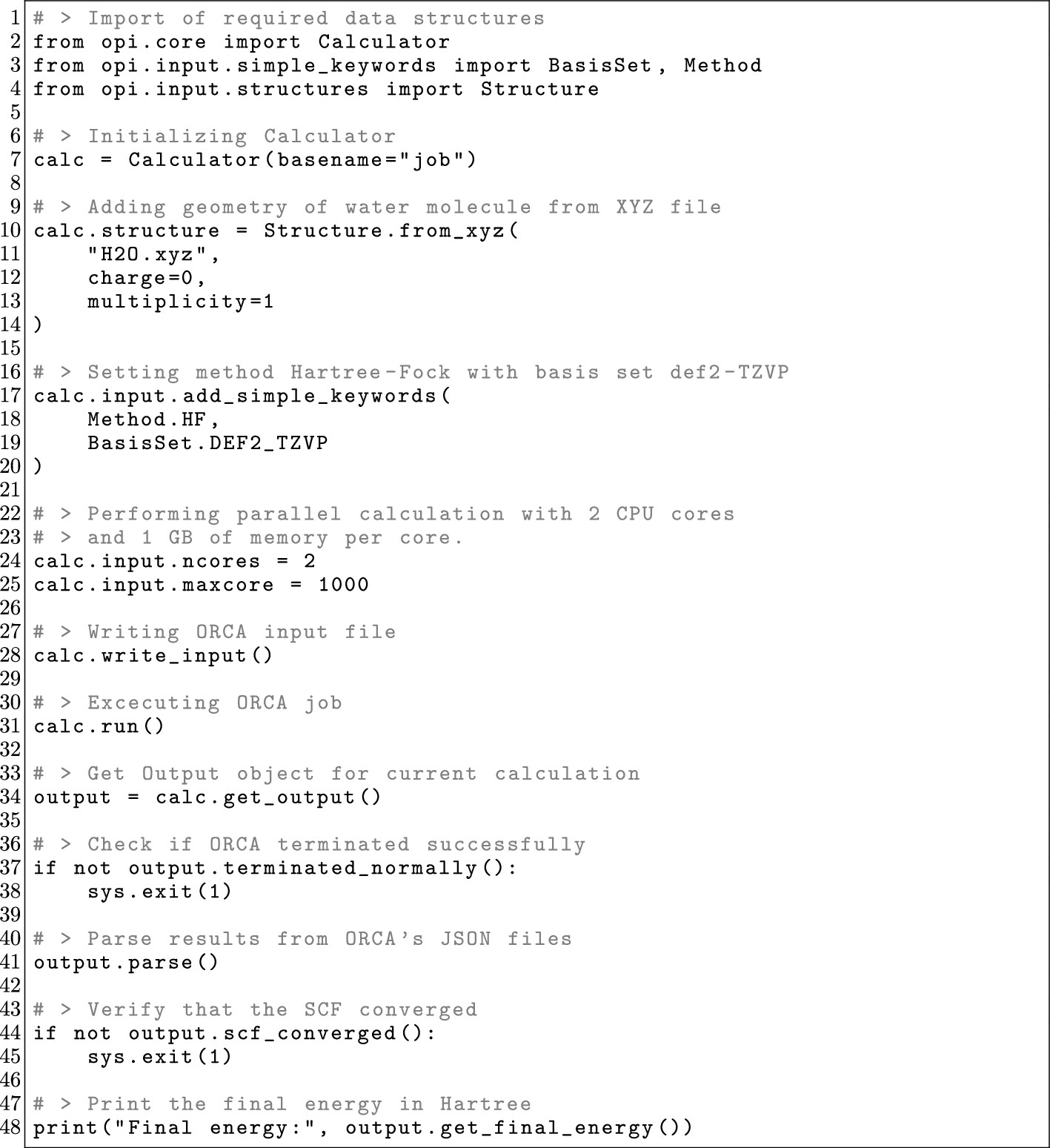
4.2. Hello Water: A First OPI Script
Figure 18
Figure 18. OPI example script of a simple energy calculation for a water molecule. It displays all steps involved: input definition, input creation, job execution, and printing of the final energy.
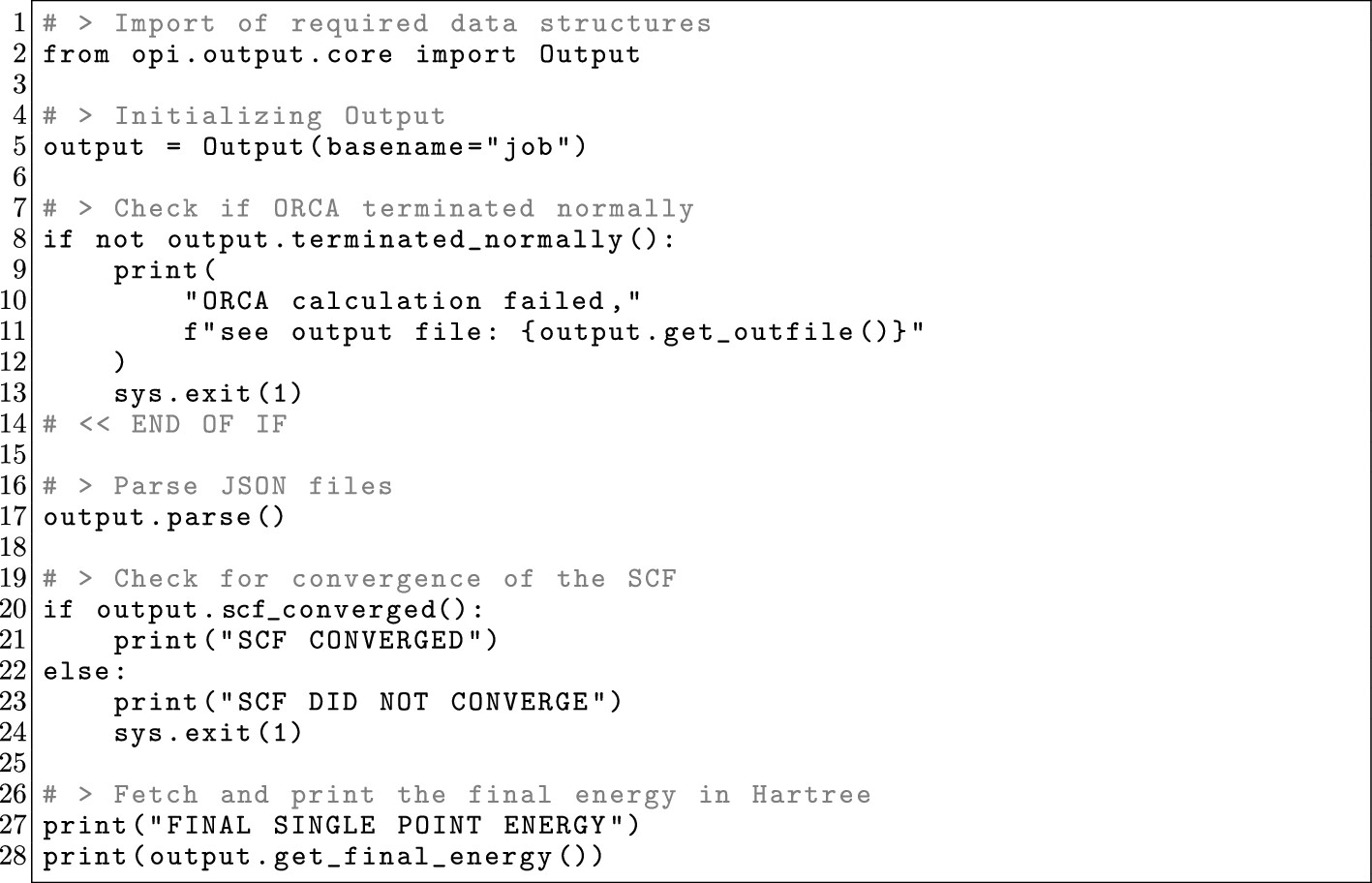
4.3. Post-Processing Existing ORCA Outputs
Figure 19
Figure 19. OPI example depicting how to only postprocess the results from a single-point calculation.
5. Examples
5.1. General ORCA Tasks
Figure 20
Figure 20. OPI example for performing a r2SCAN-3c geometry optimization followed by a frequency calculation to obtain a free energy.
Figure 21
Figure 21. OPI example of how to combine a high-level DLPNO–CCSD(T) single-point energy with thermostatistical corrections from DFT to obtain a free energy.
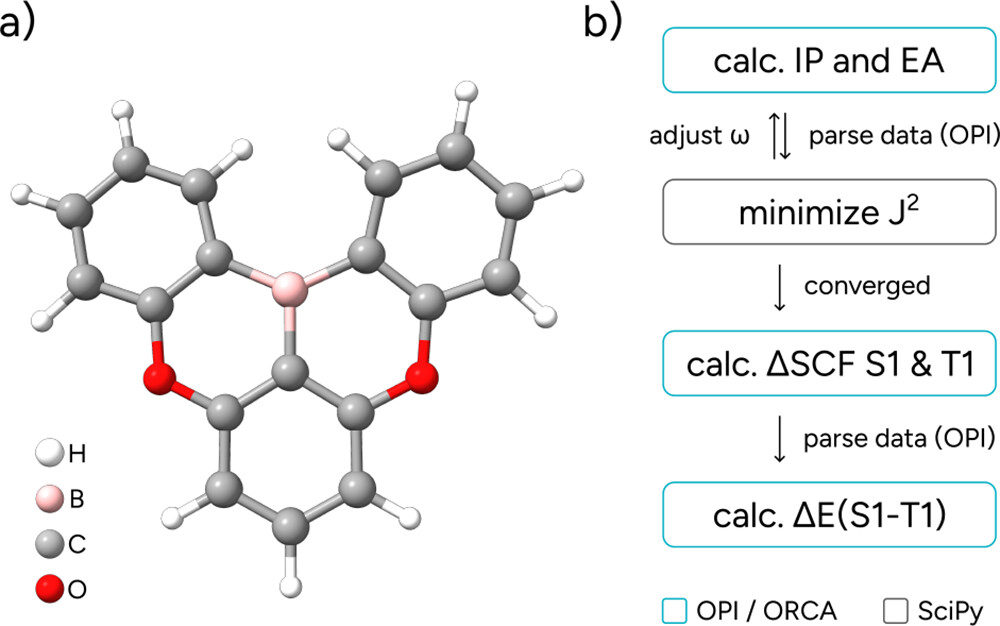
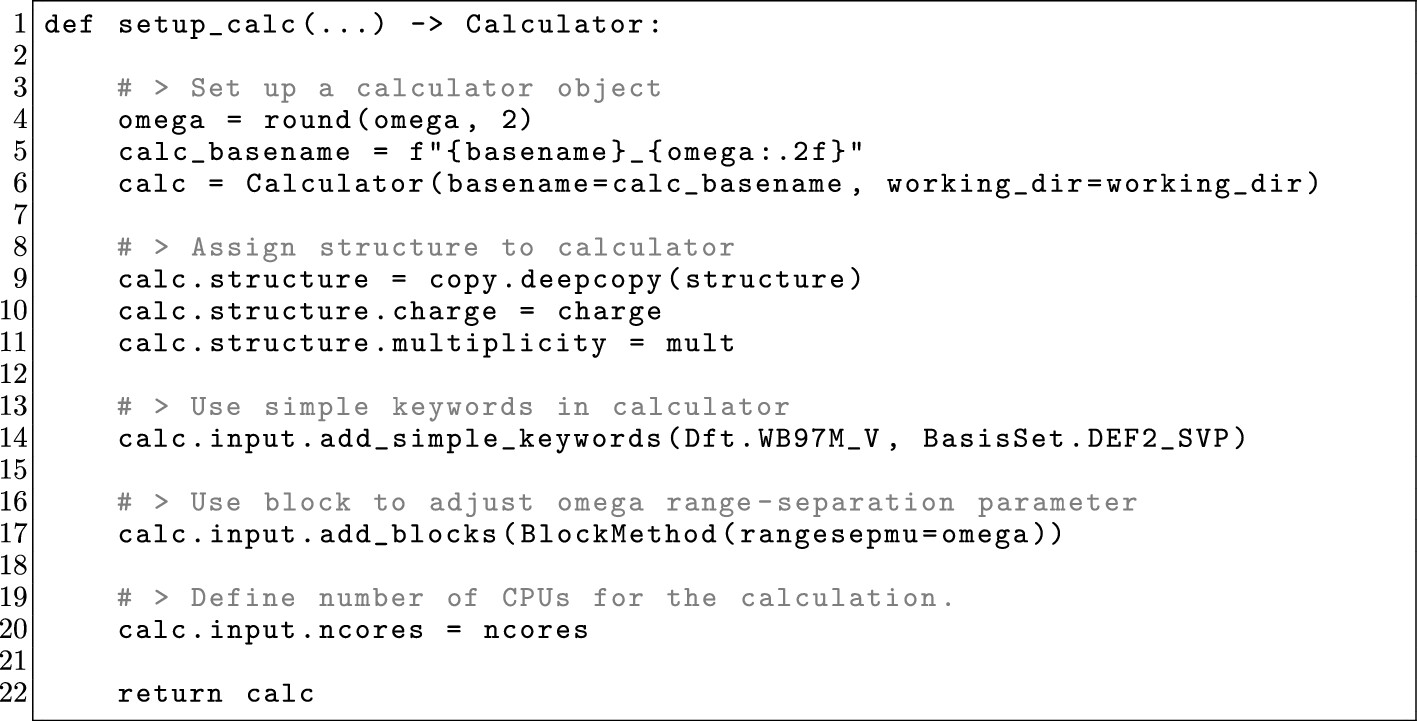
5.2. Optimal Tuning of a Range-Separated Hybrid Functional
Figure 22
Figure 22. (a) Molecular structure of the DOBNA molecule; (b) Schematic OPI workflow.
Figure 23
Figure 23. Single-point calculator setup from the OPI script to perform the optimal tuning workflow described in Section 5.2.
Figure 24
Figure 24. Function body for evaluation of J2(ω) from the OPI script to perform the optimal tuning workflow described in Section 5.2.
Figure 25
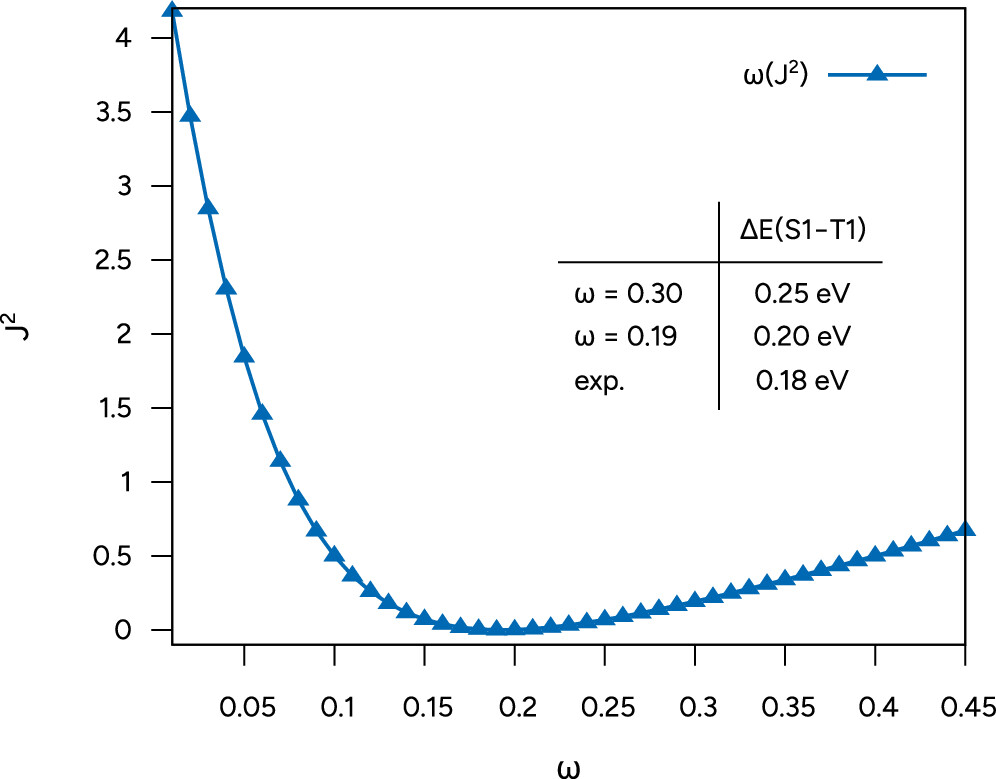
Figure 25. Plot of ω as a function of J2 and S1-T1 gaps computed with the default and optimally tuned ω in comparison to the experimental reference (cf. ref (98)).
5.3. Generating ML Training Data
Figure 26
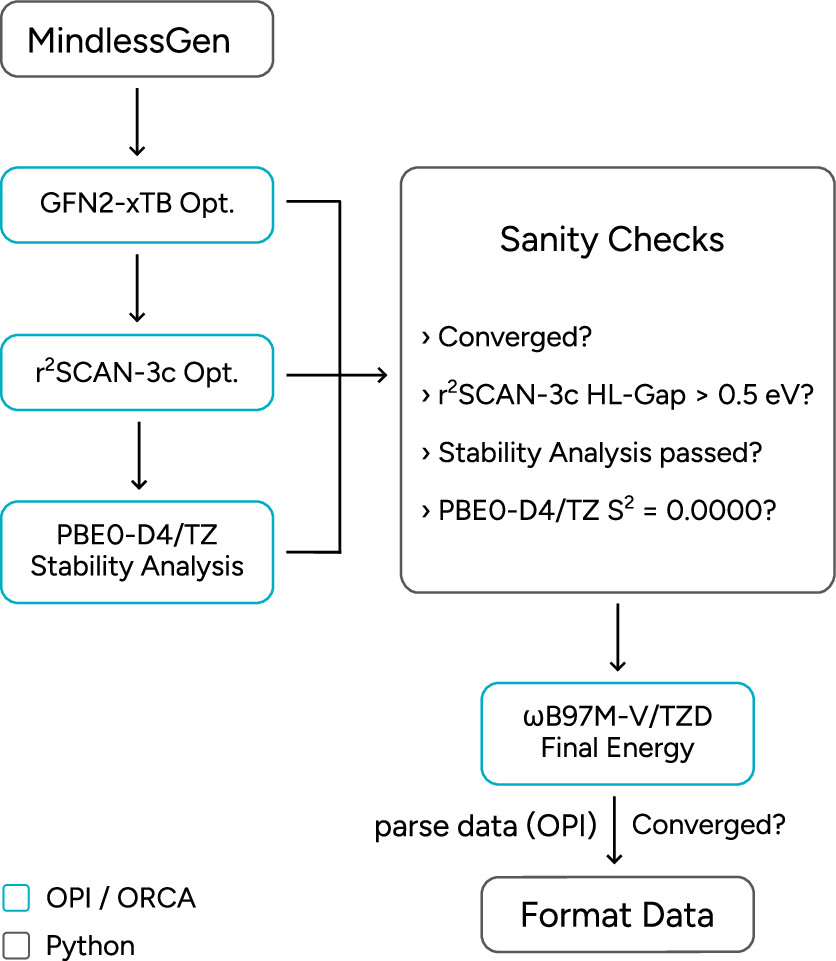
Figure 26. Schematic OPI workflow to generate new, standardized Mindless data based using MindlessGen with OPI.
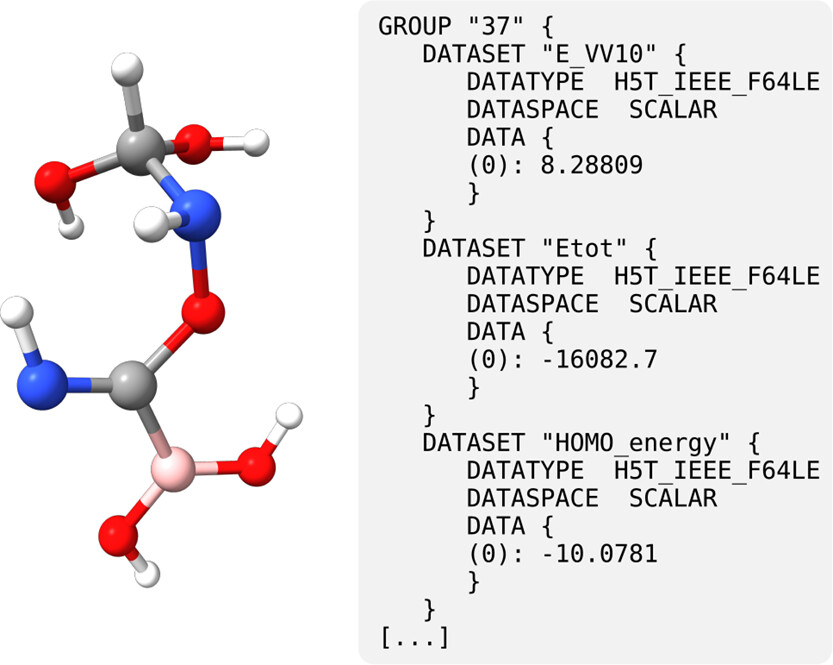
Figure 27
Figure 27. Example molecule generated with MindlessGen and a human-readable excerpt of the output generated with OPI and Python.
5.4. Density Functional Ensembles
Figure 28
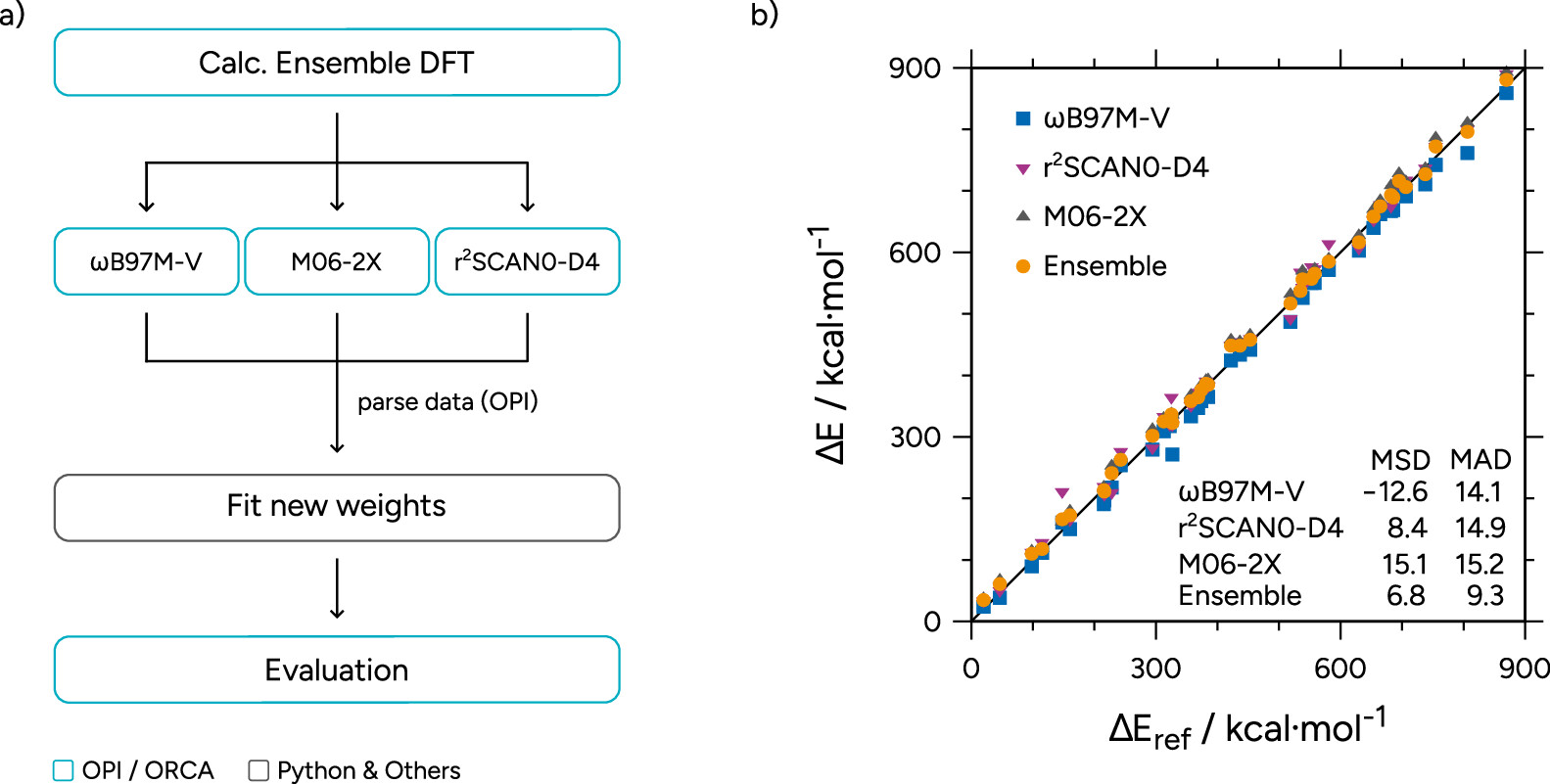
Figure 28. (a) Flowchart of the process of determining the ensemble weights. (b) Correlation plot of the tested functionals on the MB16-43 as well as the ensemble optimized on the set. MSDs and MADs are given in kcal·mol–1. One data point below 0 kcal·mol–1 and two above 900 kcal·mol–1 are omitted for clarity.
5.5. OPI in Chemical Education
Figure 29
Figure 29. Function calls for cube file generation and visualization within a Jupyter notebook.
Figure 30
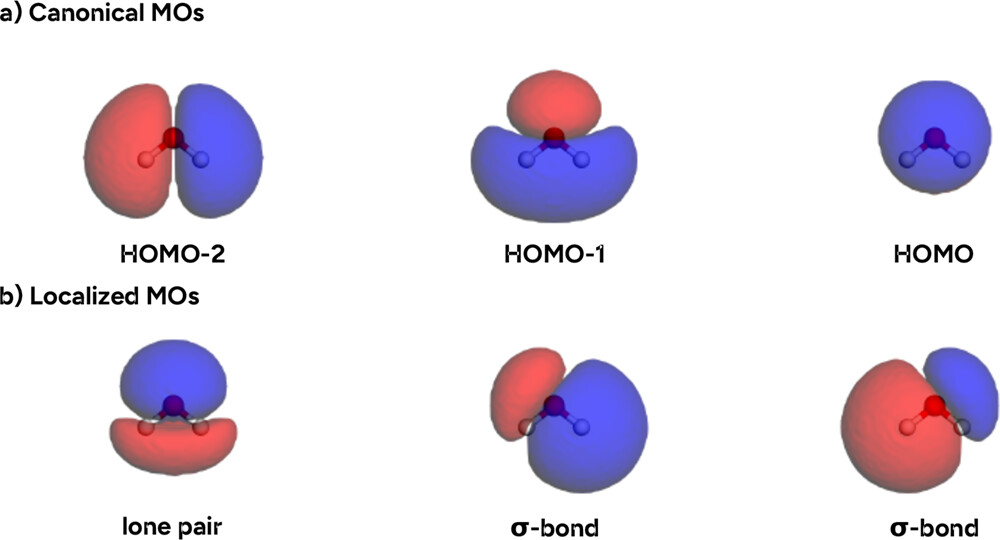
Figure 30. (a) Canonical frontier orbitals and (b) Pipek–Mezey localized orbitals of water (GFN2-xTB level) as plotted in the example notebook.
Figure 31
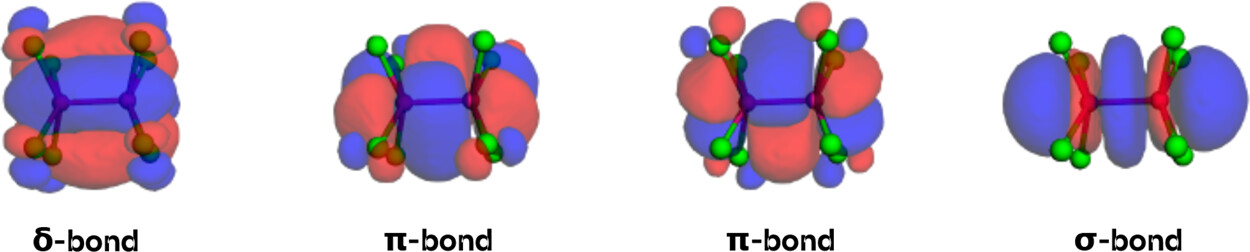
Figure 31. Bonding LMOs (PBE0/def2-SVP, Pipek-Mezey orbital localization) for [Re2Cl8]2– as plotted in the example notebook. The δ-, two π-, and the σ-bonds can be identified in line with chemical intuition.
6. Conclusions and Outlook
Data Availability
The OPI code is freely available as open source on GitHub (https://github.com/faccts/opi) and via PyPI (https://pypi.org/project/orca-pi). An online documentation of the project can be found at https://www.faccts.de/docs#opi.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.5c02141.
Computed data for the ML training data example (ZIP)
Additional overview of currently available quantities from the GBW JSON file and the property JSON file (PDF)
Computation-ready scripts and Jupyter Notebooks for all presented examples (ZIP)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
Acknowledgments
The authors thank Shiyao Ju for creating useful tutorial notebooks and initial testing of OPI and Mathieu C. Brandenburg for the initial codevelopment of OPI. The authors also thank Dr. Gregor Giesen for technical support and Dr. Georgi L. Stoychev for helpful discussions and proofreading of the manuscript. For open-source contributions to the OPI project until the time of submission, the authors thank Frederic Bender, Jackson Burns, Anselm Hahn, Jan Hamaekers, Prof. Esther Heid, Julia Kaczmarek, Matt Taylor, Anton Zamyatin, and Matthew Burn.
References
This article references 120 other publications.
- 1Houk, K. N.; Liu, F. Holy Grails for Computational Organic Chemistry and Biochemistry. Acc. Chem. Res. 2017, 50, 539– 543, DOI: 10.1021/acs.accounts.6b00532Google ScholarThere is no corresponding record for this reference.
- 2Grimme, S.; Schreiner, P. R. Computational Chemistry: The Fate of Current Methods and Future Challenges. Angew. Chem., Int. Ed. 2018, 57, 4170– 4176, DOI: 10.1002/anie.201709943Google ScholarThere is no corresponding record for this reference.
- 3Neese, F.; Atanasov, M.; Bistoni, G.; Maganas, D.; Ye, S. Chemistry and Quantum Mechanics in 2019: Give Us Insight and Numbers. J. Am. Chem. Soc. 2019, 141, 2814– 2824, DOI: 10.1021/jacs.8b13313Google ScholarThere is no corresponding record for this reference.
- 4Borges, R. M.; Colby, S. M.; Das, S.; Edison, A. S.; Fiehn, O.; Kind, T.; Lee, J.; Merrill, A. T.; Merz, K. M. J.; Metz, T. O.; Nunez, J. R.; Tantillo, D. J.; Wang, L.-P.; Wang, S.; Renslow, R. S. Quantum Chemistry Calculations for Metabolomics. Chem. Rev. 2021, 121, 5633– 5670, DOI: 10.1021/acs.chemrev.0c00901Google ScholarThere is no corresponding record for this reference.
- 5Ozaki, Y.; Beć, K. B.; Morisawa, Y.; Yamamoto, S.; Tanabe, I.; Huck, C. W.; Hofer, T. S. Advances, challenges and perspectives of quantum chemical approaches in molecular spectroscopy of the condensed phase. Chem. Soc. Rev. 2021, 50, 10917– 10954, DOI: 10.1039/D0CS01602KGoogle ScholarThere is no corresponding record for this reference.
- 6Teale, A. M.; Helgaker, T.; Savin, A.; Adamo, C.; Aradi, B.; Arbuznikov, A. V.; Ayers, P. W.; Baerends, E. J.; Barone, V.; Calaminici, P.; Cancès, E.; Carter, E. A.; Chattaraj, P. K.; Chermette, H.; Ciofini, I.; Crawford, T. D.; De Proft, F.; Dobson, J. F.; Draxl, C.; Frauenheim, T.; Fromager, E.; Fuentealba, P.; Gagliardi, L.; Galli, G.; Gao, J.; Geerlings, P.; Gidopoulos, N.; Gill, P. M. W.; Gori-Giorgi, P.; Görling, A.; Gould, T.; Grimme, S.; Gritsenko, O.; Jensen, H. J. A.; Johnson, E. R.; Jones, R. O.; Kaupp, M.; Köster, A. M.; Kronik, L.; Krylov, A. I.; Kvaal, S.; Laestadius, A.; Levy, M.; Lewin, M.; Liu, S.; Loos, P.-F.; Maitra, N. T.; Neese, F.; Perdew, J. P.; Pernal, K.; Pernot, P.; Piecuch, P.; Rebolini, E.; Reining, L.; Romaniello, P.; Ruzsinszky, A.; Salahub, D. R.; Scheffler, M.; Schwerdtfeger, P.; Staroverov, V. N.; Sun, J.; Tellgren, E.; Tozer, D. J.; Trickey, S. B.; Ullrich, C. A.; Vela, A.; Vignale, G.; Wesolowski, T. A.; Xu, X.; Yang, W. DFT exchange: sharing perspectives on the workhorse of quantum chemistry and materials science. Phys. Chem. Chem. Phys. 2022, 24, 28700– 28781, DOI: 10.1039/D2CP02827AGoogle ScholarThere is no corresponding record for this reference.
- 7Seeman, J. I.; Tantillo, D. J. Understanding chemistry: from “heuristic (soft) explanations and reasoning by analogy” to “quantum chemistry”. Chem. Sci. 2022, 13, 11461– 11486, DOI: 10.1039/D2SC02535CGoogle ScholarThere is no corresponding record for this reference.
- 8Nam, K.; Shao, Y.; Major, D. T.; Wolf-Watz, M. Perspectives on Computational Enzyme Modeling: From Mechanisms to Design and Drug Development. ACS Omega 2024, 9, 7393– 7412, DOI: 10.1021/acsomega.3c09084Google ScholarThere is no corresponding record for this reference.
- 9Pölloth, B. High School Students Experimenting with Computational Chemistry: Design-Based Research on and through the “Comp-Chem-Lab”. J. Chem. Educ. 2025, 102, 1367– 1379, DOI: 10.1021/acs.jchemed.4c01136Google ScholarThere is no corresponding record for this reference.
- 10Autschbach, J. Orbitals: Some Fiction and Some Facts. J. Chem. Educ. 2012, 89, 1032– 1040, DOI: 10.1021/ed200673wGoogle ScholarThere is no corresponding record for this reference.
- 11Grushow, A.; Reeves, M. S., Eds. Using Computational Methods to Teach Chemical Principles; ACS Symposium Series; American Chemical Society: Washington, DC, 2019; Vol. 1312.Google ScholarThere is no corresponding record for this reference.
- 12Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem., Int. Ed. 2022, 61, e202205735 DOI: 10.1002/anie.202205735Google ScholarThere is no corresponding record for this reference.
- 13Dyson, F. J. Is Science Mostly Driven by Ideas or by Tools?. Science 2012, 338, 1426– 1427, DOI: 10.1126/science.1232773Google ScholarThere is no corresponding record for this reference.
- 14Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A. T.; Wormit, M.; Kussmann, J.; Lange, A. W.; Behn, A.; Deng, J.; Feng, X.; Ghosh, D.; Goldey, M.; Horn, P. R.; Jacobson, L. D.; Kaliman, I.; Khaliullin, R. Z.; Kuś, T.; Landau, A.; Liu, J.; Proynov, E. I.; Rhee, Y. M.; Richard, R. M.; Rohrdanz, M. A.; Steele, R. P.; Sundstrom, E. J. III. H. L. W.; Zimmerman, P. M.; Zuev, D.; Albrecht, B.; Alguire, E.; Austin, B.; Beran, G. J. O.; Bernard, Y. A.; Berquist, E.; Brandhorst, K.; Bravaya, K. B.; Brown, S. T.; Casanova, D.; Chang, C.-M.; Chen, Y.; Chien, S. H.; Closser, K. D.; Crittenden, D. L.; Diedenhofen, M.Jr.R.A.D.; Do, H.; Dutoi, A. D.; Edgar, R. G.; Fatehi, S.; Fusti-Molnar, L.; Ghysels, A.; Golubeva-Zadorozhnaya, A.; Gomes, J.; Hanson-Heine, M. W.; Harbach, P. H.; Hauser, A. W.; Hohenstein, E. G.; Holden, Z. C.; Jagau, T.-C.; Ji, H.; Kaduk, B.; Khistyaev, K.; Kim, J.; Kim, J.; King, R. A.; Klunzinger, P.; Kosenkov, D.; Kowalczyk, T.; Krauter, C. M.; Lao, K. U.; Laurent, A. D.; Lawler, K. V.; Levchenko, S. V.; Lin, C. Y.; Liu, F.; Livshits, E.; Lochan, R. C.; Luenser, A.; Manohar, P.; Manzer, S. F.; Mao, S.-P.; Mardirossian, N.; Marenich, A. V.; Maurer, S. A.; Mayhall, N. J.; Neuscamman, E.; Oana, C. M.; Olivares-Amaya, R.; O’Neill, D. P.; Parkhill, J. A.; Perrine, T. M.; Peverati, R.; Prociuk, A.; Rehn, D. R.; Rosta, E.; Russ, N. J.; Sharada, S. M.; Sharma, S.; Small, D. W.; Sodt, A.; Stein, T.; Stück, D.; Su, Y.-C.; Thom, A. J.; Tsuchimochi, T.; Vanovschi, V.; Vogt, L.; Vydrov, O.; Wang, T.; Watson, M. A.; Wenzel, J.; White, A.; Williams, C. F.; Yang, J.; Yeganeh, S.; Yost, S. R.; You, Z.-Q.; Zhang, I. Y.; Zhang, X.; Zhao, Y.; Brooks, B. R.; Chan, G. K.; Chipman, D. M.; Cramer, C. J.III.W.A.G.; Gordon, M. S.; Hehre, W. J.; Klamt, A.III.H.F.S.; Schmidt, M. W.; Sherrill, C. D.; Truhlar, D. G.; Warshel, A.; Xu, X.; Aspuru-Guzik, A.; Baer, R.; Bell, A. T.; Besley, N. A.; Chai, J.-D.; Dreuw, A.; Dunietz, B. D.; Furlani, T. R.; Gwaltney, S. R.; Hsu, C.-P.; Jung, Y.; Kong, J.; Lambrecht, D. S.; Liang, W.; Ochsenfeld, C.; Rassolov, V. A.; Slipchenko, L. V.; Subotnik, J. E.; Voorhis, T. V.; Herbert, J. M.; Krylov, A. I.; Gill, P. M.; Head-Gordon, M. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184– 215, DOI: 10.1080/00268976.2014.952696Google ScholarThere is no corresponding record for this reference.
- 15Baerends, E. J.; Aguirre, N. F.; Austin, N. D.; Autschbach, J.; Bickelhaupt, F. M.; Bulo, R.; Cappelli, C.; van Duin, A. C. T.; Egidi, F.; Fonseca Guerra, C.; Förster, A.; Franchini, M.; Goumans, T. P. M.; Heine, T.; Hellström, M.; Jacob, C. R.; Jensen, L.; Krykunov, M.; van Lenthe, E.; Michalak, A.; Mitoraj, M. M.; Neugebauer, J.; Nicu, V. P.; Philipsen, P.; Ramanantoanina, H.; Rüger, R.; Schreckenbach, G.; Stener, M.; Swart, M.; Thijssen, J. M.; Trnka, T.; Visscher, L.; Yakovlev, A.; van Gisbergen, S. The Amsterdam Modeling Suite. J. Chem. Phys. 2025, 162, 162501, DOI: 10.1063/5.0258496Google ScholarThere is no corresponding record for this reference.
- 16Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165– 169, DOI: 10.1016/0009-2614(89)85118-8Google ScholarThere is no corresponding record for this reference.
- 17Balasubramani, S. G.; Chen, G. P.; Coriani, S.; Diedenhofen, M.; Frank, M. S.; Franzke, Y. J.; Furche, F.; Grotjahn, R.; Harding, M. E.; Hättig, C.; Hellweg, A.; Helmich-Paris, B.; Holzer, C.; Huniar, U.; Kaupp, M.; Marefat Khah, A.; Karbalaei Khani, S.; Müller, T.; Mack, F.; Nguyen, B. D.; Parker, S. M.; Perlt, E.; Rappoport, D.; Reiter, K.; Roy, S.; Rückert, M.; Schmitz, G.; Sierka, M.; Tapavicza, E.; Tew, D. P.; van Wüllen, C.; Voora, V. K.; Weigend, F.; Wodyński, A.; Yu, J. M. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107, DOI: 10.1063/5.0004635Google ScholarThere is no corresponding record for this reference.
- 18Franzke, Y. J.; Holzer, C.; Andersen, J. H.; Begušić, T.; Bruder, F.; Coriani, S.; Della Sala, F.; Fabiano, E.; Fedotov, D. A.; Fürst, S.; Gillhuber, S.; Grotjahn, R.; Kaupp, M.; Kehry, M.; Krstić, M.; Mack, F.; Majumdar, S.; Nguyen, B. D.; Parker, S. M.; Pauly, F.; Pausch, A.; Perlt, E.; Phun, G. S.; Rajabi, A.; Rappoport, D.; Samal, B.; Schrader, T.; Sharma, M.; Tapavicza, E.; Treß, R. S.; Voora, V.; Wodyński, A.; Yu, J. M.; Zerulla, B.; Furche, F.; Hättig, C.; Sierka, M.; Tew, D. P.; Weigend, F. TURBOMOLE: Today and Tomorrow. J. Chem. Theory Comput. 2023, 19, 6859– 6890, DOI: 10.1021/acs.jctc.3c00347Google ScholarThere is no corresponding record for this reference.
- 19Valiev, M.; Bylaska, E.; Govind, N.; Kowalski, K.; Straatsma, T.; Van Dam, H.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.; de Jong, W. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477– 1489, DOI: 10.1016/j.cpc.2010.04.018Google ScholarThere is no corresponding record for this reference.
- 20Sun, Q.; Berkelbach, T. C.; Blunt, N. S.; Booth, G. H.; Guo, S.; Li, Z.; Liu, J.; McClain, J. D.; Sayfutyarova, E. R.; Sharma, S.; Wouters, S.; Chan, G. K.-L. PySCF: the Python-based simulations of chemistry framework. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1340 DOI: 10.1002/wcms.1340Google ScholarThere is no corresponding record for this reference.
- 21Sun, Q.; Zhang, X.; Banerjee, S.; Bao, P.; Barbry, M.; Blunt, N. S.; Bogdanov, N. A.; Booth, G. H.; Chen, J.; Cui, Z.-H.; Eriksen, J. J.; Gao, Y.; Guo, S.; Hermann, J.; Hermes, M. R.; Koh, K.; Koval, P.; Lehtola, S.; Li, Z.; Liu, J.; Mardirossian, N.; McClain, J. D.; Motta, M.; Mussard, B.; Pham, H. Q.; Pulkin, A.; Purwanto, W.; Robinson, P. J.; Ronca, E.; Sayfutyarova, E. R.; Scheurer, M.; Schurkus, H. F.; Smith, J. E. T.; Sun, C.; Sun, S.-N.; Upadhyay, S.; Wagner, L. K.; Wang, X.; White, A.; Whitfield, J. D.; Williamson, M. J.; Wouters, S.; Yang, J.; Yu, J. M.; Zhu, T.; Berkelbach, T. C.; Sharma, S.; Sokolov, A. Y.; Chan, G. K.-L. Recent developments in the PySCF program package. J. Chem. Phys. 2020, 153, 024109 DOI: 10.1063/5.0006074Google ScholarThere is no corresponding record for this reference.
- 22Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G. L.; Cococcioni, M.; Dabo, I.; Dal Corso, A.; de Gironcoli, S.; Fabris, S.; Fratesi, G.; Gebauer, R.; Gerstmann, U.; Gougoussis, C.; Kokalj, A.; Lazzeri, M.; Martin-Samos, L.; Marzari, N.; Mauri, F.; Mazzarello, R.; Paolini, S.; Pasquarello, A.; Paulatto, L.; Sbraccia, C.; Scandolo, S.; Sclauzero, G.; Seitsonen, A. P.; Smogunov, A.; Umari, P.; Wentzcovitch, R. M. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 2009, 21, 395502 DOI: 10.1088/0953-8984/21/39/395502Google ScholarThere is no corresponding record for this reference.
- 23Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M. B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; Colonna, N.; Carnimeo, I.; Corso, A. D.; de Gironcoli, S.; Delugas, P.Jr.R.A.D.; Ferretti, A.; Floris, A.; Fratesi, G.; Fugallo, G.; Gebauer, R.; Gerstmann, U.; Giustino, F.; Gorni, T.; Jia, J.; Kawamura, M.; Ko, H.-Y.; Kokalj, A.; Küçükbenli, E.; Lazzeri, M.; Marsili, M.; Marzari, N.; Mauri, F.; Nguyen, N. L.; Nguyen, H.-V.; de-la Roza, A. O.; Paulatto, L.; Poncé, S.; Rocca, D.; Sabatini, R.; Santra, B.; Schlipf, M.; Seitsonen, A. P.; Smogunov, A.; Timrov, I.; Thonhauser, T.; Umari, P.; Vast, N.; Wu, X.; Baroni, S. Advanced capabilities for materials modelling with QUANTUM ESPRESSO. J. Phys.: Condens. Matter 2017, 29, 465901, DOI: 10.1088/1361-648X/aa8f79Google ScholarThere is no corresponding record for this reference.
- 24Smith, D. G. A.; Burns, L. A.; Simmonett, A. C.; Parrish, R. M.; Schieber, M. C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; James, A. M.; Lehtola, S.; Misiewicz, J. P.; Scheurer, M.; Shaw, R. A.; Schriber, J. B.; Xie, Y.; Glick, Z. L.; Sirianni, D. A.; O’Brien, J. S.; Waldrop, J. M.; Kumar, A.; Hohenstein, E. G.; Pritchard, B. P.; Brooks, B. R.; Schaefer, I.; Henry, F.; Sokolov, A. Y.; Patkowski, K.; DePrince, I.; Eugene, A.; Bozkaya, U.; King, R. A.; Evangelista, F. A.; Turney, J. M.; Crawford, T. D.; Sherrill, C. D. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108, DOI: 10.1063/5.0006002Google ScholarThere is no corresponding record for this reference.
- 25Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73– 78, DOI: 10.1002/wcms.81Google ScholarThere is no corresponding record for this reference.
- 26Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108, DOI: 10.1063/5.0004608Google ScholarThere is no corresponding record for this reference.
- 27Neese, F. Software Update: The ORCA Program System─Version 6.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2025, 15, e70019 DOI: 10.1002/wcms.70019Google ScholarThere is no corresponding record for this reference.
- 28Hagg, A.; Kirschner, K. N. Open-Source Machine Learning in Computational Chemistry. J. Chem. Inf. Model. 2023, 63, 4505– 4532, DOI: 10.1021/acs.jcim.3c00643Google ScholarThere is no corresponding record for this reference.
- 29Wilkinson, M. D.; Dumontier, M.; Aalbersberg, I. J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L. B.; Bourne, P. E.; Bouwman, J.; Brookes, A. J.; Clark, T.; Crosas, M.; Dillo, I.; Dumon, O.; Edmunds, S.; Evelo, C. T.; Finkers, R.; González-Beltrán, A.; Gray, A. J. G.; Groth, P.; Goble, C.; Grethe, J. S.; Heringa, J.; t’Hoen, P. A. C.; Hooft, R.; Kuhn, T.; Kok, R.; Kok, J.; Lusher, S. J.; Martone, M. E.; Mons, A.; Packer, A. L.; Persson, B.; Rocca-Serra, P.; Roos, M.; van Schaik, R.; Sansone, S.-A.; Schultes, E.; Sengstag, T.; Slater, T.; Strawn, G.; Swertz, M. A.; Thompson, M.; van der Lei, J.; van Mulligen, E.; Velterop, J.; Waagmeester, A.; Wittenburg, P.; Wolstencroft, K.; Zhao, J.; Mons, B. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018, DOI: 10.1038/sdata.2016.18Google ScholarThere is no corresponding record for this reference.
- 30Smith, J. S.; Isayev, O.; Roitberg, A. E. ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 2017, 8, 3192– 3203, DOI: 10.1039/C6SC05720AGoogle ScholarThere is no corresponding record for this reference.
- 31Smith, J. S.; Isayev, O.; Roitberg, A. E. ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules. Sci. Data 2017, 4, 170193, DOI: 10.1038/sdata.2017.193Google ScholarThere is no corresponding record for this reference.
- 32Smith, J. S.; Nebgen, B.; Lubbers, N.; Isayev, O.; Roitberg, A. E. Less is more: sampling chemical space with active learning. J. Chem. Phys. 2018, 148, 241733, DOI: 10.1063/1.5023802Google ScholarThere is no corresponding record for this reference.
- 33Smith, J. S.; Nebgen, B. T.; Zubatyuk, R.; Lubbers, N.; Devereux, C.; Barros, K.; Tretiak, S.; Isayev, O.; Roitberg, A. E. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 2019, 10, 2903, DOI: 10.1038/s41467-019-10827-4Google ScholarThere is no corresponding record for this reference.
- 34Smith, J. S.; Zubatyuk, R.; Nebgen, B.; Lubbers, N.; Barros, K.; Roitberg, A. E.; Isayev, O.; Tretiak, S. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci. Data 2020, 7, 134, DOI: 10.1038/s41597-020-0473-zGoogle ScholarThere is no corresponding record for this reference.
- 35Anstine, D. M.; Zubatyuk, R.; Isayev, O. AIMNet2: a neural network potential to meet your neutral, charged, organic, and elemental-organic needs. Chem. Sci. 2025, 16, 10228– 10244, DOI: 10.1039/D4SC08572HGoogle ScholarThere is no corresponding record for this reference.
- 36Wood, B. M.; Dzamba, M.; Fu, X.; Gao, M.; Shuaibi, M.; Barroso-Luque, L.; Abdelmaqsoud, K.; Gharakhanyan, V.; Kitchin, J. R.; Levine, D. S.; Michel, K.; Sriram, A.; Cohen, T.; Das, A.; Rizvi, A.; Sahoo, S. J.; Ulissi, Z. W.; Zitnick, C. L. UMA: A Family of Universal Models for Atoms . 2025; https://arxiv.org/abs/2506.23971v1.Google ScholarThere is no corresponding record for this reference.
- 37Levine, D. S.; Shuaibi, M.; Spotte-Smith, E. W. C.; Taylor, M. G.; Hasyim, M. R.; Michel, K.; Batatia, I.; Csányi, G.; Dzamba, M.; Eastman, P.; Frey, N. C.; Fu, X.; Gharakhanyan, V.; Krishnapriyan, A. S.; Rackers, J. A.; Raja, S.; Rizvi, A.; Rosen, A. S.; Ulissi, Z.; Vargas, S.; Zitnick, C. L.; Blau, S. M.; Wood, B. M. The Open Molecules 2025 (OMol25) Dataset, Evaluations, and Models. arXiv 2025. DOI: 10.48550/arXiv.2505.08762Google ScholarThere is no corresponding record for this reference.
- 38Luise, G.; Huang, C.-W.; Vogels, T.; Kooi, D. P.; Ehlert, S.; Lanius, S.; Giesbertz, K. J. H.; Karton, A.; Gunceler, D.; Stanley, M.; Bruinsma, W. P.; Huang, L.; Wei, X.; Garrido Torres, J.; Katbashev, A.; Chavez Zavaleta, R.; Máté, B.; Kaba, S.-O.; Sordillo, R.; Chen, Y.; Williams-Young, D. B.; Bishop, C. M.; Hermann, J.; van den Berg, R.; Gori-Giorgi, P. Accurate and Scalable Exchange-Correlation with Deep Learning. arXiv 2025. DOI: 10.48550/arXiv.2506.14665Google ScholarThere is no corresponding record for this reference.
- 39Ehlert, S.; Hermann, J.; Vogels, T.; Satorras, V. G.; Lanius, S.; Segler, M.; Kooi, D. P.; Takeda, K.; Huang, C.-W.; Luise, G.; van den Berg, R.; Gori-Giorgi, P.; Karton, A. Accurate Chemistry Collection: Coupled Cluster Atomization Energies for Broad Chemical Space. arXiv 2025. DOI: 10.48550/arXiv.2506.14492Google ScholarThere is no corresponding record for this reference.
- 40Hjorth Larsen, A.; Jo̷rgen Mortensen, J.; Blomqvist, J.; Castelli, I. E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M. N.; Hammer, B.; Hargus, C.; Hermes, E. D.; Jennings, P. C.; Bjerre Jensen, P.; Kermode, J.; Kitchin, J. R.; Leonhard Kolsbjerg, E.; Kubal, J.; Kaasbjerg, K.; Lysgaard, S.; Bergmann Maronsson, J.; Maxson, T.; Olsen, T.; Pastewka, L.; Peterson, A.; Rostgaard, C.; Schio̷tz, J.; Schütt, O.; Strange, M.; Thygesen, K. S.; Vegge, T.; Vilhelmsen, L.; Walter, M.; Zeng, Z.; Jacobsen, K. W. The atomic simulation environment─a Python library for working with atoms. J. Phys.: Condens. Matter 2017, 29, 273002, DOI: 10.1088/1361-648X/aa680eGoogle ScholarThere is no corresponding record for this reference.
- 41FACCTs GmbH, Germany, www.faccts.de, WEASEL 1.12.3 DOI: 10.5281/zenodo.15260476 .Google ScholarThere is no corresponding record for this reference.
- 42Grimme, S.; Bohle, F.; Hansen, A.; Pracht, P.; Spicher, S.; Stahn, M. Efficient Quantum Chemical Calculation of Structure Ensembles and Free Energies for Nonrigid Molecules. J. Phys. Chem. A 2021, 125, 4039– 4054, DOI: 10.1021/acs.jpca.1c00971Google ScholarThere is no corresponding record for this reference.
- 43Weymuth, T.; Unsleber, J. P.; Türtscher, P. L.; Steiner, M.; Sobez, J.-G.; Müller, C. H.; Mörchen, M.; Klasovita, V.; Grimmel, S. A.; Eckhoff, M.; Csizi, K.-S.; Bosia, F.; Bensberg, M.; Reiher, M. SCINE─Software for chemical interaction networks. J. Chem. Phys. 2024, 160, 222501, DOI: 10.1063/5.0206974Google ScholarThere is no corresponding record for this reference.
- 44Chen, Y.; Bannwarth, C. An Automated Intermolecular Reaction Discovery Approach Relying on Heuristic Atom-Partitioned Frontier Orbital Features. J. Chem. Inf. Model. 2025, 65, 9125– 9141, DOI: 10.1021/acs.jcim.5c00908Google ScholarThere is no corresponding record for this reference.
- 45Altun, A.; Neese, F.; Bistoni, G. LEDAW: An Integrated Software Suite with GUI for Automating Local Energy Decomposition Analysis of Molecular Interactions. J. Chem. Inf. Model. 2025, 65, 8917– 8923, DOI: 10.1021/acs.jcim.5c01561Google ScholarThere is no corresponding record for this reference.
- 46Python Software Foundation, Python (Version 3) 2025. https://www.python.org, Accessed: 12th November 2025.Google ScholarThere is no corresponding record for this reference.
- 47Perez, F.; Granger, B. E.; Hunter, J. D. Python: An Ecosystem for Scientific Computing. Comput. Sci. Eng. 2011, 13, 13– 21, DOI: 10.1109/MCSE.2010.119Google ScholarThere is no corresponding record for this reference.
- 48Sarkar, D.; Bali, R.; Sharma, T. Practical Machine Learning with Python: A Problem-Solver’s Guide to Building Real-World Intelligent Systems; Apress: Berkeley, CA, 2018; pp 67– 118 DOI: 10.1007/978-1-4842-3207-1_2 .Google ScholarThere is no corresponding record for this reference.
- 49Bommarito, E.; Bommarito, M. An Empirical Analysis of the Python Package Index (PyPI) , 2019. DOI: 10.48550/arXiv.1907.11073Google ScholarThere is no corresponding record for this reference.
- 50Berquist, E.; Dumi, A.; Upadhyay, S.; Abarbanel, O. D.; Cho, M.; Gaur, S.; Gil, R.; Hutchison, G. R.; Lee, O. S.; Rosen, A. S.; Schamnad, S.; Schneider, F. S. S.; Steinmann, C.; Stolyarchuk, M.; Vandezande, J. E.; Zak, W.; Langner, K. M. cclib 2.0: An updated architecture for interoperable computational chemistry. J. Chem. Phys. 2024, 161, 042501 DOI: 10.1063/5.0216778Google ScholarThere is no corresponding record for this reference.
- 51Ragnar Bjornsson, ASH ORCA interface, 2025. https://ash.readthedocs.io/en/latest/ORCA-interface.html, Accessed: 12th November 2025.Google ScholarThere is no corresponding record for this reference.
- 52PLAMS, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com, https://github.com/SCM-NV/PLAMS.Google ScholarThere is no corresponding record for this reference.
- 53FACCTs GmbH. ORCA Python Interface (OPI). https://github.com/faccts/opi, DOI of v1.0: DOI: 10.5281/zenodo.15688425 .Google ScholarThere is no corresponding record for this reference.
- 54FACCTs GmbH, ORCA Python Interface (OPI) Documentation. https://www.faccts.de/docs/opi/docs, Accessed: 17th November 2025.Google ScholarThere is no corresponding record for this reference.
- 55Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; Ivanov, P.; Avila, D.; Abdalla, S.; Willing, C.; Team, J. D. Jupyter Notebooks─a publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas, 2016; pp 87– 90.Google ScholarThere is no corresponding record for this reference.
- 56Jupyter, P.; Bussonnier, M.; Forde, J.; Freeman, J.; Granger, B.; Head, T.; Holdgraf, C.; Kelley, K.; Nalvarte, G.; Osheroff, A.; Pacer, M.; Panda, Y.; Perez, F.; Ragan-Kelley, B.; Willing, C.; Binder 2.0─Reproducible, Interactive, Sharable Environments for Science at Scale. In Proceedings of the 17th Python in Science Conference, 2018; pp 113– 120.Google ScholarThere is no corresponding record for this reference.
- 57FACCTs GmbH (OPI project), OPI Documentation/Tutorials (v2.0), 2025; https://www.faccts.de/docs/opi/2.0/docs/, Accessed: 2nd October 2025.Google ScholarThere is no corresponding record for this reference.
- 58Burns, J.; Zalte, A.; Green, W. Descriptor-based Foundation Models for Molecular Property Prediction , 2025; https://arxiv.org/abs/2506.15792.Google ScholarThere is no corresponding record for this reference.
- 59De Landsheere, J.; Zamyatin, A.; Karwounopoulos, J.; Heid, E. ChemTorch: A Deep Learning Framework for Benchmarking and Developing Chemical Reaction Property Prediction Models. ChemRxiv 2025, Preprint, not peer-reviewed.Google ScholarThere is no corresponding record for this reference.
- 60FACCTs GmbH, OPI Documentation/Tutorials (v2.0), How To Contribute. https://www.faccts.de/docs/opi/2.0/docs/contents/how_to_contribute.html, Accessed: 17th November 2025.Google ScholarThere is no corresponding record for this reference.
- 61FACCTs GmbH, ORCA Manual. https://www.faccts.de/docs/orca/6.1/manual/, Accessed: 17th November 2025.Google ScholarThere is no corresponding record for this reference.
- 62FACCTs GmbH, ORCA Tutorials. https://www.faccts.de/docs/orca/6.1/tutorials/, Accessed: 17th November 2025.Google ScholarThere is no corresponding record for this reference.
- 63Harris, C. R.; Millman, K. J.; van der Walt, S. J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N. J.; Kern, R.; Picus, M.; Hoyer, S.; van Kerkwijk, M. H.; Brett, M.; Haldane, A.; del Río, J. F.; Wiebe, M.; Peterson, P.; Gérard-Marchant, P.; Sheppard, K.; Reddy, T.; Weckesser, W.; Abbasi, H.; Gohlke, C.; Oliphant, T. E. Array programming with NumPy. Nature 2020, 585, 357– 362, DOI: 10.1038/s41586-020-2649-2Google ScholarThere is no corresponding record for this reference.
- 64Gábor, B.; Berman, J.; Lev, O.; Pfannschmidt, R. platformdirs. https://github.com/tox-dev/platformdirs, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 65Colvin, S.; Jolibois, E.; Ramezani, H.; Garcia Badaracco, A.; Dorsey, T.; Montague, D.; Matveenko, S.; Trylesinski, M.; Runkle, S.; Hewitt, D.; Hall, A.; Plot, V. Pydantic Validation. https://github.com/pydantic/pydantic, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 66Landrum, G. RDKit: Open-Source Cheminformatics Software , 2025. https://www.rdkit.org/, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 67Preston-Werner, T. Semantic Versioning 2.0.0 , 2025; https://semver.org/, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 68Barrois, R. python-semanticversion. https://github.com/rbarrois/python-semanticversion, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 69Astral, uv. https://github.com/astral-sh/uv, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 70Lehtosalo, J. mypy. https://www.mypy-lang.org/, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 71Flowers, A.; Nox, K. https://github.com/wntrblm/nox, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 72Astral, ruff. https://github.com/astral-sh/ruff, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 73Marchi, L. D. codespell , 2025 https://github.com/codespell-project/codespell, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 74Seipp, J. Vulture─Find Dead Code , 2025. https://github.com/jendrikseipp/vulture, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 75Krekel, H.; Oliveira, B.; Pfannschmidt, R.; Bruynooghe, F.; Laugher, B.; Bruhin, F. pytest 8.4 . 2004; https://github.com/pytest-dev/pytest, Contributors: Holger Krekel and Bruno Oliveira and Ronny Pfannschmidt and Floris Bruynooghe and Brianna Laugher and Florian Bruhin and others; Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 76MacIver, D. R. Hypothesis 6.133 . 2016; https://github.com/HypothesisWorks/hypothesis-python, Accessed: 15th October 2025.Google ScholarThere is no corresponding record for this reference.
- 77Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995– 2001, DOI: 10.1021/jp9716997Google ScholarThere is no corresponding record for this reference.
- 78Garcia-Ratés, M.; Neese, F. Effect of the Solute Cavity on the Solvation Energy and its Derivatives within the Framework of the Gaussian Charge Scheme. J. Comput. Chem. 2020, 41, 922– 939, DOI: 10.1002/jcc.26139Google ScholarThere is no corresponding record for this reference.
- 79Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378– 6396, DOI: 10.1021/jp810292nGoogle ScholarThere is no corresponding record for this reference.
- 80Gerlach, T.; Müller, S.; de Castilla, A. G.; Smirnova, I. An open source COSMO-RS implementation and parameterization supporting the efficient implementation of multiple segment descriptors. Fluid Phase Equilib. 2022, 560, 113472 DOI: 10.1016/j.fluid.2022.113472Google ScholarThere is no corresponding record for this reference.
- 81Ásgeirsson, V.; Birgisson, B. O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector Following. J. Chem. Theory Comput. 2021, 17, 4929– 4945, DOI: 10.1021/acs.jctc.1c00462Google ScholarThere is no corresponding record for this reference.
- 82de Souza, B. GOAT: A Global Optimization Algorithm for Molecules and Atomic Clusters. Angew. Chem., Int. Ed. 2025, 64, e202500393 DOI: 10.1002/anie.202500393Google ScholarThere is no corresponding record for this reference.
- 83Bistoni, G. Finding chemical concepts in the Hilbert space: Coupled cluster analyses of noncovalent interactions. WIREs Computational Molecular Science 2020, 10, e1442 DOI: 10.1002/wcms.1442Google ScholarThere is no corresponding record for this reference.
- 84FACCTs GmbH, orca-external-tools. https://github.com/faccts/orca-external-tools, Accessed: 11th October 2025.Google ScholarThere is no corresponding record for this reference.
- 85Python Software Foundation, Python 3.11.14 Documentation. https://docs.python.org/3.11/reference/lexical_analysis.html#names-identifiers-and-keywords, Accessed: 2nd February 2026.Google ScholarThere is no corresponding record for this reference.
- 86Software in the Public Interest (SPI), Open MPI: Open Source High Performance Computing. https://www.open-mpi.org/, Accessed: 2nd October 2025.Google ScholarThere is no corresponding record for this reference.
- 87Preston-Werner, T. TOML . 2021; https://toml.io/en/, Accessed: 2nd October 2025.Google ScholarThere is no corresponding record for this reference.
- 88FACCTs GmbH , 2025 https://github.com/faccts/opi/blob/release/2.0/src/opi/output/grepper/recipes.py, Accessed: 17th November 2025.Google ScholarThere is no corresponding record for this reference.
- 89Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297, DOI: 10.1039/b508541aGoogle ScholarThere is no corresponding record for this reference.
- 90Grimme, S.; Hansen, A.; Ehlert, S.; Mewes, J.-M. r2SCAN-3c: A “Swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021, 154, 064103 DOI: 10.1063/5.0040021Google ScholarThere is no corresponding record for this reference.
- 91Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106, DOI: 10.1063/1.4773581Google ScholarThere is no corresponding record for this reference.
- 92Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101, DOI: 10.1063/1.4821834Google ScholarThere is no corresponding record for this reference.
- 93Tao, Y.; Yuan, K.; Chen, T.; Xu, P.; Li, H.; Chen, R.; Zheng, C.; Zhang, L.; Huang, W. Thermally Activated Delayed Fluorescence Materials Towards the Breakthrough of Organoelectronics. Adv. Mater. 2014, 26, 7931– 7958, DOI: 10.1002/adma.201402532Google ScholarThere is no corresponding record for this reference.
- 94Kunze, L.; Hansen, A.; Grimme, S.; Mewes, J.-M. The Best of Both Worlds: ΔDFT Describes Multiresonance TADF Emitters with Wave-Function Accuracy at Density-Functional Cost. J. Phys. Chem. Lett. 2025, 16, 1114– 1125, DOI: 10.1021/acs.jpclett.4c03192Google ScholarThere is no corresponding record for this reference.
- 95Shee, J.; Head-Gordon, M. Predicting Excitation Energies of Twisted Intramolecular Charge-Transfer States with the Time-Dependent Density Functional Theory: Comparison with Experimental Measurements in the Gas Phase and Solvents Ranging from Hexanes to Acetonitrile. J. Chem. Theory Comput. 2020, 16, 6244– 6255, DOI: 10.1021/acs.jctc.0c00635Google ScholarThere is no corresponding record for this reference.
- 96Mardirossian, N.; Head-Gordon, M. ωB97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J. Chem. Phys. 2016, 144, 214110, DOI: 10.1063/1.4952647Google ScholarThere is no corresponding record for this reference.
- 97Virtanen, P.; Gommers, R.; Oliphant, T. E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; van der Walt, S. J.; Brett, M.; Wilson, J.; Millman, K. J.; Mayorov, N.; Nelson, A. R. J.; Jones, E.; Kern, R.; Larson, E.; Carey, C. J.; Polat, İ.; Feng, Y.; Moore, E. W.; VanderPlas, J.; Laxalde, D.; Perktold, J.; Cimrman, R.; Henriksen, I.; Quintero, E. A.; Harris, C. R.; Archibald, A. M.; Ribeiro, A. H.; Pedregosa, F.; van Mulbregt, P. SciPy 1.0 Contributors, SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261– 272, DOI: 10.1038/s41592-020-0772-5Google ScholarThere is no corresponding record for this reference.
- 98Ikeda, N.; Oda, S.; Matsumoto, R.; Yoshioka, M.; Fukushima, D.; Yoshiura, K.; Yasuda, N.; Hatakeyama, T. Solution-Processable Pure Green Thermally Activated Delayed Fluorescence Emitter Based on the Multiple Resonance Effect. Adv. Mater. 2020, 32, 2004072 DOI: 10.1002/adma.202004072Google ScholarThere is no corresponding record for this reference.
- 99Grimme, S.; Hansen, A. A Practicable Real-Space Measure and Visualization of Static Electron-Correlation Effects. Angew. Chem., Int. Ed. 2015, 54, 12308– 12313, DOI: 10.1002/anie.201501887Google ScholarThere is no corresponding record for this reference.
- 100Faulstich, F. M.; Kristiansen, H. E.; Csirik, M. A.; Kvaal, S.; Pedersen, T. B.; Laestadius, A. S-Diagnostic-An a Posteriori Error Assessment for Single-Reference Coupled-Cluster Methods. J. Phys. Chem. A 2023, 127, 9106– 9120, DOI: 10.1021/acs.jpca.3c01575Google ScholarThere is no corresponding record for this reference.
- 101Duan, C.; Chu, D. B. K.; Nandy, A.; Kulik, H. J. Detection of multi-reference character imbalances enables a transfer learning approach for virtual high throughput screening with coupled cluster accuracy at DFT cost. Chem. Sci. 2022, 13, 4962– 4971, DOI: 10.1039/D2SC00393GGoogle ScholarThere is no corresponding record for this reference.
- 102Gasevic, T.; Müller, M.; Schöps, J.; Lanius, S.; Hermann, J.; Grimme, S.; Hansen, A. Chemical Space Exploration with Artificial “Mindless” Molecules. J. Chem. Inf. Model. 2025, 65, 9576– 9587, DOI: 10.1021/acs.jcim.5c01364Google ScholarThere is no corresponding record for this reference.
- 103Collette, A. , e. h5py─HDF5 for Python , 2025 https://www.h5py.org/, Accessed: 17th November 2025.Google ScholarThere is no corresponding record for this reference.
- 104The HDF Group, HDF5. https://www.hdfgroup.org/solutions/hdf5/, Accessed: 17th November 2025.Google ScholarThere is no corresponding record for this reference.
- 105Rui, Y.; Chen, Y.; Ivanova, E.; Kumar, V. B.; Śmiga, S.; Grabowski, I.; Dral, P. O. The Best DFT Functional Is the Ensemble of Functionals. Adv. Sci. 2024, 11, 2408239 DOI: 10.1002/advs.202408239Google ScholarThere is no corresponding record for this reference.
- 106Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184– 32215, DOI: 10.1039/C7CP04913GGoogle ScholarThere is no corresponding record for this reference.
- 107Zhao, Y.; Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215– 241, DOI: 10.1007/s00214-007-0310-xGoogle ScholarThere is no corresponding record for this reference.
- 108Bursch, M.; Neugebauer, H.; Ehlert, S.; Grimme, S. Dispersion corrected r2SCAN based global hybrid functionals: r2SCANh, r2SCAN0, and r2SCAN50. J. Chem. Phys. 2022, 156, 134105, DOI: 10.1063/5.0086040Google ScholarThere is no corresponding record for this reference.
- 109Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105, DOI: 10.1063/1.3484283Google ScholarThere is no corresponding record for this reference.
- 110Rappoport, D. Property-optimized Gaussian basis sets for lanthanides. J. Chem. Phys. 2021, 155, 124102, DOI: 10.1063/5.0065611Google ScholarThere is no corresponding record for this reference.
- 111Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; Vanderplas, J.; Passos, A.; Cournapeau, D.; Brucher, M.; Perrot, M.; Duchesnay, E. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825– 2830Google ScholarThere is no corresponding record for this reference.
- 112Lehtola, S.; Karttunen, A. J. Free and open source software for computational chemistry education. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2022, 12, e1610 DOI: 10.1002/wcms.1610Google ScholarThere is no corresponding record for this reference.
- 113Capel, A. J.; Rimington, R. P.; Lewis, M. P.; Christie, S. D. R. 3D printing for chemical, pharmaceutical and biological applications. Nat. Rev. Chem. 2018, 2, 422– 436, DOI: 10.1038/s41570-018-0058-yGoogle ScholarThere is no corresponding record for this reference.
- 114Pinger, C. W.; Geiger, M. K.; Spence, D. M. Applications of 3D-Printing for Improving Chemistry Education. J. Chem. Educ. 2020, 97, 112– 117, DOI: 10.1021/acs.jchemed.9b00588Google ScholarThere is no corresponding record for this reference.
- 115Garcia, M.; O’Leary, F. A.; O’Leary, D. J. Do-It-Yourself 5-Color 3D Printing of Molecular Orbitals and Electron Density Surfaces. J. Chem. Educ. 2023, 100, 1648– 1658, DOI: 10.1021/acs.jchemed.2c00907Google ScholarThere is no corresponding record for this reference.
- 116Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB─An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652– 1671, DOI: 10.1021/acs.jctc.8b01176Google ScholarThere is no corresponding record for this reference.
- 117Rego, N.; Koes, D. 3Dmol.js: molecular visualization with WebGL. Bioinform. 2015, 31, 1322– 1324, DOI: 10.1093/bioinformatics/btu829Google ScholarThere is no corresponding record for this reference.
- 118Pipek, J.; Mezey, P. G. A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave functions. J. Chem. Phys. 1989, 90, 4916– 4926, DOI: 10.1063/1.456588Google ScholarThere is no corresponding record for this reference.
- 119Adamo, C.; Barone, V. Toward reliable adiabatic connection models free from adjustable parameters. Chem. Phys. Lett. 1997, 274, 242– 250, DOI: 10.1016/S0009-2614(97)00651-9Google ScholarThere is no corresponding record for this reference.
- 120Zou, Y.; Cheng, A. H.; Aldossary, A.; Bai, J.; Leong, S. X.; Campos-Gonzalez-Angulo, J. A.; Choi, C.; Ser, C. T.; Tom, G.; Wang, A.; Zhang, Z.; Yakavets, I.; Hao, H.; Crebolder, C.; Bernales, V.; Aspuru-Guzik, A. El Agente: An autonomous agent for quantum chemistry. Matter 2025, 8, 102263 DOI: 10.1016/j.matt.2025.102263Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract

Figure 1
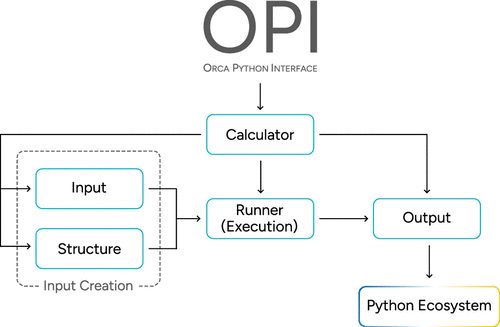
Figure 1. Schematic outline of OPI’s general structure and its five major classes: The Calculator class combines most of the functionality. The other major classes facilitate input creation (Structure and Input), job execution (Runner), and data extraction (Output). OPI benefits from Python’s ecosystem to visualize, analyze, or postprocess the data further and may be integrated into it at any point.
Figure 2

Figure 2. OPI example that shows the initialization of the Calculator class.
Figure 3

Figure 3. Example of a simple ORCA input containing all major input elements: Simple input keywords (blue), key-value options (orange), block options (purple), and the coordinates block (black).
Figure 4

Figure 4. OPI example of how to add simple keywords to the Calculator.
Figure 5
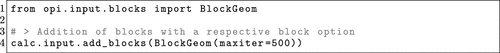
Figure 5. OPI example of how to add the maxiter options from ORCA’s %geom block to a job definition in OPI.
Figure 6

Figure 6. An example showing how to specify key-value options in OPI.
Figure 7
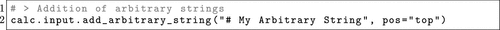
Figure 7. A code example showcasing how to add not defined or arbitrary input to the ORCA input in OPI.
Figure 8

Figure 8. Excerpt of a minimal ORCA input file showing the three principal positions for input keywords defined in OPI: top: At the very top of the file; before_coords: Right before the coordinates block; bottom: At the very end of the file.
Figure 9
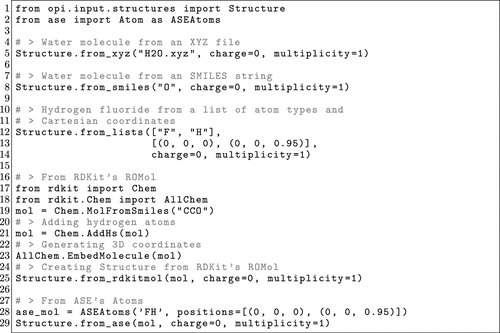
Figure 9. OPI example showcasing the different methods to create a Structure object from different sources.
Figure 10

Figure 10. Example of an OPI configuration file specifying the location of ORCA and Open MPI. The file adheres to the TOML format.
Figure 11

Figure 11. OPI example showing how to create the ORCA input and execute the job after configuring the compute resources.
Figure 12
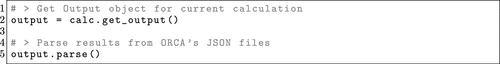
Figure 12. OPI example showing the creation of an Output from an existing Calculator object and parsing of the respective JSON files.
Figure 13
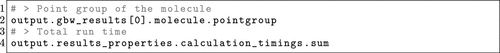
Figure 13. OPI example of how to access GBW and property file data from the Output object after parsing the files.
Figure 14
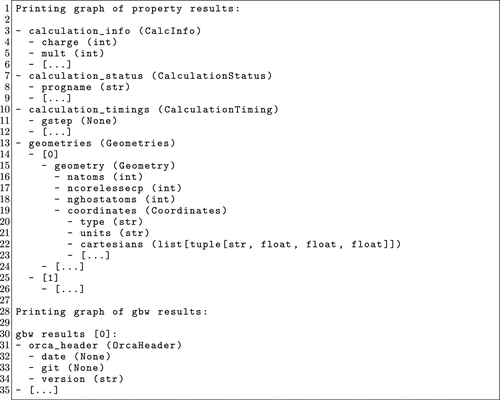
Figure 14. Shortened output example of the output.print_graph() method. Omitted parts are designated with [...].
Figure 15
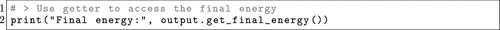
Figure 15. OPI has a series of getter methods to swiftly fetch properties from completed ORCA calculations. This example show the get_final_energy() method, which returns the final energy of the final structure.
Figure 16
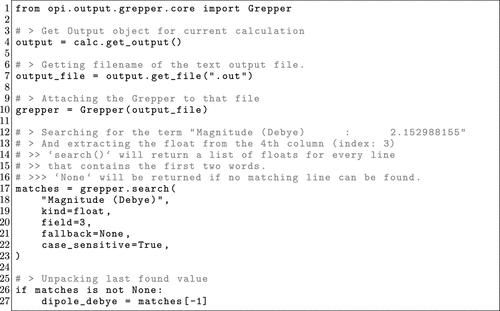
Figure 16. Minimal OPI example of the Grepper class, showcasing how to use the class to search for the line containing “Magnitude (Debye): 2.152988155” in the ORCA output of a completed calculation and extracting the value.
Figure 17

Figure 17. OPI example of predefined health-check routines.
Figure 18

Figure 18. OPI example script of a simple energy calculation for a water molecule. It displays all steps involved: input definition, input creation, job execution, and printing of the final energy.
Figure 19
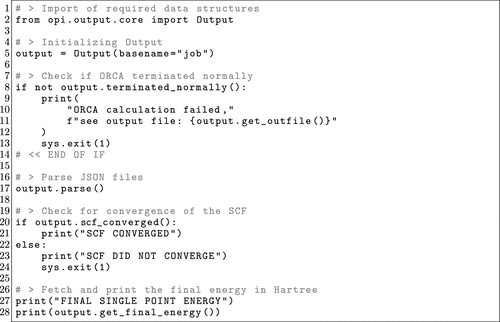
Figure 19. OPI example depicting how to only postprocess the results from a single-point calculation.
Figure 20

Figure 20. OPI example for performing a r2SCAN-3c geometry optimization followed by a frequency calculation to obtain a free energy.
Figure 21

Figure 21. OPI example of how to combine a high-level DLPNO–CCSD(T) single-point energy with thermostatistical corrections from DFT to obtain a free energy.
Figure 22
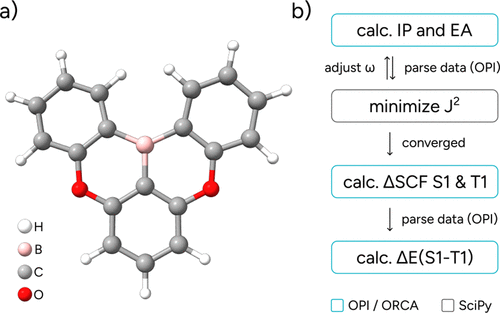
Figure 22. (a) Molecular structure of the DOBNA molecule; (b) Schematic OPI workflow.
Figure 23
Figure 23. Single-point calculator setup from the OPI script to perform the optimal tuning workflow described in Section 5.2.
Figure 24

Figure 24. Function body for evaluation of J2(ω) from the OPI script to perform the optimal tuning workflow described in Section 5.2.
Figure 25
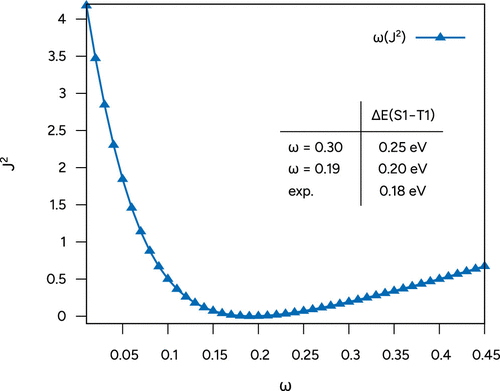
Figure 25. Plot of ω as a function of J2 and S1-T1 gaps computed with the default and optimally tuned ω in comparison to the experimental reference (cf. ref (98)).
Figure 26
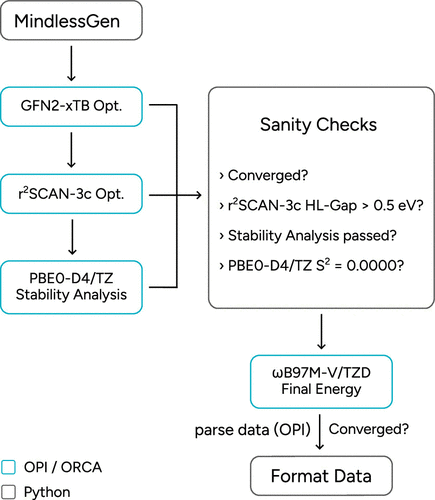
Figure 26. Schematic OPI workflow to generate new, standardized Mindless data based using MindlessGen with OPI.
Figure 27
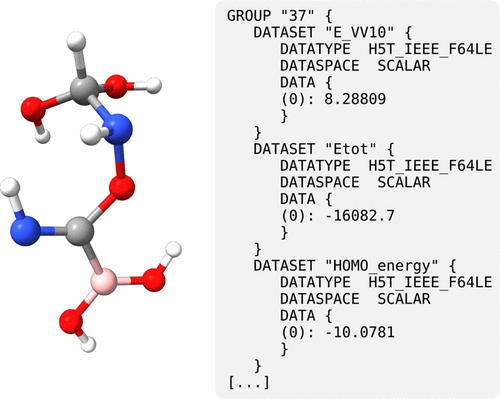
Figure 27. Example molecule generated with MindlessGen and a human-readable excerpt of the output generated with OPI and Python.
Figure 28
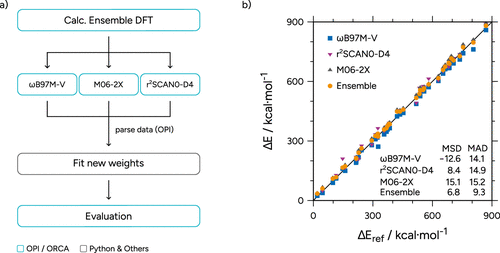
Figure 28. (a) Flowchart of the process of determining the ensemble weights. (b) Correlation plot of the tested functionals on the MB16-43 as well as the ensemble optimized on the set. MSDs and MADs are given in kcal·mol–1. One data point below 0 kcal·mol–1 and two above 900 kcal·mol–1 are omitted for clarity.
Figure 29

Figure 29. Function calls for cube file generation and visualization within a Jupyter notebook.
Figure 30
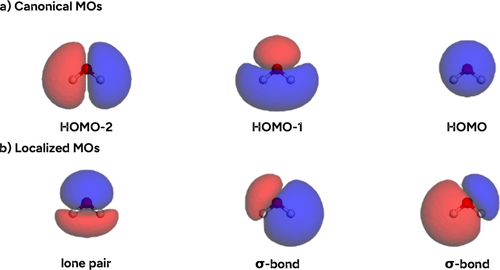
Figure 30. (a) Canonical frontier orbitals and (b) Pipek–Mezey localized orbitals of water (GFN2-xTB level) as plotted in the example notebook.
Figure 31
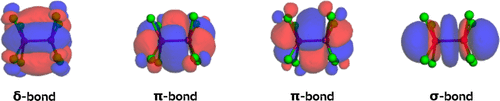
Figure 31. Bonding LMOs (PBE0/def2-SVP, Pipek-Mezey orbital localization) for [Re2Cl8]2– as plotted in the example notebook. The δ-, two π-, and the σ-bonds can be identified in line with chemical intuition.
References
This article references 120 other publications.
- 1Houk, K. N.; Liu, F. Holy Grails for Computational Organic Chemistry and Biochemistry. Acc. Chem. Res. 2017, 50, 539– 543, DOI: 10.1021/acs.accounts.6b00532There is no corresponding record for this reference.
- 2Grimme, S.; Schreiner, P. R. Computational Chemistry: The Fate of Current Methods and Future Challenges. Angew. Chem., Int. Ed. 2018, 57, 4170– 4176, DOI: 10.1002/anie.201709943There is no corresponding record for this reference.
- 3Neese, F.; Atanasov, M.; Bistoni, G.; Maganas, D.; Ye, S. Chemistry and Quantum Mechanics in 2019: Give Us Insight and Numbers. J. Am. Chem. Soc. 2019, 141, 2814– 2824, DOI: 10.1021/jacs.8b13313There is no corresponding record for this reference.
- 4Borges, R. M.; Colby, S. M.; Das, S.; Edison, A. S.; Fiehn, O.; Kind, T.; Lee, J.; Merrill, A. T.; Merz, K. M. J.; Metz, T. O.; Nunez, J. R.; Tantillo, D. J.; Wang, L.-P.; Wang, S.; Renslow, R. S. Quantum Chemistry Calculations for Metabolomics. Chem. Rev. 2021, 121, 5633– 5670, DOI: 10.1021/acs.chemrev.0c00901There is no corresponding record for this reference.
- 5Ozaki, Y.; Beć, K. B.; Morisawa, Y.; Yamamoto, S.; Tanabe, I.; Huck, C. W.; Hofer, T. S. Advances, challenges and perspectives of quantum chemical approaches in molecular spectroscopy of the condensed phase. Chem. Soc. Rev. 2021, 50, 10917– 10954, DOI: 10.1039/D0CS01602KThere is no corresponding record for this reference.
- 6Teale, A. M.; Helgaker, T.; Savin, A.; Adamo, C.; Aradi, B.; Arbuznikov, A. V.; Ayers, P. W.; Baerends, E. J.; Barone, V.; Calaminici, P.; Cancès, E.; Carter, E. A.; Chattaraj, P. K.; Chermette, H.; Ciofini, I.; Crawford, T. D.; De Proft, F.; Dobson, J. F.; Draxl, C.; Frauenheim, T.; Fromager, E.; Fuentealba, P.; Gagliardi, L.; Galli, G.; Gao, J.; Geerlings, P.; Gidopoulos, N.; Gill, P. M. W.; Gori-Giorgi, P.; Görling, A.; Gould, T.; Grimme, S.; Gritsenko, O.; Jensen, H. J. A.; Johnson, E. R.; Jones, R. O.; Kaupp, M.; Köster, A. M.; Kronik, L.; Krylov, A. I.; Kvaal, S.; Laestadius, A.; Levy, M.; Lewin, M.; Liu, S.; Loos, P.-F.; Maitra, N. T.; Neese, F.; Perdew, J. P.; Pernal, K.; Pernot, P.; Piecuch, P.; Rebolini, E.; Reining, L.; Romaniello, P.; Ruzsinszky, A.; Salahub, D. R.; Scheffler, M.; Schwerdtfeger, P.; Staroverov, V. N.; Sun, J.; Tellgren, E.; Tozer, D. J.; Trickey, S. B.; Ullrich, C. A.; Vela, A.; Vignale, G.; Wesolowski, T. A.; Xu, X.; Yang, W. DFT exchange: sharing perspectives on the workhorse of quantum chemistry and materials science. Phys. Chem. Chem. Phys. 2022, 24, 28700– 28781, DOI: 10.1039/D2CP02827AThere is no corresponding record for this reference.
- 7Seeman, J. I.; Tantillo, D. J. Understanding chemistry: from “heuristic (soft) explanations and reasoning by analogy” to “quantum chemistry”. Chem. Sci. 2022, 13, 11461– 11486, DOI: 10.1039/D2SC02535CThere is no corresponding record for this reference.
- 8Nam, K.; Shao, Y.; Major, D. T.; Wolf-Watz, M. Perspectives on Computational Enzyme Modeling: From Mechanisms to Design and Drug Development. ACS Omega 2024, 9, 7393– 7412, DOI: 10.1021/acsomega.3c09084There is no corresponding record for this reference.
- 9Pölloth, B. High School Students Experimenting with Computational Chemistry: Design-Based Research on and through the “Comp-Chem-Lab”. J. Chem. Educ. 2025, 102, 1367– 1379, DOI: 10.1021/acs.jchemed.4c01136There is no corresponding record for this reference.
- 10Autschbach, J. Orbitals: Some Fiction and Some Facts. J. Chem. Educ. 2012, 89, 1032– 1040, DOI: 10.1021/ed200673wThere is no corresponding record for this reference.
- 11Grushow, A.; Reeves, M. S., Eds. Using Computational Methods to Teach Chemical Principles; ACS Symposium Series; American Chemical Society: Washington, DC, 2019; Vol. 1312.There is no corresponding record for this reference.
- 12Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem., Int. Ed. 2022, 61, e202205735 DOI: 10.1002/anie.202205735There is no corresponding record for this reference.
- 13Dyson, F. J. Is Science Mostly Driven by Ideas or by Tools?. Science 2012, 338, 1426– 1427, DOI: 10.1126/science.1232773There is no corresponding record for this reference.
- 14Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A. T.; Wormit, M.; Kussmann, J.; Lange, A. W.; Behn, A.; Deng, J.; Feng, X.; Ghosh, D.; Goldey, M.; Horn, P. R.; Jacobson, L. D.; Kaliman, I.; Khaliullin, R. Z.; Kuś, T.; Landau, A.; Liu, J.; Proynov, E. I.; Rhee, Y. M.; Richard, R. M.; Rohrdanz, M. A.; Steele, R. P.; Sundstrom, E. J. III. H. L. W.; Zimmerman, P. M.; Zuev, D.; Albrecht, B.; Alguire, E.; Austin, B.; Beran, G. J. O.; Bernard, Y. A.; Berquist, E.; Brandhorst, K.; Bravaya, K. B.; Brown, S. T.; Casanova, D.; Chang, C.-M.; Chen, Y.; Chien, S. H.; Closser, K. D.; Crittenden, D. L.; Diedenhofen, M.Jr.R.A.D.; Do, H.; Dutoi, A. D.; Edgar, R. G.; Fatehi, S.; Fusti-Molnar, L.; Ghysels, A.; Golubeva-Zadorozhnaya, A.; Gomes, J.; Hanson-Heine, M. W.; Harbach, P. H.; Hauser, A. W.; Hohenstein, E. G.; Holden, Z. C.; Jagau, T.-C.; Ji, H.; Kaduk, B.; Khistyaev, K.; Kim, J.; Kim, J.; King, R. A.; Klunzinger, P.; Kosenkov, D.; Kowalczyk, T.; Krauter, C. M.; Lao, K. U.; Laurent, A. D.; Lawler, K. V.; Levchenko, S. V.; Lin, C. Y.; Liu, F.; Livshits, E.; Lochan, R. C.; Luenser, A.; Manohar, P.; Manzer, S. F.; Mao, S.-P.; Mardirossian, N.; Marenich, A. V.; Maurer, S. A.; Mayhall, N. J.; Neuscamman, E.; Oana, C. M.; Olivares-Amaya, R.; O’Neill, D. P.; Parkhill, J. A.; Perrine, T. M.; Peverati, R.; Prociuk, A.; Rehn, D. R.; Rosta, E.; Russ, N. J.; Sharada, S. M.; Sharma, S.; Small, D. W.; Sodt, A.; Stein, T.; Stück, D.; Su, Y.-C.; Thom, A. J.; Tsuchimochi, T.; Vanovschi, V.; Vogt, L.; Vydrov, O.; Wang, T.; Watson, M. A.; Wenzel, J.; White, A.; Williams, C. F.; Yang, J.; Yeganeh, S.; Yost, S. R.; You, Z.-Q.; Zhang, I. Y.; Zhang, X.; Zhao, Y.; Brooks, B. R.; Chan, G. K.; Chipman, D. M.; Cramer, C. J.III.W.A.G.; Gordon, M. S.; Hehre, W. J.; Klamt, A.III.H.F.S.; Schmidt, M. W.; Sherrill, C. D.; Truhlar, D. G.; Warshel, A.; Xu, X.; Aspuru-Guzik, A.; Baer, R.; Bell, A. T.; Besley, N. A.; Chai, J.-D.; Dreuw, A.; Dunietz, B. D.; Furlani, T. R.; Gwaltney, S. R.; Hsu, C.-P.; Jung, Y.; Kong, J.; Lambrecht, D. S.; Liang, W.; Ochsenfeld, C.; Rassolov, V. A.; Slipchenko, L. V.; Subotnik, J. E.; Voorhis, T. V.; Herbert, J. M.; Krylov, A. I.; Gill, P. M.; Head-Gordon, M. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184– 215, DOI: 10.1080/00268976.2014.952696There is no corresponding record for this reference.
- 15Baerends, E. J.; Aguirre, N. F.; Austin, N. D.; Autschbach, J.; Bickelhaupt, F. M.; Bulo, R.; Cappelli, C.; van Duin, A. C. T.; Egidi, F.; Fonseca Guerra, C.; Förster, A.; Franchini, M.; Goumans, T. P. M.; Heine, T.; Hellström, M.; Jacob, C. R.; Jensen, L.; Krykunov, M.; van Lenthe, E.; Michalak, A.; Mitoraj, M. M.; Neugebauer, J.; Nicu, V. P.; Philipsen, P.; Ramanantoanina, H.; Rüger, R.; Schreckenbach, G.; Stener, M.; Swart, M.; Thijssen, J. M.; Trnka, T.; Visscher, L.; Yakovlev, A.; van Gisbergen, S. The Amsterdam Modeling Suite. J. Chem. Phys. 2025, 162, 162501, DOI: 10.1063/5.0258496There is no corresponding record for this reference.
- 16Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165– 169, DOI: 10.1016/0009-2614(89)85118-8There is no corresponding record for this reference.
- 17Balasubramani, S. G.; Chen, G. P.; Coriani, S.; Diedenhofen, M.; Frank, M. S.; Franzke, Y. J.; Furche, F.; Grotjahn, R.; Harding, M. E.; Hättig, C.; Hellweg, A.; Helmich-Paris, B.; Holzer, C.; Huniar, U.; Kaupp, M.; Marefat Khah, A.; Karbalaei Khani, S.; Müller, T.; Mack, F.; Nguyen, B. D.; Parker, S. M.; Perlt, E.; Rappoport, D.; Reiter, K.; Roy, S.; Rückert, M.; Schmitz, G.; Sierka, M.; Tapavicza, E.; Tew, D. P.; van Wüllen, C.; Voora, V. K.; Weigend, F.; Wodyński, A.; Yu, J. M. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107, DOI: 10.1063/5.0004635There is no corresponding record for this reference.
- 18Franzke, Y. J.; Holzer, C.; Andersen, J. H.; Begušić, T.; Bruder, F.; Coriani, S.; Della Sala, F.; Fabiano, E.; Fedotov, D. A.; Fürst, S.; Gillhuber, S.; Grotjahn, R.; Kaupp, M.; Kehry, M.; Krstić, M.; Mack, F.; Majumdar, S.; Nguyen, B. D.; Parker, S. M.; Pauly, F.; Pausch, A.; Perlt, E.; Phun, G. S.; Rajabi, A.; Rappoport, D.; Samal, B.; Schrader, T.; Sharma, M.; Tapavicza, E.; Treß, R. S.; Voora, V.; Wodyński, A.; Yu, J. M.; Zerulla, B.; Furche, F.; Hättig, C.; Sierka, M.; Tew, D. P.; Weigend, F. TURBOMOLE: Today and Tomorrow. J. Chem. Theory Comput. 2023, 19, 6859– 6890, DOI: 10.1021/acs.jctc.3c00347There is no corresponding record for this reference.
- 19Valiev, M.; Bylaska, E.; Govind, N.; Kowalski, K.; Straatsma, T.; Van Dam, H.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.; de Jong, W. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477– 1489, DOI: 10.1016/j.cpc.2010.04.018There is no corresponding record for this reference.
- 20Sun, Q.; Berkelbach, T. C.; Blunt, N. S.; Booth, G. H.; Guo, S.; Li, Z.; Liu, J.; McClain, J. D.; Sayfutyarova, E. R.; Sharma, S.; Wouters, S.; Chan, G. K.-L. PySCF: the Python-based simulations of chemistry framework. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1340 DOI: 10.1002/wcms.1340There is no corresponding record for this reference.
- 21Sun, Q.; Zhang, X.; Banerjee, S.; Bao, P.; Barbry, M.; Blunt, N. S.; Bogdanov, N. A.; Booth, G. H.; Chen, J.; Cui, Z.-H.; Eriksen, J. J.; Gao, Y.; Guo, S.; Hermann, J.; Hermes, M. R.; Koh, K.; Koval, P.; Lehtola, S.; Li, Z.; Liu, J.; Mardirossian, N.; McClain, J. D.; Motta, M.; Mussard, B.; Pham, H. Q.; Pulkin, A.; Purwanto, W.; Robinson, P. J.; Ronca, E.; Sayfutyarova, E. R.; Scheurer, M.; Schurkus, H. F.; Smith, J. E. T.; Sun, C.; Sun, S.-N.; Upadhyay, S.; Wagner, L. K.; Wang, X.; White, A.; Whitfield, J. D.; Williamson, M. J.; Wouters, S.; Yang, J.; Yu, J. M.; Zhu, T.; Berkelbach, T. C.; Sharma, S.; Sokolov, A. Y.; Chan, G. K.-L. Recent developments in the PySCF program package. J. Chem. Phys. 2020, 153, 024109 DOI: 10.1063/5.0006074There is no corresponding record for this reference.
- 22Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G. L.; Cococcioni, M.; Dabo, I.; Dal Corso, A.; de Gironcoli, S.; Fabris, S.; Fratesi, G.; Gebauer, R.; Gerstmann, U.; Gougoussis, C.; Kokalj, A.; Lazzeri, M.; Martin-Samos, L.; Marzari, N.; Mauri, F.; Mazzarello, R.; Paolini, S.; Pasquarello, A.; Paulatto, L.; Sbraccia, C.; Scandolo, S.; Sclauzero, G.; Seitsonen, A. P.; Smogunov, A.; Umari, P.; Wentzcovitch, R. M. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 2009, 21, 395502 DOI: 10.1088/0953-8984/21/39/395502There is no corresponding record for this reference.
- 23Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M. B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; Colonna, N.; Carnimeo, I.; Corso, A. D.; de Gironcoli, S.; Delugas, P.Jr.R.A.D.; Ferretti, A.; Floris, A.; Fratesi, G.; Fugallo, G.; Gebauer, R.; Gerstmann, U.; Giustino, F.; Gorni, T.; Jia, J.; Kawamura, M.; Ko, H.-Y.; Kokalj, A.; Küçükbenli, E.; Lazzeri, M.; Marsili, M.; Marzari, N.; Mauri, F.; Nguyen, N. L.; Nguyen, H.-V.; de-la Roza, A. O.; Paulatto, L.; Poncé, S.; Rocca, D.; Sabatini, R.; Santra, B.; Schlipf, M.; Seitsonen, A. P.; Smogunov, A.; Timrov, I.; Thonhauser, T.; Umari, P.; Vast, N.; Wu, X.; Baroni, S. Advanced capabilities for materials modelling with QUANTUM ESPRESSO. J. Phys.: Condens. Matter 2017, 29, 465901, DOI: 10.1088/1361-648X/aa8f79There is no corresponding record for this reference.
- 24Smith, D. G. A.; Burns, L. A.; Simmonett, A. C.; Parrish, R. M.; Schieber, M. C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; James, A. M.; Lehtola, S.; Misiewicz, J. P.; Scheurer, M.; Shaw, R. A.; Schriber, J. B.; Xie, Y.; Glick, Z. L.; Sirianni, D. A.; O’Brien, J. S.; Waldrop, J. M.; Kumar, A.; Hohenstein, E. G.; Pritchard, B. P.; Brooks, B. R.; Schaefer, I.; Henry, F.; Sokolov, A. Y.; Patkowski, K.; DePrince, I.; Eugene, A.; Bozkaya, U.; King, R. A.; Evangelista, F. A.; Turney, J. M.; Crawford, T. D.; Sherrill, C. D. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108, DOI: 10.1063/5.0006002There is no corresponding record for this reference.
- 25Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73– 78, DOI: 10.1002/wcms.81There is no corresponding record for this reference.
- 26Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108, DOI: 10.1063/5.0004608There is no corresponding record for this reference.
- 27Neese, F. Software Update: The ORCA Program System─Version 6.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2025, 15, e70019 DOI: 10.1002/wcms.70019There is no corresponding record for this reference.
- 28Hagg, A.; Kirschner, K. N. Open-Source Machine Learning in Computational Chemistry. J. Chem. Inf. Model. 2023, 63, 4505– 4532, DOI: 10.1021/acs.jcim.3c00643There is no corresponding record for this reference.
- 29Wilkinson, M. D.; Dumontier, M.; Aalbersberg, I. J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L. B.; Bourne, P. E.; Bouwman, J.; Brookes, A. J.; Clark, T.; Crosas, M.; Dillo, I.; Dumon, O.; Edmunds, S.; Evelo, C. T.; Finkers, R.; González-Beltrán, A.; Gray, A. J. G.; Groth, P.; Goble, C.; Grethe, J. S.; Heringa, J.; t’Hoen, P. A. C.; Hooft, R.; Kuhn, T.; Kok, R.; Kok, J.; Lusher, S. J.; Martone, M. E.; Mons, A.; Packer, A. L.; Persson, B.; Rocca-Serra, P.; Roos, M.; van Schaik, R.; Sansone, S.-A.; Schultes, E.; Sengstag, T.; Slater, T.; Strawn, G.; Swertz, M. A.; Thompson, M.; van der Lei, J.; van Mulligen, E.; Velterop, J.; Waagmeester, A.; Wittenburg, P.; Wolstencroft, K.; Zhao, J.; Mons, B. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018, DOI: 10.1038/sdata.2016.18There is no corresponding record for this reference.
- 30Smith, J. S.; Isayev, O.; Roitberg, A. E. ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 2017, 8, 3192– 3203, DOI: 10.1039/C6SC05720AThere is no corresponding record for this reference.
- 31Smith, J. S.; Isayev, O.; Roitberg, A. E. ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules. Sci. Data 2017, 4, 170193, DOI: 10.1038/sdata.2017.193There is no corresponding record for this reference.
- 32Smith, J. S.; Nebgen, B.; Lubbers, N.; Isayev, O.; Roitberg, A. E. Less is more: sampling chemical space with active learning. J. Chem. Phys. 2018, 148, 241733, DOI: 10.1063/1.5023802There is no corresponding record for this reference.
- 33Smith, J. S.; Nebgen, B. T.; Zubatyuk, R.; Lubbers, N.; Devereux, C.; Barros, K.; Tretiak, S.; Isayev, O.; Roitberg, A. E. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 2019, 10, 2903, DOI: 10.1038/s41467-019-10827-4There is no corresponding record for this reference.
- 34Smith, J. S.; Zubatyuk, R.; Nebgen, B.; Lubbers, N.; Barros, K.; Roitberg, A. E.; Isayev, O.; Tretiak, S. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci. Data 2020, 7, 134, DOI: 10.1038/s41597-020-0473-zThere is no corresponding record for this reference.
- 35Anstine, D. M.; Zubatyuk, R.; Isayev, O. AIMNet2: a neural network potential to meet your neutral, charged, organic, and elemental-organic needs. Chem. Sci. 2025, 16, 10228– 10244, DOI: 10.1039/D4SC08572HThere is no corresponding record for this reference.
- 36Wood, B. M.; Dzamba, M.; Fu, X.; Gao, M.; Shuaibi, M.; Barroso-Luque, L.; Abdelmaqsoud, K.; Gharakhanyan, V.; Kitchin, J. R.; Levine, D. S.; Michel, K.; Sriram, A.; Cohen, T.; Das, A.; Rizvi, A.; Sahoo, S. J.; Ulissi, Z. W.; Zitnick, C. L. UMA: A Family of Universal Models for Atoms . 2025; https://arxiv.org/abs/2506.23971v1.There is no corresponding record for this reference.
- 37Levine, D. S.; Shuaibi, M.; Spotte-Smith, E. W. C.; Taylor, M. G.; Hasyim, M. R.; Michel, K.; Batatia, I.; Csányi, G.; Dzamba, M.; Eastman, P.; Frey, N. C.; Fu, X.; Gharakhanyan, V.; Krishnapriyan, A. S.; Rackers, J. A.; Raja, S.; Rizvi, A.; Rosen, A. S.; Ulissi, Z.; Vargas, S.; Zitnick, C. L.; Blau, S. M.; Wood, B. M. The Open Molecules 2025 (OMol25) Dataset, Evaluations, and Models. arXiv 2025. DOI: 10.48550/arXiv.2505.08762There is no corresponding record for this reference.
- 38Luise, G.; Huang, C.-W.; Vogels, T.; Kooi, D. P.; Ehlert, S.; Lanius, S.; Giesbertz, K. J. H.; Karton, A.; Gunceler, D.; Stanley, M.; Bruinsma, W. P.; Huang, L.; Wei, X.; Garrido Torres, J.; Katbashev, A.; Chavez Zavaleta, R.; Máté, B.; Kaba, S.-O.; Sordillo, R.; Chen, Y.; Williams-Young, D. B.; Bishop, C. M.; Hermann, J.; van den Berg, R.; Gori-Giorgi, P. Accurate and Scalable Exchange-Correlation with Deep Learning. arXiv 2025. DOI: 10.48550/arXiv.2506.14665There is no corresponding record for this reference.
- 39Ehlert, S.; Hermann, J.; Vogels, T.; Satorras, V. G.; Lanius, S.; Segler, M.; Kooi, D. P.; Takeda, K.; Huang, C.-W.; Luise, G.; van den Berg, R.; Gori-Giorgi, P.; Karton, A. Accurate Chemistry Collection: Coupled Cluster Atomization Energies for Broad Chemical Space. arXiv 2025. DOI: 10.48550/arXiv.2506.14492There is no corresponding record for this reference.
- 40Hjorth Larsen, A.; Jo̷rgen Mortensen, J.; Blomqvist, J.; Castelli, I. E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M. N.; Hammer, B.; Hargus, C.; Hermes, E. D.; Jennings, P. C.; Bjerre Jensen, P.; Kermode, J.; Kitchin, J. R.; Leonhard Kolsbjerg, E.; Kubal, J.; Kaasbjerg, K.; Lysgaard, S.; Bergmann Maronsson, J.; Maxson, T.; Olsen, T.; Pastewka, L.; Peterson, A.; Rostgaard, C.; Schio̷tz, J.; Schütt, O.; Strange, M.; Thygesen, K. S.; Vegge, T.; Vilhelmsen, L.; Walter, M.; Zeng, Z.; Jacobsen, K. W. The atomic simulation environment─a Python library for working with atoms. J. Phys.: Condens. Matter 2017, 29, 273002, DOI: 10.1088/1361-648X/aa680eThere is no corresponding record for this reference.
- 41FACCTs GmbH, Germany, www.faccts.de, WEASEL 1.12.3 DOI: 10.5281/zenodo.15260476 .There is no corresponding record for this reference.
- 42Grimme, S.; Bohle, F.; Hansen, A.; Pracht, P.; Spicher, S.; Stahn, M. Efficient Quantum Chemical Calculation of Structure Ensembles and Free Energies for Nonrigid Molecules. J. Phys. Chem. A 2021, 125, 4039– 4054, DOI: 10.1021/acs.jpca.1c00971There is no corresponding record for this reference.
- 43Weymuth, T.; Unsleber, J. P.; Türtscher, P. L.; Steiner, M.; Sobez, J.-G.; Müller, C. H.; Mörchen, M.; Klasovita, V.; Grimmel, S. A.; Eckhoff, M.; Csizi, K.-S.; Bosia, F.; Bensberg, M.; Reiher, M. SCINE─Software for chemical interaction networks. J. Chem. Phys. 2024, 160, 222501, DOI: 10.1063/5.0206974There is no corresponding record for this reference.
- 44Chen, Y.; Bannwarth, C. An Automated Intermolecular Reaction Discovery Approach Relying on Heuristic Atom-Partitioned Frontier Orbital Features. J. Chem. Inf. Model. 2025, 65, 9125– 9141, DOI: 10.1021/acs.jcim.5c00908There is no corresponding record for this reference.
- 45Altun, A.; Neese, F.; Bistoni, G. LEDAW: An Integrated Software Suite with GUI for Automating Local Energy Decomposition Analysis of Molecular Interactions. J. Chem. Inf. Model. 2025, 65, 8917– 8923, DOI: 10.1021/acs.jcim.5c01561There is no corresponding record for this reference.
- 46Python Software Foundation, Python (Version 3) 2025. https://www.python.org, Accessed: 12th November 2025.There is no corresponding record for this reference.
- 47Perez, F.; Granger, B. E.; Hunter, J. D. Python: An Ecosystem for Scientific Computing. Comput. Sci. Eng. 2011, 13, 13– 21, DOI: 10.1109/MCSE.2010.119There is no corresponding record for this reference.
- 48Sarkar, D.; Bali, R.; Sharma, T. Practical Machine Learning with Python: A Problem-Solver’s Guide to Building Real-World Intelligent Systems; Apress: Berkeley, CA, 2018; pp 67– 118 DOI: 10.1007/978-1-4842-3207-1_2 .There is no corresponding record for this reference.
- 49Bommarito, E.; Bommarito, M. An Empirical Analysis of the Python Package Index (PyPI) , 2019. DOI: 10.48550/arXiv.1907.11073There is no corresponding record for this reference.
- 50Berquist, E.; Dumi, A.; Upadhyay, S.; Abarbanel, O. D.; Cho, M.; Gaur, S.; Gil, R.; Hutchison, G. R.; Lee, O. S.; Rosen, A. S.; Schamnad, S.; Schneider, F. S. S.; Steinmann, C.; Stolyarchuk, M.; Vandezande, J. E.; Zak, W.; Langner, K. M. cclib 2.0: An updated architecture for interoperable computational chemistry. J. Chem. Phys. 2024, 161, 042501 DOI: 10.1063/5.0216778There is no corresponding record for this reference.
- 51Ragnar Bjornsson, ASH ORCA interface, 2025. https://ash.readthedocs.io/en/latest/ORCA-interface.html, Accessed: 12th November 2025.There is no corresponding record for this reference.
- 52PLAMS, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com, https://github.com/SCM-NV/PLAMS.There is no corresponding record for this reference.
- 53FACCTs GmbH. ORCA Python Interface (OPI). https://github.com/faccts/opi, DOI of v1.0: DOI: 10.5281/zenodo.15688425 .There is no corresponding record for this reference.
- 54FACCTs GmbH, ORCA Python Interface (OPI) Documentation. https://www.faccts.de/docs/opi/docs, Accessed: 17th November 2025.There is no corresponding record for this reference.
- 55Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; Ivanov, P.; Avila, D.; Abdalla, S.; Willing, C.; Team, J. D. Jupyter Notebooks─a publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas, 2016; pp 87– 90.There is no corresponding record for this reference.
- 56Jupyter, P.; Bussonnier, M.; Forde, J.; Freeman, J.; Granger, B.; Head, T.; Holdgraf, C.; Kelley, K.; Nalvarte, G.; Osheroff, A.; Pacer, M.; Panda, Y.; Perez, F.; Ragan-Kelley, B.; Willing, C.; Binder 2.0─Reproducible, Interactive, Sharable Environments for Science at Scale. In Proceedings of the 17th Python in Science Conference, 2018; pp 113– 120.There is no corresponding record for this reference.
- 57FACCTs GmbH (OPI project), OPI Documentation/Tutorials (v2.0), 2025; https://www.faccts.de/docs/opi/2.0/docs/, Accessed: 2nd October 2025.There is no corresponding record for this reference.
- 58Burns, J.; Zalte, A.; Green, W. Descriptor-based Foundation Models for Molecular Property Prediction , 2025; https://arxiv.org/abs/2506.15792.There is no corresponding record for this reference.
- 59De Landsheere, J.; Zamyatin, A.; Karwounopoulos, J.; Heid, E. ChemTorch: A Deep Learning Framework for Benchmarking and Developing Chemical Reaction Property Prediction Models. ChemRxiv 2025, Preprint, not peer-reviewed.There is no corresponding record for this reference.
- 60FACCTs GmbH, OPI Documentation/Tutorials (v2.0), How To Contribute. https://www.faccts.de/docs/opi/2.0/docs/contents/how_to_contribute.html, Accessed: 17th November 2025.There is no corresponding record for this reference.
- 61FACCTs GmbH, ORCA Manual. https://www.faccts.de/docs/orca/6.1/manual/, Accessed: 17th November 2025.There is no corresponding record for this reference.
- 62FACCTs GmbH, ORCA Tutorials. https://www.faccts.de/docs/orca/6.1/tutorials/, Accessed: 17th November 2025.There is no corresponding record for this reference.
- 63Harris, C. R.; Millman, K. J.; van der Walt, S. J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N. J.; Kern, R.; Picus, M.; Hoyer, S.; van Kerkwijk, M. H.; Brett, M.; Haldane, A.; del Río, J. F.; Wiebe, M.; Peterson, P.; Gérard-Marchant, P.; Sheppard, K.; Reddy, T.; Weckesser, W.; Abbasi, H.; Gohlke, C.; Oliphant, T. E. Array programming with NumPy. Nature 2020, 585, 357– 362, DOI: 10.1038/s41586-020-2649-2There is no corresponding record for this reference.
- 64Gábor, B.; Berman, J.; Lev, O.; Pfannschmidt, R. platformdirs. https://github.com/tox-dev/platformdirs, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 65Colvin, S.; Jolibois, E.; Ramezani, H.; Garcia Badaracco, A.; Dorsey, T.; Montague, D.; Matveenko, S.; Trylesinski, M.; Runkle, S.; Hewitt, D.; Hall, A.; Plot, V. Pydantic Validation. https://github.com/pydantic/pydantic, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 66Landrum, G. RDKit: Open-Source Cheminformatics Software , 2025. https://www.rdkit.org/, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 67Preston-Werner, T. Semantic Versioning 2.0.0 , 2025; https://semver.org/, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 68Barrois, R. python-semanticversion. https://github.com/rbarrois/python-semanticversion, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 69Astral, uv. https://github.com/astral-sh/uv, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 70Lehtosalo, J. mypy. https://www.mypy-lang.org/, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 71Flowers, A.; Nox, K. https://github.com/wntrblm/nox, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 72Astral, ruff. https://github.com/astral-sh/ruff, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 73Marchi, L. D. codespell , 2025 https://github.com/codespell-project/codespell, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 74Seipp, J. Vulture─Find Dead Code , 2025. https://github.com/jendrikseipp/vulture, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 75Krekel, H.; Oliveira, B.; Pfannschmidt, R.; Bruynooghe, F.; Laugher, B.; Bruhin, F. pytest 8.4 . 2004; https://github.com/pytest-dev/pytest, Contributors: Holger Krekel and Bruno Oliveira and Ronny Pfannschmidt and Floris Bruynooghe and Brianna Laugher and Florian Bruhin and others; Accessed: 15th October 2025.There is no corresponding record for this reference.
- 76MacIver, D. R. Hypothesis 6.133 . 2016; https://github.com/HypothesisWorks/hypothesis-python, Accessed: 15th October 2025.There is no corresponding record for this reference.
- 77Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995– 2001, DOI: 10.1021/jp9716997There is no corresponding record for this reference.
- 78Garcia-Ratés, M.; Neese, F. Effect of the Solute Cavity on the Solvation Energy and its Derivatives within the Framework of the Gaussian Charge Scheme. J. Comput. Chem. 2020, 41, 922– 939, DOI: 10.1002/jcc.26139There is no corresponding record for this reference.
- 79Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378– 6396, DOI: 10.1021/jp810292nThere is no corresponding record for this reference.
- 80Gerlach, T.; Müller, S.; de Castilla, A. G.; Smirnova, I. An open source COSMO-RS implementation and parameterization supporting the efficient implementation of multiple segment descriptors. Fluid Phase Equilib. 2022, 560, 113472 DOI: 10.1016/j.fluid.2022.113472There is no corresponding record for this reference.
- 81Ásgeirsson, V.; Birgisson, B. O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector Following. J. Chem. Theory Comput. 2021, 17, 4929– 4945, DOI: 10.1021/acs.jctc.1c00462There is no corresponding record for this reference.
- 82de Souza, B. GOAT: A Global Optimization Algorithm for Molecules and Atomic Clusters. Angew. Chem., Int. Ed. 2025, 64, e202500393 DOI: 10.1002/anie.202500393There is no corresponding record for this reference.
- 83Bistoni, G. Finding chemical concepts in the Hilbert space: Coupled cluster analyses of noncovalent interactions. WIREs Computational Molecular Science 2020, 10, e1442 DOI: 10.1002/wcms.1442There is no corresponding record for this reference.
- 84FACCTs GmbH, orca-external-tools. https://github.com/faccts/orca-external-tools, Accessed: 11th October 2025.There is no corresponding record for this reference.
- 85Python Software Foundation, Python 3.11.14 Documentation. https://docs.python.org/3.11/reference/lexical_analysis.html#names-identifiers-and-keywords, Accessed: 2nd February 2026.There is no corresponding record for this reference.
- 86Software in the Public Interest (SPI), Open MPI: Open Source High Performance Computing. https://www.open-mpi.org/, Accessed: 2nd October 2025.There is no corresponding record for this reference.
- 87Preston-Werner, T. TOML . 2021; https://toml.io/en/, Accessed: 2nd October 2025.There is no corresponding record for this reference.
- 88FACCTs GmbH , 2025 https://github.com/faccts/opi/blob/release/2.0/src/opi/output/grepper/recipes.py, Accessed: 17th November 2025.There is no corresponding record for this reference.
- 89Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297, DOI: 10.1039/b508541aThere is no corresponding record for this reference.
- 90Grimme, S.; Hansen, A.; Ehlert, S.; Mewes, J.-M. r2SCAN-3c: A “Swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021, 154, 064103 DOI: 10.1063/5.0040021There is no corresponding record for this reference.
- 91Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106, DOI: 10.1063/1.4773581There is no corresponding record for this reference.
- 92Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101, DOI: 10.1063/1.4821834There is no corresponding record for this reference.
- 93Tao, Y.; Yuan, K.; Chen, T.; Xu, P.; Li, H.; Chen, R.; Zheng, C.; Zhang, L.; Huang, W. Thermally Activated Delayed Fluorescence Materials Towards the Breakthrough of Organoelectronics. Adv. Mater. 2014, 26, 7931– 7958, DOI: 10.1002/adma.201402532There is no corresponding record for this reference.
- 94Kunze, L.; Hansen, A.; Grimme, S.; Mewes, J.-M. The Best of Both Worlds: ΔDFT Describes Multiresonance TADF Emitters with Wave-Function Accuracy at Density-Functional Cost. J. Phys. Chem. Lett. 2025, 16, 1114– 1125, DOI: 10.1021/acs.jpclett.4c03192There is no corresponding record for this reference.
- 95Shee, J.; Head-Gordon, M. Predicting Excitation Energies of Twisted Intramolecular Charge-Transfer States with the Time-Dependent Density Functional Theory: Comparison with Experimental Measurements in the Gas Phase and Solvents Ranging from Hexanes to Acetonitrile. J. Chem. Theory Comput. 2020, 16, 6244– 6255, DOI: 10.1021/acs.jctc.0c00635There is no corresponding record for this reference.
- 96Mardirossian, N.; Head-Gordon, M. ωB97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J. Chem. Phys. 2016, 144, 214110, DOI: 10.1063/1.4952647There is no corresponding record for this reference.
- 97Virtanen, P.; Gommers, R.; Oliphant, T. E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; van der Walt, S. J.; Brett, M.; Wilson, J.; Millman, K. J.; Mayorov, N.; Nelson, A. R. J.; Jones, E.; Kern, R.; Larson, E.; Carey, C. J.; Polat, İ.; Feng, Y.; Moore, E. W.; VanderPlas, J.; Laxalde, D.; Perktold, J.; Cimrman, R.; Henriksen, I.; Quintero, E. A.; Harris, C. R.; Archibald, A. M.; Ribeiro, A. H.; Pedregosa, F.; van Mulbregt, P. SciPy 1.0 Contributors, SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261– 272, DOI: 10.1038/s41592-020-0772-5There is no corresponding record for this reference.
- 98Ikeda, N.; Oda, S.; Matsumoto, R.; Yoshioka, M.; Fukushima, D.; Yoshiura, K.; Yasuda, N.; Hatakeyama, T. Solution-Processable Pure Green Thermally Activated Delayed Fluorescence Emitter Based on the Multiple Resonance Effect. Adv. Mater. 2020, 32, 2004072 DOI: 10.1002/adma.202004072There is no corresponding record for this reference.
- 99Grimme, S.; Hansen, A. A Practicable Real-Space Measure and Visualization of Static Electron-Correlation Effects. Angew. Chem., Int. Ed. 2015, 54, 12308– 12313, DOI: 10.1002/anie.201501887There is no corresponding record for this reference.
- 100Faulstich, F. M.; Kristiansen, H. E.; Csirik, M. A.; Kvaal, S.; Pedersen, T. B.; Laestadius, A. S-Diagnostic-An a Posteriori Error Assessment for Single-Reference Coupled-Cluster Methods. J. Phys. Chem. A 2023, 127, 9106– 9120, DOI: 10.1021/acs.jpca.3c01575There is no corresponding record for this reference.
- 101Duan, C.; Chu, D. B. K.; Nandy, A.; Kulik, H. J. Detection of multi-reference character imbalances enables a transfer learning approach for virtual high throughput screening with coupled cluster accuracy at DFT cost. Chem. Sci. 2022, 13, 4962– 4971, DOI: 10.1039/D2SC00393GThere is no corresponding record for this reference.
- 102Gasevic, T.; Müller, M.; Schöps, J.; Lanius, S.; Hermann, J.; Grimme, S.; Hansen, A. Chemical Space Exploration with Artificial “Mindless” Molecules. J. Chem. Inf. Model. 2025, 65, 9576– 9587, DOI: 10.1021/acs.jcim.5c01364There is no corresponding record for this reference.
- 103Collette, A. , e. h5py─HDF5 for Python , 2025 https://www.h5py.org/, Accessed: 17th November 2025.There is no corresponding record for this reference.
- 104The HDF Group, HDF5. https://www.hdfgroup.org/solutions/hdf5/, Accessed: 17th November 2025.There is no corresponding record for this reference.
- 105Rui, Y.; Chen, Y.; Ivanova, E.; Kumar, V. B.; Śmiga, S.; Grabowski, I.; Dral, P. O. The Best DFT Functional Is the Ensemble of Functionals. Adv. Sci. 2024, 11, 2408239 DOI: 10.1002/advs.202408239There is no corresponding record for this reference.
- 106Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184– 32215, DOI: 10.1039/C7CP04913GThere is no corresponding record for this reference.
- 107Zhao, Y.; Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215– 241, DOI: 10.1007/s00214-007-0310-xThere is no corresponding record for this reference.
- 108Bursch, M.; Neugebauer, H.; Ehlert, S.; Grimme, S. Dispersion corrected r2SCAN based global hybrid functionals: r2SCANh, r2SCAN0, and r2SCAN50. J. Chem. Phys. 2022, 156, 134105, DOI: 10.1063/5.0086040There is no corresponding record for this reference.
- 109Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105, DOI: 10.1063/1.3484283There is no corresponding record for this reference.
- 110Rappoport, D. Property-optimized Gaussian basis sets for lanthanides. J. Chem. Phys. 2021, 155, 124102, DOI: 10.1063/5.0065611There is no corresponding record for this reference.
- 111Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; Vanderplas, J.; Passos, A.; Cournapeau, D.; Brucher, M.; Perrot, M.; Duchesnay, E. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825– 2830There is no corresponding record for this reference.
- 112Lehtola, S.; Karttunen, A. J. Free and open source software for computational chemistry education. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2022, 12, e1610 DOI: 10.1002/wcms.1610There is no corresponding record for this reference.
- 113Capel, A. J.; Rimington, R. P.; Lewis, M. P.; Christie, S. D. R. 3D printing for chemical, pharmaceutical and biological applications. Nat. Rev. Chem. 2018, 2, 422– 436, DOI: 10.1038/s41570-018-0058-yThere is no corresponding record for this reference.
- 114Pinger, C. W.; Geiger, M. K.; Spence, D. M. Applications of 3D-Printing for Improving Chemistry Education. J. Chem. Educ. 2020, 97, 112– 117, DOI: 10.1021/acs.jchemed.9b00588There is no corresponding record for this reference.
- 115Garcia, M.; O’Leary, F. A.; O’Leary, D. J. Do-It-Yourself 5-Color 3D Printing of Molecular Orbitals and Electron Density Surfaces. J. Chem. Educ. 2023, 100, 1648– 1658, DOI: 10.1021/acs.jchemed.2c00907There is no corresponding record for this reference.
- 116Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB─An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652– 1671, DOI: 10.1021/acs.jctc.8b01176There is no corresponding record for this reference.
- 117Rego, N.; Koes, D. 3Dmol.js: molecular visualization with WebGL. Bioinform. 2015, 31, 1322– 1324, DOI: 10.1093/bioinformatics/btu829There is no corresponding record for this reference.
- 118Pipek, J.; Mezey, P. G. A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave functions. J. Chem. Phys. 1989, 90, 4916– 4926, DOI: 10.1063/1.456588There is no corresponding record for this reference.
- 119Adamo, C.; Barone, V. Toward reliable adiabatic connection models free from adjustable parameters. Chem. Phys. Lett. 1997, 274, 242– 250, DOI: 10.1016/S0009-2614(97)00651-9There is no corresponding record for this reference.
- 120Zou, Y.; Cheng, A. H.; Aldossary, A.; Bai, J.; Leong, S. X.; Campos-Gonzalez-Angulo, J. A.; Choi, C.; Ser, C. T.; Tom, G.; Wang, A.; Zhang, Z.; Yakavets, I.; Hao, H.; Crebolder, C.; Bernales, V.; Aspuru-Guzik, A. El Agente: An autonomous agent for quantum chemistry. Matter 2025, 8, 102263 DOI: 10.1016/j.matt.2025.102263There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.5c02141.
Computed data for the ML training data example (ZIP)
Additional overview of currently available quantities from the GBW JSON file and the property JSON file (PDF)
Computation-ready scripts and Jupyter Notebooks for all presented examples (ZIP)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.