



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Fine-Tuning of Label-Free Single-Cell Proteomics WorkflowsClick to copy article linkArticle link copied!
- Pauline Perdu-AlloyPauline Perdu-AlloyLaboratoire de Spectrométrie de Masse BioOrganique (LSMBO), IPHC UMR7178, CNRS, Université de Strasbourg, 25 Rue Becquerel, Strasbourg, Grand Est 67087, FranceInfrastructure Nationale de Protéomique ProFI, UAR2048, Strasbourg 67087, FranceMore by Pauline Perdu-Alloy
- Charline KellerCharline KellerLaboratoire de Spectrométrie de Masse BioOrganique (LSMBO), IPHC UMR7178, CNRS, Université de Strasbourg, 25 Rue Becquerel, Strasbourg, Grand Est 67087, FranceInfrastructure Nationale de Protéomique ProFI, UAR2048, Strasbourg 67087, FranceMore by Charline Keller
- Anjali SethAnjali SethCellenion SASU, 60 Avenue Rockefeller, Bioserra2, Lyon, Auvergne-Rhône-Alpes 69008, FranceMore by Anjali Seth
- Christine Carapito*Christine Carapito*Email: [email protected]Laboratoire de Spectrométrie de Masse BioOrganique (LSMBO), IPHC UMR7178, CNRS, Université de Strasbourg, 25 Rue Becquerel, Strasbourg, Grand Est 67087, FranceInfrastructure Nationale de Protéomique ProFI, UAR2048, Strasbourg 67087, FranceMore by Christine Carapito
Abstract
Mass spectrometry-based single-cell proteomics emerges as the most promising method for studying cellular heterogeneity at the global proteome level with unprecedented depth and coverage. Its widespread application remains limited due to robustness, reproducibility, and throughput requirements, still difficult to meet as analyzing large cohorts of single cells is necessary to ensure statistical confidence. In this context, we conducted method optimizations at three levels. First, we benchmarked three distinct workflows compatible with the nanoElute2 platform using different sample collection/preparation plate supports (EVO96 oil-free, LF48 oil-based, and LF48 oil-free, a streamlined automated sample resuspension, and direct injection protocol). Then, we compared the optimized EVO96 workflow on nanoElute2 with Evosep-based separations operating at two analytical throughputs (80 and 120 samples per day). Subsequently, we evaluated digestion efficiency using a range of enzyme/protein ratios (1:1; 10:1; 20:1; 50:1) to maximize peptide recovery. Finally, the chromatographic setup was refined to determine the best compromise between throughput and robustness. Altogether, these optimizations allowed to establish a robust workflow quantifying up to 5000 proteins in 10 min gradient time per single HeLa cell at a 55 samples-per-day throughput.
This publication is licensed for personal use by The American Chemical Society.
1. Introduction
2. Experimental Procedures
2.1. Single Cells’ Preparation on the CellenONE System
2.1.1. Nanoliquid Chromatography
2.1.2. Tandem Mass Spectrometry
2.2. Data Treatment
3. Results and Discussion
3.1. Sample Preparation Plate Support Optimization
Option 1─The LF48 oil-based support, the original cellenOne protocol, for the collection of 48 single cells per chip, integrating a thin hexadecane layer requiring a final manual sample transfer.
Options 2 and 3─The EVO96 support, an oil-free proteoCHIP for the single-cell collection (of up to 96), with hands-free peptide transfer via a short centrifugation step. Although originally developed for coupling with the Evosep system (Option 2), we also attempted its adaptation for nanoElute injections (Option 3).
Option 4─The LF48 oil-free support enabling automated resuspension and injection, using a custom-designed proteoCHIP holder compatible with the nanoElute2 autosampler. In this workflow, the proteoCHIP was transferred from the cellenONE to the nanoElute autosampler as soon as the cell isolation/digestion was completed (no storage, no freezing), with each single-cell digest in approximately 300 nL (corresponding to the Mastermix volume). Each single-cell sample was automatically resuspended and rehydrated by the nanoElute system in 1.5 μL of 2% ACN, 0.015% DDM, and 0.1% FA (final concentrations) right before injection.
Figure 1
Figure 1. Four single-cell sample preparation protocols benchmarked using the cellenONE sorter/liquid dispenser. Lysis and digestion, followed by incubation with automated rehydration cycles are common steps to all three workflows. Then, option 1 relies on a LF48 oil-based plate support involving a manual dilution step in liquid oil, followed by a manual peptide transfer after oil solidification (Tf (hexadecane) = 18 °C); option 2 uses an EVO96 oil-free plate support, involving manual dilution followed by peptide loading onto preconditioned EVOTIPs after centrifugation for injection on the Evosep system; option 3 employs the same EVO96 oil-free support, repurposed for nanoElute injection, where peptides are transferred into a 96-well injection plate via centrifugation; and option 4 relies on a LF48 oil-free plate support suited for automated resuspension and direct injection on a nanoElute 2 system.
Figure 2
Figure 2. Benchmarking of three single-cell proteomics workflows (EVO96 (blue), LF48 oil-based (green), and LF48 oil-free (orange)) run on nanoElute 2 on 96 single-cell replicates per workflow. (A) Violin plots representing the number of protein groups identified per single cell for each tested workflow using DIA-NN with MBR (left) and without MBR (right). (B) Violin plots representing the number of unique peptides identified per single cell for each tested workflow using DIA-NN with MBR (left) and without MBR (right). (C) Venn diagram showing proteins’ overlap across the three workflows. (D) Coefficients of variation (CVs) density distributions across the three sample preparation workflows. (E) Protein abundance ranking plot comparing the dynamic range of protein groups across the three workflows. Black dots correspond to the shared proteins across the conditions, whereas the colored ones correspond to unique proteins per condition.
Figure 3
Figure 3. Comparison of SCP EVO96-based workflows across two chromatographic systems. EVO96_nE was analyzed on a nanoElute 2 system (N = 96 single-cells) compared to two Evosep workflows that were acquired using two sample throughputs (80 and 120SPD with N = 40 single-cell replicates each). (A) Violin plots representing the number of protein groups identified per single cell for each workflow (EVO96_nE in blue, EVOSEP80SPD in gray, and EVOSEP120SPD in purple). (B) Venn diagram showing the overlap of identified protein groups across the workflows. (C) Violin plots representing the number of unique peptides identified per single cell for each workflow (EVO96_nE in blue, EVOSEP_80SPD in gray, and EVOSEP_120SPD in purple). (D) Density distributions of protein abundance coefficients of variation (CVs) across the three workflows.
3.2. Comparison of the EVO96 Workflow on Two Chromatographic Platforms
3.3. Enzyme-To-Protein Ratio Optimization
Figure 4
Figure 4. Evaluation of Trypsin-LysC digestion ratios. This figure presents a comprehensive analysis of HeLa single cells (N = 96 single-cell replicates per condition), comparing two digestion ratios (10:1 and 20:1). (A) Violin plots showing the distributions of protein groups identified per single-cell replicates under each digestion ratio, with or without MBR processing. (B) Violin plots showing the distributions of unique peptides identified per single-cell replicates under each digestion ratio, with or without MBR processing. (C) Venn diagrams illustrate the overlap in identified precursors (left), protein groups (right), and single-peptide hit proteins (down) between the two digestion ratios. (D) Density plots represent the distribution of coefficient of variation (CV) values across single cells.
3.4. LC–MS/MS Method Optimization
Figure 5
Figure 5. Gradient time and column length optimizations. (A) Average and standard deviation of the number of proteins identified per single-cell replicate (pink, N = 10) and per ten cells’ replicates (gray, N = 2), analyzed using 5 and 25 cm IonOpticks separation columns. (B) Union of all proteins identified across all 5 cm column replicates (blue) versus the union of all proteins identified across all 25 cm column replicates (orange). (C) The blue and orange dashed lines indicate the mean CV values calculated for 250 pg HeLa QC lysates (N = 3) run on a 5 cm (average 3753 protein group IDs) versus 25 cm (average 3585 protein group IDs) column, respectively.
4. Conclusion
Data Availability
Complete data set has been deposited in the ProteomeXchange Consortium via the PRIDE partner17 repository with the data set identifier PXD067019. This research did not involve human or animal participants.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.5c01075.
Outliers representation for the oil-free LF48 direct injection workflow on the 96 single-cell replicates; missing values and single-peptide hits’ distributions for EVO96, LF48 oil-free, and LF48 oil-based workflows;pairwise protein abundance correlations for common proteins identified across EVO96/LF48 oil-free/LF48 oil-based and nanoElute_EVO96/EVOSEP_80SPD/EVOSEP_120SPD workflows; repartition and distribution of GRAVY scores for EVO96, LF48 oil-free, and LF48 oil-based workflows; distribution of methionine oxidations identified across workflows; and impact of enzyme-to-protein ratios (10:1 and 20:1) on peptide and protein identifications (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Christine Carapito - Laboratoire de Spectrométrie de Masse BioOrganique (LSMBO), IPHC UMR7178, CNRS, Université de Strasbourg, 25 Rue Becquerel, Strasbourg, Grand Est 67087, France; Infrastructure Nationale de Protéomique ProFI, UAR2048, Strasbourg 67087, France;
 https://orcid.org/0000-0002-0079-319X;
https://orcid.org/0000-0002-0079-319X;
This work was supported by the Agence Nationale de la Recherche via the French Proteomic Infrastructure (ProFI UAR2048; ANR-10-INBS08-03 and ANR-24-INBS-0015), by the Region Grand-Est (SC-Proteomics project) and by the ITMO Cancer of Aviesan within the framework of the 2021–2030 Cancer Control Strategy, on funds administered by Inserm (ProteomiSC project), for equipment funds. It was also supported by the French Ministry of Higher Education and Research for the PhD fellowship of P.P.A and by the Interdisciplinary Thematic Institute IMS, the drug discovery and development institute, as part of the ITI 2021-2028 program of the University of Strasbourg, CNRS and Inserm, supported by IdEx Unistra (ANR-10-IDEX-0002), and by SFRI-STRAT’US project (ANR-20-SFRI-0012) for the PhD fellowship of C.K. C.C. is further supported by the European Union’s Horizon Europe MSCA PROHITS project under grant agreement (no. 101119980) and the CHIST-ERA project ODEEP-EU (ANR-23-CHRO-0005).
Acknowledgments
The authors thank Dr. Christoph Krisp and Pierre-Olivier Schmit from Bruker Daltonics for their support and insightful advices on the instrumental settings.
References
This article references 25 other publications.
- 1Laisné, M.; Lupien, M.; Vallot, C. Epigenomic Heterogeneity as a Source of Tumour Evolution. Nat. Rev. Cancer 2025, 25 (1), 7– 26, DOI: 10.1038/s41568-024-00757-9Google ScholarThere is no corresponding record for this reference.
- 2Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass Spectrometry of Single Mammalian Cells Quantifies Proteome Heterogeneity during Cell Differentiation. Genome Biol. 2018, 19 (1), 161, DOI: 10.1186/s13059-018-1547-5Google ScholarThere is no corresponding record for this reference.
- 3Minakshi, P.; Kumar, R.; Ghosh, M.; Saini, H. M.; Ranjan, K.; Brar, B.; Prasad, G. Single-Cell Proteomics: Technology and Applications. In Single-Cell Omics; Barh, D., Azevedo, V., Eds.; Elsevier, 2019; pp 283– 318.Google ScholarThere is no corresponding record for this reference.
- 4Tian, Y.; Li, Q.; Yang, Z.; Zhang, S.; Xu, J.; Wang, Z.; Bai, H.; Duan, J.; Zheng, B.; Li, W.; Cui, Y.; Wang, X.; Wan, R.; Fei, K.; Zhong, J.; Gao, S.; He, J.; Gay, C. M.; Zhang, J.; Wang, J.; Tang, F. Single-Cell Transcriptomic Profiling Reveals the Tumor Heterogeneity of Small-Cell Lung Cancer. Signal Transduct. Target Ther. 2022, 7 (1), 346, DOI: 10.1038/s41392-022-01150-4Google ScholarThere is no corresponding record for this reference.
- 5Nalla, L. V.; Kanukolanu, A.; Yeduvaka, M.; Gajula, S. N. R. Advancements in Single-Cell Proteomics and Mass Spectrometry-Based Techniques for Unmasking Cellular Diversity in Triple Negative Breast Cancer. Proteomics Clin. Appl. 2025, 19 (1), e202400101 DOI: 10.1002/prca.202400101Google ScholarThere is no corresponding record for this reference.
- 6Marino, F. Z.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular Heterogeneity in Lung Cancer: From Mechanisms of Origin to Clinical Implications. Int. J. Med. Sci. 2019, 16 (7), 981– 989, DOI: 10.7150/ijms.34739Google ScholarThere is no corresponding record for this reference.
- 7Black, G. S.; Huang, X.; Qiao, Y.; Moos, P.; Sampath, D.; Stephens, D. M.; Woyach, J. A.; Marth, G. T. Long-Read Single-Cell RNA Sequencing Enables the Study of Cancer Subclone-Specific Genotypes and Phenotypes in Chronic Lymphocytic Leukemia. Genome Res. 2025, 35 (4), 686– 697, DOI: 10.1101/gr.279049.124Google ScholarThere is no corresponding record for this reference.
- 8Mund, A.; Coscia, F.; Kriston, A.; Hollandi, R.; Kovács, F.; Brunner, A.-D.; Migh, E.; Schweizer, L.; Santos, A.; Bzorek, M.; Naimy, S.; Rahbek-Gjerdrum, L. M.; Dyring-Andersen, B.; Bulkescher, J.; Lukas, C.; Eckert, M. A.; Lengyel, E.; Gnann, C.; Lundberg, E.; Horvath, P.; Mann, M. Deep Visual Proteomics Defines Single-Cell Identity and Heterogeneity. Nat. Biotechnol. 2022, 40 (8), 1231– 1240, DOI: 10.1038/s41587-022-01302-5Google ScholarThere is no corresponding record for this reference.
- 9Gerniers, A.; Bricard, O.; Dupont, P. MicroCellClust: Mining Rare and Highly Specific Subpopulations from Single-Cell Expression Data. Bioinformatics 2021, 37 (19), 3220– 3227, DOI: 10.1093/bioinformatics/btab239Google ScholarThere is no corresponding record for this reference.
- 10Heide, T.; Househam, J.; Cresswell, G. D.; Spiteri, I.; Lynn, C.; Kimberley, C.; Mossner, M.; Zapata, L.; Gabbutt, C.; Ramazzotti, D.; Chen, B.; Fernandez-Mateos, J.; James, C.; Vinceti, A.; Berner, A.; Schmidt, M.; Lakatos, E.; Baker, A.-M.; Nichol, D.; Costa, H.; Mitchinson, M.; Werner, B.; Iorio, F.; Jansen, M.; Barnes, C.; Caravagna, G.; Shibata, D.; Bridgewater, J.; Rodriguez-Justo, M.; Magnani, L.; Graham, T. A.; Sottoriva, A. Assessment of the Evolutionary Consequence of Putative Driver Mutations in Colorectal Cancer with Spatial Multiomic Data. bioRxiv 2021, 451265, DOI: 10.1101/2021.07.14.451265Google ScholarThere is no corresponding record for this reference.
- 11Zhang, Z.; Mathew, D.; Lim, T.; Mason, K.; Martinez, C. M.; Huang, S.; Wherry, E. J.; Susztak, K.; Minn, A. J.; Ma, Z.; Zhang, N. R. Signal Recovery in Single Cell Batch Integration. bioRxiv 2023, 539614, DOI: 10.1101/2023.05.05.539614Google ScholarThere is no corresponding record for this reference.
- 12Fritzsch, F. S. O.; Dusny, C.; Frick, O.; Schmid, A. Single-Cell Analysis in Biotechnology, Systems Biology, and Biocatalysis. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 129– 155, DOI: 10.1146/annurev-chembioeng-062011-081056Google ScholarThere is no corresponding record for this reference.
- 13Dodds, J. N.; Baker, E. S.; Lubman, D. M. Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead. J. Am. Soc. Mass Spectrom. 2019, 30 (11), 2185– 2195, DOI: 10.1007/s13361-019-02288-2Google ScholarThere is no corresponding record for this reference.
- 14Fernandez-Lima, F. A.; Kaplan, D. A.; Park, M. A. Note: Integration of Trapped Ion Mobility Spectrometry with Mass Spectrometry. Rev. Sci. Instrum. 2011, 82 (12), 126106, DOI: 10.1063/1.3665933Google ScholarThere is no corresponding record for this reference.
- 15Meier, F.; Brunner, A.-D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M. A.; Bache, N.; Hoerning, O.; Cox, J.; Räther, O.; Mann, M. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteomics 2018, 17 (12), 2534– 2545, DOI: 10.1074/mcp.TIR118.000900Google ScholarThere is no corresponding record for this reference.
- 16Ctortecka, C.; Hartlmayr, D.; Seth, A.; Mendjan, S.; Tourniaire, G.; Udeshi, N. D.; Carr, S. A.; Mechtler, K. An Automated Nanowell-Array Workflow for Quantitative Multiplexed Single-Cell Proteomics Sample Preparation at High Sensitivity. Mol. Cell. Proteomics 2023, 22 (12), 100665, DOI: 10.1016/j.mcpro.2023.100665Google ScholarThere is no corresponding record for this reference.
- 17Perez-Riverol, Y.; Bandla, C.; Kundu, D. J.; Kamatchinathan, S.; Bai, J.; Hewapathirana, S.; John, N. S.; Prakash, A.; Walzer, M.; Wang, S.; Vizcaíno, J. A. The PRIDE Database at 20 Years: 2025 Update. Nucleic Acids Res. 2025, 53 (D1), D543– D553, DOI: 10.1093/nar/gkae1011Google ScholarThere is no corresponding record for this reference.
- 18Demichev, V.; Messner, C. B.; Vernardis, S. I.; Lilley, K. S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41– 44, DOI: 10.1038/s41592-019-0638-xGoogle ScholarThere is no corresponding record for this reference.
- 19Demichev, V.; Messner, C. B.; Vernardis, S. I.; Lilley, K. S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41– 44, DOI: 10.1038/s41592-019-0638-xGoogle ScholarThere is no corresponding record for this reference.
- 20Kyte, J.; Doolittle, R. F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157 (1), 105– 132, DOI: 10.1016/0022-2836(82)90515-0Google ScholarThere is no corresponding record for this reference.
- 21Wang, Y.; Guan, Z.-Y.; Shi, S.-W.; Jiang, Y.-R.; Zhang, J.; Yang, Y.; Wu, Q.; Wu, J.; Chen, J.-B.; Ying, W.-X.; Xu, Q.-Q.; Fan, Q.-X.; Wang, H.-F.; Zhou, L.; Wang, L.; Fang, J.; Pan, J.-Z.; Fang, Q. Pick-up Single-Cell Proteomic Analysis for Quantifying up to 3000 Proteins in a Mammalian Cell. Nat. Commun. 2024, 15 (1), 1279, DOI: 10.1038/s41467-024-45659-4Google ScholarThere is no corresponding record for this reference.
- 22Ctortecka, C.; Mechtler, K. The Rise of Single-Cell Proteomics. Anal. Sci. Adv. 2021, 2 (3–4), 84– 94, DOI: 10.1002/ansa.202000152Google ScholarThere is no corresponding record for this reference.
- 23Mansuri, M. S.; Bathla, S.; Lam, T. T.; Nairn, A. C.; Williams, K. R. Optimal Conditions for Carrying out Trypsin Digestions on Complex Proteomes: From Bulk Samples to Single Cells. J. Proteomics 2024, 297, 105109, DOI: 10.1016/j.jprot.2024.105109Google ScholarThere is no corresponding record for this reference.
- 24Woessmann, J.; Petrosius, V.; Üresin, N.; Kotol, D.; Aragon-Fernandez, P.; Hober, A.; Auf Dem Keller, U.; Edfors, F.; Schoof, E. M. Assessing the Role of Trypsin in Quantitative Plasma and Single-Cell Proteomics toward Clinical Application. Anal. Chem. 2023, 95 (36), 13649– 13658, DOI: 10.1021/acs.analchem.3c02543Google ScholarThere is no corresponding record for this reference.
- 25Slavov, N. Single-Cell Protein Analysis by Mass Spectrometry. Curr. Opin. Chem. Biol. 2021, 60, 1– 9, DOI: 10.1016/j.cbpa.2020.04.018Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
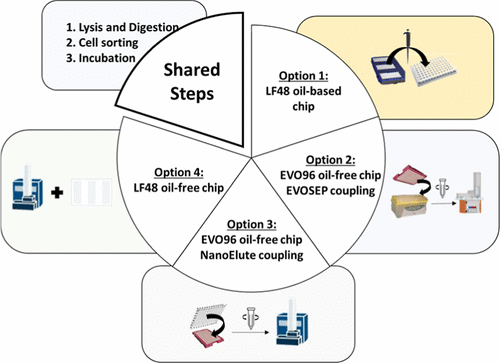
Figure 1
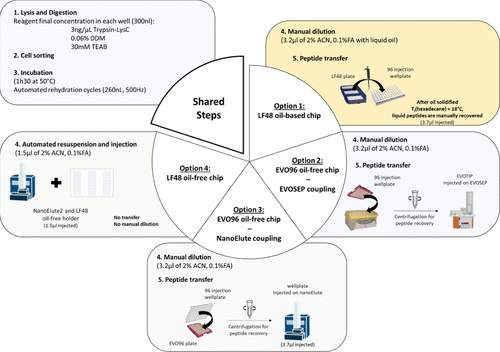
Figure 1. Four single-cell sample preparation protocols benchmarked using the cellenONE sorter/liquid dispenser. Lysis and digestion, followed by incubation with automated rehydration cycles are common steps to all three workflows. Then, option 1 relies on a LF48 oil-based plate support involving a manual dilution step in liquid oil, followed by a manual peptide transfer after oil solidification (Tf (hexadecane) = 18 °C); option 2 uses an EVO96 oil-free plate support, involving manual dilution followed by peptide loading onto preconditioned EVOTIPs after centrifugation for injection on the Evosep system; option 3 employs the same EVO96 oil-free support, repurposed for nanoElute injection, where peptides are transferred into a 96-well injection plate via centrifugation; and option 4 relies on a LF48 oil-free plate support suited for automated resuspension and direct injection on a nanoElute 2 system.
Figure 2
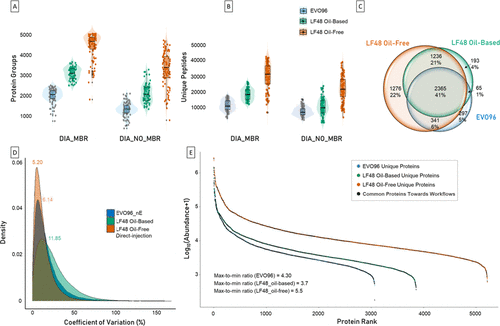
Figure 2. Benchmarking of three single-cell proteomics workflows (EVO96 (blue), LF48 oil-based (green), and LF48 oil-free (orange)) run on nanoElute 2 on 96 single-cell replicates per workflow. (A) Violin plots representing the number of protein groups identified per single cell for each tested workflow using DIA-NN with MBR (left) and without MBR (right). (B) Violin plots representing the number of unique peptides identified per single cell for each tested workflow using DIA-NN with MBR (left) and without MBR (right). (C) Venn diagram showing proteins’ overlap across the three workflows. (D) Coefficients of variation (CVs) density distributions across the three sample preparation workflows. (E) Protein abundance ranking plot comparing the dynamic range of protein groups across the three workflows. Black dots correspond to the shared proteins across the conditions, whereas the colored ones correspond to unique proteins per condition.
Figure 3
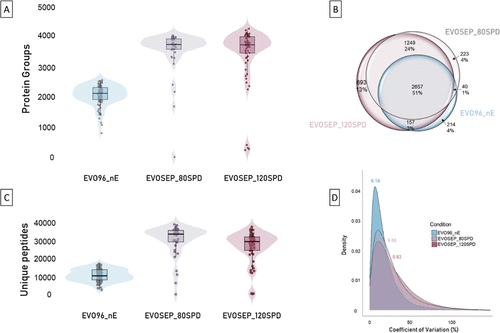
Figure 3. Comparison of SCP EVO96-based workflows across two chromatographic systems. EVO96_nE was analyzed on a nanoElute 2 system (N = 96 single-cells) compared to two Evosep workflows that were acquired using two sample throughputs (80 and 120SPD with N = 40 single-cell replicates each). (A) Violin plots representing the number of protein groups identified per single cell for each workflow (EVO96_nE in blue, EVOSEP80SPD in gray, and EVOSEP120SPD in purple). (B) Venn diagram showing the overlap of identified protein groups across the workflows. (C) Violin plots representing the number of unique peptides identified per single cell for each workflow (EVO96_nE in blue, EVOSEP_80SPD in gray, and EVOSEP_120SPD in purple). (D) Density distributions of protein abundance coefficients of variation (CVs) across the three workflows.
Figure 4
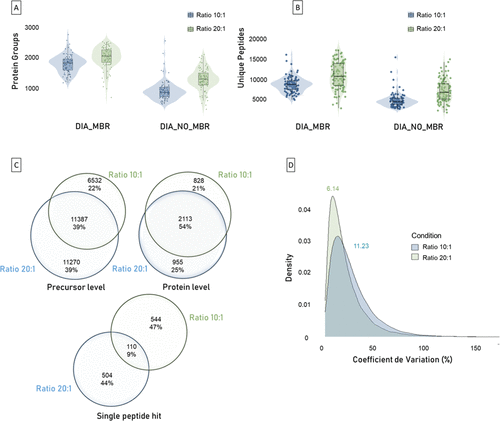
Figure 4. Evaluation of Trypsin-LysC digestion ratios. This figure presents a comprehensive analysis of HeLa single cells (N = 96 single-cell replicates per condition), comparing two digestion ratios (10:1 and 20:1). (A) Violin plots showing the distributions of protein groups identified per single-cell replicates under each digestion ratio, with or without MBR processing. (B) Violin plots showing the distributions of unique peptides identified per single-cell replicates under each digestion ratio, with or without MBR processing. (C) Venn diagrams illustrate the overlap in identified precursors (left), protein groups (right), and single-peptide hit proteins (down) between the two digestion ratios. (D) Density plots represent the distribution of coefficient of variation (CV) values across single cells.
Figure 5
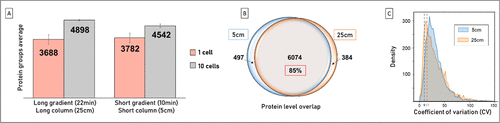
Figure 5. Gradient time and column length optimizations. (A) Average and standard deviation of the number of proteins identified per single-cell replicate (pink, N = 10) and per ten cells’ replicates (gray, N = 2), analyzed using 5 and 25 cm IonOpticks separation columns. (B) Union of all proteins identified across all 5 cm column replicates (blue) versus the union of all proteins identified across all 25 cm column replicates (orange). (C) The blue and orange dashed lines indicate the mean CV values calculated for 250 pg HeLa QC lysates (N = 3) run on a 5 cm (average 3753 protein group IDs) versus 25 cm (average 3585 protein group IDs) column, respectively.
References
This article references 25 other publications.
- 1Laisné, M.; Lupien, M.; Vallot, C. Epigenomic Heterogeneity as a Source of Tumour Evolution. Nat. Rev. Cancer 2025, 25 (1), 7– 26, DOI: 10.1038/s41568-024-00757-9There is no corresponding record for this reference.
- 2Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass Spectrometry of Single Mammalian Cells Quantifies Proteome Heterogeneity during Cell Differentiation. Genome Biol. 2018, 19 (1), 161, DOI: 10.1186/s13059-018-1547-5There is no corresponding record for this reference.
- 3Minakshi, P.; Kumar, R.; Ghosh, M.; Saini, H. M.; Ranjan, K.; Brar, B.; Prasad, G. Single-Cell Proteomics: Technology and Applications. In Single-Cell Omics; Barh, D., Azevedo, V., Eds.; Elsevier, 2019; pp 283– 318.There is no corresponding record for this reference.
- 4Tian, Y.; Li, Q.; Yang, Z.; Zhang, S.; Xu, J.; Wang, Z.; Bai, H.; Duan, J.; Zheng, B.; Li, W.; Cui, Y.; Wang, X.; Wan, R.; Fei, K.; Zhong, J.; Gao, S.; He, J.; Gay, C. M.; Zhang, J.; Wang, J.; Tang, F. Single-Cell Transcriptomic Profiling Reveals the Tumor Heterogeneity of Small-Cell Lung Cancer. Signal Transduct. Target Ther. 2022, 7 (1), 346, DOI: 10.1038/s41392-022-01150-4There is no corresponding record for this reference.
- 5Nalla, L. V.; Kanukolanu, A.; Yeduvaka, M.; Gajula, S. N. R. Advancements in Single-Cell Proteomics and Mass Spectrometry-Based Techniques for Unmasking Cellular Diversity in Triple Negative Breast Cancer. Proteomics Clin. Appl. 2025, 19 (1), e202400101 DOI: 10.1002/prca.202400101There is no corresponding record for this reference.
- 6Marino, F. Z.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular Heterogeneity in Lung Cancer: From Mechanisms of Origin to Clinical Implications. Int. J. Med. Sci. 2019, 16 (7), 981– 989, DOI: 10.7150/ijms.34739There is no corresponding record for this reference.
- 7Black, G. S.; Huang, X.; Qiao, Y.; Moos, P.; Sampath, D.; Stephens, D. M.; Woyach, J. A.; Marth, G. T. Long-Read Single-Cell RNA Sequencing Enables the Study of Cancer Subclone-Specific Genotypes and Phenotypes in Chronic Lymphocytic Leukemia. Genome Res. 2025, 35 (4), 686– 697, DOI: 10.1101/gr.279049.124There is no corresponding record for this reference.
- 8Mund, A.; Coscia, F.; Kriston, A.; Hollandi, R.; Kovács, F.; Brunner, A.-D.; Migh, E.; Schweizer, L.; Santos, A.; Bzorek, M.; Naimy, S.; Rahbek-Gjerdrum, L. M.; Dyring-Andersen, B.; Bulkescher, J.; Lukas, C.; Eckert, M. A.; Lengyel, E.; Gnann, C.; Lundberg, E.; Horvath, P.; Mann, M. Deep Visual Proteomics Defines Single-Cell Identity and Heterogeneity. Nat. Biotechnol. 2022, 40 (8), 1231– 1240, DOI: 10.1038/s41587-022-01302-5There is no corresponding record for this reference.
- 9Gerniers, A.; Bricard, O.; Dupont, P. MicroCellClust: Mining Rare and Highly Specific Subpopulations from Single-Cell Expression Data. Bioinformatics 2021, 37 (19), 3220– 3227, DOI: 10.1093/bioinformatics/btab239There is no corresponding record for this reference.
- 10Heide, T.; Househam, J.; Cresswell, G. D.; Spiteri, I.; Lynn, C.; Kimberley, C.; Mossner, M.; Zapata, L.; Gabbutt, C.; Ramazzotti, D.; Chen, B.; Fernandez-Mateos, J.; James, C.; Vinceti, A.; Berner, A.; Schmidt, M.; Lakatos, E.; Baker, A.-M.; Nichol, D.; Costa, H.; Mitchinson, M.; Werner, B.; Iorio, F.; Jansen, M.; Barnes, C.; Caravagna, G.; Shibata, D.; Bridgewater, J.; Rodriguez-Justo, M.; Magnani, L.; Graham, T. A.; Sottoriva, A. Assessment of the Evolutionary Consequence of Putative Driver Mutations in Colorectal Cancer with Spatial Multiomic Data. bioRxiv 2021, 451265, DOI: 10.1101/2021.07.14.451265There is no corresponding record for this reference.
- 11Zhang, Z.; Mathew, D.; Lim, T.; Mason, K.; Martinez, C. M.; Huang, S.; Wherry, E. J.; Susztak, K.; Minn, A. J.; Ma, Z.; Zhang, N. R. Signal Recovery in Single Cell Batch Integration. bioRxiv 2023, 539614, DOI: 10.1101/2023.05.05.539614There is no corresponding record for this reference.
- 12Fritzsch, F. S. O.; Dusny, C.; Frick, O.; Schmid, A. Single-Cell Analysis in Biotechnology, Systems Biology, and Biocatalysis. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 129– 155, DOI: 10.1146/annurev-chembioeng-062011-081056There is no corresponding record for this reference.
- 13Dodds, J. N.; Baker, E. S.; Lubman, D. M. Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead. J. Am. Soc. Mass Spectrom. 2019, 30 (11), 2185– 2195, DOI: 10.1007/s13361-019-02288-2There is no corresponding record for this reference.
- 14Fernandez-Lima, F. A.; Kaplan, D. A.; Park, M. A. Note: Integration of Trapped Ion Mobility Spectrometry with Mass Spectrometry. Rev. Sci. Instrum. 2011, 82 (12), 126106, DOI: 10.1063/1.3665933There is no corresponding record for this reference.
- 15Meier, F.; Brunner, A.-D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M. A.; Bache, N.; Hoerning, O.; Cox, J.; Räther, O.; Mann, M. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteomics 2018, 17 (12), 2534– 2545, DOI: 10.1074/mcp.TIR118.000900There is no corresponding record for this reference.
- 16Ctortecka, C.; Hartlmayr, D.; Seth, A.; Mendjan, S.; Tourniaire, G.; Udeshi, N. D.; Carr, S. A.; Mechtler, K. An Automated Nanowell-Array Workflow for Quantitative Multiplexed Single-Cell Proteomics Sample Preparation at High Sensitivity. Mol. Cell. Proteomics 2023, 22 (12), 100665, DOI: 10.1016/j.mcpro.2023.100665There is no corresponding record for this reference.
- 17Perez-Riverol, Y.; Bandla, C.; Kundu, D. J.; Kamatchinathan, S.; Bai, J.; Hewapathirana, S.; John, N. S.; Prakash, A.; Walzer, M.; Wang, S.; Vizcaíno, J. A. The PRIDE Database at 20 Years: 2025 Update. Nucleic Acids Res. 2025, 53 (D1), D543– D553, DOI: 10.1093/nar/gkae1011There is no corresponding record for this reference.
- 18Demichev, V.; Messner, C. B.; Vernardis, S. I.; Lilley, K. S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41– 44, DOI: 10.1038/s41592-019-0638-xThere is no corresponding record for this reference.
- 19Demichev, V.; Messner, C. B.; Vernardis, S. I.; Lilley, K. S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41– 44, DOI: 10.1038/s41592-019-0638-xThere is no corresponding record for this reference.
- 20Kyte, J.; Doolittle, R. F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157 (1), 105– 132, DOI: 10.1016/0022-2836(82)90515-0There is no corresponding record for this reference.
- 21Wang, Y.; Guan, Z.-Y.; Shi, S.-W.; Jiang, Y.-R.; Zhang, J.; Yang, Y.; Wu, Q.; Wu, J.; Chen, J.-B.; Ying, W.-X.; Xu, Q.-Q.; Fan, Q.-X.; Wang, H.-F.; Zhou, L.; Wang, L.; Fang, J.; Pan, J.-Z.; Fang, Q. Pick-up Single-Cell Proteomic Analysis for Quantifying up to 3000 Proteins in a Mammalian Cell. Nat. Commun. 2024, 15 (1), 1279, DOI: 10.1038/s41467-024-45659-4There is no corresponding record for this reference.
- 22Ctortecka, C.; Mechtler, K. The Rise of Single-Cell Proteomics. Anal. Sci. Adv. 2021, 2 (3–4), 84– 94, DOI: 10.1002/ansa.202000152There is no corresponding record for this reference.
- 23Mansuri, M. S.; Bathla, S.; Lam, T. T.; Nairn, A. C.; Williams, K. R. Optimal Conditions for Carrying out Trypsin Digestions on Complex Proteomes: From Bulk Samples to Single Cells. J. Proteomics 2024, 297, 105109, DOI: 10.1016/j.jprot.2024.105109There is no corresponding record for this reference.
- 24Woessmann, J.; Petrosius, V.; Üresin, N.; Kotol, D.; Aragon-Fernandez, P.; Hober, A.; Auf Dem Keller, U.; Edfors, F.; Schoof, E. M. Assessing the Role of Trypsin in Quantitative Plasma and Single-Cell Proteomics toward Clinical Application. Anal. Chem. 2023, 95 (36), 13649– 13658, DOI: 10.1021/acs.analchem.3c02543There is no corresponding record for this reference.
- 25Slavov, N. Single-Cell Protein Analysis by Mass Spectrometry. Curr. Opin. Chem. Biol. 2021, 60, 1– 9, DOI: 10.1016/j.cbpa.2020.04.018There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.5c01075.
Outliers representation for the oil-free LF48 direct injection workflow on the 96 single-cell replicates; missing values and single-peptide hits’ distributions for EVO96, LF48 oil-free, and LF48 oil-based workflows;pairwise protein abundance correlations for common proteins identified across EVO96/LF48 oil-free/LF48 oil-based and nanoElute_EVO96/EVOSEP_80SPD/EVOSEP_120SPD workflows; repartition and distribution of GRAVY scores for EVO96, LF48 oil-free, and LF48 oil-based workflows; distribution of methionine oxidations identified across workflows; and impact of enzyme-to-protein ratios (10:1 and 20:1) on peptide and protein identifications (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.