



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Trapping Polymer Entanglements via Prolonged InitiationClick to copy article linkArticle link copied!
- Suyoung LeeSuyoung LeeDepartment of Mechanical Engineering, Northwestern University, Evanston, Illinois 60208, United StatesMore by Suyoung Lee
- Yan HuangYan HuangDepartment of Mechanical Engineering, Northwestern University, Evanston, Illinois 60208, United StatesMore by Yan Huang
- Jinyue DaiJinyue DaiDepartment of Mechanical Engineering, Northwestern University, Evanston, Illinois 60208, United StatesMore by Jinyue Dai
- Haeji KimHaeji KimDepartment of Mechanical Engineering, Northwestern University, Evanston, Illinois 60208, United StatesMore by Haeji Kim
- Junsoo Kim*Junsoo Kim*Email: [email protected]. Phone: +1 (847) 491-4322.Department of Mechanical Engineering, Northwestern University, Evanston, Illinois 60208, United StatesMore by Junsoo Kim
Abstract
Entanglements in polymer networks can be either trapped or transient, and their ratio is crucial to the mechanical properties of soft materials. Traditionally, entanglement formation has been mostly linked to chain length─polymers entangle when their lengths exceed the entanglement molecular weight─without independent control of the ratio between the two. Here, we demonstrate that the initiation rate significantly affects the fraction of trapped entanglements and the resulting mechanical properties. We hypothesize that more monomers grow in the presence of existing polymers as the initiation rate decreases, forming more trapped entanglements. To demonstrate this, we synthesize UV-curable, highly entangled polyacrylamide hydrogels in which the initiation rate varies with UV intensity. Upon swelling, transient entanglements can detangle, whereas trapped entanglements cannot, by which we characterize the fraction of entanglements. We observe that the swelling ratio of the same precursor decreases significantly as UV intensity decreases, indicating a higher fraction of trapped entanglements. Additionally, such hydrogels with many trapped entanglements exhibit superior fracture resistance due to their swell-resistance. Our work offers a kinetic approach to network topology design, expanding the material property space of polymer networks.
This publication is licensed for personal use by The American Chemical Society.
1. Introduction
2. Hypothesis
Figure 1
Figure 1. Control the fraction of trapped entanglements via prolonged initiation. (A) A precursor for highly entangled long-chain polymer networks. (B) Simultaneous triggering of initiators. (C) The fully cured polymer network with fast initiation. Entanglements are either trapped or transient. (D) A low fraction of trapped entanglements results in significant swelling. (E, F) Gradual triggering of initiators. Monomers are present inside the free volume. (G) Monomers inside the free volume polymerize in the presence of polymers, forming trapped entanglements. The monomer concentration at the overlapping region decreases, allowing additional monomers to diffuse in. (H) The resulting polymer network has the same concentration as (C) but a different fraction of trapped entanglements. (I) Limited swelling due to a high fraction of trapped entanglements.
3. Materials and Experimental Methods
3.1. Materials
3.2. Preparation of the Precursor of Highly Entangled PAAm Hydrogel
3.3. Photopolymerization
3.4. Mechanical Tests for Hydrogels
3.5. DLS Analysis to Measure the Length of Synthesized Polymer Backbones
3.6. Measurement of the Heat Flux from Gelation of PAAm Hydrogel
4. Results and Discussion
4.1. Model Material System
Figure 2
Figure 2. Measurement of the heat flux from hydrogel gelation. (A) The experiment setup for the heat flux measurement. (B) The temperature profile by polymerization and UV exposure. (C) The temperature of the precursor, Tm, over time upon UV exposure. (D) The heat flux generated by polymerization over time at different UV intensities.
Figure 3
Figure 3. Validation of the model material. (A) Polymer chain size at different UV intensities measured by DLS. (B) Gel fraction at different UV intensities.
4.2. Swelling Ratio
Figure 4
Figure 4. Swelling ratio of PAAm hydrogels synthesized at different UV intensities. (A) The swelling ratio as a function of the exposure time at various UV Intensities. (B) The swelling ratio of the fully cured samples as a function of the UV Intensity. (C) The swelling ratio as a function of the exposed energy. All samples were prepared with a precursor of W = 2, C = 10–5, and I = 4 × 10–6.
4.3. Elastic and Fracture Properties
Figure 5
Figure 5. Stress–strain curves of fully swollen, fully cured PAAm hydrogels at various UV intensities. (A) Stress–strain curves at different UV intensities. (B) Young’s modulus as a function of the UV intensity. (C) Strength as a function of the UV intensity. (D) Work of fracture as a function of the UV intensity. (E) Toughness at different UV intensities.
4.4. Rheological Properties
Figure 6
Figure 6. Rheological properties of PAAm hydrogels synthesized at various UV intensities. (A) Storage (filled dots) and loss modulus (open dots), (B) Tan δ for various PAAm hydrogels synthesized at three different UV intensities.
4.5. Stoichiometry of Precursors
Figure 7
Figure 7. Swelling ratio of PAAm hydrogels depending on the precursor conditions. Swelling ratio as a function of (A) W, (B) C, and (C) I for various PAAm hydrogels synthesized at different UV intensities.
5. Conclusions
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.macromol.5c03018.
Raw data of the graphs in the main figures; Heat flux from hydrogel gelation at various UV intensities; Temperature profiles in the heat flux measurement; Toughness of PAAm hydrogels divided by φ2/3 at different UV intensities; Heat flux analysis (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Junsoo Kim - Department of Mechanical Engineering, Northwestern University, Evanston, Illinois 60208, United States;
 https://orcid.org/0000-0001-7340-2770;
https://orcid.org/0000-0001-7340-2770;
Suyoung Lee: Hydrogel fabrication, Formal Analysis, Investigation, Validation, Writing─drafting, reviewing and editing. Yan Huang: Toughness measurement, Gel fraction measurement Jinyue Dai: DLS analysis Haeji Kim: Hydrogel fabrication Junsoo Kim: Supervision, Conceptualization, Methodology, Formal Analysis, Validation, Writing─drafting, reviewing and editing.
Acknowledgments
This work was primarily supported by the National Science Foundation’s MRSEC program (DMR-2308691) at the Materials Research Center of Northwestern University. This work was also supported by start-up funds from Northwestern University.
References
This article references 47 other publications.
- 1Rubinstein, M.; Colby, R. H. Polymer Physics; Oxford University Press, 2003. DOI: 10.1093/oso/9780198520597.001.0001 .Google ScholarThere is no corresponding record for this reference.
- 2Edwards, S. F. Statistical Mechanics with Topological Constraints: I. Proc. Phys. Soc. 1967, 91 (3), 513, DOI: 10.1088/0370-1328/91/3/301Google ScholarThere is no corresponding record for this reference.
- 3Rubinstein, M.; Panyukov, S. Nonaffine Deformation and Elasticity of Polymer Networks. Macromolecules 1997, 30 (25), 8036– 8044, DOI: 10.1021/ma970364kGoogle ScholarThere is no corresponding record for this reference.
- 4Norioka, C.; Inamoto, Y.; Hajime, C.; Kawamura, A.; Miyata, T. A Universal Method to Easily Design Tough and Stretchable Hydrogels. NPG Asia Mater. 2021, 13 (1), 34 DOI: 10.1038/s41427-021-00302-2Google ScholarThere is no corresponding record for this reference.
- 5Kim, J.; Zhang, G.; Shi, M.; Suo, Z. Fracture, Fatigue, and Friction of Polymers in Which Entanglements Greatly Outnumber Cross-Links. Science 2021, 374 (6564), 212– 216, DOI: 10.1126/science.abg6320Google ScholarThere is no corresponding record for this reference.
- 6Nian, G.; Kim, J.; Bao, X.; Suo, Z. Making Highly Elastic and Tough Hydrogels from Doughs. Adv. Mater. 2022, 34 (50), 2206577 DOI: 10.1002/adma.202206577Google ScholarThere is no corresponding record for this reference.
- 7Steck, J.; Kim, J.; Kutsovsky, Y.; Suo, Z. Multiscale Stress Deconcentration Amplifies Fatigue Resistance of Rubber. Nature 2023, 624 (7991), 303– 308, DOI: 10.1038/s41586-023-06782-2Google ScholarThere is no corresponding record for this reference.
- 8Fu, L.; Li, L.; Bian, Q.; Xue, B.; Jin, J.; Li, J.; Cao, Y.; Jiang, Q.; Li, H. Cartilage-like Protein Hydrogels Engineered via Entanglement. Nature 2023, 618 (7966), 740– 747, DOI: 10.1038/s41586-023-06037-0Google ScholarThere is no corresponding record for this reference.
- 9Dhand, A. P.; Davidson, M. D.; Zlotnick, H. M.; Kolibaba, T. J.; Killgore, J. P.; Burdick, J. A. Additive Manufacturing of Highly Entangled Polymer Networks. Science 2024, 385 (6708), 566– 572, DOI: 10.1126/science.adn6925Google ScholarThere is no corresponding record for this reference.
- 10Shi, M.; Kim, J.; Nian, G.; Suo, Z. Highly Entangled Hydrogels with Degradable Crosslinks. Extreme Mech. Lett. 2023, 59, 101953 DOI: 10.1016/j.eml.2022.101953Google ScholarThere is no corresponding record for this reference.
- 11Wang, F.-S.; Kosovsky, L. M.; Krist, E. C.; Kruse, B. J.; Zhukhovitskiy, A. V. Trapped Entanglements in Polymer Networks: Formation and Characterization. Trends Chem. 2024, 6 (8), 447– 458, DOI: 10.1016/j.trechm.2024.05.005Google ScholarThere is no corresponding record for this reference.
- 12Yamamoto, T.; Campbell, J. A.; Panyukov, S.; Rubinstein, M. Scaling Theory of Swelling and Deswelling of Polymer Networks. Macromolecules 2022, 55 (9), 3588– 3601, DOI: 10.1021/acs.macromol.1c02553Google ScholarThere is no corresponding record for this reference.
- 13Agudelo, D. C.; Roth, L. E.; Vega, D. A.; Vallés, E. M.; Villar, M. A. Dynamic Response of Transiently Trapped Entanglements in Polymer Networks. Polymer 2014, 55 (4), 1061– 1069, DOI: 10.1016/j.polymer.2014.01.010Google ScholarThere is no corresponding record for this reference.
- 14Albers, P. T. M.; van der Ven, L. G. J.; van Benthem, R. A. T. M.; Esteves, A. C. C.; de With, G. Water Swelling Behavior of Poly(Ethylene Glycol)-Based Polyurethane Networks. Macromolecules 2020, 53 (3), 862– 874, DOI: 10.1021/acs.macromol.9b02275Google ScholarThere is no corresponding record for this reference.
- 15Lang, M.; Scholz, R.; Löser, L.; Bunk, C.; Fribiczer, N.; Seiffert, S.; Böhme, F.; Saalwächter, K. Swelling and Residual Bond Orientations of Polymer Model Gels: The Entanglement-Free Limit. Macromolecules 2022, 55 (14), 5997– 6014, DOI: 10.1021/acs.macromol.2c00589Google ScholarThere is no corresponding record for this reference.
- 16Rubinstein, M. Dynamics of Ring Polymers in the Presence of Fixed Obstacles. Phys. Rev. Lett. 1986, 57 (24), 3023– 3026, DOI: 10.1103/PhysRevLett.57.3023Google ScholarThere is no corresponding record for this reference.
- 17Obukhov, S. P.; Rubinstein, M.; Colby, R. H. Network Modulus and Superelasticity. Macromolecules 1994, 27 (12), 3191– 3198, DOI: 10.1021/ma00090a012Google ScholarThere is no corresponding record for this reference.
- 18Rajeev, M.; Helms, C. C. A Study of the Relationship between Polymer Solution Entanglement and Electrospun PCL Fiber Mechanics. Polymers 2023, 15 (23), 4555, DOI: 10.3390/polym15234555Google ScholarThere is no corresponding record for this reference.
- 19Zhao, K.; Khan, H. U.; Li, R.; Su, Y.; Amassian, A. Entanglement of Conjugated Polymer Chains Influences Molecular Self-Assembly and Carrier Transport. Adv. Funct. Mater. 2013, 23 (48), 6024– 6035, DOI: 10.1002/adfm.201301007Google ScholarThere is no corresponding record for this reference.
- 20Fetters, L. J. Determination of the Intermolecular Entanglement Coupling Spacings in Polyisoprene by Viscosity Measurements. J. Res. Natl. Bur. Stand. Sect. A: Phys. Chem. 1965, 69A (1), 33– 37, DOI: 10.6028/jres.069A.006Google ScholarThere is no corresponding record for this reference.
- 21Porter, R. S.; Johnson, J. F. The Entanglement Concept in Polymer Systems. Chem. Rev. 1966, 66 (1), 1– 27, DOI: 10.1021/cr60239a001Google ScholarThere is no corresponding record for this reference.
- 22Wool, R. P. Polymer Entanglements. Macromolecules 1993, 26 (7), 1564– 1569, DOI: 10.1021/ma00059a012Google ScholarThere is no corresponding record for this reference.
- 23Irvine, G.; Myronidis, K.; Pinto, F.; Kopeć, M. Highly Entangled Hydrogels by Photoiniferter-Mediated Polymerization. Angew. Chem., Int. Ed. 2025, 64 (17), e202421970 DOI: 10.1002/anie.202421970Google ScholarThere is no corresponding record for this reference.
- 24Zhu, R.; Zheng, Z.; Zhu, D.; Wang, X. Hydrogels with High Sacrifice Efficiency of Sacrificial Bonds and with High Strength and Toughness Due to Dense Entanglements of Polymer Chains. J. Colloid Interface Sci. 2025, 677, 687– 696, DOI: 10.1016/j.jcis.2024.08.008Google ScholarThere is no corresponding record for this reference.
- 25Trigg, E. B.; Wiegart, L.; Fluerasu, A.; Koerner, H. Dynamics of Polymerization and Gelation in Epoxy Nanocomposites via X-Ray Photon Correlation Spectroscopy. Macromolecules 2021, 54 (13), 6575– 6584, DOI: 10.1021/acs.macromol.1c00727Google ScholarThere is no corresponding record for this reference.
- 26Gao, H.; Huang, X.; Arockiyasamy, A. R.; Zhou, X.; Ding, X.; Zheng, L.; Liu, Y.; Ye, F.; Weng, Z.; Wu, L. Vat Photopolymerization of Stretchable Foam with Highly Entangled and Crosslinked Structures. Nat. Commun. 2025, 16 (1), 4756 DOI: 10.1038/s41467-025-60087-8Google ScholarThere is no corresponding record for this reference.
- 27Zhu, R.; Zhu, D.; Zheng, Z.; Wang, X. Tough Double Network Hydrogels with Rapid Self-Reinforcement and Low Hysteresis Based on Highly Entangled Networks. Nat. Commun. 2024, 15 (1), 1344 DOI: 10.1038/s41467-024-45485-8Google ScholarThere is no corresponding record for this reference.
- 28Puza, F.; Zheng, Y.; Han, L.; Xue, L.; Cui, J. Physical Entanglement Hydrogels: Ultrahigh Water Content but Good Toughness and Stretchability. Polym. Chem. 2020, 11 (13), 2339– 2345, DOI: 10.1039/D0PY00294AGoogle ScholarThere is no corresponding record for this reference.
- 29Kong, D.-C.; Yang, M.-H.; Zhang, X.-S.; Du, Z.-C.; Fu, Q.; Gao, X.-Q.; Gong, J.-W. Control of Polymer Properties by Entanglement: A Review. Macromol. Mater. Eng. 2021, 306 (12), 2100536 DOI: 10.1002/mame.202100536Google ScholarThere is no corresponding record for this reference.
- 30Luu, T. T. H.; Jia, Z.; Kanaev, A.; Museur, L. Effect of Light Intensity on the Free-Radical Photopolymerization Kinetics of 2-Hydroxyethyl Methacrylate: Experiments and Simulations. J. Phys. Chem. B 2020, 124 (31), 6857– 6866, DOI: 10.1021/acs.jpcb.0c03140Google ScholarThere is no corresponding record for this reference.
- 31Li, L.; Lee, L. J. Photopolymerization of HEMA/DEGDMA Hydrogels in Solution. Polymer 2005, 46 (25), 11540– 11547, DOI: 10.1016/j.polymer.2005.10.051Google ScholarThere is no corresponding record for this reference.
- 32Sheth, S.; Jain, E.; Karadaghy, A.; Syed, S.; Stevenson, H.; Zustiak, S. P. UV Dose Governs UV-Polymerized Polyacrylamide Hydrogel Modulus. Int. J. Polym. Sci. 2017, 2017 (1), 5147482 DOI: 10.1155/2017/5147482Google ScholarThere is no corresponding record for this reference.
- 33Wei, A.; Wang, Q.; Liu, J.; Huang, Y.; Li, H.; Zhu, Z.; Wang, T.; Yu, Y. Co-Initiating-System Dual-Mechanism Drives the Design of Printable Entangled Polymer Multinetworks. Nat. Commun. 2025, 16 (1), 4407 DOI: 10.1038/s41467-025-59669-3Google ScholarThere is no corresponding record for this reference.
- 34Dookhith, A. Z.; Lynd, N. A.; Creton, C.; Sanoja, G. E. Controlling Architecture and Mechanical Properties of Polyether Networks with Organoaluminum Catalysts. Macromolecules 2022, 55 (13), 5601– 5609, DOI: 10.1021/acs.macromol.2c00602Google ScholarThere is no corresponding record for this reference.
- 35Patel, S. K.; Rodriguez, F.; Cohen, C. Mechanical and Swelling Properties of Polyacrylamide Gel Spheres. Polymer 1989, 30 (12), 2198– 2203, DOI: 10.1016/0032-3861(89)90249-8Google ScholarThere is no corresponding record for this reference.
- 36Augé, A.; Zhao, Y. What Determines the Volume Transition Temperature of UCST Acrylamide–Acrylonitrile Hydrogels?. RSC Adv. 2016, 6 (74), 70616– 70623, DOI: 10.1039/C6RA12720GGoogle ScholarThere is no corresponding record for this reference.
- 37Kopač, T. Mathematical Model for Characterization of Temperature-Responsive Polymers: A Study on the Rheological Behavior of Gelatin and Poly(N-Isopropylacrylamide). Polym. Test. 2024, 133, 108402 DOI: 10.1016/j.polymertesting.2024.108402Google ScholarThere is no corresponding record for this reference.
- 38D’Oria, G.; Zeng, X.; Limbach, H. J.; Hartmann, C.; Ahrné, L.; Gunes, D. Z. Effect of Stirring Speed on Low Acyl Gellan Gum Fluid Gels’ Rheology, Particle Morphology and Physical Ageing. Food Hydrocolloids 2024, 149, 109614 DOI: 10.1016/j.foodhyd.2023.109614Google ScholarThere is no corresponding record for this reference.
- 39Suslick, B. A.; Hemmer, J.; Groce, B. R.; Stawiasz, K. J.; Geubelle, P. H.; Malucelli, G.; Mariani, A.; Moore, J. S.; Pojman, J. A.; Sottos, N. R. Frontal Polymerizations: From Chemical Perspectives to Macroscopic Properties and Applications. Chem. Rev. 2023, 123 (6), 3237– 3298, DOI: 10.1021/acs.chemrev.2c00686Google ScholarThere is no corresponding record for this reference.
- 40Hale, G. M.; Querry, M. R. Optical Constants of Water in the 200-Nm to 200-Μm Wavelength Region. Appl. Opt. 1973, 12 (3), 555– 563, DOI: 10.1364/AO.12.000555Google ScholarThere is no corresponding record for this reference.
- 41Kitamura, R.; Pilon, L.; Jonasz, M. Optical Constants of Silica Glass from Extreme Ultraviolet to Far Infrared at near Room Temperature. Appl. Opt. 2007, 46 (33), 8118– 8133, DOI: 10.1364/AO.46.008118Google ScholarThere is no corresponding record for this reference.
- 42Yang, C.; Yin, T.; Suo, Z. Polyacrylamide Hydrogels. I. Network Imperfection. J. Mech. Phys. Solids 2019, 131, 43– 55, DOI: 10.1016/j.jmps.2019.06.018Google ScholarThere is no corresponding record for this reference.
- 43Graessley, W. W. The Entanglement Concept in Polymer Rheology. In The Entanglement Concept in Polymer Rheology; Springer: Berlin, Heidelberg, 1974; pp 1– 179 DOI: 10.1007/BFb0031037 .Google ScholarThere is no corresponding record for this reference.
- 44Wang, S.-Q.; Ravindranath, S.; Wang, Y.; Boukany, P. New Theoretical Considerations in Polymer Rheology: Elastic Breakdown of Chain Entanglement Network. J. Chem. Phys. 2007, 127 (6), 064903 DOI: 10.1063/1.2753156Google ScholarThere is no corresponding record for this reference.
- 45Cao, X.-Z.; Forest, M. G. Rheological Tuning of Entangled Polymer Networks by Transient Cross-Links. J. Phys. Chem. B 2019, 123 (5), 974– 982, DOI: 10.1021/acs.jpcb.8b09357Google ScholarThere is no corresponding record for this reference.
- 46Feng, Y.; Wu, D.; Li, R.; Akcora, P. Rheological Properties of Crosslinked Unentangled and Entangled Poly(Methyl Acrylate) Nanocomposite Networks. Polymer 2022, 255, 125150 DOI: 10.1016/j.polymer.2022.125150Google ScholarThere is no corresponding record for this reference.
- 47Pawlak, A.; Krajenta, J. Entanglements of Macromolecules and Their Influence on Rheological and Mechanical Properties of Polymers. Molecules 2024, 29 (14), 3410, DOI: 10.3390/molecules29143410Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract

Figure 1
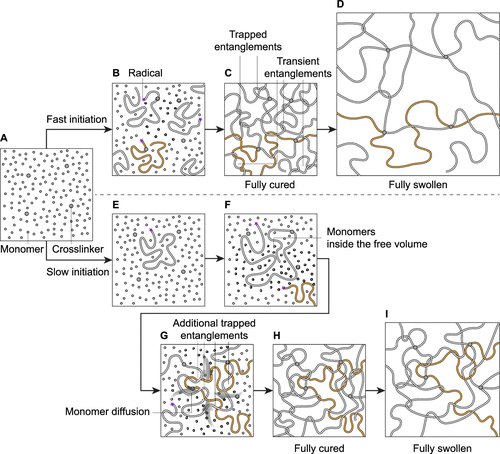
Figure 1. Control the fraction of trapped entanglements via prolonged initiation. (A) A precursor for highly entangled long-chain polymer networks. (B) Simultaneous triggering of initiators. (C) The fully cured polymer network with fast initiation. Entanglements are either trapped or transient. (D) A low fraction of trapped entanglements results in significant swelling. (E, F) Gradual triggering of initiators. Monomers are present inside the free volume. (G) Monomers inside the free volume polymerize in the presence of polymers, forming trapped entanglements. The monomer concentration at the overlapping region decreases, allowing additional monomers to diffuse in. (H) The resulting polymer network has the same concentration as (C) but a different fraction of trapped entanglements. (I) Limited swelling due to a high fraction of trapped entanglements.
Figure 2
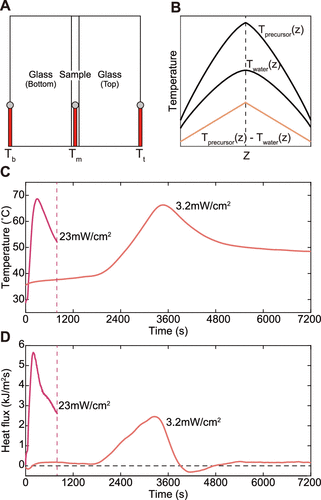
Figure 2. Measurement of the heat flux from hydrogel gelation. (A) The experiment setup for the heat flux measurement. (B) The temperature profile by polymerization and UV exposure. (C) The temperature of the precursor, Tm, over time upon UV exposure. (D) The heat flux generated by polymerization over time at different UV intensities.
Figure 3

Figure 3. Validation of the model material. (A) Polymer chain size at different UV intensities measured by DLS. (B) Gel fraction at different UV intensities.
Figure 4
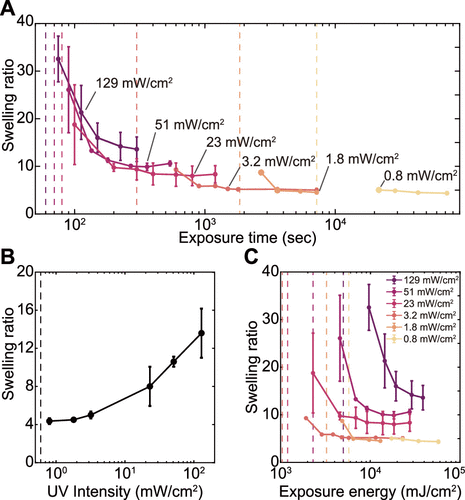
Figure 4. Swelling ratio of PAAm hydrogels synthesized at different UV intensities. (A) The swelling ratio as a function of the exposure time at various UV Intensities. (B) The swelling ratio of the fully cured samples as a function of the UV Intensity. (C) The swelling ratio as a function of the exposed energy. All samples were prepared with a precursor of W = 2, C = 10–5, and I = 4 × 10–6.
Figure 5

Figure 5. Stress–strain curves of fully swollen, fully cured PAAm hydrogels at various UV intensities. (A) Stress–strain curves at different UV intensities. (B) Young’s modulus as a function of the UV intensity. (C) Strength as a function of the UV intensity. (D) Work of fracture as a function of the UV intensity. (E) Toughness at different UV intensities.
Figure 6

Figure 6. Rheological properties of PAAm hydrogels synthesized at various UV intensities. (A) Storage (filled dots) and loss modulus (open dots), (B) Tan δ for various PAAm hydrogels synthesized at three different UV intensities.
Figure 7
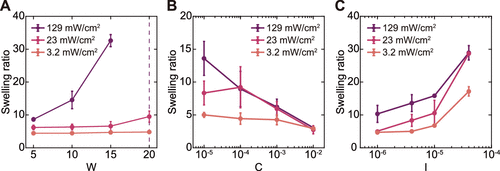
Figure 7. Swelling ratio of PAAm hydrogels depending on the precursor conditions. Swelling ratio as a function of (A) W, (B) C, and (C) I for various PAAm hydrogels synthesized at different UV intensities.
References
This article references 47 other publications.
- 1Rubinstein, M.; Colby, R. H. Polymer Physics; Oxford University Press, 2003. DOI: 10.1093/oso/9780198520597.001.0001 .There is no corresponding record for this reference.
- 2Edwards, S. F. Statistical Mechanics with Topological Constraints: I. Proc. Phys. Soc. 1967, 91 (3), 513, DOI: 10.1088/0370-1328/91/3/301There is no corresponding record for this reference.
- 3Rubinstein, M.; Panyukov, S. Nonaffine Deformation and Elasticity of Polymer Networks. Macromolecules 1997, 30 (25), 8036– 8044, DOI: 10.1021/ma970364kThere is no corresponding record for this reference.
- 4Norioka, C.; Inamoto, Y.; Hajime, C.; Kawamura, A.; Miyata, T. A Universal Method to Easily Design Tough and Stretchable Hydrogels. NPG Asia Mater. 2021, 13 (1), 34 DOI: 10.1038/s41427-021-00302-2There is no corresponding record for this reference.
- 5Kim, J.; Zhang, G.; Shi, M.; Suo, Z. Fracture, Fatigue, and Friction of Polymers in Which Entanglements Greatly Outnumber Cross-Links. Science 2021, 374 (6564), 212– 216, DOI: 10.1126/science.abg6320There is no corresponding record for this reference.
- 6Nian, G.; Kim, J.; Bao, X.; Suo, Z. Making Highly Elastic and Tough Hydrogels from Doughs. Adv. Mater. 2022, 34 (50), 2206577 DOI: 10.1002/adma.202206577There is no corresponding record for this reference.
- 7Steck, J.; Kim, J.; Kutsovsky, Y.; Suo, Z. Multiscale Stress Deconcentration Amplifies Fatigue Resistance of Rubber. Nature 2023, 624 (7991), 303– 308, DOI: 10.1038/s41586-023-06782-2There is no corresponding record for this reference.
- 8Fu, L.; Li, L.; Bian, Q.; Xue, B.; Jin, J.; Li, J.; Cao, Y.; Jiang, Q.; Li, H. Cartilage-like Protein Hydrogels Engineered via Entanglement. Nature 2023, 618 (7966), 740– 747, DOI: 10.1038/s41586-023-06037-0There is no corresponding record for this reference.
- 9Dhand, A. P.; Davidson, M. D.; Zlotnick, H. M.; Kolibaba, T. J.; Killgore, J. P.; Burdick, J. A. Additive Manufacturing of Highly Entangled Polymer Networks. Science 2024, 385 (6708), 566– 572, DOI: 10.1126/science.adn6925There is no corresponding record for this reference.
- 10Shi, M.; Kim, J.; Nian, G.; Suo, Z. Highly Entangled Hydrogels with Degradable Crosslinks. Extreme Mech. Lett. 2023, 59, 101953 DOI: 10.1016/j.eml.2022.101953There is no corresponding record for this reference.
- 11Wang, F.-S.; Kosovsky, L. M.; Krist, E. C.; Kruse, B. J.; Zhukhovitskiy, A. V. Trapped Entanglements in Polymer Networks: Formation and Characterization. Trends Chem. 2024, 6 (8), 447– 458, DOI: 10.1016/j.trechm.2024.05.005There is no corresponding record for this reference.
- 12Yamamoto, T.; Campbell, J. A.; Panyukov, S.; Rubinstein, M. Scaling Theory of Swelling and Deswelling of Polymer Networks. Macromolecules 2022, 55 (9), 3588– 3601, DOI: 10.1021/acs.macromol.1c02553There is no corresponding record for this reference.
- 13Agudelo, D. C.; Roth, L. E.; Vega, D. A.; Vallés, E. M.; Villar, M. A. Dynamic Response of Transiently Trapped Entanglements in Polymer Networks. Polymer 2014, 55 (4), 1061– 1069, DOI: 10.1016/j.polymer.2014.01.010There is no corresponding record for this reference.
- 14Albers, P. T. M.; van der Ven, L. G. J.; van Benthem, R. A. T. M.; Esteves, A. C. C.; de With, G. Water Swelling Behavior of Poly(Ethylene Glycol)-Based Polyurethane Networks. Macromolecules 2020, 53 (3), 862– 874, DOI: 10.1021/acs.macromol.9b02275There is no corresponding record for this reference.
- 15Lang, M.; Scholz, R.; Löser, L.; Bunk, C.; Fribiczer, N.; Seiffert, S.; Böhme, F.; Saalwächter, K. Swelling and Residual Bond Orientations of Polymer Model Gels: The Entanglement-Free Limit. Macromolecules 2022, 55 (14), 5997– 6014, DOI: 10.1021/acs.macromol.2c00589There is no corresponding record for this reference.
- 16Rubinstein, M. Dynamics of Ring Polymers in the Presence of Fixed Obstacles. Phys. Rev. Lett. 1986, 57 (24), 3023– 3026, DOI: 10.1103/PhysRevLett.57.3023There is no corresponding record for this reference.
- 17Obukhov, S. P.; Rubinstein, M.; Colby, R. H. Network Modulus and Superelasticity. Macromolecules 1994, 27 (12), 3191– 3198, DOI: 10.1021/ma00090a012There is no corresponding record for this reference.
- 18Rajeev, M.; Helms, C. C. A Study of the Relationship between Polymer Solution Entanglement and Electrospun PCL Fiber Mechanics. Polymers 2023, 15 (23), 4555, DOI: 10.3390/polym15234555There is no corresponding record for this reference.
- 19Zhao, K.; Khan, H. U.; Li, R.; Su, Y.; Amassian, A. Entanglement of Conjugated Polymer Chains Influences Molecular Self-Assembly and Carrier Transport. Adv. Funct. Mater. 2013, 23 (48), 6024– 6035, DOI: 10.1002/adfm.201301007There is no corresponding record for this reference.
- 20Fetters, L. J. Determination of the Intermolecular Entanglement Coupling Spacings in Polyisoprene by Viscosity Measurements. J. Res. Natl. Bur. Stand. Sect. A: Phys. Chem. 1965, 69A (1), 33– 37, DOI: 10.6028/jres.069A.006There is no corresponding record for this reference.
- 21Porter, R. S.; Johnson, J. F. The Entanglement Concept in Polymer Systems. Chem. Rev. 1966, 66 (1), 1– 27, DOI: 10.1021/cr60239a001There is no corresponding record for this reference.
- 22Wool, R. P. Polymer Entanglements. Macromolecules 1993, 26 (7), 1564– 1569, DOI: 10.1021/ma00059a012There is no corresponding record for this reference.
- 23Irvine, G.; Myronidis, K.; Pinto, F.; Kopeć, M. Highly Entangled Hydrogels by Photoiniferter-Mediated Polymerization. Angew. Chem., Int. Ed. 2025, 64 (17), e202421970 DOI: 10.1002/anie.202421970There is no corresponding record for this reference.
- 24Zhu, R.; Zheng, Z.; Zhu, D.; Wang, X. Hydrogels with High Sacrifice Efficiency of Sacrificial Bonds and with High Strength and Toughness Due to Dense Entanglements of Polymer Chains. J. Colloid Interface Sci. 2025, 677, 687– 696, DOI: 10.1016/j.jcis.2024.08.008There is no corresponding record for this reference.
- 25Trigg, E. B.; Wiegart, L.; Fluerasu, A.; Koerner, H. Dynamics of Polymerization and Gelation in Epoxy Nanocomposites via X-Ray Photon Correlation Spectroscopy. Macromolecules 2021, 54 (13), 6575– 6584, DOI: 10.1021/acs.macromol.1c00727There is no corresponding record for this reference.
- 26Gao, H.; Huang, X.; Arockiyasamy, A. R.; Zhou, X.; Ding, X.; Zheng, L.; Liu, Y.; Ye, F.; Weng, Z.; Wu, L. Vat Photopolymerization of Stretchable Foam with Highly Entangled and Crosslinked Structures. Nat. Commun. 2025, 16 (1), 4756 DOI: 10.1038/s41467-025-60087-8There is no corresponding record for this reference.
- 27Zhu, R.; Zhu, D.; Zheng, Z.; Wang, X. Tough Double Network Hydrogels with Rapid Self-Reinforcement and Low Hysteresis Based on Highly Entangled Networks. Nat. Commun. 2024, 15 (1), 1344 DOI: 10.1038/s41467-024-45485-8There is no corresponding record for this reference.
- 28Puza, F.; Zheng, Y.; Han, L.; Xue, L.; Cui, J. Physical Entanglement Hydrogels: Ultrahigh Water Content but Good Toughness and Stretchability. Polym. Chem. 2020, 11 (13), 2339– 2345, DOI: 10.1039/D0PY00294AThere is no corresponding record for this reference.
- 29Kong, D.-C.; Yang, M.-H.; Zhang, X.-S.; Du, Z.-C.; Fu, Q.; Gao, X.-Q.; Gong, J.-W. Control of Polymer Properties by Entanglement: A Review. Macromol. Mater. Eng. 2021, 306 (12), 2100536 DOI: 10.1002/mame.202100536There is no corresponding record for this reference.
- 30Luu, T. T. H.; Jia, Z.; Kanaev, A.; Museur, L. Effect of Light Intensity on the Free-Radical Photopolymerization Kinetics of 2-Hydroxyethyl Methacrylate: Experiments and Simulations. J. Phys. Chem. B 2020, 124 (31), 6857– 6866, DOI: 10.1021/acs.jpcb.0c03140There is no corresponding record for this reference.
- 31Li, L.; Lee, L. J. Photopolymerization of HEMA/DEGDMA Hydrogels in Solution. Polymer 2005, 46 (25), 11540– 11547, DOI: 10.1016/j.polymer.2005.10.051There is no corresponding record for this reference.
- 32Sheth, S.; Jain, E.; Karadaghy, A.; Syed, S.; Stevenson, H.; Zustiak, S. P. UV Dose Governs UV-Polymerized Polyacrylamide Hydrogel Modulus. Int. J. Polym. Sci. 2017, 2017 (1), 5147482 DOI: 10.1155/2017/5147482There is no corresponding record for this reference.
- 33Wei, A.; Wang, Q.; Liu, J.; Huang, Y.; Li, H.; Zhu, Z.; Wang, T.; Yu, Y. Co-Initiating-System Dual-Mechanism Drives the Design of Printable Entangled Polymer Multinetworks. Nat. Commun. 2025, 16 (1), 4407 DOI: 10.1038/s41467-025-59669-3There is no corresponding record for this reference.
- 34Dookhith, A. Z.; Lynd, N. A.; Creton, C.; Sanoja, G. E. Controlling Architecture and Mechanical Properties of Polyether Networks with Organoaluminum Catalysts. Macromolecules 2022, 55 (13), 5601– 5609, DOI: 10.1021/acs.macromol.2c00602There is no corresponding record for this reference.
- 35Patel, S. K.; Rodriguez, F.; Cohen, C. Mechanical and Swelling Properties of Polyacrylamide Gel Spheres. Polymer 1989, 30 (12), 2198– 2203, DOI: 10.1016/0032-3861(89)90249-8There is no corresponding record for this reference.
- 36Augé, A.; Zhao, Y. What Determines the Volume Transition Temperature of UCST Acrylamide–Acrylonitrile Hydrogels?. RSC Adv. 2016, 6 (74), 70616– 70623, DOI: 10.1039/C6RA12720GThere is no corresponding record for this reference.
- 37Kopač, T. Mathematical Model for Characterization of Temperature-Responsive Polymers: A Study on the Rheological Behavior of Gelatin and Poly(N-Isopropylacrylamide). Polym. Test. 2024, 133, 108402 DOI: 10.1016/j.polymertesting.2024.108402There is no corresponding record for this reference.
- 38D’Oria, G.; Zeng, X.; Limbach, H. J.; Hartmann, C.; Ahrné, L.; Gunes, D. Z. Effect of Stirring Speed on Low Acyl Gellan Gum Fluid Gels’ Rheology, Particle Morphology and Physical Ageing. Food Hydrocolloids 2024, 149, 109614 DOI: 10.1016/j.foodhyd.2023.109614There is no corresponding record for this reference.
- 39Suslick, B. A.; Hemmer, J.; Groce, B. R.; Stawiasz, K. J.; Geubelle, P. H.; Malucelli, G.; Mariani, A.; Moore, J. S.; Pojman, J. A.; Sottos, N. R. Frontal Polymerizations: From Chemical Perspectives to Macroscopic Properties and Applications. Chem. Rev. 2023, 123 (6), 3237– 3298, DOI: 10.1021/acs.chemrev.2c00686There is no corresponding record for this reference.
- 40Hale, G. M.; Querry, M. R. Optical Constants of Water in the 200-Nm to 200-Μm Wavelength Region. Appl. Opt. 1973, 12 (3), 555– 563, DOI: 10.1364/AO.12.000555There is no corresponding record for this reference.
- 41Kitamura, R.; Pilon, L.; Jonasz, M. Optical Constants of Silica Glass from Extreme Ultraviolet to Far Infrared at near Room Temperature. Appl. Opt. 2007, 46 (33), 8118– 8133, DOI: 10.1364/AO.46.008118There is no corresponding record for this reference.
- 42Yang, C.; Yin, T.; Suo, Z. Polyacrylamide Hydrogels. I. Network Imperfection. J. Mech. Phys. Solids 2019, 131, 43– 55, DOI: 10.1016/j.jmps.2019.06.018There is no corresponding record for this reference.
- 43Graessley, W. W. The Entanglement Concept in Polymer Rheology. In The Entanglement Concept in Polymer Rheology; Springer: Berlin, Heidelberg, 1974; pp 1– 179 DOI: 10.1007/BFb0031037 .There is no corresponding record for this reference.
- 44Wang, S.-Q.; Ravindranath, S.; Wang, Y.; Boukany, P. New Theoretical Considerations in Polymer Rheology: Elastic Breakdown of Chain Entanglement Network. J. Chem. Phys. 2007, 127 (6), 064903 DOI: 10.1063/1.2753156There is no corresponding record for this reference.
- 45Cao, X.-Z.; Forest, M. G. Rheological Tuning of Entangled Polymer Networks by Transient Cross-Links. J. Phys. Chem. B 2019, 123 (5), 974– 982, DOI: 10.1021/acs.jpcb.8b09357There is no corresponding record for this reference.
- 46Feng, Y.; Wu, D.; Li, R.; Akcora, P. Rheological Properties of Crosslinked Unentangled and Entangled Poly(Methyl Acrylate) Nanocomposite Networks. Polymer 2022, 255, 125150 DOI: 10.1016/j.polymer.2022.125150There is no corresponding record for this reference.
- 47Pawlak, A.; Krajenta, J. Entanglements of Macromolecules and Their Influence on Rheological and Mechanical Properties of Polymers. Molecules 2024, 29 (14), 3410, DOI: 10.3390/molecules29143410There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.macromol.5c03018.
Raw data of the graphs in the main figures; Heat flux from hydrogel gelation at various UV intensities; Temperature profiles in the heat flux measurement; Toughness of PAAm hydrogels divided by φ2/3 at different UV intensities; Heat flux analysis (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.