



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Selectivity for Exhaustive Cross-Coupling of Dihaloarenes Is Affected by the Interplay between the Halide Byproduct, Solvent, and LigandClick to copy article linkArticle link copied!
- Nathaniel G. LarsonNathaniel G. LarsonDepartment of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, United StatesMore by Nathaniel G. Larson
- Matthew P. SandinMatthew P. SandinDepartment of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, United StatesMore by Matthew P. Sandin
- Sharon R. Neufeldt*Sharon R. Neufeldt*Email: [email protected]Department of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, United StatesMore by Sharon R. Neufeldt
Abstract
In dihaloarene cross-couplings, the step(s) that take place after reductive elimination and before the start of the next cycle influence selectivity for monofunctionalization versus difunctionalization. When palladium is supported by a bulky ligand “L”, a competition exists between a second oxidative addition (leading to difunctionalization) and bimolecular displacement of Pd0 from the monofunctionalized product by a second smaller ligand. Because the oxidative addition of Ar–Br is faster than that of Ar–Cl, more difunctionalization might be expected with dibromoarenes compared to dichloroarenes. However, the opposite has been reported for some Suzuki–Miyaura cross-couplings. Here, we report that the selectivity outcome is closely tied to solvents: dibromoarenes tend to give less diarylation than dichloroarenes in oxygen-containing solvents of at least moderate polarity (e.g., THF), whereas a high selectivity for diarylation can be achieved in most aromatic and chlorinated solvents. The results suggest that, in polar oxygen-containing solvents, the bromide anion byproduct displaces LPd0 from the monocross-coupled product. The rate of this process is competitive with the rate of intramolecular oxidative addition en route to the diarylated product. In contrast, the analogous displacement of Pd0 by chloride (the byproduct when using dichloroarenes) appears to be a much slower process if it occurs at all.
This publication is licensed for personal use by The American Chemical Society.
Introduction
Scheme 1
Results and Discussion
Solvent Effects on Selectivity with Pd/IPent
Figure 1
Figure 1. Solvents exert different effects on the selectivity for monoarylation of (A) a dichloroarene versus (B) a dibromoarene. “Pd/IPent” = (η3-1-tBu-indenyl)Pd(IPent)(Cl). Average of ≥ 2 trials. The area of the bubbles correlates with the combined % yields of mono- and diarylated products with GC % yields calibrated against undecane as the internal standard and calculated based on 2 as the limiting reagent. Due to the stoichiometry of the coupling partner, the maximum possible % yield is only ∼50% if 4 is exclusively formed and ∼100% if 3 is exclusively formed.
Relevance of the Bromide Anion to Selectivity
Figure 2
Figure 2. In acetone and THF, the cross-coupling of 2-Br becomes more monoselective after the first few catalytic turnovers, while the reaction in benzene gives exclusively diarylation throughout the course of the reaction. “Pd/IPent” = (η3-1-tBu-indenyl)Pd(IPent)(Cl). Results based on GC yields calibrated against undecane as an internal standard.

| entry | 2 | solvent | Ag2O (equiv) | 3 | 4 | 3:4 |
|---|---|---|---|---|---|---|
| 1 | 2-Br | acetone | 0 | 23 | 34 | 1:1.5 |
| 2 | 2-Br | 2 | n.d | 52 | 1: >99 | |
| 3 | 2-Br | THF | 0 | 22 | 38 | 1:1.7 |
| 4 | 2-Br | 2 | n.d | 56 | 1: >99 | |
| 5 | 2-Cl | THF | 0 | 9 | 50 | 1:5.4 |
| 6 | 2-Cl | 2 | 9 | 49 | 1:5.6 |
GC % yields calibrated against undecane as the internal standard and calculated based on 2 as the limiting reagent. Average of ≥2 trials except entry 6 (1 trial). Due to the stoichiometry of the coupling partner, the maximum possible % yield is only ∼50% if 4 is exclusively formed and ∼100% if 3 is exclusively formed. Mass balances slightly over the theoretical maximum are presumed due to boroxine impurity in the PhB(OH)2 reagent. “Pd/IPent” = (η3-1-tBu-indenyl)Pd(IPent)(Cl). “n.d” = not detected.

| entry | solvent | 2 | 3-Cl (%) | 3-Br (%) | 4 (%) | 3:4 |
|---|---|---|---|---|---|---|
| 1 | THF | 2-Cl | 9 | -- | 50 | 1:5.4 |
| 2 | 2-BrCl | 46 | n.d | 29 | 1.6:1 | |
| 3 | 2-Br | -- | 22 | 38 | 1:1.7 | |
| 4 | benzene | 2-Cl | 52 | -- | 29 | 1.8:1 |
| 5 | 2-BrCl | 64 | n.d | 24 | 2.6:1 | |
| 6 | 2-Br | n.d | -- | 53 | 1: >99 |
GC % yields calibrated against undecane as the internal standard and calculated based on 2 as the limiting reagent. Average of ≥2 trials. Due to the stoichiometry of the coupling partner, the maximum possible % yield is only ∼50% if 4 is exclusively formed and ∼100% if 3 is exclusively formed. Mass balances slightly over the theoretical maximum are presumed due to boroxine impurity in the PhB(OH)2 reagent. “Pd/IPent” = (η3-1-tBu-indenyl)Pd(IPent)(Cl). “n.d” = not detected; “--” = not applicable.
An Updated Description of the Competing Pathways Available after Reductive Elimination
Figure 3
Figure 3. π-Complex between (IPent)Pd0 and the monocross-coupled product (shaded box) can proceed through divergent paths, leading to diarylation (path (i)) or to release of the monoarylated product (paths ii–iv). Selectivity depends on the relative rate of oxidative addition (path (i)) versus displacement of Pd by the substrate (path ii, relevant when X = Cl), by the solvent (path iii, relevant when X = Cl), or by the halide (path iv, relevant when X = Br).
Selectivity with Other Bulky Ligands
Figure 4
Figure 4. Solvent has a different effect on selectivity for NHC ligands compared to phosphine ligands. GC % yields were calibrated against undecane as the internal standard and calculated based on 2 as the limiting reagent. Average of ≥2 trials. Due to the stoichiometry of the coupling partner, the maximum possible % yield is only ∼50% if 4 is exclusively formed and ∼100% if 3-Br is exclusively formed. Mass balances slightly over the theoretical maximum are presumed due to boroxine impurity in the PhB(OH)2 reagent.
Figure 5
Figure 5. Ag(I) suppresses monoarylation in THF for NHC ligands but not for phosphine ligands. GC % yields were calibrated against undecane as the internal standard and calculated based on 2 as the limiting reagent. Average of ≥2 trials. Due to the stoichiometry of the coupling partner, the maximum possible % yield is only ∼50% if 4 is exclusively formed and ∼100% if 3-Br is exclusively formed. Mass balances slightly over the theoretical maximum are presumed to be due to boroxine impurity in the PhB(OH)2 reagent.
Methods
Representative Procedure
Conclusions
Figure 6
Figure 6. Evaluating the conditions from this work that provided the highest selectivity in either direction using (A) dihalofluorene (NMR yields) and (B) ortho- and para-dibromobenzene (calibrated GC yields assume that the products have a response factor similar to 3-Br and 4).
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.5c00423.
Experimental details and NMR spectra (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Sharon R. Neufeldt - Department of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, United States;
 https://orcid.org/0000-0001-7995-3995;
https://orcid.org/0000-0001-7995-3995;
- Nathaniel G. Larson - Department of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, United States;https://orcid.org/0000-0002-7631-3332
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (NIGMS) of the NIH under Award Number R35GM137971. Support for MSU’s NMR Center was provided by the NSF (Grant Nos. NSF-MRI:CHE-2018388 and NSF-MRI:DBI-1532078), MSU, and the Murdock Charitable Trust Foundation (2015066:MNL). We are grateful to Umicore for gifts of (η3-1-tBu-indenyl)2(μ-Cl)2Pd2, (η3-1-tBu-indenyl)Pd(IPr)(Cl), and (η3-1-tBu-indenyl)Pd(IPent)(Cl). We also thank Colin McLeod, who was supported by an NSF REU (CHE-2349748), for assistance with solubility studies.
References
This article references 24 other publications.
- 1United States Food and Drug Administration, 2024, https://www.fda.gov/drugs/novel-drug-approvals-fda/novel-drug-approvals-2024 (accessed on Sep 15, 2025).Google ScholarThere is no corresponding record for this reference.
- 2Zani, L.; Dessi, A.; Franchi, D.; Calamante, M.; Reginato, G.; Mordini, A. Transition metal-catalyzed cross-coupling methodologies for the engineering of small molecules with applications in organic electronics and photovoltaics. Coord. Chem. Rev. 2019, 392, 177– 236, DOI: 10.1016/j.ccr.2019.04.007Google ScholarThere is no corresponding record for this reference.
- 3(a) Shaughnessy, K. H. Development of Palladium Precatalysts That Efficiently Generate LPd(0) Active Species. Isr. J. Chem. 2020, 60, 180– 194, DOI: 10.1002/ijch.201900067Google ScholarThere is no corresponding record for this reference.(b) Firsan, S. J.; Sivakumar, V.; Colacot, T. J. Emerging Trends in Cross-Coupling: Twelve-Electron-Based L1Pd(0) Catalysts, Their Mechanism of Action, and Selected Applications. Chem. Rev. 2022, 122, 16983– 17027, DOI: 10.1021/acs.chemrev.2c00204Google ScholarThere is no corresponding record for this reference.
- 4(a) Dong, C.-G.; Hu, Q.-S. Preferential Oxidative Addition in Palladium(0)-Catalyzed Suzuki Cross-Coupling Reactions of Dihaloarenes with Arylboronic Acids. J. Am. Chem. Soc. 2005, 127 (28), 10006– 10007, DOI: 10.1021/ja052547pGoogle ScholarThere is no corresponding record for this reference.(b) Weber, S. K.; Galbrecht, F.; Scherf, U. Preferential Oxidative Addition in Suzuki Cross-Coupling Reactions Across One Fluorene Unit. Org. Lett. 2006, 8, 4039– 4041, DOI: 10.1021/ol061476bGoogle ScholarThere is no corresponding record for this reference.(c) Dai, X.; Chen, Y.; Garrell, S.; Liu, H.; Zhang, L.-K.; Palani, A.; Hughes, G.; Nargund, R. Ligand-Dependent Site-Selective Suzuki Cross-Coupling of 3,5-Dichloropyridazines. J. Org. Chem. 2013, 78, 7758– 7763, DOI: 10.1021/jo401096uGoogle ScholarThere is no corresponding record for this reference.(d) Norman, J. P.; Larson, N. G.; Entz, E. D.; Neufeldt, S. R. Unconventional Site-Selectivity in Palladium-Catalyzed Cross-Couplings of Dichloroheteroarenes under Ligand-Controlled and Ligand-Free Systems. J. Org. Chem. 2022, 87, 7414– 7421, DOI: 10.1021/acs.joc.2c00665Google ScholarThere is no corresponding record for this reference.(e) Norman, J. P.; Larson, N. G.; Neufeldt, S. R. Different Oxidative Addition Mechanisms for 12- and 14-Electron Palladium(0) Explain Ligand-Controlled Divergent Site Selectivity. ACS Catal. 2022, 12, 8822– 8828, DOI: 10.1021/acscatal.2c01698Google ScholarThere is no corresponding record for this reference.(f) Zhu, Y. X.; Li, E.-C.; Shen, K.; Hang, X.; Bonnesen, P. V.; Hong, K.; Zhang, H.-H.; Huang, W. Intramolecular Catalyst Transfer over Sterically Hindered Arenes in Suzuki Cross-Coupling Reactions. Asian J. Org. Chem. 2019, 8, 1506– 1512, DOI: 10.1002/ajoc.201900228Google ScholarThere is no corresponding record for this reference.(g) Deem, M. C.; Derasp, J. S.; Malig, T. C.; Legard, K.; Berlinguette, C. P.; Hein, J. E. Ring walking as a regioselectivity control element in Pd-catalyzed C-N cross-coupling. Nature. Commun. 2022, 13, 2869, DOI: 10.1038/s41467-022-30255-1Google ScholarThere is no corresponding record for this reference.(h) Sun, K.-X.; He, Q.-W.; Xu, B.-B.; Wu, X.-T.; Lu, J.-M. Synthesis of N-Heterocyclic Carbene–PdII–2-Methyl-4,5- dihydrooxazole Complexes and Their Application Toward Highly Chemoselective Mono-Suzuki–Miyaura Coupling of Dichlorobenzenes. Asian J. Org. Chem. 2018, 7, 781– 787, DOI: 10.1002/ajoc.201800001Google ScholarThere is no corresponding record for this reference.(i) Larrosa, I.; Somoza, C.; Banquy, A.; Goldup, S. M. Two Flavors of PEPPSI-IPr: Activation and Diffusion Control in a Single NHC-Ligated Pd Catalyst?. Org. Lett. 2011, 13, 146– 149, DOI: 10.1021/ol1027283Google ScholarThere is no corresponding record for this reference.(j) Yang, M.; Chen, J.; He, C.; Hu, X.; Ding, Y.; Kuang, Y.; Liu, J.; Huang, Q. Palladium-Catalyzed C-4 Selective Coupling of 2,4-Dichloropyridines and Synthesis of Pyridine-Based Dyes for Live-Cell Imaging. J. Org. Chem. 2020, 85, 6498– 6508, DOI: 10.1021/acs.joc.0c00449Google ScholarThere is no corresponding record for this reference.(k) Yang, M.; Chen, J.; He, C.; Hu, X.; Ding, Y.; Kuang, Y.; Liu, J.; Huang, Q. Palladium-Catalyzed C-4 Selective Coupling of 2,4-Dichloropyridines and Synthesis of Pyridine-Based Dyes for Live-Cell Imaging. J. Org. Chem. 2020, 85, 6498– 6508, DOI: 10.1021/acs.joc.0c00449Google ScholarThere is no corresponding record for this reference.(l) Groombridge, B. J.; Goldup, S. M.; Larrosa, I. Selective and general exhaustive cross-coupling of dichloroarenes with a deficit of nucleophiles mediated by a Pd–NHC complex. Chem. Commun. 2015, 51, 3832– 3834, DOI: 10.1039/C4CC08920KGoogle ScholarThere is no corresponding record for this reference.(m) Kosaka, K.; Uchida, T.; Mikami, K.; Ohta, Y.; Yokozawa, T. AmPhos Pd-Catalyzed Suzuki–Miyaura Catalyst-Transfer Condensation Polymerization: Narrower Dispersity by Mixing the Catalyst and Base Prior to Polymerization. Macromolecules 2018, 51, 364– 369, DOI: 10.1021/acs.macromol.7b01990Google ScholarThere is no corresponding record for this reference.(n) Bryan, Z. J.; Smith, M. L.; McNeil, A. J. Chain-Growth Polymerization of Aryl Grignards Initiated by A Stabilized NHC-Pd Precatalyst. Macromol. Rapid Commun. 2012, 33, 842– 847, DOI: 10.1002/marc.201200096Google ScholarThere is no corresponding record for this reference.(o) Leone, A. K.; Mueller, E. A.; McNeil, A. J. The History of Palladium-Catalyzed Cross-Coupling Schould Inspire the Future of Catalyst-Transfer Polymerization. J. Am. Chem. Soc. 2018, 140, 15126– 15139, DOI: 10.1021/jacs.8b09103Google ScholarThere is no corresponding record for this reference.
- 5(a) Larson, N. G.; Norman, J. P.; Neufeldt, S. R. Mechanistic Origin of Ligand Effects on Exhaustive Functionalization During Pd-Catalyzed Cross-Coupling of Dihaloarenes. ACS Catal. 2024, 14, 7127– 7135, DOI: 10.1021/acscatal.4c00646Google ScholarThere is no corresponding record for this reference.(b) Larson, N.; Sandin, M.; Neufeldt, S. Selectivity for Exhaustive Cross-Coupling of Dihaloarenes is Affected by the Interplay Between Halide Byproduct, Solvent, and Ligand. ChemRxiv 2025, chemrxiv-2025-kzb89, DOI: 10.26434/chemrxiv-2025-kzb89Google ScholarThere is no corresponding record for this reference.
- 6Review:Reeves, E. K.; Entz, E. D.; Neufeldt, S. R. Chemodivergence between Electrophiles in Cross-Coupling Reactions. Chem.─Eur. J. 2021, 27, 6161– 6177, DOI: 10.1002/chem.202004437Google ScholarThere is no corresponding record for this reference.
- 7Peng, Y.-Q.; Li, Y.-Q.; Liu, M.-M.; Ni, C.; Cao, Y.-C. Unexpectedly superior efficiency of chloride-directed double Suzuki–Miyaura cross-coupling reactions to bromide-directed reactions for the synthesis of sterically hindered 2,7-diaryl fluorenes. New J. Chem. 2024, 48, 12130– 12137, DOI: 10.1039/D4NJ00718BGoogle ScholarThere is no corresponding record for this reference.
- 8(a) Reeves, E. K.; Bauman, O. R.; Mitchem, G. B.; Neufeldt, S. R. Solvent Effects on the Selectivity of Palladium-Catalyzed Suzuki-Miyaura Couplings. Isr. J. Chem. 2020, 60, 406– 409, DOI: 10.1002/ijch.201900082Google ScholarThere is no corresponding record for this reference.(b) Elias, E. K.; Rehbein, S. M.; Neufeldt, S. R. Solvent coordination to palladium can invert the selectivity of oxidative addition. Chem. Sci. 2022, 13, 1618– 1628, DOI: 10.1039/D1SC05862BGoogle ScholarThere is no corresponding record for this reference.
- 9Semeniuchenko, V.; Sharif, S.; Rana, N.; Chandrasoma, N.; Braje, W. M.; Baker, R. T.; Manthorpe, J. M.; Pietro, W. J.; Organ, M. G. Experimental Evidence for Zerovalent Pd(NHC) as a Competent Catalyst in C–N Cross-Coupling (NHC = DiMeIHeptCl). J. Am. Chem. Soc. 2024, 146, 29224– 29236, DOI: 10.1021/jacs.4c12203Google ScholarThere is no corresponding record for this reference.
- 10(a) Young, S. J.; Kellenberger, B.; Reibenspies, J. H.; Himmel, S. E.; Manning, M.; Anderson, O. P.; Stille, J. K. Synthesis and Reactions of Dinuclear Palladium Complexes Containing Methyls and Hydride on Adjacent Palladium Centers: Reductive Elimination and Carbonylation Reactions. J. Am. Chem. Soc. 1988, 110, 5744– 5753, DOI: 10.1021/ja00225a026Google ScholarThere is no corresponding record for this reference.(b) Lumbreras, E., Jr.; Sisler, E. M.; Shelby, Q. D. Synthesis, X-ray Crystal Structure, and Reactivity of Pd2(μ-dotpm)2 (dotpm = bis(di-ortho-tolylphosphino)methane). J. Organomet. Chem. 2010, 695, 201– 205, DOI: 10.1016/j.jorganchem.2009.10.010Google ScholarThere is no corresponding record for this reference.
- 11
Dichloromethane has been observed as a ligand for Ag(I), which is isoelectronic with Pd(0); see
(a) Newbound, T. D.; Colsman, M. R.; Miller, M. M.; Wulfsberg, G. P.; Anderson, O. P.; Strauss, S. H. Dichloromethane is a Coordinating Solvent. J. Am. Chem. Soc. 1989, 111, 3762– 3764, DOI: 10.1021/ja00192a052Google ScholarThere is no corresponding record for this reference.(b) Colsman, M. R.; Newbound, T. D.; Marshall, L. J.; Noirot, M. D.; Miller, M. M.; Wulfsberg, G. P.; Frye, J. S.; Anderson, O. P.; Strauss, S. H. Silver(I) Complexes of Dichloromethane and 1,2-Dichloroethane. J. Am. Chem. Soc. 1990, 112, 2349– 2362, DOI: 10.1021/ja00162a040Google ScholarThere is no corresponding record for this reference. - 12
It is not clear how MeCN and DMF fit into the trends with 2-Br, as the yields were low in these solvents (19% and 8%, respectively), suggesting that coordination of solvent to Pd inhibits catalysis. DMSO and nitromethane were also tested with 2-Br, but 0% yield was obtained in both cases (see Supporting Information).
There is no corresponding record for this reference. - 13
In further experiments, the effect of adding substoichiometric quantities of bromide salts (NBu4Br or KBr) to the cross-coupling of 2-Cl was examined. As expected, both additives increase the proportion of monoarylation in THF and acetone, but the results are complicated by low yields, reflecting an inhibitory effect of these additives on catalysis (see Supporting Information).
There is no corresponding record for this reference. - 15
For example, in the reaction of 1-Cl, the ratio of mono:di increased from about 1:4 to about 1:2 when the starting concentration of substration was doubled, and this effect remains even when the data are normalized to account for the competition between dichloroarene and monocross-coupled product as substrates for Pd. See ref (5)a for details.
There is no corresponding record for this reference. - 16Review:Chernyshev, V. M.; Denisova, E.; Eremin, D. B.; Ananikov, V. P. The key role of R–NHC coupling (R = C, H, heteroatom) and M–NHC bond cleavage in the evolution of M/NHC complexes and formation of catalytically active species. Chem. Sci. 2020, 11, 6957– 6977, DOI: 10.1039/D0SC02629HGoogle ScholarThere is no corresponding record for this reference.
- 17Semeniuchenko, V.; Sharif, S.; Rana, N.; Chandrasoma, N.; Braje, W. M.; Baker, R. T.; Manthorpe, J. M.; Pietro, W. J.; Organ, M. G. Unexpected Deactivation of PdCl(cinnamyl)(NHC Cl) Precatalysts Mediated by Alkylamines. Organometallics 2025, 44, 2654– 2662, DOI: 10.1021/acs.organomet.5c00318Google ScholarThere is no corresponding record for this reference.
- 18Newman-Stonebraker, S. H.; Smith, S. R.; Borowski, J. E.; Peters, E.; Gensch, T.; Johnson, H. C.; Sigman, M. S.; Doyle, A. G. Univariate classification of phosphine ligation state and reactivity in cross-coupling catalysis. Science 2021, 374, 301– 308, DOI: 10.1126/science.abj4213Google ScholarThere is no corresponding record for this reference.
- 19Gensch, T.; dos Passos Gomes, G.; Friederich, P.; Peters, E.; Gaudin, T.; Pollice, R.; Jorner, K.; Nigam, A.; Lindner-D’Addario, M.; Sigman, M. S.; Aspuru-Guzik, A. A Comprehensive Discovery Platform for Organophosphorus Ligands for Catalysis. J. Am. Chem. Soc. 2022, 144, 1205– 1217, DOI: 10.1021/jacs.1c09718Google ScholarThere is no corresponding record for this reference.
- 20
If silver oxidizes PtBu3, the observed selectivity would no longer reflect the selectivity of Pd(PtBu3) catalyst. Indeed, in the absence of any phosphine (or NHC) ligand at all, the reaction favors monoarylation (see Supporting Information).
There is no corresponding record for this reference. - 21
For selected reviews of cross-couplings catalyzed by Pd/NHC complexes, see:
(a) Marion, N.; Nolan, S. P. Well-Defined N-Heterocyclic Carbenes–Palladium(II) Precatalysts for Cross-Coupling Reactions. Acc. Chem. Res. 2008, 41, 1440– 1449, DOI: 10.1021/ar800020yGoogle ScholarThere is no corresponding record for this reference.(b) Fortman, G. C.; Nolan, S. P. N-Heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: a perfect union. Chem. Soc. Rev. 2011, 40, 5151– 5169, DOI: 10.1039/c1cs15088jGoogle ScholarThere is no corresponding record for this reference.(c) Valente, C.; Calimsiz, S.; Hoi, K. H.; Mallik, D.; Sayah, M.; Organ, M. G. The Development of Bulky Palladium NHC Complexes for the Most-Challenging Cross-Coupling Reactions. Angew. Chem., Int. Ed. 2012, 51, 3314– 3332, DOI: 10.1002/anie.201106131Google ScholarThere is no corresponding record for this reference.(d) Froese, R. D. J.; Lombardi, C.; Pompeo, M.; Rucker, R. P.; Organ, M. G. Designing Pd N-Heterocyclic Carbene Complexes for High Reactivity and Selectivity for Cross-Coupling Applications. Acc. Chem. Res. 2017, 50, 2244– 2253, DOI: 10.1021/acs.accounts.7b00249Google ScholarThere is no corresponding record for this reference.(e) Yang, S.; Zhou, T.; Yu, X.; Nolan, S. P.; Szostak, M. [Pd(NHC)(μ-Cl)Cl]2: The Highly Reactive Air- and Moisture-Stable, Well-Defined Pd(II)-N-Heterocyclic Carbene (NHC) Complexes for Cross-Coupling Reactions. Acc. Chem. Res. 2024, 57, 3343– 3355, DOI: 10.1021/acs.accounts.4c00549Google ScholarThere is no corresponding record for this reference.(f) Bera, S. S.; Utecht-Jarzynska, G.; Yang, S.; Nolan, S. P.; Szostak, M. Metal–N-Heterocyclic Carbene Complexes in Buchwald–Hartwig Amination Reactions. Chem. Rev. 2025, 125, 5349– 5435, DOI: 10.1021/acs.chemrev.5c00088Google ScholarThere is no corresponding record for this reference. - 22
For selected reviews of Pd-catalyzed cross-couplings using bulky phosphine ligands, see:
(a) Fu, G. C. The Development of Versatile Methods for Palladium-Catalyzed Coupling Reactions of Aryl Electrophiles through the Use of P(t-Bu)3 and PCy3 as Ligands. Acc. Chem. Res. 2008, 41, 1555– 1564, DOI: 10.1021/ar800148fGoogle ScholarThere is no corresponding record for this reference.(b) Martin, R.; Buchwald, S. L. Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands. Acc. Chem. Res. 2008, 41, 1461– 1473, DOI: 10.1021/ar800036sGoogle ScholarThere is no corresponding record for this reference.(c) Fleckenstein, C. A.; Plenio, H. Sterically demanding trialkylphosphines for palladium-catalyzed cross coupling reactions─alternatives to PtBu3. Chem. Soc. Rev. 2010, 39, 694– 711, DOI: 10.1039/B903646FGoogle ScholarThere is no corresponding record for this reference.(d) Bruno, N. C.; Tudge, M. T.; Buchwald, S. L. Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem. Sci. 2013, 4, 916– 920, DOI: 10.1039/C2SC20903AGoogle ScholarThere is no corresponding record for this reference.(e) Ruiz-Castillo, P.; Buchwald, S. L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564– 12649, DOI: 10.1021/acs.chemrev.6b00512Google ScholarThere is no corresponding record for this reference. - 23Review:Durand, D. J.; Fey, N. Computational Ligand Descriptors for Catalyst Design. Chem. Rev. 2019, 119, 6561– 6594, DOI: 10.1021/acs.chemrev.8b00588Google ScholarThere is no corresponding record for this reference.
- 24(a) Antonova, N. S.; Carbó, J. J.; Poblet, J. M. Quantifying the Donor-Acceptor Properties of Phosphine and N-Heterocyclic Carbene Ligands in Grubbs’ Catalysts Using a Modified EDA Procedure Based on Orbital Deletion. Organometallics 2009, 28, 4283– 4287, DOI: 10.1021/om900180mGoogle ScholarThere is no corresponding record for this reference.(b) Vummaleti, S. V. C.; Nelson, D. J.; Poater, A.; Gómez-Suárez, A.; Cordes, D. B.; Slawin, A. M. Z.; Nolan, S. P.; Cavallo, L. What can NMR spectroscopy of selenoureas and phosphinidenes teach us about the π-accepting abilities of N-heterocyclic carbenes?. Chem. Sci. 2015, 6, 1895– 1904, DOI: 10.1039/C4SC03264KGoogle ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
Scheme 1
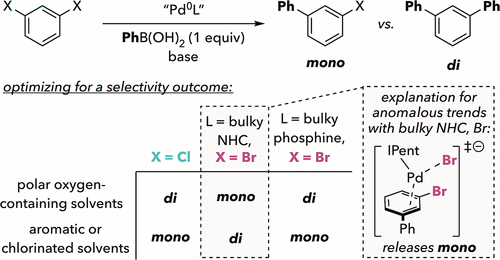
 Scheme 1. Prior Work: (A) Competing Pathways Leading to Di- versus Monoarylation in Suzuki–Miyaura Coupling of Dichloroarenes; (B,C) Dibromoarenes Unexpectedly Give Less Diarylation than Dichloroarenes
Scheme 1. Prior Work: (A) Competing Pathways Leading to Di- versus Monoarylation in Suzuki–Miyaura Coupling of Dichloroarenes; (B,C) Dibromoarenes Unexpectedly Give Less Diarylation than DichloroarenesFigure 1

Figure 1. Solvents exert different effects on the selectivity for monoarylation of (A) a dichloroarene versus (B) a dibromoarene. “Pd/IPent” = (η3-1-tBu-indenyl)Pd(IPent)(Cl). Average of ≥ 2 trials. The area of the bubbles correlates with the combined % yields of mono- and diarylated products with GC % yields calibrated against undecane as the internal standard and calculated based on 2 as the limiting reagent. Due to the stoichiometry of the coupling partner, the maximum possible % yield is only ∼50% if 4 is exclusively formed and ∼100% if 3 is exclusively formed.
Figure 2

Figure 2. In acetone and THF, the cross-coupling of 2-Br becomes more monoselective after the first few catalytic turnovers, while the reaction in benzene gives exclusively diarylation throughout the course of the reaction. “Pd/IPent” = (η3-1-tBu-indenyl)Pd(IPent)(Cl). Results based on GC yields calibrated against undecane as an internal standard.
Figure 3

Figure 3. π-Complex between (IPent)Pd0 and the monocross-coupled product (shaded box) can proceed through divergent paths, leading to diarylation (path (i)) or to release of the monoarylated product (paths ii–iv). Selectivity depends on the relative rate of oxidative addition (path (i)) versus displacement of Pd by the substrate (path ii, relevant when X = Cl), by the solvent (path iii, relevant when X = Cl), or by the halide (path iv, relevant when X = Br).
Figure 4
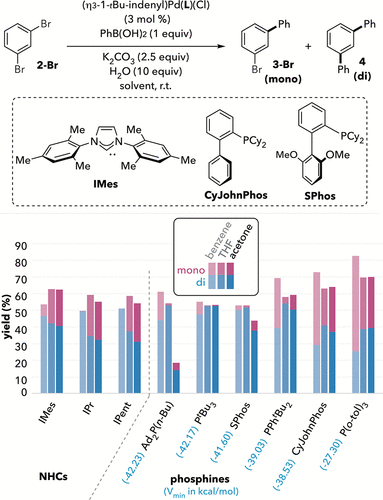
Figure 4. Solvent has a different effect on selectivity for NHC ligands compared to phosphine ligands. GC % yields were calibrated against undecane as the internal standard and calculated based on 2 as the limiting reagent. Average of ≥2 trials. Due to the stoichiometry of the coupling partner, the maximum possible % yield is only ∼50% if 4 is exclusively formed and ∼100% if 3-Br is exclusively formed. Mass balances slightly over the theoretical maximum are presumed due to boroxine impurity in the PhB(OH)2 reagent.
Figure 5

Figure 5. Ag(I) suppresses monoarylation in THF for NHC ligands but not for phosphine ligands. GC % yields were calibrated against undecane as the internal standard and calculated based on 2 as the limiting reagent. Average of ≥2 trials. Due to the stoichiometry of the coupling partner, the maximum possible % yield is only ∼50% if 4 is exclusively formed and ∼100% if 3-Br is exclusively formed. Mass balances slightly over the theoretical maximum are presumed to be due to boroxine impurity in the PhB(OH)2 reagent.
Figure 6

Figure 6. Evaluating the conditions from this work that provided the highest selectivity in either direction using (A) dihalofluorene (NMR yields) and (B) ortho- and para-dibromobenzene (calibrated GC yields assume that the products have a response factor similar to 3-Br and 4).
References
This article references 24 other publications.
- 1United States Food and Drug Administration, 2024, https://www.fda.gov/drugs/novel-drug-approvals-fda/novel-drug-approvals-2024 (accessed on Sep 15, 2025).There is no corresponding record for this reference.
- 2Zani, L.; Dessi, A.; Franchi, D.; Calamante, M.; Reginato, G.; Mordini, A. Transition metal-catalyzed cross-coupling methodologies for the engineering of small molecules with applications in organic electronics and photovoltaics. Coord. Chem. Rev. 2019, 392, 177– 236, DOI: 10.1016/j.ccr.2019.04.007There is no corresponding record for this reference.
- 3(a) Shaughnessy, K. H. Development of Palladium Precatalysts That Efficiently Generate LPd(0) Active Species. Isr. J. Chem. 2020, 60, 180– 194, DOI: 10.1002/ijch.201900067There is no corresponding record for this reference.(b) Firsan, S. J.; Sivakumar, V.; Colacot, T. J. Emerging Trends in Cross-Coupling: Twelve-Electron-Based L1Pd(0) Catalysts, Their Mechanism of Action, and Selected Applications. Chem. Rev. 2022, 122, 16983– 17027, DOI: 10.1021/acs.chemrev.2c00204There is no corresponding record for this reference.
- 4(a) Dong, C.-G.; Hu, Q.-S. Preferential Oxidative Addition in Palladium(0)-Catalyzed Suzuki Cross-Coupling Reactions of Dihaloarenes with Arylboronic Acids. J. Am. Chem. Soc. 2005, 127 (28), 10006– 10007, DOI: 10.1021/ja052547pThere is no corresponding record for this reference.(b) Weber, S. K.; Galbrecht, F.; Scherf, U. Preferential Oxidative Addition in Suzuki Cross-Coupling Reactions Across One Fluorene Unit. Org. Lett. 2006, 8, 4039– 4041, DOI: 10.1021/ol061476bThere is no corresponding record for this reference.(c) Dai, X.; Chen, Y.; Garrell, S.; Liu, H.; Zhang, L.-K.; Palani, A.; Hughes, G.; Nargund, R. Ligand-Dependent Site-Selective Suzuki Cross-Coupling of 3,5-Dichloropyridazines. J. Org. Chem. 2013, 78, 7758– 7763, DOI: 10.1021/jo401096uThere is no corresponding record for this reference.(d) Norman, J. P.; Larson, N. G.; Entz, E. D.; Neufeldt, S. R. Unconventional Site-Selectivity in Palladium-Catalyzed Cross-Couplings of Dichloroheteroarenes under Ligand-Controlled and Ligand-Free Systems. J. Org. Chem. 2022, 87, 7414– 7421, DOI: 10.1021/acs.joc.2c00665There is no corresponding record for this reference.(e) Norman, J. P.; Larson, N. G.; Neufeldt, S. R. Different Oxidative Addition Mechanisms for 12- and 14-Electron Palladium(0) Explain Ligand-Controlled Divergent Site Selectivity. ACS Catal. 2022, 12, 8822– 8828, DOI: 10.1021/acscatal.2c01698There is no corresponding record for this reference.(f) Zhu, Y. X.; Li, E.-C.; Shen, K.; Hang, X.; Bonnesen, P. V.; Hong, K.; Zhang, H.-H.; Huang, W. Intramolecular Catalyst Transfer over Sterically Hindered Arenes in Suzuki Cross-Coupling Reactions. Asian J. Org. Chem. 2019, 8, 1506– 1512, DOI: 10.1002/ajoc.201900228There is no corresponding record for this reference.(g) Deem, M. C.; Derasp, J. S.; Malig, T. C.; Legard, K.; Berlinguette, C. P.; Hein, J. E. Ring walking as a regioselectivity control element in Pd-catalyzed C-N cross-coupling. Nature. Commun. 2022, 13, 2869, DOI: 10.1038/s41467-022-30255-1There is no corresponding record for this reference.(h) Sun, K.-X.; He, Q.-W.; Xu, B.-B.; Wu, X.-T.; Lu, J.-M. Synthesis of N-Heterocyclic Carbene–PdII–2-Methyl-4,5- dihydrooxazole Complexes and Their Application Toward Highly Chemoselective Mono-Suzuki–Miyaura Coupling of Dichlorobenzenes. Asian J. Org. Chem. 2018, 7, 781– 787, DOI: 10.1002/ajoc.201800001There is no corresponding record for this reference.(i) Larrosa, I.; Somoza, C.; Banquy, A.; Goldup, S. M. Two Flavors of PEPPSI-IPr: Activation and Diffusion Control in a Single NHC-Ligated Pd Catalyst?. Org. Lett. 2011, 13, 146– 149, DOI: 10.1021/ol1027283There is no corresponding record for this reference.(j) Yang, M.; Chen, J.; He, C.; Hu, X.; Ding, Y.; Kuang, Y.; Liu, J.; Huang, Q. Palladium-Catalyzed C-4 Selective Coupling of 2,4-Dichloropyridines and Synthesis of Pyridine-Based Dyes for Live-Cell Imaging. J. Org. Chem. 2020, 85, 6498– 6508, DOI: 10.1021/acs.joc.0c00449There is no corresponding record for this reference.(k) Yang, M.; Chen, J.; He, C.; Hu, X.; Ding, Y.; Kuang, Y.; Liu, J.; Huang, Q. Palladium-Catalyzed C-4 Selective Coupling of 2,4-Dichloropyridines and Synthesis of Pyridine-Based Dyes for Live-Cell Imaging. J. Org. Chem. 2020, 85, 6498– 6508, DOI: 10.1021/acs.joc.0c00449There is no corresponding record for this reference.(l) Groombridge, B. J.; Goldup, S. M.; Larrosa, I. Selective and general exhaustive cross-coupling of dichloroarenes with a deficit of nucleophiles mediated by a Pd–NHC complex. Chem. Commun. 2015, 51, 3832– 3834, DOI: 10.1039/C4CC08920KThere is no corresponding record for this reference.(m) Kosaka, K.; Uchida, T.; Mikami, K.; Ohta, Y.; Yokozawa, T. AmPhos Pd-Catalyzed Suzuki–Miyaura Catalyst-Transfer Condensation Polymerization: Narrower Dispersity by Mixing the Catalyst and Base Prior to Polymerization. Macromolecules 2018, 51, 364– 369, DOI: 10.1021/acs.macromol.7b01990There is no corresponding record for this reference.(n) Bryan, Z. J.; Smith, M. L.; McNeil, A. J. Chain-Growth Polymerization of Aryl Grignards Initiated by A Stabilized NHC-Pd Precatalyst. Macromol. Rapid Commun. 2012, 33, 842– 847, DOI: 10.1002/marc.201200096There is no corresponding record for this reference.(o) Leone, A. K.; Mueller, E. A.; McNeil, A. J. The History of Palladium-Catalyzed Cross-Coupling Schould Inspire the Future of Catalyst-Transfer Polymerization. J. Am. Chem. Soc. 2018, 140, 15126– 15139, DOI: 10.1021/jacs.8b09103There is no corresponding record for this reference.
- 5(a) Larson, N. G.; Norman, J. P.; Neufeldt, S. R. Mechanistic Origin of Ligand Effects on Exhaustive Functionalization During Pd-Catalyzed Cross-Coupling of Dihaloarenes. ACS Catal. 2024, 14, 7127– 7135, DOI: 10.1021/acscatal.4c00646There is no corresponding record for this reference.(b) Larson, N.; Sandin, M.; Neufeldt, S. Selectivity for Exhaustive Cross-Coupling of Dihaloarenes is Affected by the Interplay Between Halide Byproduct, Solvent, and Ligand. ChemRxiv 2025, chemrxiv-2025-kzb89, DOI: 10.26434/chemrxiv-2025-kzb89There is no corresponding record for this reference.
- 6Review:Reeves, E. K.; Entz, E. D.; Neufeldt, S. R. Chemodivergence between Electrophiles in Cross-Coupling Reactions. Chem.─Eur. J. 2021, 27, 6161– 6177, DOI: 10.1002/chem.202004437There is no corresponding record for this reference.
- 7Peng, Y.-Q.; Li, Y.-Q.; Liu, M.-M.; Ni, C.; Cao, Y.-C. Unexpectedly superior efficiency of chloride-directed double Suzuki–Miyaura cross-coupling reactions to bromide-directed reactions for the synthesis of sterically hindered 2,7-diaryl fluorenes. New J. Chem. 2024, 48, 12130– 12137, DOI: 10.1039/D4NJ00718BThere is no corresponding record for this reference.
- 8(a) Reeves, E. K.; Bauman, O. R.; Mitchem, G. B.; Neufeldt, S. R. Solvent Effects on the Selectivity of Palladium-Catalyzed Suzuki-Miyaura Couplings. Isr. J. Chem. 2020, 60, 406– 409, DOI: 10.1002/ijch.201900082There is no corresponding record for this reference.(b) Elias, E. K.; Rehbein, S. M.; Neufeldt, S. R. Solvent coordination to palladium can invert the selectivity of oxidative addition. Chem. Sci. 2022, 13, 1618– 1628, DOI: 10.1039/D1SC05862BThere is no corresponding record for this reference.
- 9Semeniuchenko, V.; Sharif, S.; Rana, N.; Chandrasoma, N.; Braje, W. M.; Baker, R. T.; Manthorpe, J. M.; Pietro, W. J.; Organ, M. G. Experimental Evidence for Zerovalent Pd(NHC) as a Competent Catalyst in C–N Cross-Coupling (NHC = DiMeIHeptCl). J. Am. Chem. Soc. 2024, 146, 29224– 29236, DOI: 10.1021/jacs.4c12203There is no corresponding record for this reference.
- 10(a) Young, S. J.; Kellenberger, B.; Reibenspies, J. H.; Himmel, S. E.; Manning, M.; Anderson, O. P.; Stille, J. K. Synthesis and Reactions of Dinuclear Palladium Complexes Containing Methyls and Hydride on Adjacent Palladium Centers: Reductive Elimination and Carbonylation Reactions. J. Am. Chem. Soc. 1988, 110, 5744– 5753, DOI: 10.1021/ja00225a026There is no corresponding record for this reference.(b) Lumbreras, E., Jr.; Sisler, E. M.; Shelby, Q. D. Synthesis, X-ray Crystal Structure, and Reactivity of Pd2(μ-dotpm)2 (dotpm = bis(di-ortho-tolylphosphino)methane). J. Organomet. Chem. 2010, 695, 201– 205, DOI: 10.1016/j.jorganchem.2009.10.010There is no corresponding record for this reference.
- 11
Dichloromethane has been observed as a ligand for Ag(I), which is isoelectronic with Pd(0); see
(a) Newbound, T. D.; Colsman, M. R.; Miller, M. M.; Wulfsberg, G. P.; Anderson, O. P.; Strauss, S. H. Dichloromethane is a Coordinating Solvent. J. Am. Chem. Soc. 1989, 111, 3762– 3764, DOI: 10.1021/ja00192a052There is no corresponding record for this reference.(b) Colsman, M. R.; Newbound, T. D.; Marshall, L. J.; Noirot, M. D.; Miller, M. M.; Wulfsberg, G. P.; Frye, J. S.; Anderson, O. P.; Strauss, S. H. Silver(I) Complexes of Dichloromethane and 1,2-Dichloroethane. J. Am. Chem. Soc. 1990, 112, 2349– 2362, DOI: 10.1021/ja00162a040There is no corresponding record for this reference. - 12
It is not clear how MeCN and DMF fit into the trends with 2-Br, as the yields were low in these solvents (19% and 8%, respectively), suggesting that coordination of solvent to Pd inhibits catalysis. DMSO and nitromethane were also tested with 2-Br, but 0% yield was obtained in both cases (see Supporting Information).
There is no corresponding record for this reference. - 13
In further experiments, the effect of adding substoichiometric quantities of bromide salts (NBu4Br or KBr) to the cross-coupling of 2-Cl was examined. As expected, both additives increase the proportion of monoarylation in THF and acetone, but the results are complicated by low yields, reflecting an inhibitory effect of these additives on catalysis (see Supporting Information).
There is no corresponding record for this reference. - 15
For example, in the reaction of 1-Cl, the ratio of mono:di increased from about 1:4 to about 1:2 when the starting concentration of substration was doubled, and this effect remains even when the data are normalized to account for the competition between dichloroarene and monocross-coupled product as substrates for Pd. See ref (5)a for details.
There is no corresponding record for this reference. - 16Review:Chernyshev, V. M.; Denisova, E.; Eremin, D. B.; Ananikov, V. P. The key role of R–NHC coupling (R = C, H, heteroatom) and M–NHC bond cleavage in the evolution of M/NHC complexes and formation of catalytically active species. Chem. Sci. 2020, 11, 6957– 6977, DOI: 10.1039/D0SC02629HThere is no corresponding record for this reference.
- 17Semeniuchenko, V.; Sharif, S.; Rana, N.; Chandrasoma, N.; Braje, W. M.; Baker, R. T.; Manthorpe, J. M.; Pietro, W. J.; Organ, M. G. Unexpected Deactivation of PdCl(cinnamyl)(NHC Cl) Precatalysts Mediated by Alkylamines. Organometallics 2025, 44, 2654– 2662, DOI: 10.1021/acs.organomet.5c00318There is no corresponding record for this reference.
- 18Newman-Stonebraker, S. H.; Smith, S. R.; Borowski, J. E.; Peters, E.; Gensch, T.; Johnson, H. C.; Sigman, M. S.; Doyle, A. G. Univariate classification of phosphine ligation state and reactivity in cross-coupling catalysis. Science 2021, 374, 301– 308, DOI: 10.1126/science.abj4213There is no corresponding record for this reference.
- 19Gensch, T.; dos Passos Gomes, G.; Friederich, P.; Peters, E.; Gaudin, T.; Pollice, R.; Jorner, K.; Nigam, A.; Lindner-D’Addario, M.; Sigman, M. S.; Aspuru-Guzik, A. A Comprehensive Discovery Platform for Organophosphorus Ligands for Catalysis. J. Am. Chem. Soc. 2022, 144, 1205– 1217, DOI: 10.1021/jacs.1c09718There is no corresponding record for this reference.
- 20
If silver oxidizes PtBu3, the observed selectivity would no longer reflect the selectivity of Pd(PtBu3) catalyst. Indeed, in the absence of any phosphine (or NHC) ligand at all, the reaction favors monoarylation (see Supporting Information).
There is no corresponding record for this reference. - 21
For selected reviews of cross-couplings catalyzed by Pd/NHC complexes, see:
(a) Marion, N.; Nolan, S. P. Well-Defined N-Heterocyclic Carbenes–Palladium(II) Precatalysts for Cross-Coupling Reactions. Acc. Chem. Res. 2008, 41, 1440– 1449, DOI: 10.1021/ar800020yThere is no corresponding record for this reference.(b) Fortman, G. C.; Nolan, S. P. N-Heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: a perfect union. Chem. Soc. Rev. 2011, 40, 5151– 5169, DOI: 10.1039/c1cs15088jThere is no corresponding record for this reference.(c) Valente, C.; Calimsiz, S.; Hoi, K. H.; Mallik, D.; Sayah, M.; Organ, M. G. The Development of Bulky Palladium NHC Complexes for the Most-Challenging Cross-Coupling Reactions. Angew. Chem., Int. Ed. 2012, 51, 3314– 3332, DOI: 10.1002/anie.201106131There is no corresponding record for this reference.(d) Froese, R. D. J.; Lombardi, C.; Pompeo, M.; Rucker, R. P.; Organ, M. G. Designing Pd N-Heterocyclic Carbene Complexes for High Reactivity and Selectivity for Cross-Coupling Applications. Acc. Chem. Res. 2017, 50, 2244– 2253, DOI: 10.1021/acs.accounts.7b00249There is no corresponding record for this reference.(e) Yang, S.; Zhou, T.; Yu, X.; Nolan, S. P.; Szostak, M. [Pd(NHC)(μ-Cl)Cl]2: The Highly Reactive Air- and Moisture-Stable, Well-Defined Pd(II)-N-Heterocyclic Carbene (NHC) Complexes for Cross-Coupling Reactions. Acc. Chem. Res. 2024, 57, 3343– 3355, DOI: 10.1021/acs.accounts.4c00549There is no corresponding record for this reference.(f) Bera, S. S.; Utecht-Jarzynska, G.; Yang, S.; Nolan, S. P.; Szostak, M. Metal–N-Heterocyclic Carbene Complexes in Buchwald–Hartwig Amination Reactions. Chem. Rev. 2025, 125, 5349– 5435, DOI: 10.1021/acs.chemrev.5c00088There is no corresponding record for this reference. - 22
For selected reviews of Pd-catalyzed cross-couplings using bulky phosphine ligands, see:
(a) Fu, G. C. The Development of Versatile Methods for Palladium-Catalyzed Coupling Reactions of Aryl Electrophiles through the Use of P(t-Bu)3 and PCy3 as Ligands. Acc. Chem. Res. 2008, 41, 1555– 1564, DOI: 10.1021/ar800148fThere is no corresponding record for this reference.(b) Martin, R.; Buchwald, S. L. Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands. Acc. Chem. Res. 2008, 41, 1461– 1473, DOI: 10.1021/ar800036sThere is no corresponding record for this reference.(c) Fleckenstein, C. A.; Plenio, H. Sterically demanding trialkylphosphines for palladium-catalyzed cross coupling reactions─alternatives to PtBu3. Chem. Soc. Rev. 2010, 39, 694– 711, DOI: 10.1039/B903646FThere is no corresponding record for this reference.(d) Bruno, N. C.; Tudge, M. T.; Buchwald, S. L. Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem. Sci. 2013, 4, 916– 920, DOI: 10.1039/C2SC20903AThere is no corresponding record for this reference.(e) Ruiz-Castillo, P.; Buchwald, S. L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564– 12649, DOI: 10.1021/acs.chemrev.6b00512There is no corresponding record for this reference. - 23Review:Durand, D. J.; Fey, N. Computational Ligand Descriptors for Catalyst Design. Chem. Rev. 2019, 119, 6561– 6594, DOI: 10.1021/acs.chemrev.8b00588There is no corresponding record for this reference.
- 24(a) Antonova, N. S.; Carbó, J. J.; Poblet, J. M. Quantifying the Donor-Acceptor Properties of Phosphine and N-Heterocyclic Carbene Ligands in Grubbs’ Catalysts Using a Modified EDA Procedure Based on Orbital Deletion. Organometallics 2009, 28, 4283– 4287, DOI: 10.1021/om900180mThere is no corresponding record for this reference.(b) Vummaleti, S. V. C.; Nelson, D. J.; Poater, A.; Gómez-Suárez, A.; Cordes, D. B.; Slawin, A. M. Z.; Nolan, S. P.; Cavallo, L. What can NMR spectroscopy of selenoureas and phosphinidenes teach us about the π-accepting abilities of N-heterocyclic carbenes?. Chem. Sci. 2015, 6, 1895– 1904, DOI: 10.1039/C4SC03264KThere is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.5c00423.
Experimental details and NMR spectra (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.