



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Synthesis of Cobaltocenes Bearing 4-(2,6-Dimethylpyridin-1-ium-4-yl)phenyl Moiety and Their Stoichiometric and Catalytic Reactivity toward Ammonia FormationClick to copy article linkArticle link copied!
- Hiroki OtsukaHiroki OtsukaDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, JapanMore by Hiroki Otsuka
- Kazuya ArashibaKazuya ArashibaDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, JapanMore by Kazuya Arashiba
- Taiji NakamuraTaiji NakamuraFukui Institute for Fundamental Chemistry, Kyoto University, Takano-Nishibiraki-cho 34-4, Sakyo-ku, Kyoto 606-8103, JapanMore by Taiji Nakamura
- Hiromasa TanakaHiromasa TanakaSchool of Liberal Arts and Sciences, Daido University, Takiharu-cho, Minami-ku, Nagoya 457-8530, JapanMore by Hiromasa Tanaka
- Kazunari Yoshizawa*Kazunari Yoshizawa*Email: [email protected]Fukui Institute for Fundamental Chemistry, Kyoto University, Takano-Nishibiraki-cho 34-4, Sakyo-ku, Kyoto 606-8103, JapanMore by Kazunari Yoshizawa
- Yoshiaki Nishibayashi*Yoshiaki Nishibayashi*Email: [email protected]Department of Applied Chemistry, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, JapanMore by Yoshiaki Nishibayashi
Abstract
Novel cobaltocenium compounds bearing a 4-(2,6-dimethylpyridin-4-yl)phenyl group are designed and synthesized for the use of proton-coupled electron transfer (PCET) reagents. Reduction and protonation of these compounds afford the corresponding cobaltocenes bearing a 4-(2,6-dimethylpyridin-1-ium-4-yl)phenyl moiety with bond dissociation free energies (BDFEs) of 34.4–43.0 kcal/mol. A superstoichiometric amount of ammonia is formed from the catalytic reduction of dinitrogen with these cobaltocenes in the presence of a molybdenum-nitride complex under ambient reaction conditions.
This publication is licensed for personal use by The American Chemical Society.
Introduction
Figure 1
Figure 1. (a) Catalytic cycle for ammonia production from dinitrogen and water using [MoI3(PCP)] as a catalyst. (b) Stepwise conversion of [Mo(N)I(PCP)] into [Mo(NH3)I(PCP)] via reduction and protonation. (c) Catalytic ammonia formation reaction using the combination of SmI2 and water and that of cobaltocene and pyridinium salt.
Figure 2
Figure 2. (a) Previous work by Peters and co-workers: cobaltocene derivative bearing N,N-dimethylanilinium. (17,18) (b) Our previous work: iron complexes bearing π-phenol ligand. (19,20) (c) Our previous work: samarium complexes bearing cyclopentadienyl-amine chelate ligands. (21) (d) This work: cobaltocenes bearing 4-(2,6-dimethylpyridin-1-ium-4-yl)phenyl moiety [1-H]+.
Results and Discussion
DFT Calculations

| cobaltocenes | pKaa | E(CoII/III) (V)b | BDFEexp (kcal/mol)c | BDFEcalcd (kcal/mol)d |
|---|---|---|---|---|
| [1a-H]+ | 9.0 | –1.64 | 34.4 | 32.9 |
| [1b-H]+ | 8.8 | –1.52 | 37.0 | 37.9 |
| [1c-H]+ | 8.9 | –1.26 | 43.0 | 47.6 |
For [1-H](OTf)2 measured in THF using UV–vis titration.
For [1]OTf in the presence of 10 equiv of [ColH]OTf as a proton source in THF (vs Fc/Fc+, Figure S20).
Estimated from eq 1 with each pKa and E(CoII/III) of [1-H]2+.
Determined by DFT calculations.
Preparation of Cobaltocenes Bearing 4-(2,6-Dimethylpyridin-1-ium-4-yl)phenyl Moiety
Scheme 1
Figure 3
Figure 3. ORTEP drawings of the cationic part of (a) [1a]OTf and (b) [1a-H](OTf)2. Hydrogen atoms except for a NH hydrogen atom in [1a-H](OTf)2 were omitted for clarity.
Scheme 2
Figure 4
Figure 4. X-band EPR spectra of [1a] (black) and [1a-H]OTf (observed: red, simulated: blue) in 2-MeTHF at −196 °C. giso = 1.99911, line width = 4.65 mT.
Figure 5
Figure 5. Absorption spectra of [1a] (0.05 mM) in THF at −78 °C by the addition of [PicH]OTf as the proton source.
Property of Cobaltocenes Bearing 4-(2,6-Dimethylpyridin-1-ium-4-yl)phenyl Moiety
Reactivity of Cobaltocenes Bearing 4-(2,6-Dimethylpyridin-1-ium-4-yl)phenyl Moiety with Molybdenum-Nitride Complex

| NH3 | H2 | ||
|---|---|---|---|
| entry | cobaltocenes | (%/Mo) | (%/[1-H]+) |
| 1 | 1a | 71 | 16 |
| 2 | 1b | 70 | 37 |
| 3 | 1c | 18 | 12 |
| entry | cobaltocenes | proton source | NH3 (equiv)a | NH3 (%)b | H2 (equiv)a | H2 (%)b |
|---|---|---|---|---|---|---|
| 1 | 1a | [PicH]OTf | 1.9 ± 0.0 | 19 ± 0 | 0.3 ± 0.0 | 2 ± 0 |
| 2 | 1a | [ColH]OTf | 4.9 ± 0.2 | 49 ± 2 | 0.3 ± 0.0 | 2 ± 0 |
| 3 | 1b | [ColH]OTf | 6.1 ± 0.3 | 61 ± 3 | 1.4 ± 0.2 | 9 ± 1 |
| 4 | [Cp*2Co] | [ColH]OTf | 2.7 ± 0.1 | 27 ± 1 | 8.2 ± 0.0 | 55 ± 0 |
Equiv based on [Mo(N)I(PCP)].
Yield based on the Co atom of cobaltocenes.
Conclusions
Experimental Section
General Methods
Synthesis of [(η5-C5Me5)Co(η5-C5Me4(C6H4–C5H2Me2N))]OTf ([1a]OTf)
Synthesis of [(η5-C5Me5)Co(η5-C5Me4(C6H4–C5H2Me2NH))](OTf)2 ([1a-H](OTf)2)
Synthesis of [(η5-C5Me5)Co(η5-C5Me4(C6H4–C5H2Me2N))] ([1a])
EPR Observation of [1a-H]OTf
UV–Vis Observation of [1a-H]OTf
Stoichiometric Reaction of Molybdenum-Nitride Complex with [1a-H]OTf
Reactions of N2 (1 atm) with Molybdenum-Nitride Complex as Catalyst and [1a-H]OTf
X-ray Crystallography
Computational Details
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.5c00492.
Detailed experiment procedures and computational details, Synthesis and reactivity of cobaltocenes, X-ray crystallography, EPR study, UV–vis absorption spectroscopy, cyclic voltammetry, NMR and IR spectra, and DFT calculations (PDF)
Cartesian coordinates of all optimized geometries (XYZ)
Deposition Numbers 2512896–2512902 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via the joint Cambridge Crystallographic Data Centre (CCDC) and Fachinformationszentrum Karlsruhe Access Structures service.
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Kazunari Yoshizawa - Fukui Institute for Fundamental Chemistry, Kyoto University, Takano-Nishibiraki-cho 34-4, Sakyo-ku, Kyoto 606-8103, Japan;
 https://orcid.org/0000-0002-6279-9722;
https://orcid.org/0000-0002-6279-9722;
- Yoshiaki Nishibayashi - Department of Applied Chemistry, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan;https://orcid.org/0000-0001-9739-9588;
- Taiji Nakamura - Fukui Institute for Fundamental Chemistry, Kyoto University, Takano-Nishibiraki-cho 34-4, Sakyo-ku, Kyoto 606-8103, Japan;https://orcid.org/0000-0002-7301-9752
- Hiromasa Tanaka - School of Liberal Arts and Sciences, Daido University, Takiharu-cho, Minami-ku, Nagoya 457-8530, Japan;https://orcid.org/0000-0001-8516-8280
Acknowledgments
We acknowledge Grants-in-Aid for Scientific Research (20H05671, 22K19041, 24H00049, 24H01834, 24K21245, and 24K21778) from JSPS and MEXT. This paper is based on results obtained from a project, JPNP21020, commissioned by the New Energy and Industrial Technology Development Organization (NEDO).
References
This article references 60 other publications.
- 1Liu, H. Ammonia Synthesis Catalyst 100 Years: Practice, Enlightenment and Challenge. Chin. J. Catal. 2014, 35, 1619– 1640, DOI: 10.1016/S1872-2067(14)60118-2Google ScholarThere is no corresponding record for this reference.
- 2Wang, X.; Fan, W.; Chen, J.; Feng, G.; Zhang, X. Experimental Study and Kinetic Analysis of the Impact of Ammonia Co-Firing Ratio on Products Formation Characteristics in Ammonia/Coal Co-Firing Process. Fuel 2022, 329, 125496 DOI: 10.1016/j.fuel.2022.125496Google ScholarThere is no corresponding record for this reference.
- 3Guo, J.; Chen, P. Catalyst: NH3 as an Energy Carrier. Chem 2017, 3, 709– 712, DOI: 10.1016/j.chempr.2017.10.004Google ScholarThere is no corresponding record for this reference.
- 4Levi, P. G.; Cullen, J. M. Mapping Global Flows of Chemicals: From Fossil Fuel Feedstocks to Chemical Products. Environ. Sci. Technol. 2018, 52, 1725– 1734, DOI: 10.1021/acs.est.7b04573Google ScholarThere is no corresponding record for this reference.
- 5Boerner, L. K. Taking the CO2 out of NH3. Chem. Eng. News 2019, 97, 18– 21Google ScholarThere is no corresponding record for this reference.
- 6Bezdek, M. J.; Pappas, I.; Chirik, P. J. Determining and Understanding N-H Bond Strengths in Synthetic Nitrogen Fixation Cycles. Top. Organomet. Chem. 2017, 60, 1– 21, DOI: 10.1007/3418_2016_8Google ScholarThere is no corresponding record for this reference.
- 7Bruch, Q. J.; Connor, G. P.; McMillion, N. D.; Goldman, A. S.; Hasanayn, F.; Holland, P. L.; Miller, A. J. M. Considering Electrocatalytic Ammonia Synthesis via Bimetallic Dinitrogen Cleavage. ACS Catal. 2020, 10, 10826– 10846, DOI: 10.1021/acscatal.0c02606Google ScholarThere is no corresponding record for this reference.
- 8Tanabe, Y.; Nishibayashi, Y. Catalytic Nitrogen Fixation Using Well-Defined Molecular Catalysts under Ambient or Mild Reaction Conditions. Angew. Chem., Int. Ed. 2024, 63, e202406404 DOI: 10.1002/anie.202406404Google ScholarThere is no corresponding record for this reference.
- 9Chalkley, M. J.; Drover, M. W.; Peters, J. C. Catalytic N2-to-NH3 (or -N2H4) Conversion by Well-Defined Molecular Coordination Complexes. Chem. Rev. 2020, 120, 5582– 5636, DOI: 10.1021/acs.chemrev.9b00638Google ScholarThere is no corresponding record for this reference.
- 10(a) Eizawa, A.; Arashiba, K.; Tanaka, H.; Kuriyama, S.; Matsuo, Y.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Remarkable Catalytic Activity of Dinitrogen-Bridged Dimolybdenum Complexes Bearing NHC-Based PCP-Pincer Ligands toward Nitrogen Fixation. Nat. Commun. 2017, 8, 14874 DOI: 10.1038/ncomms14874Google ScholarThere is no corresponding record for this reference.(b) Eizawa, A.; Arashiba, K.; Egi, A.; Tanaka, H.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Reactivity of Molybdenum–Trihalide Complexes Bearing PCP-Type Pincer Ligands. Chem. - Asian J. 2019, 14, 2091– 2096, DOI: 10.1002/asia.201900496Google ScholarThere is no corresponding record for this reference.
- 11Nakamura, T.; Tsuruta, Y.; Egi, A.; Tanaka, H.; Nishibayashi, Y.; Yoshizawa, K. Theoretical Study of Imide Formation in Nitrogen Fixation Catalyzed by Molybdenum Complex Bearing PCP-Type Pincer Ligand with Metallocenes. Inorg. Chem. 2025, 64, 9124– 9136, DOI: 10.1021/acs.inorgchem.5c00695Google ScholarThere is no corresponding record for this reference.
- 12Arashiba, K.; Eizawa, A.; Tanaka, H.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Nitrogen Fixation via Direct Cleavage of Nitrogen–Nitrogen Triple Bond of Molecular Dinitrogen under Ambient Reaction Conditions. Bull. Chem. Soc. Jpn. 2017, 90, 1111– 1118, DOI: 10.1246/bcsj.20170197Google ScholarThere is no corresponding record for this reference.
- 13Arashiba, K.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Cycling between Molybdenum-Dinitrogen and -Nitride Complexes to Support the Reaction Pathway for Catalytic Formation of Ammonia from Dinitrogen. Chem. - Eur. J. 2020, 26, 13383– 13389, DOI: 10.1002/chem.202002200Google ScholarThere is no corresponding record for this reference.
- 14(a) Ashida, Y.; Arashiba, K.; Nakajima, K.; Nishibayashi, Y. Molybdenum-Catalysed Ammonia Production with Samarium Diiodide and Alcohols or Water. Nature 2019, 568, 536– 540, DOI: 10.1038/s41586-019-1134-2Google ScholarThere is no corresponding record for this reference.(b) Ashida, Y.; Mizushima, T.; Arashiba, K.; Egi, A.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Production of Ammonia from Dinitrogen Employing Molybdenum Complexes Bearing N-Heterocyclic Carbene-Based PCP-Type Pincer Ligands. Nat. Synth. 2023, 2, 635– 644, DOI: 10.1038/s44160-023-00292-9Google ScholarThere is no corresponding record for this reference.
- 15Suginome, S.; Murota, K.; Yamamoto, A.; Yoshida, H.; Nishibayashi, Y. Mechanochemical Nitrogen Fixation Catalysed by Molybdenum Complexes. Nat. Synth. 2025, 4, 243– 251, DOI: 10.1038/s44160-024-00661-yGoogle ScholarThere is no corresponding record for this reference.
- 16Kolmar, S. S.; Mayer, J. M. SmI2(H2O)n Reduction of Electron Rich Enamines by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2017, 139, 10687– 10692, DOI: 10.1021/jacs.7b03667Google ScholarThere is no corresponding record for this reference.
- 17Chalkley, M. J.; Garrido-Barros, P.; Peters, J. C. A Molecular Mediator for Reductive Concerted Proton-Electron Transfers via Electrocatalysis. Science 2020, 369, 850– 854, DOI: 10.1126/science.abc1607Google ScholarThere is no corresponding record for this reference.
- 18Garrido-Barros, P.; Derosa, J.; Chalkley, M. J.; Peters, J. C. Tandem Electrocatalytic N2 Fixation via Proton-Coupled Electron Transfer. Nature 2022, 609, 71– 76, DOI: 10.1038/s41586-022-05011-6Google ScholarThere is no corresponding record for this reference.
- 19Kuriyama, S.; Zhang, Y.; Tanaka, H.; Konomi, A.; Egi, A.; Yoshizawa, K.; Nishibayashi, Y. Synthesis and Reactivity of Iron-Oxocyclohexadienyl Complexes toward Proton-Coupled Electron Transfer. Organometallics 2024, 43, 2736– 2746, DOI: 10.1021/acs.organomet.4c00286Google ScholarThere is no corresponding record for this reference.
- 20Tanabe, Y.; Yamamoto, S.; Kuriyama, S.; Nishibayashi, Y. Preparation and Reactivity of Anionic Iron Sandwich Complex Bearing π-Phenol Ligand Acting as Proton-Coupled Electron Transfer Reagent. Chem. -Asian J. 2026, 21, e00951 DOI: 10.1002/asia.202500951Google ScholarThere is no corresponding record for this reference.
- 21Mitsumoto, T.; Nakamura, T.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Cooperative Ammonia Formation Catalyzed by Molybdenum and Samarium Complexes. Angew. Chem., Int. Ed. 2025, 64, e202507061 DOI: 10.1002/anie.202507061Google ScholarThere is no corresponding record for this reference.
- 22Feng, R.; Zhang, L.; Ruan, H.; Zhao, Y.; Tan, G.; Wang, X. A Main-Group Element Radical Based One-Dimensional Magnetic Chain. Angew. Chem., Int. Ed. 2019, 58, 6084– 6088, DOI: 10.1002/anie.201901177Google ScholarThere is no corresponding record for this reference.
- 23Ammeter, J. H. EPR of orbitally degenerate sandwich compounds. J. Magn. Reson. 1978, 30, 299– 325, DOI: 10.1016/0022-2364(78)90103-8Google ScholarThere is no corresponding record for this reference.
- 24Robbins, J. L.; Edelstein, N.; Spencer, B.; Smart, J. C. Synthesis and Electronic Structure of Decamethylmetallocenes. J. Am. Chem. Soc. 1982, 104, 1882– 1893, DOI: 10.1021/ja00371a017Google ScholarThere is no corresponding record for this reference.
- 25Garrido, G.; Rosés, M.; Ràfols, C.; Bosch, E. Acidity of Several Anilinium Derivatives in Pure Tetrahydrofuran. J. Solution Chem. 2008, 37, 689– 700, DOI: 10.1007/s10953-008-9262-6Google ScholarThere is no corresponding record for this reference.
- 26Bordwell, F. G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456– 463, DOI: 10.1021/ar00156a004Google ScholarThere is no corresponding record for this reference.
- 27Agarwal, R. G.; Coste, S. C.; Groff, B. D.; Heuer, A. M.; Noh, H.; Parada, G. A.; Wise, C. F.; Nichols, E. M.; Warren, J. J.; Mayer, J. M. Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2022, 122, 1– 49, DOI: 10.1021/acs.chemrev.1c00521Google ScholarThere is no corresponding record for this reference.
- 28
From absorption spectroscopic studies, half of [1] was initially converted to [1-H]OTf in this condition. ([1a-H]OTf: 45%, [1b-H]OTf: 47%, Figures S12 and S13)
There is no corresponding record for this reference. - 29Evans, D. F. 400. The Determination of the Paramagnetic Susceptibility of Substances in Solution by Nuclear Magnetic Resonance. J. Chem. Soc. 1959, 2003– 2005, DOI: 10.1039/jr9590002003Google ScholarThere is no corresponding record for this reference.
- 30Live, D. H.; Chan, S. I. Bulk Susceptibility Corrections in Nuclear Magnetic Resonance Experiments Using Superconducting Solenoids. Anal. Chem. 1970, 42, 791– 792, DOI: 10.1021/ac60289a028Google ScholarThere is no corresponding record for this reference.
- 31Bain, G. A.; Berry, J. F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532, DOI: 10.1021/ed085p532Google ScholarThere is no corresponding record for this reference.
- 32Stoll, S.; Schweiger, A. EasySpin, a comprehensive softwarepackage for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42– 55, DOI: 10.1016/j.jmr.2005.08.013Google ScholarThere is no corresponding record for this reference.
- 33Molton, F. Simultispin: A versatile graphical user interface for the simulation of solid-state continuous wave EPR spectra. Magn. Reson. Chem. 2020, 58, 718– 726, DOI: 10.1002/mrc.5019Google ScholarThere is no corresponding record for this reference.
- 34Martinez, V.; Blais, J.-C.; Astruc, D. Coupling Multiple Benzylic Activation of Simple Arenes by CpFe+ with Multiple Alkene Metathesis Using Grubbs Catalysts: An Efficient Carbon–Carbon Bond Formation Strategy Leading to Polycycles, Cyclophanes, Capsules, and Polymeric Compounds and Their CpFe+ Complexes. Organometallics 2004, 23, 861– 874, DOI: 10.1021/om030623wGoogle ScholarThere is no corresponding record for this reference.
- 35Chmely, S. C.; Hanusa, T. P.; Rheingold, A. L. Influence of Ring Methylation in Group 15 Tetramethylcyclopentadienyl Complexes, M(C5Me4H)nI3–n (M = As, Sb). Organometallics 2010, 29, 5551– 5557, DOI: 10.1021/om100474mGoogle ScholarThere is no corresponding record for this reference.
- 36Bunel, E. E.; Valle, L.; Manriquez, J. M. Pentamethylcyclopentadienyl Acetylacetonate Complexes of Iron(II), Cobalt(II), and Nickel(II). Convenient Synthetic Entries to Mono-η5-C5Me5 Derivatives. Organometallics 1985, 4, 1680– 1682, DOI: 10.1021/om00128a034Google ScholarThere is no corresponding record for this reference.
- 37Tanabe, Y.; Nakajima, K.; Nishibayashi, Y. Phosphine Oxidation with Water and Ferrocenium(III) Cation Induced by Visible Light Irradiation. Chem. - Eur. J. 2018, 24, 18618– 18622, DOI: 10.1002/chem.201805129Google ScholarThere is no corresponding record for this reference.
- 38Weitz, I. S.; Rabinovitz, M. The application of C8K for organic synthesis: reduction of substituted naphthalenes. J. Chem. Soc., Perkin Trans. 1993, 1, 117– 120, DOI: 10.1039/p19930000117Google ScholarThere is no corresponding record for this reference.
- 39Weatherburn, M. W. Phenol-Hypochlorite Reaction for Determination of Ammonia. Anal. Chem. 1967, 39, 971– 974, DOI: 10.1021/ac60252a045Google ScholarThere is no corresponding record for this reference.
- 40CrysAlisPro: Data Collection and Processing Software; RigakuCorporation: Tokyo, Japan, 2015.Google ScholarThere is no corresponding record for this reference.
- 41Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339– 341, DOI: 10.1107/S0021889808042726Google ScholarThere is no corresponding record for this reference.
- 42Sheldrick, G. SHELXT – Integrated space-group and crystal structure determination. Acta Crystallogr., Sect. A:Found. Adv. 2015, 71, 3– 8, DOI: 10.1107/S2053273314026370Google ScholarThere is no corresponding record for this reference.
- 43Sheldrick, G. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C:Struct. Chem. 2015, 71, 3– 8, DOI: 10.1107/S2053229614024218Google ScholarThere is no corresponding record for this reference.
- 44van der Sluis, P.; Spek, A. L. BYPASS: an effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. 1990, 46, 194– 201, DOI: 10.1107/S0108767389011189Google ScholarThere is no corresponding record for this reference.
- 45Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Cioslowski, J.; Fox, D. J. Gaussian 16; Gaussian, Inc.: Wallingford, CT, 2016.Google ScholarThere is no corresponding record for this reference.
- 46Becke, A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098– 3100, DOI: 10.1103/PhysRevA.38.3098Google ScholarThere is no corresponding record for this reference.
- 47Becke, A. D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648– 5652, DOI: 10.1063/1.464913Google ScholarThere is no corresponding record for this reference.
- 48Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the ElectronDensity. Phys. Rev. B 1988, 37, 785– 789, DOI: 10.1103/PhysRevB.37.785Google ScholarThere is no corresponding record for this reference.
- 49Vosko, S. H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200– 1211, DOI: 10.1139/p80-159Google ScholarThere is no corresponding record for this reference.
- 50Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104 DOI: 10.1063/1.3382344Google ScholarThere is no corresponding record for this reference.
- 51Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted Ab Initio Pseudopotentials for the First Row Transition Elements. J. Chem. Phys. 1987, 86, 866– 872, DOI: 10.1063/1.452288Google ScholarThere is no corresponding record for this reference.
- 52Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjusted ab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123– 141, DOI: 10.1007/BF01114537Google ScholarThere is no corresponding record for this reference.
- 53Ditchfield, R.; Hehre, W. J.; Pople, J. A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724– 728, DOI: 10.1063/1.1674902Google ScholarThere is no corresponding record for this reference.
- 54Hehre, W. J.; Ditchfield, R.; Pople, J. A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257– 2261, DOI: 10.1063/1.1677527Google ScholarThere is no corresponding record for this reference.
- 55Hariharan, P. C.; Pople, J. A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213– 222, DOI: 10.1007/BF00533485Google ScholarThere is no corresponding record for this reference.
- 56Francl, M. M.; Pietro, W. J.; Hehre, W. J.; Binkley, J. S.; Gordon, M. S.; DeFrees, D. J.; Pople, J. A. Self-consistent Molecular Orbital Methods. XXIII. A Polarization-type Basis Set for Second-row Elements. J. Chem. Phys. 1982, 77, 3654– 3665, DOI: 10.1063/1.444267Google ScholarThere is no corresponding record for this reference.
- 57Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. Self-consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650– 654, DOI: 10.1063/1.438955Google ScholarThere is no corresponding record for this reference.
- 58McLean, A. D.; Chandler, G. S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639– 5648, DOI: 10.1063/1.438980Google ScholarThere is no corresponding record for this reference.
- 59Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, P. V. R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3–21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294– 301, DOI: 10.1002/jcc.540040303Google ScholarThere is no corresponding record for this reference.
- 60Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999– 3094, DOI: 10.1021/cr9904009Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract

Figure 1

Figure 1. (a) Catalytic cycle for ammonia production from dinitrogen and water using [MoI3(PCP)] as a catalyst. (b) Stepwise conversion of [Mo(N)I(PCP)] into [Mo(NH3)I(PCP)] via reduction and protonation. (c) Catalytic ammonia formation reaction using the combination of SmI2 and water and that of cobaltocene and pyridinium salt.
Figure 2

Figure 2. (a) Previous work by Peters and co-workers: cobaltocene derivative bearing N,N-dimethylanilinium. (17,18) (b) Our previous work: iron complexes bearing π-phenol ligand. (19,20) (c) Our previous work: samarium complexes bearing cyclopentadienyl-amine chelate ligands. (21) (d) This work: cobaltocenes bearing 4-(2,6-dimethylpyridin-1-ium-4-yl)phenyl moiety [1-H]+.
Scheme 1
 Scheme 1. Synthesis of [1a]OTf, [1a], and [1a-H](OTf)2
Scheme 1. Synthesis of [1a]OTf, [1a], and [1a-H](OTf)2Figure 3

Figure 3. ORTEP drawings of the cationic part of (a) [1a]OTf and (b) [1a-H](OTf)2. Hydrogen atoms except for a NH hydrogen atom in [1a-H](OTf)2 were omitted for clarity.
Scheme 2
 Scheme 2. Formation of [1a-H]OTf
Scheme 2. Formation of [1a-H]OTfFigure 4
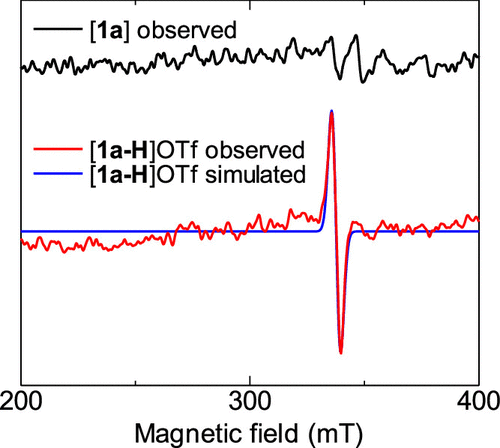
Figure 4. X-band EPR spectra of [1a] (black) and [1a-H]OTf (observed: red, simulated: blue) in 2-MeTHF at −196 °C. giso = 1.99911, line width = 4.65 mT.
Figure 5

Figure 5. Absorption spectra of [1a] (0.05 mM) in THF at −78 °C by the addition of [PicH]OTf as the proton source.
References
This article references 60 other publications.
- 1Liu, H. Ammonia Synthesis Catalyst 100 Years: Practice, Enlightenment and Challenge. Chin. J. Catal. 2014, 35, 1619– 1640, DOI: 10.1016/S1872-2067(14)60118-2There is no corresponding record for this reference.
- 2Wang, X.; Fan, W.; Chen, J.; Feng, G.; Zhang, X. Experimental Study and Kinetic Analysis of the Impact of Ammonia Co-Firing Ratio on Products Formation Characteristics in Ammonia/Coal Co-Firing Process. Fuel 2022, 329, 125496 DOI: 10.1016/j.fuel.2022.125496There is no corresponding record for this reference.
- 3Guo, J.; Chen, P. Catalyst: NH3 as an Energy Carrier. Chem 2017, 3, 709– 712, DOI: 10.1016/j.chempr.2017.10.004There is no corresponding record for this reference.
- 4Levi, P. G.; Cullen, J. M. Mapping Global Flows of Chemicals: From Fossil Fuel Feedstocks to Chemical Products. Environ. Sci. Technol. 2018, 52, 1725– 1734, DOI: 10.1021/acs.est.7b04573There is no corresponding record for this reference.
- 5Boerner, L. K. Taking the CO2 out of NH3. Chem. Eng. News 2019, 97, 18– 21There is no corresponding record for this reference.
- 6Bezdek, M. J.; Pappas, I.; Chirik, P. J. Determining and Understanding N-H Bond Strengths in Synthetic Nitrogen Fixation Cycles. Top. Organomet. Chem. 2017, 60, 1– 21, DOI: 10.1007/3418_2016_8There is no corresponding record for this reference.
- 7Bruch, Q. J.; Connor, G. P.; McMillion, N. D.; Goldman, A. S.; Hasanayn, F.; Holland, P. L.; Miller, A. J. M. Considering Electrocatalytic Ammonia Synthesis via Bimetallic Dinitrogen Cleavage. ACS Catal. 2020, 10, 10826– 10846, DOI: 10.1021/acscatal.0c02606There is no corresponding record for this reference.
- 8Tanabe, Y.; Nishibayashi, Y. Catalytic Nitrogen Fixation Using Well-Defined Molecular Catalysts under Ambient or Mild Reaction Conditions. Angew. Chem., Int. Ed. 2024, 63, e202406404 DOI: 10.1002/anie.202406404There is no corresponding record for this reference.
- 9Chalkley, M. J.; Drover, M. W.; Peters, J. C. Catalytic N2-to-NH3 (or -N2H4) Conversion by Well-Defined Molecular Coordination Complexes. Chem. Rev. 2020, 120, 5582– 5636, DOI: 10.1021/acs.chemrev.9b00638There is no corresponding record for this reference.
- 10(a) Eizawa, A.; Arashiba, K.; Tanaka, H.; Kuriyama, S.; Matsuo, Y.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Remarkable Catalytic Activity of Dinitrogen-Bridged Dimolybdenum Complexes Bearing NHC-Based PCP-Pincer Ligands toward Nitrogen Fixation. Nat. Commun. 2017, 8, 14874 DOI: 10.1038/ncomms14874There is no corresponding record for this reference.(b) Eizawa, A.; Arashiba, K.; Egi, A.; Tanaka, H.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Reactivity of Molybdenum–Trihalide Complexes Bearing PCP-Type Pincer Ligands. Chem. - Asian J. 2019, 14, 2091– 2096, DOI: 10.1002/asia.201900496There is no corresponding record for this reference.
- 11Nakamura, T.; Tsuruta, Y.; Egi, A.; Tanaka, H.; Nishibayashi, Y.; Yoshizawa, K. Theoretical Study of Imide Formation in Nitrogen Fixation Catalyzed by Molybdenum Complex Bearing PCP-Type Pincer Ligand with Metallocenes. Inorg. Chem. 2025, 64, 9124– 9136, DOI: 10.1021/acs.inorgchem.5c00695There is no corresponding record for this reference.
- 12Arashiba, K.; Eizawa, A.; Tanaka, H.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Nitrogen Fixation via Direct Cleavage of Nitrogen–Nitrogen Triple Bond of Molecular Dinitrogen under Ambient Reaction Conditions. Bull. Chem. Soc. Jpn. 2017, 90, 1111– 1118, DOI: 10.1246/bcsj.20170197There is no corresponding record for this reference.
- 13Arashiba, K.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Cycling between Molybdenum-Dinitrogen and -Nitride Complexes to Support the Reaction Pathway for Catalytic Formation of Ammonia from Dinitrogen. Chem. - Eur. J. 2020, 26, 13383– 13389, DOI: 10.1002/chem.202002200There is no corresponding record for this reference.
- 14(a) Ashida, Y.; Arashiba, K.; Nakajima, K.; Nishibayashi, Y. Molybdenum-Catalysed Ammonia Production with Samarium Diiodide and Alcohols or Water. Nature 2019, 568, 536– 540, DOI: 10.1038/s41586-019-1134-2There is no corresponding record for this reference.(b) Ashida, Y.; Mizushima, T.; Arashiba, K.; Egi, A.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Production of Ammonia from Dinitrogen Employing Molybdenum Complexes Bearing N-Heterocyclic Carbene-Based PCP-Type Pincer Ligands. Nat. Synth. 2023, 2, 635– 644, DOI: 10.1038/s44160-023-00292-9There is no corresponding record for this reference.
- 15Suginome, S.; Murota, K.; Yamamoto, A.; Yoshida, H.; Nishibayashi, Y. Mechanochemical Nitrogen Fixation Catalysed by Molybdenum Complexes. Nat. Synth. 2025, 4, 243– 251, DOI: 10.1038/s44160-024-00661-yThere is no corresponding record for this reference.
- 16Kolmar, S. S.; Mayer, J. M. SmI2(H2O)n Reduction of Electron Rich Enamines by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2017, 139, 10687– 10692, DOI: 10.1021/jacs.7b03667There is no corresponding record for this reference.
- 17Chalkley, M. J.; Garrido-Barros, P.; Peters, J. C. A Molecular Mediator for Reductive Concerted Proton-Electron Transfers via Electrocatalysis. Science 2020, 369, 850– 854, DOI: 10.1126/science.abc1607There is no corresponding record for this reference.
- 18Garrido-Barros, P.; Derosa, J.; Chalkley, M. J.; Peters, J. C. Tandem Electrocatalytic N2 Fixation via Proton-Coupled Electron Transfer. Nature 2022, 609, 71– 76, DOI: 10.1038/s41586-022-05011-6There is no corresponding record for this reference.
- 19Kuriyama, S.; Zhang, Y.; Tanaka, H.; Konomi, A.; Egi, A.; Yoshizawa, K.; Nishibayashi, Y. Synthesis and Reactivity of Iron-Oxocyclohexadienyl Complexes toward Proton-Coupled Electron Transfer. Organometallics 2024, 43, 2736– 2746, DOI: 10.1021/acs.organomet.4c00286There is no corresponding record for this reference.
- 20Tanabe, Y.; Yamamoto, S.; Kuriyama, S.; Nishibayashi, Y. Preparation and Reactivity of Anionic Iron Sandwich Complex Bearing π-Phenol Ligand Acting as Proton-Coupled Electron Transfer Reagent. Chem. -Asian J. 2026, 21, e00951 DOI: 10.1002/asia.202500951There is no corresponding record for this reference.
- 21Mitsumoto, T.; Nakamura, T.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Cooperative Ammonia Formation Catalyzed by Molybdenum and Samarium Complexes. Angew. Chem., Int. Ed. 2025, 64, e202507061 DOI: 10.1002/anie.202507061There is no corresponding record for this reference.
- 22Feng, R.; Zhang, L.; Ruan, H.; Zhao, Y.; Tan, G.; Wang, X. A Main-Group Element Radical Based One-Dimensional Magnetic Chain. Angew. Chem., Int. Ed. 2019, 58, 6084– 6088, DOI: 10.1002/anie.201901177There is no corresponding record for this reference.
- 23Ammeter, J. H. EPR of orbitally degenerate sandwich compounds. J. Magn. Reson. 1978, 30, 299– 325, DOI: 10.1016/0022-2364(78)90103-8There is no corresponding record for this reference.
- 24Robbins, J. L.; Edelstein, N.; Spencer, B.; Smart, J. C. Synthesis and Electronic Structure of Decamethylmetallocenes. J. Am. Chem. Soc. 1982, 104, 1882– 1893, DOI: 10.1021/ja00371a017There is no corresponding record for this reference.
- 25Garrido, G.; Rosés, M.; Ràfols, C.; Bosch, E. Acidity of Several Anilinium Derivatives in Pure Tetrahydrofuran. J. Solution Chem. 2008, 37, 689– 700, DOI: 10.1007/s10953-008-9262-6There is no corresponding record for this reference.
- 26Bordwell, F. G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456– 463, DOI: 10.1021/ar00156a004There is no corresponding record for this reference.
- 27Agarwal, R. G.; Coste, S. C.; Groff, B. D.; Heuer, A. M.; Noh, H.; Parada, G. A.; Wise, C. F.; Nichols, E. M.; Warren, J. J.; Mayer, J. M. Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2022, 122, 1– 49, DOI: 10.1021/acs.chemrev.1c00521There is no corresponding record for this reference.
- 28
From absorption spectroscopic studies, half of [1] was initially converted to [1-H]OTf in this condition. ([1a-H]OTf: 45%, [1b-H]OTf: 47%, Figures S12 and S13)
There is no corresponding record for this reference. - 29Evans, D. F. 400. The Determination of the Paramagnetic Susceptibility of Substances in Solution by Nuclear Magnetic Resonance. J. Chem. Soc. 1959, 2003– 2005, DOI: 10.1039/jr9590002003There is no corresponding record for this reference.
- 30Live, D. H.; Chan, S. I. Bulk Susceptibility Corrections in Nuclear Magnetic Resonance Experiments Using Superconducting Solenoids. Anal. Chem. 1970, 42, 791– 792, DOI: 10.1021/ac60289a028There is no corresponding record for this reference.
- 31Bain, G. A.; Berry, J. F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532, DOI: 10.1021/ed085p532There is no corresponding record for this reference.
- 32Stoll, S.; Schweiger, A. EasySpin, a comprehensive softwarepackage for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42– 55, DOI: 10.1016/j.jmr.2005.08.013There is no corresponding record for this reference.
- 33Molton, F. Simultispin: A versatile graphical user interface for the simulation of solid-state continuous wave EPR spectra. Magn. Reson. Chem. 2020, 58, 718– 726, DOI: 10.1002/mrc.5019There is no corresponding record for this reference.
- 34Martinez, V.; Blais, J.-C.; Astruc, D. Coupling Multiple Benzylic Activation of Simple Arenes by CpFe+ with Multiple Alkene Metathesis Using Grubbs Catalysts: An Efficient Carbon–Carbon Bond Formation Strategy Leading to Polycycles, Cyclophanes, Capsules, and Polymeric Compounds and Their CpFe+ Complexes. Organometallics 2004, 23, 861– 874, DOI: 10.1021/om030623wThere is no corresponding record for this reference.
- 35Chmely, S. C.; Hanusa, T. P.; Rheingold, A. L. Influence of Ring Methylation in Group 15 Tetramethylcyclopentadienyl Complexes, M(C5Me4H)nI3–n (M = As, Sb). Organometallics 2010, 29, 5551– 5557, DOI: 10.1021/om100474mThere is no corresponding record for this reference.
- 36Bunel, E. E.; Valle, L.; Manriquez, J. M. Pentamethylcyclopentadienyl Acetylacetonate Complexes of Iron(II), Cobalt(II), and Nickel(II). Convenient Synthetic Entries to Mono-η5-C5Me5 Derivatives. Organometallics 1985, 4, 1680– 1682, DOI: 10.1021/om00128a034There is no corresponding record for this reference.
- 37Tanabe, Y.; Nakajima, K.; Nishibayashi, Y. Phosphine Oxidation with Water and Ferrocenium(III) Cation Induced by Visible Light Irradiation. Chem. - Eur. J. 2018, 24, 18618– 18622, DOI: 10.1002/chem.201805129There is no corresponding record for this reference.
- 38Weitz, I. S.; Rabinovitz, M. The application of C8K for organic synthesis: reduction of substituted naphthalenes. J. Chem. Soc., Perkin Trans. 1993, 1, 117– 120, DOI: 10.1039/p19930000117There is no corresponding record for this reference.
- 39Weatherburn, M. W. Phenol-Hypochlorite Reaction for Determination of Ammonia. Anal. Chem. 1967, 39, 971– 974, DOI: 10.1021/ac60252a045There is no corresponding record for this reference.
- 40CrysAlisPro: Data Collection and Processing Software; RigakuCorporation: Tokyo, Japan, 2015.There is no corresponding record for this reference.
- 41Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339– 341, DOI: 10.1107/S0021889808042726There is no corresponding record for this reference.
- 42Sheldrick, G. SHELXT – Integrated space-group and crystal structure determination. Acta Crystallogr., Sect. A:Found. Adv. 2015, 71, 3– 8, DOI: 10.1107/S2053273314026370There is no corresponding record for this reference.
- 43Sheldrick, G. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C:Struct. Chem. 2015, 71, 3– 8, DOI: 10.1107/S2053229614024218There is no corresponding record for this reference.
- 44van der Sluis, P.; Spek, A. L. BYPASS: an effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. 1990, 46, 194– 201, DOI: 10.1107/S0108767389011189There is no corresponding record for this reference.
- 45Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Cioslowski, J.; Fox, D. J. Gaussian 16; Gaussian, Inc.: Wallingford, CT, 2016.There is no corresponding record for this reference.
- 46Becke, A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098– 3100, DOI: 10.1103/PhysRevA.38.3098There is no corresponding record for this reference.
- 47Becke, A. D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648– 5652, DOI: 10.1063/1.464913There is no corresponding record for this reference.
- 48Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the ElectronDensity. Phys. Rev. B 1988, 37, 785– 789, DOI: 10.1103/PhysRevB.37.785There is no corresponding record for this reference.
- 49Vosko, S. H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200– 1211, DOI: 10.1139/p80-159There is no corresponding record for this reference.
- 50Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104 DOI: 10.1063/1.3382344There is no corresponding record for this reference.
- 51Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted Ab Initio Pseudopotentials for the First Row Transition Elements. J. Chem. Phys. 1987, 86, 866– 872, DOI: 10.1063/1.452288There is no corresponding record for this reference.
- 52Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjusted ab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123– 141, DOI: 10.1007/BF01114537There is no corresponding record for this reference.
- 53Ditchfield, R.; Hehre, W. J.; Pople, J. A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724– 728, DOI: 10.1063/1.1674902There is no corresponding record for this reference.
- 54Hehre, W. J.; Ditchfield, R.; Pople, J. A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257– 2261, DOI: 10.1063/1.1677527There is no corresponding record for this reference.
- 55Hariharan, P. C.; Pople, J. A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213– 222, DOI: 10.1007/BF00533485There is no corresponding record for this reference.
- 56Francl, M. M.; Pietro, W. J.; Hehre, W. J.; Binkley, J. S.; Gordon, M. S.; DeFrees, D. J.; Pople, J. A. Self-consistent Molecular Orbital Methods. XXIII. A Polarization-type Basis Set for Second-row Elements. J. Chem. Phys. 1982, 77, 3654– 3665, DOI: 10.1063/1.444267There is no corresponding record for this reference.
- 57Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. Self-consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650– 654, DOI: 10.1063/1.438955There is no corresponding record for this reference.
- 58McLean, A. D.; Chandler, G. S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639– 5648, DOI: 10.1063/1.438980There is no corresponding record for this reference.
- 59Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, P. V. R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3–21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294– 301, DOI: 10.1002/jcc.540040303There is no corresponding record for this reference.
- 60Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999– 3094, DOI: 10.1021/cr9904009There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.5c00492.
Detailed experiment procedures and computational details, Synthesis and reactivity of cobaltocenes, X-ray crystallography, EPR study, UV–vis absorption spectroscopy, cyclic voltammetry, NMR and IR spectra, and DFT calculations (PDF)
Cartesian coordinates of all optimized geometries (XYZ)
Deposition Numbers 2512896–2512902 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via the joint Cambridge Crystallographic Data Centre (CCDC) and Fachinformationszentrum Karlsruhe Access Structures service.
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.