



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Electrochemical or Photoredox Activation of Latent Electrophiles: Three-Component Mumm Rearrangement Cascade Reactions from AlkoxyaminesClick to copy article linkArticle link copied!
- Tan N. HuynhTan N. HuynhInstitute for Nanoscale Science and Technology, Flinders University, Adelaide, South Australia 5042, AustraliaMore by Tan N. Huynh
- Philip L. NorcottPhilip L. NorcottInstitute for Nanoscale Science and Technology, Flinders University, Adelaide, South Australia 5042, AustraliaMore by Philip L. Norcott
- Michelle L. Coote*Michelle L. Coote*E-mail for M.L.C.: [email protected]Institute for Nanoscale Science and Technology, Flinders University, Adelaide, South Australia 5042, AustraliaMore by Michelle L. Coote
- Alex C. Bissember*Alex C. Bissember*E-mail for A.C.B.: [email protected]School of Natural Sciences − Chemistry, University of Tasmania, Hobart, Tasmania 7001, AustraliaMore by Alex C. Bissember
Abstract
This report exploits either electrochemical or photoredox triggers to promote the oxidative mesolytic cleavage of alkoxyamines to establish multicomponent Mumm rearrangement cascade reactions for the synthesis of imides and imidates. Readily accessible, bench-, air-, and moisture-stable alkoxyamines derived from 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) serve as masked carbocations in this approach.
This publication is licensed for personal use by The American Chemical Society.
Alkoxyamines, such as those derived from 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), are bench-, air-, and moisture-stable compounds. They are directly accessible from a broad range of chemical feedstocks, including activated alkanes, (1) aldehydes, (2,3) alkenes, (4) organohalides, (5) and carboxylic acids (Figure 1A). (6−8) TEMPO-based alkoxyamines (TAs) are most commonly exploited synthetically in polymer chemistry, where they serve as radical initiators or mediators. (9) In these reactions, heating at elevated temperatures facilitates homolytic bond cleavage, which affords a carbon-centered radical and the persistent radical TEMPO. (10) In contrast, the broader applications of TAs in small molecule synthesis remain largely unrealized. (11)
Figure 1
Figure 1. (A) Representative synthetic routes to TAs. (B) Overview of previously reported photoredox- and electrochemically-triggered alkylations using TAs. (C) Overview of previously reported methods for electrochemical Mumm rearrangement cascades.
In 2006, Braslau and O’Brya discovered that cerium ammonium nitrate mediated oxidative cleavage of alkoxyamines which enabled nucleophilic substitution reactions at 70 °C. (12) A decade later, the Knowles group reported that the photoredox-catalyzed one-electron oxidation of certain TAs (A) at ambient temperature generates transient cationic radicals A+• that undergo mesolytic cleavage to form carbocations (Figure 1B). (13) Exploiting this mode of activation allowed trapping of these electrophiles by an array of nucleophiles, facilitating substitution reactions under mild conditions. (13) In 2018, we combined cyclic voltammetry, EPR spectroscopy and theoretical calculations to demonstrate that mesolytic cleavage of TAs affords carbocations under mild electrochemical conditions. (14) We, subsequently, showed that additional modes of reactivity are possible, including mesolytic cleavage to carbon-centered radicals or SN2 reactions with weak nucleophiles including common solvents and electrolytes. (2,15) In 2019, we exploited electrochemical activation to reveal the previously inaccessible latent electrophilicity of TEMPO–Me, developing a new methylating agent (Figure 1B). (16) Notably, this reactivity was exclusively restricted to electrosynthesis and was unattainable by photoirradiation or chemical oxidation.
Although photoredox catalysis has enabled alkylations employing TAs with a relatively large number of nucleophiles, (4,13,17−19) the scope of analogous electrochemical methods is confined to carboxylates (Figure 1B). (16,20,21) Nevertheless, cyclic voltammetry indicates that a much wider scope of reactivity is accessible under electrochemical conditions. (2,15) Expanding the range of nucleophiles is therefore crucial for fully realizing the potential of TAs in electrosynthesis and organic synthesis more generally.
The Mumm rearrangement is a multicomponent cascade reaction in which nucleophilic trapping of a carbocation by a nitrile (e.g., CH3CN) forms a nitrilium ion that is then captured, in situ, by a carboxylic acid. Subsequent rearrangement affords an imide product (Figure 1C). (22) Recently, a handful of Mumm rearrangement protocols have been reported in which anodic oxidation is utilized to access carbocations from activated alkenes, benzylic C(sp3)–H substrates, or via decarboxylation of carboxylic acids. (23−27) In this proof of concept study, we demonstrate that electrochemical triggers enable TAs to serve as synthetically useful, latent carbocations in three-component Mumm rearrangement cascade reactions to generate functionalized imides and imidates. Our work emphasizes that electrochemical activation of TAs can facilitate transformations in which multiple bonds are formed in a single operation─i.e., reactivity beyond the direct alkylation of carboxylic acids. (28) In addition, we investigate and develop analogous photoredox-catalyzed methodology and demonstrate that TAs can serve as masked amides, which can be selectively revealed under relatively mild conditions.
Our investigation commenced with an optimization study examining the reaction of alkoxyamine (1a) and benzoic acid (2a) in an appropriate electrolyte (Table 1). We determined that the reaction of these substrates (1:1.5 ratio of 2a/1a) in an anhydrous electrolyte solution (nBu4NPF6 and CH3CN) employing an undivided cell equipped with graphite working electrodes operating at 10 mA under an argon atmosphere delivered imide 4a in 88% yield after 16 h (Table 1, entry 1). These optimal conditions required very high electron equivalents (29.8 F/mol; current density: ∼5 mA/cm2). (29,30) Shorter reaction times provided lower product yields due to incomplete conversion (41% yield after 4 h; and 74% yield after 12 h), while reaction in air afforded imide 4a in 80% yield (entries 2–4). When we performed the transformation using a nonanhydrous, bench-grade electrolyte in the presence of activated molecular sieves 4Å and air, we obtained a 59% yield (entry 5). (31) Rapid alternating polarity electrolysis (RAP) at 25 Hz (every 0.04 s) did not promote the reaction and returned unreacted starting materials (entry 6). Galvanostatic electrolysis at either 5 or 15 mA did not provide enhanced results (entries 7 and 8). Potentiostatic electrolysis applied at either 1.6 and 1.8 V vs Ag/AgCl afforded product 4a in 40% and 64% yields, respectively (entries 9 and 10). The substitution of nBu4NPF6 for either nBu4NOAc or nBu4NBF4 gave lower yields (entries 11 and 12), while a blended solvent system (3:1 CH2Cl2/CH3CN) provided product 4a in 75% yield (entry 13). We found that employing either 1:1 or 1:1.5 ratios of 1a/2a resulted in diminished efficiency (entries 14 and 15). Finally, reaction in a divided cell using a protocol in which compounds 1a and 2a were initially introduced into the anodic chamber delivered imide 4a in 73% yield (entry 16).
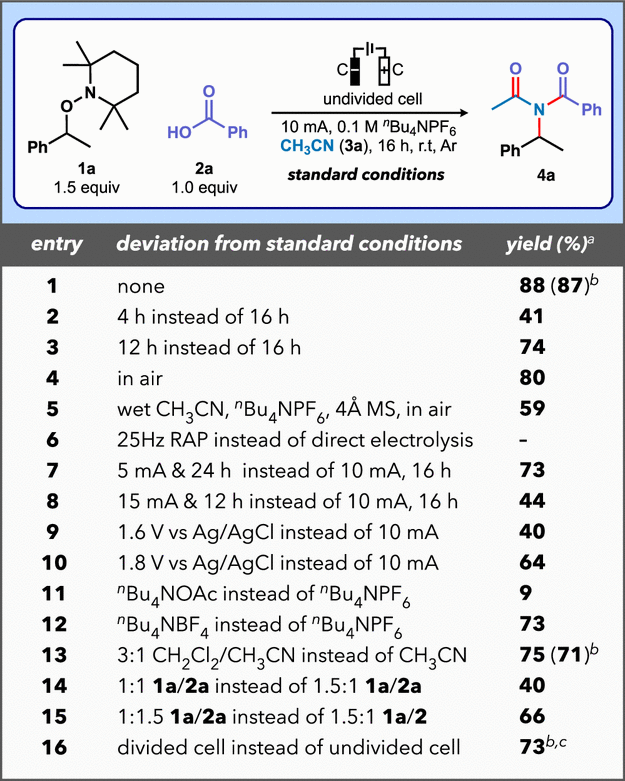
1H NMR yield of 4a using 1,3,5-trimethoxybenzene as internal standard.
Isolated yield.
1 mmol scale, 3 h.
Standard conditions: 0.2 mmol 2a, 1.5 equiv 1a, nBu4NPF6 (4 mL of a 0.1 M anhydrous CH3CN solution), undivided cell equipped with graphite working electrodes, 10 mA, 16 h, Ar.
With the optimized reaction conditions in hand, we initially explored the substrate scope with respect to the carboxylic acid partner (Figure 2). The more sterically encumbered reactant 2-ethylbenzoic acid provided imide 4b in 74% yield. Electronically diverse para-substituted benzoic acids bearing either electron-donating (methoxy) or electron-withdrawing (benzoyl, bromo, and nitro) groups were tolerated under the reaction conditions (4c–4f). Unsurprisingly, we found that employing a divided cell considerably improved the yield of nitro-containing imide 4f. meta-Disubstituted benzoic acids were compatible (4g and 4h), and 2-naphthoic acid afforded the corresponding imide 4i in 74% yield. Notably, pivalic acid and adamantane carboxylic acid were tolerated by our protocol (4j and 4k). The structure of 4k was confirmed by X-ray crystal diffraction. These findings are significant because previously reported electrochemically promoted Mumm rearrangement methods have been typically restricted to aromatic acid substrates. (23−27)
Figure 2
Figure 2. Electrochemically-promoted Mumm rearrangement cascades: representative substrate scope. Legend: a Divided cell, 2.0 equiv 1a, 6 h, 1 mmol scale; b nBu4NPF6 (4 mL of a 0.1 M anhydrous 3:1 CH2Cl2/nitrile 3 solution); c 10 equiv nitrile 3; nBu4NPF6 (4 mL of a 0.1 M CH2Cl2 solution). brsm: based on recovered starting material.
The alkoxyamine scope was also examined. TAs bearing para-fluoro- and para-phenyl-substituted groups efficiently delivered respective imides 4l and 4m (Figure 2). 4-Chlorobenzyl-substituted TA, which represents an activated primary electrophile afforded imide 4n in a low 31% yield. The generation of tertiary carbocations, such as those derived from cumenyl, tert-butyl and 1-adamantyl systems, gave the corresponding imides (4o–4q) in synthetically useful yields. A cyclohexene-containing TA also engaged in the transformation to furnish imide 4r in 63% yield. In comparison, the cyclohexyl-based TA analogue gave product 4s in a poor yield at low conversion in an undivided cell (12% yield; 96% yield brsm). This improved to a 28% yield of imide 4s when the reaction was performed in a divided cell. Interestingly, imide 4t could not be prepared under undivided cell conditions and isopropyl benzoate was formed in preference (19% NMR yield). However, imide 4t was obtained in a 45% yield under divided cell conditions. Poor conversion was observed in the synthesis of imide 4u. Our approach also furnished derivatives of the drugs ibuprofen and flurbiprofen (4v and 4w) in 74% and 44% (85% brsm) yields, respectively.
Based on the alkoxyamine scope, supported by cyclic voltammetry on representative substrates (Supporting Information), we conclude that our protocol is generally tolerated by alkoxyamines that undergo facile oxidative cleavage to carbocations. Even the cyclohexyl-based TA substrate which, based on cyclic voltammetry (see Figure S7, Supporting Information), undergoes slow cleavage to carbocations, nonetheless gives high selectivity for product 4s (96% yield brsm under undivided cell conditions). In this case, we posit that low conversion arises from the partially reversible nature of the alkoxyamine oxidation. The protocol fails for carbocations that are especially prone to decomposition, such as from 2-THF-containing substrate 1ac (Figure 3). Yields are probably also somewhat diminished by competing E1 elimination from the respective tertiary carbocation intermediates that lead to 4o and 4p. As anticipated, the protocol fails for substrates bearing electron-withdrawing groups (such as 1ad and 1ae). Based on our previous studies, (14) these systems are expected to undergo oxidative cleavage to carbon-centered radicals.
Figure 3
Figure 3. Other TA substrates examined in this study.
Consistent with our synthetic experiments and cyclic voltammetry studies, we propose a mechanism in which reaction is initiated via one-electron oxidation of the TA at the anode leading to mesolytic cleavage that generates a carbocation. TEMPO is formed as a byproduct of this process. We suspect that the relatively low Faradaic efficiency we often observe in this chemistry principally derives from the unproductive cycling of TEMPO/TEMPO+ across electrodes. Thus, we anticipate that reduction of nitroxide species serve as the primary cathodic processes. Consistent with this, we observed vast differences in the Faradaic efficiency of experiments performed in undivided and divided cells using 1a, 2a (1.0 mmol), and CH3CN: 18.9 vs 1.11 F/mol, respectively (see: p S-13, Supporting Information). The undivided cell experiment required a reaction time of 50 h, in comparison to 3 h for the equivalent process performed in a divided cell.
We examined the nitrile scope using a predominantly dichloromethane-based electrolyte. Acetonitrile-d3 afforded imide 4x in 58% yield under these conditions (Figure 2). Reactions employing aromatic nitriles, such as benzonitrile, 4-bromobenzonitrile, 4-methoxybenzonitrile, and 2-thiophenecarbonitrile, were slower and provided respective products 4y, 4z, 4aa, and 4ab in low yields at low conversion. In all cases, the yields calculated relative to the recovered starting material are high, indicating good selectivity. The lower conversions compared with the analogous reactions with acetonitrile are attributed to changes in solution conductivity arising from the use of mixed nitrile/dichloromethane solvents; yields for these may be improved through further optimization of the reaction conditions. (32)
As noted earlier, photoredox activation of alkoxyamines has also been exploited to generate carbocations under mild conditions. (13,17,18) This prompted us to examine the utility of this alterative strategy to facilitate Mumm rearrangement cascades from alkoxyamines. To this end, irradiating an acetonitrile solution of TA 1 and aryl carboxylic acid 2 with blue light in the presence of iridium photoredox catalyst B afforded imides 4a, 4d, 4p, and 4q in excellent yields (Figure 4). In contrast to the electrochemical conditions, product 4n could not be prepared from 4-chlorobenzyl-substituted TA under photoredox activation (no conversion of the starting material was observed). We also noted that photoredox activation enabled efficient capture of nitrilium with hexafluoroisopropanol (HFIP), affording somewhat unstable imidate 4af. Limited formation of product 4af was observed via electrosynthesis (∼16% yield, as judged by 1H NMR spectroscopy), in comparison. These results emphasize that, although carbocations can be generated from TAs via either photochemical or electrochemical triggers, notable differences remain in observed reaction outcomes depending on the method used to promote the initial redox event. This discovery suggests interesting opportunities for complementary or divergent reactivity are possible in these manifolds.
Figure 4
Figure 4. Photoredox-catalyzed Mumm rearrangement cascades. Reaction conditions: 0.1 mmol 2, 1.5 equiv 1, 2 mol % [Ir(dF(CF3)ppy)2(5,5′-dCF3bpy)]PF6 (B), anhydrous CH3CN (2 mL), 16 h, under Ar. Legend: a0.1 mmol 1, 10 equiv HFIP, 2 mol % B.
Consistent with previous reports, (24) we could selectively convert imide 4a to either benzamide 5 or acetamide 6 by judicious selection of reaction conditions (eq 1). This demonstrates that TAs can serve as masked amides that can be selectively revealed under relatively mild conditions over 2 steps.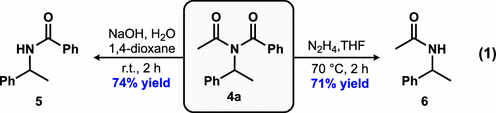
In summary, our work highlights that either electrochemical or photoredox triggers can induce oxidative mesolytic cleavage of TAs 1 under mild conditions to establish three-component Mumm rearrangement cascade reactions. These new protocols broaden the scope of alkoxyamines in chemical synthesis and the extension of these strategies to more complex systems remain ongoing in our laboratories.
Data Availability
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.5c05049.
All experimental procedures, characterization data, X-ray crystallographic data for 4k, and NMR spectra (PDF)
Deposition Number 2494351 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via the joint Cambridge Crystallographic Data Centre (CCDC) and Fachinformationszentrum Karlsruhe Access Structures service.
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Michelle L. Coote - Institute for Nanoscale Science and Technology, Flinders University, Adelaide, South Australia 5042, Australia;
 https://orcid.org/0000-0003-0828-7053;
https://orcid.org/0000-0003-0828-7053;
- Alex C. Bissember - School of Natural Sciences − Chemistry, University of Tasmania, Hobart, Tasmania 7001, Australia;https://orcid.org/0000-0001-5515-2878;
- Tan N. Huynh - Institute for Nanoscale Science and Technology, Flinders University, Adelaide, South Australia 5042, Australia;https://orcid.org/0000-0003-4373-6483
- Philip L. Norcott - Institute for Nanoscale Science and Technology, Flinders University, Adelaide, South Australia 5042, Australia;https://orcid.org/0000-0003-4082-2079
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Acknowledgments
The authors thank the Australian Research Council (DP250100285 and FT200100049) for funding. A/Prof. Lorenzo White (Jinan University), Muhammad Saqib (University of Tasmania) and Dr Curtis C. Ho (University of Tasmania) are thanked for useful discussions and assistance with preliminary experiments. Dr Wit Bloch and Steven Tsoukatos from Flinders University are thanked for X-ray crystallography experiments. This research was undertaken in part using the MX1 beamline at the Australian Synchrotron, part of ANSTO.
References
This article references 32 other publications.
- 1Li, L.; Yu, Z.; Shen, Z. Copper-Catalyzed Aminoxylation of Different Types of Hydrocarbons with TEMPO: A Concise Route to N-Alkoxyamine Derivatives. Adv. Synth. Catal. 2015, 357, 3495– 3500, DOI: 10.1002/adsc.201500544Google ScholarThere is no corresponding record for this reference.
- 2Hammill, C. L.; Noble, B. B.; Norcott, P. L.; Ciampi, S.; Coote, M. L. Effect of Chemical Structure on the Electrochemical Cleavage of Alkoxyamines. J. Phys. Chem. C 2019, 123, 5273– 5281, DOI: 10.1021/acs.jpcc.8b12545Google ScholarThere is no corresponding record for this reference.
- 3Schoening, K.-U.; Fischer, W.; Hauck, S.; Dichtl, A.; Kuepfert, M. Synthetic Studies on N-Alkoxyamines: A Mild and Broadly Applicable Route Starting from Nitroxide Radicals and Aldehydes. J. Org. Chem. 2009, 74, 1567– 1573, DOI: 10.1021/jo802403jGoogle ScholarThere is no corresponding record for this reference.
- 4Yao, C.; Aliyu, M. A.; Wan, Y.; Norton, J. R. Formation of TEMPO Adducts with Hydrogen Atom Transfer: An Alternative Pathway to Versatile Hydrofunctionalizations of Olefins. Chem. Eur. J. 2025, 31, e202403899 DOI: 10.1002/chem.202403899Google ScholarThere is no corresponding record for this reference.
- 5Matyjaszewski, K.; Woodworth, B. E.; Zhang, X.; Gaynor, S. G.; Metzner, Z. Simple and Efficient Synthesis of Various Alkoxyamines for Stable Free Radical Polymerization. Macromolecules 1998, 31, 5955– 5957, DOI: 10.1021/ma9807264Google ScholarThere is no corresponding record for this reference.
- 6Schulz, G.; Kirschning, A. Metal free decarboxylative aminoxylation of carboxylic acids using a biphasic solvent system. Org. Biomol. Chem. 2021, 19, 273– 278, DOI: 10.1039/D0OB01773FGoogle ScholarThere is no corresponding record for this reference.
- 7Innocent, M.; Tanguy, C.; Gavelle, S.; Aubineau, T.; Guérinot, A. Iron-Catalyzed, Light-Driven Decarboxylative Alkoxyamination. Chem. Eur. J. 2024, 30, e202401252 DOI: 10.1002/chem.202401252Google ScholarThere is no corresponding record for this reference.
- 8Denkler, L. M.; Aladahalli Shekar, M.; Ngan, T. S. J.; Wylie, L.; Abdullin, D.; Engeser, M.; Schnakenburg, G.; Hett, T.; Pilz, F. H.; Kirchner, B.; Schiemann, O.; Kielb, P.; Bunescu, A. General Iron-Catalyzed Decarboxylative Oxygenation of Aliphatic Carboxylic Acids. Angew. Chem., Int. Ed. 2024, 63, e202403292 DOI: 10.1002/anie.202403292Google ScholarThere is no corresponding record for this reference.
- 9Hawker, C. J.; Bosman, A. W.; Harth, E. New Polymer Synthesis by Nitroxide Mediated Living Radical Polymerizations. Chem. Rev. 2001, 101, 3661– 3688, DOI: 10.1021/cr990119uGoogle ScholarThere is no corresponding record for this reference.
- 10Audran, G.; Bagryanskaya, E. G.; Bikanga, R.; Coote, M. L.; Guselnikova, O.; Hammill, C. L.; Marque, S. R. A.; Mellet, P.; Postnikov, P. S. Dynamic Covalent Bond: Modes of Activation of the C-ON Bond in Alkoxyamines. Prog. Polym. Sci. 2023, 144, 101726, DOI: 10.1016/j.progpolymsci.2023.101726Google ScholarThere is no corresponding record for this reference.
- 11Lee, K.; Yom, C.; Gilberti, D.; Tahsin, S.; Meza, J.; Stewart, J.; Boone, D.; Xing, Y. Oxidative Cleavage of Alkoxyamines for Nucleophilic Substitution Reactions. Eur. J. Org. Chem. 2024, 27, e202400845 DOI: 10.1002/ejoc.202400845Google ScholarThere is no corresponding record for this reference.
- 12O’Brya, G.; Braslau, R. Terminal Functionalization of Polymers via Single Electron Oxidation of N-Alkoxyamines. Macromolecules 2006, 39, 9010– 9017, DOI: 10.1021/ma061940sGoogle ScholarThere is no corresponding record for this reference.
- 13Zhu, Q.; Gentry, E. C.; Knowles, R. R. Catalytic Carbocation Generation Enabled by the Mesolytic Cleavage of Alkoxyamine Radical Cations. Angew. Chem., Int. Ed. 2016, 55, 9969– 9973, DOI: 10.1002/anie.201604619Google ScholarThere is no corresponding record for this reference.
- 14Zhang, L.; Laborda, E.; Darwish, N.; Noble, B. B.; Tyrell, J. H.; Pluczyk, S.; Le Brun, A. P.; Wallace, G. G.; Gonzalez, J.; Coote, M. L.; Ciampi, S. Electrochemical and Electrostatic Cleavage of Alkoxyamines. J. Am. Chem. Soc. 2018, 140, 766– 774, DOI: 10.1021/jacs.7b11628Google ScholarThere is no corresponding record for this reference.
- 15Noble, B. B.; Norcott, P. L.; Hammill, C. L.; Ciampi, S.; Coote, M. L. Mechanism of Oxidative Alkoxyamine Cleavage: The Surprising Role of the Solvent and Supporting Electrolyte. J. Phys. Chem. C 2019, 123, 10300– 10305, DOI: 10.1021/acs.jpcc.9b01832Google ScholarThere is no corresponding record for this reference.
- 16Norcott, P. L.; Hammill, C. L.; Noble, B. B.; Robertson, J. C.; Olding, A.; Bissember, A. C.; Coote, M. L. TEMPO–Me: An Electrochemically Activated Methylating Agent. J. Am. Chem. Soc. 2019, 141, 15450– 15455, DOI: 10.1021/jacs.9b08634Google ScholarThere is no corresponding record for this reference.
- 17Ortalli, S.; Ford, J.; Trabanco, A. A.; Tredwell, M.; Gouverneur, V. Photoredox Nucleophilic (Radio)fluorination of Alkoxyamines. J. Am. Chem. Soc. 2024, 146, 11599– 11604, DOI: 10.1021/jacs.4c02474Google ScholarThere is no corresponding record for this reference.
- 18Gentry, E. C.; Rono, L. J.; Hale, M. E.; Matsuura, R.; Knowles, R. R. Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products. J. Am. Chem. Soc. 2018, 140, 3394– 3402, DOI: 10.1021/jacs.7b13616Google ScholarThere is no corresponding record for this reference.
- 19Wen, P.; Crich, D. Blue Light Photocatalytic Glycosylation without Electrophilic Additives. Org. Lett. 2017, 19, 2402– 2405, DOI: 10.1021/acs.orglett.7b00932Google ScholarThere is no corresponding record for this reference.
- 20Lee, K.; Gilberti, D.; Yom, C.; Meza, J.; Stewart, J.; Lee, M.; Alvarez, J.; Hart, A.; Zheng, J.; Xing, Y. Synthetic Electrochemistry Enabled Esterification via Oxidative Mesolytic Cleavage of Alkoxyamines. J. Org. Chem. 2023, 88, 9523– 9529, DOI: 10.1021/acs.joc.3c00617Google ScholarThere is no corresponding record for this reference.
- 21Hawkins, B. C.; Chalker, J. M.; Coote, M. L.; Bissember, A. C. Electrochemically Generated Carbocations in Organic Synthesis. Angew. Chem., Int. Ed. 2024, 63, e202407207 DOI: 10.1002/anie.202407207Google ScholarThere is no corresponding record for this reference.
- 22Mei, Y.; Liao, P.; Zhang, Y. Advances in electrochemical reactions involving acetonitrile. Org. Biomol. Chem. 2025, 23, 9752, DOI: 10.1039/D5OB00647CGoogle ScholarThere is no corresponding record for this reference.
- 23Zhang, X.; Cui, T.; Zhao, X.; Liu, P.; Sun, P. Electrochemical Difunctionalization of Alkenes by a Four-Component Reaction Cascade Mumm Rearrangement: Rapid Access to Functionalized Imides. Angew. Chem., Int. Ed. 2020, 59, 3465– 3469, DOI: 10.1002/anie.201913332Google ScholarThere is no corresponding record for this reference.
- 24Chu, Q.; Feng, Z.; Zhang, S.; Liu, P.; Sun, P. Three-component reaction for the synthesis of imides enabled by electrochemical C(sp3)–H functionalization. Green Chem. 2023, 25, 6728– 6732, DOI: 10.1039/D3GC02174BGoogle ScholarThere is no corresponding record for this reference.
- 25Qian, P.; Zhu, D.; Wang, X.; Sun, Q.; Zhang, S. Electrochemical Benzylic C(sp3)-H Imidation Enabled by Benzoic Acid Derived Radicals. J. Org. Chem. 2024, 89, 6395– 6404, DOI: 10.1021/acs.joc.4c00425Google ScholarThere is no corresponding record for this reference.
- 26Yu, M.; Gao, Y.; Zhang, L.; Zhang, Y.; Zhang, Y.; Yi, H.; Huang, Z.; Lei, A. Electrochemical-induced benzyl C–H amination towards the synthesis of isoindolinones via aroyloxy radical-mediated C–H activation. Green Chem. 2022, 24, 1445– 1450, DOI: 10.1039/D1GC04676DGoogle ScholarThere is no corresponding record for this reference.
- 27Jiang, P.; Liang, C.; He, T.; Liu, R.; Meng, X.; Zheng, Y.; Huang, S. Electrochemical Decarboxylation/Mumm Rearrangement towards Imides. Eur. J. Org. Chem. 2024, 27, e202400469 DOI: 10.1002/ejoc.202400469Google ScholarThere is no corresponding record for this reference.
- 28
For respective examples of electrochemically-promoted eliminations or deaminations of TAs see:
(a) Lu, Z.; Ju, M.; Wang, Y.; Meinhardt, J. M.; Martinez Alvarado, J. I.; Villemure, E.; Terrett, J. A.; Lin, S. Regioselective aliphatic C–H functionalization using frustrated radical pairs. Nature 2023, 619, 514– 520, DOI: 10.1038/s41586-023-06131-3Google ScholarThere is no corresponding record for this reference.(b) Romero-Ibañez, J.; Cruz-Gregorio, E.; Cruz-Gregorio, S.; Quintero, L.; Bernès, S.; González-Perea, M.; Sartillo-Piscil, F. Electrochemical deamination of alkoxyamine lactams. Tetrahedron Lett. 2020, 61, 152279, DOI: 10.1016/j.tetlet.2020.152279Google ScholarThere is no corresponding record for this reference. - 29
The current density was approximated by estimating the surface area of the electrode immersed in the electrolyte at the beginning of the reaction.
There is no corresponding record for this reference. - 30
Product 4a was prepared in 86% yield when this reaction was scaled up (1.0 mmol 2a).
There is no corresponding record for this reference. - 31
In a separate experiment, we performed the reaction shown in Table 1, entry 1, but instead, used anhydrous MeCN and bench-grade nBuNPF6 to prepare the electrolyte and added all reactants in air. The cell was then sparged with nitrogen for 3 minutes prior to electrolysis. This variation afforded product 4a in 88% yield.
There is no corresponding record for this reference. - 32
In undivided cell experiments employing pyridine-2-carbonitrile we only observed decomposition, while the reaction of 4-methoxybenzonitrile only proceeded at low conversion. Based on our cyclic voltammetry studies examining these nitrile substrates, we suspect these results may derive from undesirable cathodic processes. Although this could, in principle, be addressed using divided cell conditions, we found this to be problematic for nitrile substrates such as these, due to the high resistance encountered when using inherently non-polar CH2Cl2-based electrolytes in divided cells.
There is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
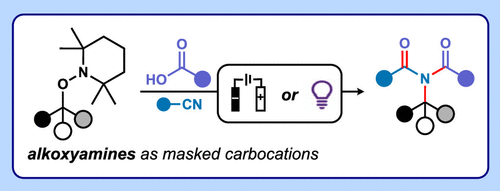
Figure 1

Figure 1. (A) Representative synthetic routes to TAs. (B) Overview of previously reported photoredox- and electrochemically-triggered alkylations using TAs. (C) Overview of previously reported methods for electrochemical Mumm rearrangement cascades.
Figure 2
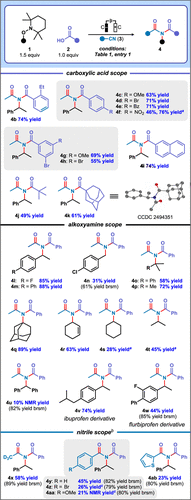
Figure 2. Electrochemically-promoted Mumm rearrangement cascades: representative substrate scope. Legend: a Divided cell, 2.0 equiv 1a, 6 h, 1 mmol scale; b nBu4NPF6 (4 mL of a 0.1 M anhydrous 3:1 CH2Cl2/nitrile 3 solution); c 10 equiv nitrile 3; nBu4NPF6 (4 mL of a 0.1 M CH2Cl2 solution). brsm: based on recovered starting material.
Figure 3

Figure 3. Other TA substrates examined in this study.
Figure 4
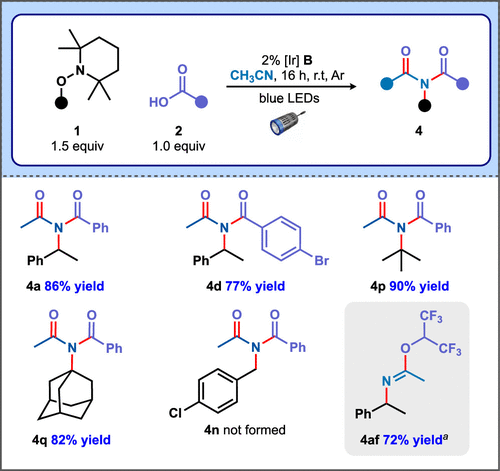
Figure 4. Photoredox-catalyzed Mumm rearrangement cascades. Reaction conditions: 0.1 mmol 2, 1.5 equiv 1, 2 mol % [Ir(dF(CF3)ppy)2(5,5′-dCF3bpy)]PF6 (B), anhydrous CH3CN (2 mL), 16 h, under Ar. Legend: a0.1 mmol 1, 10 equiv HFIP, 2 mol % B.
References
This article references 32 other publications.
- 1Li, L.; Yu, Z.; Shen, Z. Copper-Catalyzed Aminoxylation of Different Types of Hydrocarbons with TEMPO: A Concise Route to N-Alkoxyamine Derivatives. Adv. Synth. Catal. 2015, 357, 3495– 3500, DOI: 10.1002/adsc.201500544There is no corresponding record for this reference.
- 2Hammill, C. L.; Noble, B. B.; Norcott, P. L.; Ciampi, S.; Coote, M. L. Effect of Chemical Structure on the Electrochemical Cleavage of Alkoxyamines. J. Phys. Chem. C 2019, 123, 5273– 5281, DOI: 10.1021/acs.jpcc.8b12545There is no corresponding record for this reference.
- 3Schoening, K.-U.; Fischer, W.; Hauck, S.; Dichtl, A.; Kuepfert, M. Synthetic Studies on N-Alkoxyamines: A Mild and Broadly Applicable Route Starting from Nitroxide Radicals and Aldehydes. J. Org. Chem. 2009, 74, 1567– 1573, DOI: 10.1021/jo802403jThere is no corresponding record for this reference.
- 4Yao, C.; Aliyu, M. A.; Wan, Y.; Norton, J. R. Formation of TEMPO Adducts with Hydrogen Atom Transfer: An Alternative Pathway to Versatile Hydrofunctionalizations of Olefins. Chem. Eur. J. 2025, 31, e202403899 DOI: 10.1002/chem.202403899There is no corresponding record for this reference.
- 5Matyjaszewski, K.; Woodworth, B. E.; Zhang, X.; Gaynor, S. G.; Metzner, Z. Simple and Efficient Synthesis of Various Alkoxyamines for Stable Free Radical Polymerization. Macromolecules 1998, 31, 5955– 5957, DOI: 10.1021/ma9807264There is no corresponding record for this reference.
- 6Schulz, G.; Kirschning, A. Metal free decarboxylative aminoxylation of carboxylic acids using a biphasic solvent system. Org. Biomol. Chem. 2021, 19, 273– 278, DOI: 10.1039/D0OB01773FThere is no corresponding record for this reference.
- 7Innocent, M.; Tanguy, C.; Gavelle, S.; Aubineau, T.; Guérinot, A. Iron-Catalyzed, Light-Driven Decarboxylative Alkoxyamination. Chem. Eur. J. 2024, 30, e202401252 DOI: 10.1002/chem.202401252There is no corresponding record for this reference.
- 8Denkler, L. M.; Aladahalli Shekar, M.; Ngan, T. S. J.; Wylie, L.; Abdullin, D.; Engeser, M.; Schnakenburg, G.; Hett, T.; Pilz, F. H.; Kirchner, B.; Schiemann, O.; Kielb, P.; Bunescu, A. General Iron-Catalyzed Decarboxylative Oxygenation of Aliphatic Carboxylic Acids. Angew. Chem., Int. Ed. 2024, 63, e202403292 DOI: 10.1002/anie.202403292There is no corresponding record for this reference.
- 9Hawker, C. J.; Bosman, A. W.; Harth, E. New Polymer Synthesis by Nitroxide Mediated Living Radical Polymerizations. Chem. Rev. 2001, 101, 3661– 3688, DOI: 10.1021/cr990119uThere is no corresponding record for this reference.
- 10Audran, G.; Bagryanskaya, E. G.; Bikanga, R.; Coote, M. L.; Guselnikova, O.; Hammill, C. L.; Marque, S. R. A.; Mellet, P.; Postnikov, P. S. Dynamic Covalent Bond: Modes of Activation of the C-ON Bond in Alkoxyamines. Prog. Polym. Sci. 2023, 144, 101726, DOI: 10.1016/j.progpolymsci.2023.101726There is no corresponding record for this reference.
- 11Lee, K.; Yom, C.; Gilberti, D.; Tahsin, S.; Meza, J.; Stewart, J.; Boone, D.; Xing, Y. Oxidative Cleavage of Alkoxyamines for Nucleophilic Substitution Reactions. Eur. J. Org. Chem. 2024, 27, e202400845 DOI: 10.1002/ejoc.202400845There is no corresponding record for this reference.
- 12O’Brya, G.; Braslau, R. Terminal Functionalization of Polymers via Single Electron Oxidation of N-Alkoxyamines. Macromolecules 2006, 39, 9010– 9017, DOI: 10.1021/ma061940sThere is no corresponding record for this reference.
- 13Zhu, Q.; Gentry, E. C.; Knowles, R. R. Catalytic Carbocation Generation Enabled by the Mesolytic Cleavage of Alkoxyamine Radical Cations. Angew. Chem., Int. Ed. 2016, 55, 9969– 9973, DOI: 10.1002/anie.201604619There is no corresponding record for this reference.
- 14Zhang, L.; Laborda, E.; Darwish, N.; Noble, B. B.; Tyrell, J. H.; Pluczyk, S.; Le Brun, A. P.; Wallace, G. G.; Gonzalez, J.; Coote, M. L.; Ciampi, S. Electrochemical and Electrostatic Cleavage of Alkoxyamines. J. Am. Chem. Soc. 2018, 140, 766– 774, DOI: 10.1021/jacs.7b11628There is no corresponding record for this reference.
- 15Noble, B. B.; Norcott, P. L.; Hammill, C. L.; Ciampi, S.; Coote, M. L. Mechanism of Oxidative Alkoxyamine Cleavage: The Surprising Role of the Solvent and Supporting Electrolyte. J. Phys. Chem. C 2019, 123, 10300– 10305, DOI: 10.1021/acs.jpcc.9b01832There is no corresponding record for this reference.
- 16Norcott, P. L.; Hammill, C. L.; Noble, B. B.; Robertson, J. C.; Olding, A.; Bissember, A. C.; Coote, M. L. TEMPO–Me: An Electrochemically Activated Methylating Agent. J. Am. Chem. Soc. 2019, 141, 15450– 15455, DOI: 10.1021/jacs.9b08634There is no corresponding record for this reference.
- 17Ortalli, S.; Ford, J.; Trabanco, A. A.; Tredwell, M.; Gouverneur, V. Photoredox Nucleophilic (Radio)fluorination of Alkoxyamines. J. Am. Chem. Soc. 2024, 146, 11599– 11604, DOI: 10.1021/jacs.4c02474There is no corresponding record for this reference.
- 18Gentry, E. C.; Rono, L. J.; Hale, M. E.; Matsuura, R.; Knowles, R. R. Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products. J. Am. Chem. Soc. 2018, 140, 3394– 3402, DOI: 10.1021/jacs.7b13616There is no corresponding record for this reference.
- 19Wen, P.; Crich, D. Blue Light Photocatalytic Glycosylation without Electrophilic Additives. Org. Lett. 2017, 19, 2402– 2405, DOI: 10.1021/acs.orglett.7b00932There is no corresponding record for this reference.
- 20Lee, K.; Gilberti, D.; Yom, C.; Meza, J.; Stewart, J.; Lee, M.; Alvarez, J.; Hart, A.; Zheng, J.; Xing, Y. Synthetic Electrochemistry Enabled Esterification via Oxidative Mesolytic Cleavage of Alkoxyamines. J. Org. Chem. 2023, 88, 9523– 9529, DOI: 10.1021/acs.joc.3c00617There is no corresponding record for this reference.
- 21Hawkins, B. C.; Chalker, J. M.; Coote, M. L.; Bissember, A. C. Electrochemically Generated Carbocations in Organic Synthesis. Angew. Chem., Int. Ed. 2024, 63, e202407207 DOI: 10.1002/anie.202407207There is no corresponding record for this reference.
- 22Mei, Y.; Liao, P.; Zhang, Y. Advances in electrochemical reactions involving acetonitrile. Org. Biomol. Chem. 2025, 23, 9752, DOI: 10.1039/D5OB00647CThere is no corresponding record for this reference.
- 23Zhang, X.; Cui, T.; Zhao, X.; Liu, P.; Sun, P. Electrochemical Difunctionalization of Alkenes by a Four-Component Reaction Cascade Mumm Rearrangement: Rapid Access to Functionalized Imides. Angew. Chem., Int. Ed. 2020, 59, 3465– 3469, DOI: 10.1002/anie.201913332There is no corresponding record for this reference.
- 24Chu, Q.; Feng, Z.; Zhang, S.; Liu, P.; Sun, P. Three-component reaction for the synthesis of imides enabled by electrochemical C(sp3)–H functionalization. Green Chem. 2023, 25, 6728– 6732, DOI: 10.1039/D3GC02174BThere is no corresponding record for this reference.
- 25Qian, P.; Zhu, D.; Wang, X.; Sun, Q.; Zhang, S. Electrochemical Benzylic C(sp3)-H Imidation Enabled by Benzoic Acid Derived Radicals. J. Org. Chem. 2024, 89, 6395– 6404, DOI: 10.1021/acs.joc.4c00425There is no corresponding record for this reference.
- 26Yu, M.; Gao, Y.; Zhang, L.; Zhang, Y.; Zhang, Y.; Yi, H.; Huang, Z.; Lei, A. Electrochemical-induced benzyl C–H amination towards the synthesis of isoindolinones via aroyloxy radical-mediated C–H activation. Green Chem. 2022, 24, 1445– 1450, DOI: 10.1039/D1GC04676DThere is no corresponding record for this reference.
- 27Jiang, P.; Liang, C.; He, T.; Liu, R.; Meng, X.; Zheng, Y.; Huang, S. Electrochemical Decarboxylation/Mumm Rearrangement towards Imides. Eur. J. Org. Chem. 2024, 27, e202400469 DOI: 10.1002/ejoc.202400469There is no corresponding record for this reference.
- 28
For respective examples of electrochemically-promoted eliminations or deaminations of TAs see:
(a) Lu, Z.; Ju, M.; Wang, Y.; Meinhardt, J. M.; Martinez Alvarado, J. I.; Villemure, E.; Terrett, J. A.; Lin, S. Regioselective aliphatic C–H functionalization using frustrated radical pairs. Nature 2023, 619, 514– 520, DOI: 10.1038/s41586-023-06131-3There is no corresponding record for this reference.(b) Romero-Ibañez, J.; Cruz-Gregorio, E.; Cruz-Gregorio, S.; Quintero, L.; Bernès, S.; González-Perea, M.; Sartillo-Piscil, F. Electrochemical deamination of alkoxyamine lactams. Tetrahedron Lett. 2020, 61, 152279, DOI: 10.1016/j.tetlet.2020.152279There is no corresponding record for this reference. - 29
The current density was approximated by estimating the surface area of the electrode immersed in the electrolyte at the beginning of the reaction.
There is no corresponding record for this reference. - 30
Product 4a was prepared in 86% yield when this reaction was scaled up (1.0 mmol 2a).
There is no corresponding record for this reference. - 31
In a separate experiment, we performed the reaction shown in Table 1, entry 1, but instead, used anhydrous MeCN and bench-grade nBuNPF6 to prepare the electrolyte and added all reactants in air. The cell was then sparged with nitrogen for 3 minutes prior to electrolysis. This variation afforded product 4a in 88% yield.
There is no corresponding record for this reference. - 32
In undivided cell experiments employing pyridine-2-carbonitrile we only observed decomposition, while the reaction of 4-methoxybenzonitrile only proceeded at low conversion. Based on our cyclic voltammetry studies examining these nitrile substrates, we suspect these results may derive from undesirable cathodic processes. Although this could, in principle, be addressed using divided cell conditions, we found this to be problematic for nitrile substrates such as these, due to the high resistance encountered when using inherently non-polar CH2Cl2-based electrolytes in divided cells.
There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.5c05049.
All experimental procedures, characterization data, X-ray crystallographic data for 4k, and NMR spectra (PDF)
Deposition Number 2494351 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via the joint Cambridge Crystallographic Data Centre (CCDC) and Fachinformationszentrum Karlsruhe Access Structures service.
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.