



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
A Unique Case of the “Goldilocks Rule” in Solid-State Electrolytes: Two Are Good, Four Are Too ManyClick to copy article linkArticle link copied!
- Bingning WangBingning WangChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesDepartment of Chemical & Biomolecular Engineering, Institute of Materials Science, University of Connecticut, Storrs, Connecticut 06269, United StatesMore by Bingning Wang
- Yan QinYan QinChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Yan Qin
- Ying ChenYing ChenPacific Northwest National Laboratory, PO Box 999, Richland, Washington 99352, United StatesEnergy Storage Research Alliance, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Ying Chen
- Sungil HongSungil HongMaterials Sciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesEnergy Storage Research Alliance, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Sungil Hong
- Lihong GaoLihong GaoChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Lihong Gao
- Andrew N. JansenAndrew N. JansenChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Andrew N. Jansen
- Rajeev Surendran AssaryRajeev Surendran AssaryMaterials Sciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesEnergy Storage Research Alliance, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Rajeev Surendran Assary
- Kin Lung See ThoKin Lung See ThoChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesEnergy Storage Research Alliance, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Kin Lung See Tho
- Jiyu CaiJiyu CaiChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Jiyu Cai
- Zonghai ChenZonghai ChenChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Zonghai Chen
- Yang QinYang QinDepartment of Chemical & Biomolecular Engineering, Institute of Materials Science, University of Connecticut, Storrs, Connecticut 06269, United StatesMore by Yang Qin
- Zhengcheng ZhangZhengcheng ZhangChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Zhengcheng Zhang
- Wenquan Lu*Wenquan Lu*Email: [email protected]Chemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Wenquan Lu
- Chen Liao*Chen Liao*Email: [email protected]Chemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesEnergy Storage Research Alliance, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United StatesMore by Chen Liao
Abstract
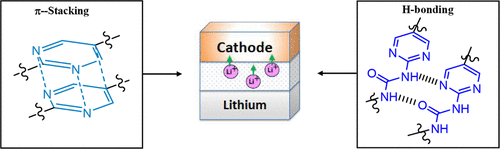
We report the syntheses of two new series of methacrylate monomers with different backbones: ureidopyrimidinone (PU) and boron-substituted urea pyrimidine (U), which enhance both the mechanical and electrochemical properties of the solid-state electrolyte (SSE) while improving the cycle life of lithium iron phosphate (LiFePO4, LFP) cathodes. The PU backbone is characterized by four hydrogen bonds (H-bonds), while the U backbone bears only two. Importantly, our research reveals that two H-bonds in these monomers are optimal; in contrast, four are excessive. The exceptional mechanical properties and processability of the SSE with the U series additives, resulting from the optimal H-bonds, were unexpectedly achieved. This leads to the establishment of a “Goldilocks rule” for additive design. The key strategies include: 1) reducing hydrogen-bonding (H-bonding) sites by changing pyrimidinone to pyrimidine and 2) shifting from intermolecular to intramolecular H-bonding and π–π bonding. This reduction in H-bonding also offers significant advantages in processability. The advancement can be extended to electrode fabrication, making the manufacturing of all-solid-state batteries more practical and efficient.
This publication is licensed for personal use by The American Chemical Society.
1. Introduction
Scheme 1
2. Results
2.1. Syntheses
2.1.1. Syntheses of the H-Bonding Cross-Linkers
Scheme 2
2.1.2. Syntheses of the Deep Eutectic Solvent (DES) and Polymer Cross-Linking Systems
Figure 1
Figure 1. Electrolyte composition of SSE with lithium trifluoromethylsulfonyl imide (LiTFSI), NMU, PEGDA (polyethylene glycol diacrylate), and additives (using UB as an example) that are capable of H-bonding. The H-bonding mechanism shows how UB can exceptionally improve both the mechanical strength of the polymeric systems.
2.2. Computational Chemistry Calculations
2.2.1. Computational Methods
Figure 2
Figure 2. Molecular structures of two conformers (Conf.) of PU-CMe, U–H, UB, and UB–OH monomers, which are generated with two different dihedral angles of O═C─N─H (ϕO═C─N─H) of the urea scaffold (highlighted in green). Intramolecular H-bonds are highlighted by blue circles. Gibbs free energy differences between two conformers (ΔG = GConf. 2 – GConf. 1) are reported in eV.
2.2.2. Conformers of Urea Pyrimidine Monomers
3. Discussion
3.1. Influence of Chemical Structure Groups
3.2. Electrochemical and Mechanical Properties
| Name | LiTFSI | NMU | PC | NMU:PC | PEGDAb | Additive amount |
|---|---|---|---|---|---|---|
| UB-1/4 | 33.2 | 6.65 | 26.4 | 1/4 | 33.1 | 0.7 |
| UB-2/3 | 33.1 | 13.4 | 19.9 | 2/3 | 32.9 | 0.7 |
| A-1 | 33.3 | 16.6 | 16.5 | 1 | 32.9 | 0 |
| UB-1 | 33.3 | 16.6 | 16.5 | 1 | 32.9 | 0.7 |
| UB-1-B | 32.86 | 16.69 | 16.88 | 1 | 30.29 | 3.28 |
| UB-3/2 | 32.7 | 19.9 | 13.4 | 3/2 | 32.7 | 0.7 |
| UB–OH-1 | 33.3 | 16.6 | 16.5 | 1 | 32.9 | 0.7 |
| U–H-1 | 33.1 | 16.6 | 16.4 | 1 | 32.9 | 0.7 |
All chemical composition values reported here are given in weight percent (wt %).
PEGDA refers to poly(ethylene glycol) diacrylate, Mn = 600.
3.2.1. Ionic Conductivity
Figure 3
Figure 3. Ionic conductivity of baseline A-1 and the UB series with various ratios of NMU:PC, UB with extra additive concentration, and pure UB–OH.
3.2.2. Lithium Plating/Stripping and Transference Number
Figure 4
Figure 4. (a) Critical current density (CCD) determination of SSE with baseline A-1 and additive (UB-1) via DC polarization (0.1, 0.4, and 0.8 mA/cm2). (b) Voltage profile comparisons of A-1 and UB-1 at 0.1, 0.4, and 0.8 mA/cm2.
3.2.3. Optimization of Electrolytes
Figure 5
Figure 5. (a) Optical images of flexible SSE prepared by cutting with a round-shaped cutter. (b) Specific capacity of the LFP//Li at 45 °C in the baseline A-1 and UB series with various PC:NMU amounts, with a ratio of NMU:PC from 0.67 to 1.5.
Increasing amounts of NMU:PC ratio enhance performance, but only up to an optimal ratio of 1:1. As can be seen in Figure 5b, the amount of NMU varies from a ratio of NMU: PC from 0.67 to 1.5. An SSE with a low NMU:PC ratio of 0.25 was prepared, but it showed no cycling performance. At 45 °C, UB-1 has the best performance, with an initial and final specific capacity of 141.8 and 123.95 mAh g–1, respectively. The baseline A-1 exhibits rapid degradation, with significant performance loss observed after just 10 cycles. With varying amounts NMU:PC, the performance varies, following the order of UB-1 > UB-3/2 > UB-2/3. The optimal NMU:PC ratio is 1:1, and beyond this point, further increases can deteriorate electrochemical performance.
Temperature plays a critical role on electrochemical performance, as demonstrated by comparing UB-1 and U–H-1. At 30 °C, cycling performance is maximized in UB-1, following the order: UB-1 > UB-OH-1 > U–H-1, as shown in Figure 6a. However, increasing the temperature to 45 °C significantly enhances their performance, bringing it to an optimized level. Since most additives perform better at higher temperatures, a higher temperature (45 °C) was selected to test all the U series additives in the next section. Note that the rate performance rate of A-1 is similar to that of UB-1 (Supporting Information, Figure S10), and the performance rank is changed to (Figure 6b).
Figure 6
Figure 6. Specific capacity of the LFP//Li at (a) 30 and (b) 45 °C in baseline A-1 and UB-1.
3.2.4. Properties of SSE Containing Various U Series Additives
Figure 7
Figure 7. Cycling performance comparison of LFP//Li using SSE with different additives at 45 °C.
3.2.5. Mechanical Properties
Figure 8
Figure 8. Nanoindentation process shows the maximum indentation load vs the depth at maximum load. UB-1 shows a 4-fold increase in strength than that of A-1.
3.3. Intermolecular Interactions
Figure 9
Figure 9. Molecular structures of two interacting monomers of (a) PU-CMe, (b) U–H, (c) UB, and (d) UB–OH, with corresponding interaction Gibbs free energies in eV. (a) For PU-CMe, both conformers (Conf. 1 and 2) in a side-by-side alignment are considered since they are isoenergetic (Figure 2). For (c) UB and (d) UB-OH, two modes of interactions, side-by-side (Mode 1) and π- π stacking (Mode 2), are presented..
Figure 10
Figure 10. Summary of the role of functionalization in their intermolecular interaction modes and corresponding interaction Gibbs free energies from DFT calculations in eV.
3.4. NMR
Figure 11
Figure 11. Restricted ion diffusion within the polymer membranes revealed by PFG-NMR. Measured diffusion coefficients (a-c) and estimated diffusion lengths (d-f) of Li+ and TFSI– within the membranes as a function of diffusion time at 30 °C (a,d), 45 °C (b,e), and 60 °C (c,f). The insets in (b) and (c) show examples of 7Li and 19F NMR spectra during PFG measurements.
4. Conclusion
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsaem.5c02674.
Materials, syntheses, preparation of polymer electrolyte precursor solution, physical and electrochemical properties, and electrochemical characterization (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Chen Liao - Chemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United States; Energy Storage Research Alliance, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United States;
 https://orcid.org/0000-0001-5168-6493;
https://orcid.org/0000-0001-5168-6493;
- Ying Chen - Pacific Northwest National Laboratory, PO Box 999, Richland, Washington 99352, United States; Energy Storage Research Alliance, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United States;https://orcid.org/0000-0001-7417-0991
- Sungil Hong - Materials Sciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United States; Energy Storage Research Alliance, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United States;https://orcid.org/0000-0001-8729-0861
- Rajeev Surendran Assary - Materials Sciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United States; Energy Storage Research Alliance, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United States;https://orcid.org/0000-0002-9571-3307
- Jiyu Cai - Chemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United States;https://orcid.org/0000-0003-2283-2281
- Yang Qin - Department of Chemical & Biomolecular Engineering, Institute of Materials Science, University of Connecticut, Storrs, Connecticut 06269, United States;https://orcid.org/0000-0002-5764-8137
- Zhengcheng Zhang - Chemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, United States;https://orcid.org/0000-0002-0467-5801
B.W. and Y.Q. contributed equally. B.W.: Data collection and analysis, Writing─review and editing. Y.Q.: Experiment design, Data collection and analysis, Writing─review and editing. Y.C.: Data collection and analysis, Writing─original draft and editing. S.H.: Data collection and analysis, Writing─original draft and editing. L.G.: Experiment design, Writing─review and editing. A.N.J.: Funding, Writing─review and editing. R.S.A.: Writing─review and editing. K.L.S.T.: Writing─review and editing. J.C.: Data collection and analysis, Writing─review and editing. Z.C.: Writing─review and editing. Yg.Q.: Writing─review and editing. Z.Z.: Funding, Writing─review and editing. W.L.: Supervision, Writing─review and editing. C.L.: Experiment design, Supervision, Conceptualization, Funding, Writing─original draft, Writing─review and editing.
Acknowledgments
The syntheses of the PU and U series and solid-state electrolyte performance and properties made by B.W., L.G., K.L.S.T., and C.L.; the NMR measurement by Y.C.; and the computational work by S.H. and R.S.A. were supported by the Energy Storage Research Alliance “ESRA”, an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences. The development and measurement of SSE by Y.Q., A.N.J., and W.L. was supported by the Vehicle Technologies Office (VTO), Haiyan Croft and Brian Cunningham at the U.S. Department of Energy. The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract No. DE-AC02-06CH11357.
References
This article references 32 other publications.
- 1Johnshon, P. Toyota confirms 750 mi range solid-state EV battery plans to catch up to Tesla, but when?; Electrek, 2024.Google ScholarThere is no corresponding record for this reference.
- 2Sahore, R.; Dogan, F.; Bloom, I. D. Identification of Electrolyte-Soluble Organic Cross-Talk Species in a Lithium-Ion Battery via a Two-Compartment Cell. Chem. Mater. 2019, 31 (8), 2884– 2891, DOI: 10.1021/acs.chemmater.9b00063Google ScholarThere is no corresponding record for this reference.
- 3Murugan, R.; Thangadurai, V.; Weppner, W. Fast Lithium Ion Conduction in Garnet-Type Li7La3Zr2O12. Angew. Chem., Int. Ed. 2007, 46 (41), 7778– 7781, DOI: 10.1002/anie.200701144Google ScholarThere is no corresponding record for this reference.
- 4Lee, Y.-G.; Fujiki, S.; Jung, C.; Suzuki, N.; Yashiro, N.; Omoda, R.; Ko, D.-S.; Shiratsuchi, T.; Sugimoto, T.; Ryu, S.; Ku, J. H.; Watanabe, T.; Park, Y.; Aihara, Y.; Im, D.; Han, I. T. High-energy long-cycling all-solid-state lithium metal batteries enabled by silver–carbon composite anodes. Nat. Energy 2020, 5 (4), 299– 308, DOI: 10.1038/s41560-020-0575-zGoogle ScholarThere is no corresponding record for this reference.
- 5Janek, J.; Zeier, W. G. Challenges in speeding up solid-state battery development. Nat. Energy 2023, 8 (3), 230– 240, DOI: 10.1038/s41560-023-01208-9Google ScholarThere is no corresponding record for this reference.
- 6CATL, BYD, others unite in China for solid-state battery breakthrough; https://asia.nikkei.com/Business/Technology/CATL-BYD-others-unite-in-China-for-solid-state-battery-breakthrough. (Accessed 12 February 2024).Google ScholarThere is no corresponding record for this reference.
- 7Jing, M.-X.; Yang, H.; Han, C.; Chen, F.; Yuan, W.-Y.; Ju, B.-W.; Tu, F.-Y.; Shen, X.-Q.; Qin, S.-B. Improving room-temperature electrochemical performance of solid-state lithium battery by using electrospun La2Zr2O7 fibers-filled composite solid electrolyte. Ceram. Int. 2019, 45 (15), 18614– 18622, DOI: 10.1016/j.ceramint.2019.06.085Google ScholarThere is no corresponding record for this reference.
- 8Zhang, L.-K.; Jing, M.-X.; Yang, H.; Liu, Q.-Y.; Chen, F.; Yuan, W.-Y.; Liu, M.-Q.; Ji, Y.-S.; Shen, X.-Q. Highly Efficient Interface Modification between Poly(Propylene Carbonate)-Based Solid Electrolytes and a Lithium Anode by Facile Graphite Coating. ACS Sustainable Chem. Eng. 2020, 8 (46), 17106– 17115, DOI: 10.1021/acssuschemeng.0c05153Google ScholarThere is no corresponding record for this reference.
- 9Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451 (7181), 977– 980, DOI: 10.1038/nature06669Google ScholarThere is no corresponding record for this reference.
- 10Tee, B. C.-K.; Wang, C.; Allen, R.; Bao, Z. An electrically and mechanically self-healing composite with pressure- and flexion-sensitive properties for electronic skin applications. Nat. Nanotechnol. 2012, 7 (12), 825– 832, DOI: 10.1038/nnano.2012.192Google ScholarThere is no corresponding record for this reference.
- 11Wang, C.; Wu, H.; Chen, Z.; McDowell, M. T.; Cui, Y.; Bao, Z. Self-healing chemistry enables the stable operation of silicon microparticle anodes for high-energy lithium-ion batteries. Nat. Chem. 2013, 5 (12), 1042– 1048, DOI: 10.1038/nchem.1802Google ScholarThere is no corresponding record for this reference.
- 12Manthiram, A.; Yu, X.; Wang, S. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2017, 2 (4), 16103, DOI: 10.1038/natrevmats.2016.103Google ScholarThere is no corresponding record for this reference.
- 13Gracia, I.; Armand, M.; Shanmukaraj, D. Li Metal Polymer Batteries. In Solid Electrolytes for Advanced Applications: Garnets and Competitors; Murugan, R.; Weppner, W., Eds.; Springer International Publishing: Cham, 2019, pp. 347– 373.Google ScholarThere is no corresponding record for this reference.
- 14Wang, H.; Song, J.; Zhang, K.; Fang, Q.; Zuo, Y.; Yang, T.; Yang, Y.; Gao, C.; Wang, X.; Pang, Q.; Xia, D. A strongly complexed solid polymer electrolyte enables a stable solid state high-voltage lithium metal battery. Energy Environ. Sci. 2022, 15 (12), 5149– 5158, DOI: 10.1039/D2EE02904AGoogle ScholarThere is no corresponding record for this reference.
- 15Streipert, B.; Janßen, P.; Cao, X.; Kasnatscheew, J.; Wagner, R.; Cekic-Laskovic, I.; Winter, M.; Placke, T. Evaluation of Allylboronic Acid Pinacol Ester as Effective Shutdown Overcharge Additive for Lithium Ion Cells. J. Electro. Soc. 2017, 164 (2), A168, DOI: 10.1149/2.0711702jesGoogle ScholarThere is no corresponding record for this reference.
- 16Kelchtermans, A.-S.; De Sloovere, D.; Mercken, J.; Vranken, T.; Mangione, G.; Joos, B.; Vercruysse, W.; Vandamme, D.; Hamed, H.; Safari, M.; Derveaux, E.; Adriaensens, P.; Van Bael, M. K.; Hardy, A. Superconcentration Strategy Allows Sodium Metal Compatibility in Deep Eutectic Solvents for Sodium-Ion Batteries. ACS Omega 2024, 9 (41), 42343– 42352, DOI: 10.1021/acsomega.4c02896Google ScholarThere is no corresponding record for this reference.
- 17Boisset, A.; Menne, S.; Jacquemin, J.; Balducci, A.; Anouti, M. Deep eutectic solvents based on N-methylacetamide and a lithium salt as suitable electrolytes for lithium-ion batteries. Phys. Chem. Chem. Phys. 2013, 15 (46), 20054– 20063, DOI: 10.1039/c3cp53406eGoogle ScholarThere is no corresponding record for this reference.
- 18Meles Neguse, S.; Yoon, S.; Lim, H.; Jang, J.; Baek, S.; Jöckel, D. M.; Widenmeyer, M.; Balke-Grünewald, B.; Weidenkaff, A. The Pitfalls of Deep Eutectic Solvents in the Recycling of Lithium-Ion Batteries. Energy Technol. 2024, 12 (4), 2301213, DOI: 10.1002/ente.202301213Google ScholarThere is no corresponding record for this reference.
- 19Minkiewicz, J.; Jones, G. M.; Ghanizadeh, S.; Bostanchi, S.; Wasely, T. J.; Yamini, S. A.; Nekouie, V. Large-scale manufacturing of solid-state electrolytes: Challenges, progress, and prospects. Open Ceram. 2023, 16, 100497, DOI: 10.1016/j.oceram.2023.100497Google ScholarThere is no corresponding record for this reference.
- 20Bae, J.; Zhu, Z.; Yan, J.; Kim, D.-M.; Ko, Y.; Jain, A.; Helms, B. A. Closed-loop cathode recycling in solid-state batteries enabled by supramolecular electrolytes. Sci. Adv. 2023, 9 (32), eadh9020 DOI: 10.1126/sciadv.adh9020Google ScholarThere is no corresponding record for this reference.
- 21Stewart, J. J. P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19 (1), 1– 32, DOI: 10.1007/s00894-012-1667-xGoogle ScholarThere is no corresponding record for this reference.
- 22Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10 (44), 6615– 6620, DOI: 10.1039/b810189bGoogle ScholarThere is no corresponding record for this reference.
- 23Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Gaussian 16; Revision, C; Gaussian, Inc: Wallingford CT, 2016.Google ScholarThere is no corresponding record for this reference.
- 24Liu, K.; Li, X.; Cai, J.; Yang, Z.; Chen, Z.; Key, B.; Zhang, Z.; Dzwiniel, T. L.; Liao, C. Design of High-Voltage Stable Hybrid Electrolyte with an Ultrahigh Li Transference Number. ACS Energy Lett. 2021, 6 (4), 1315– 1323, DOI: 10.1021/acsenergylett.0c02559Google ScholarThere is no corresponding record for this reference.
- 25Matsumi, N.; Sugai, K.; Ohno, H. Selective Ion Transport in Organoboron Polymer Electrolytes Bearing a Mesitylboron Unit. Macromolecules 2002, 35 (15), 5731– 5733, DOI: 10.1021/ma0121666Google ScholarThere is no corresponding record for this reference.
- 26Matsumi, N.; Sugai, K.; Ohno, H. Ion Conductive Characteristics of Alkylborane Type and Boric Ester Type Polymer Electrolytes Derived from Mesitylborane. Macromolecules 2003, 36 (7), 2321– 2326, DOI: 10.1021/ma021734uGoogle ScholarThere is no corresponding record for this reference.
- 27Nishihara, Y.; Miyazaki, M.; Tomita, Y.; Kadono, Y.; Takagi, K. Synthesis and ion conductive characteristics of inorganic–organic hybrid polymers bearing a tetraarylpentaborate unit. J. Polym. Sci., Part A: Polym. Chem. 2008, 46 (23), 7913– 7918, DOI: 10.1002/pola.23070Google ScholarThere is no corresponding record for this reference.
- 28Yuan, C.; Xu, J. Quantitative regulation of electrochemical-mechanical performance of composite cathode in all-solid-state batteries. Nano Energy 2024, 121, 109193, DOI: 10.1016/j.nanoen.2023.109193Google ScholarThere is no corresponding record for this reference.
- 29Yuan, C.; Lu, W.; Xu, J. Electrochemical-mechanical coupling failure mechanism of composite cathode in all-solid-state batteries. Energy Storage Mater. 2023, 60, 102834, DOI: 10.1016/j.ensm.2023.102834Google ScholarThere is no corresponding record for this reference.
- 30Orisekeh, D.; Roh, B.-M.; Xiao, X. Solid-to-Solid Manufacturing Processes for High-Performance Li-Ion Solid-State Batteries. Polymer 2025, 17 (13), 1788, DOI: 10.3390/polym17131788Google ScholarThere is no corresponding record for this reference.
- 31Monroe, C.; Newman, J. The Impact of Elastic Deformation on Deposition Kinetics at Lithium/Polymer Interfaces. J. Electro. Soc. 2005, 152 (2), A396, DOI: 10.1149/1.1850854Google ScholarThere is no corresponding record for this reference.
- 32Xu, R.; Cheng, X.-B.; Yan, C.; Zhang, X.-Q.; Xiao, Y.; Zhao, C.-Z.; Huang, J.-Q.; Zhang, Q. Artificial Interphases for Highly Stable Lithium Metal Anode. Matter 2019, 1 (2), 317– 344, DOI: 10.1016/j.matt.2019.05.016Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
Scheme 1
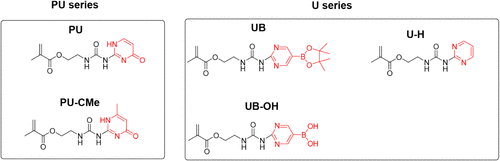 Scheme 1. Molecular Structures of the Series of Ureidopyrimidinone Monomers (the PU Series with PU and PU-CMe) and the Urea Pyrimidine Monomers (the U Series with UB, U–H, UB–OH)
Scheme 1. Molecular Structures of the Series of Ureidopyrimidinone Monomers (the PU Series with PU and PU-CMe) and the Urea Pyrimidine Monomers (the U Series with UB, U–H, UB–OH)Scheme 2
 Scheme 2. (a) Synthetic Procedure for the PU Series from the Literature; (14) (b) Synthetic Procedure Using Increased Temperature for the U Series with Either Boronic Acid Pinacol Ester Pyrimidinylurea (UB and UB–OH) or Simply Pyrimidine (U–H); (c) Spontaneous Hydrolysis of UB to UB–OH
Scheme 2. (a) Synthetic Procedure for the PU Series from the Literature; (14) (b) Synthetic Procedure Using Increased Temperature for the U Series with Either Boronic Acid Pinacol Ester Pyrimidinylurea (UB and UB–OH) or Simply Pyrimidine (U–H); (c) Spontaneous Hydrolysis of UB to UB–OHFigure 1

Figure 1. Electrolyte composition of SSE with lithium trifluoromethylsulfonyl imide (LiTFSI), NMU, PEGDA (polyethylene glycol diacrylate), and additives (using UB as an example) that are capable of H-bonding. The H-bonding mechanism shows how UB can exceptionally improve both the mechanical strength of the polymeric systems.
Figure 2
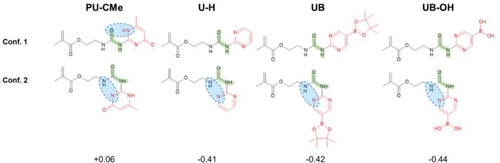
Figure 2. Molecular structures of two conformers (Conf.) of PU-CMe, U–H, UB, and UB–OH monomers, which are generated with two different dihedral angles of O═C─N─H (ϕO═C─N─H) of the urea scaffold (highlighted in green). Intramolecular H-bonds are highlighted by blue circles. Gibbs free energy differences between two conformers (ΔG = GConf. 2 – GConf. 1) are reported in eV.
Figure 3

Figure 3. Ionic conductivity of baseline A-1 and the UB series with various ratios of NMU:PC, UB with extra additive concentration, and pure UB–OH.
Figure 4
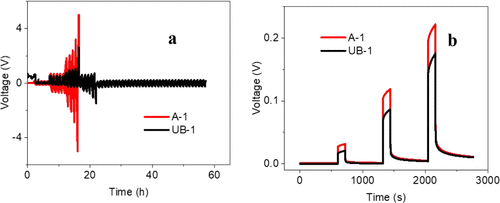
Figure 4. (a) Critical current density (CCD) determination of SSE with baseline A-1 and additive (UB-1) via DC polarization (0.1, 0.4, and 0.8 mA/cm2). (b) Voltage profile comparisons of A-1 and UB-1 at 0.1, 0.4, and 0.8 mA/cm2.
Figure 5

Figure 5. (a) Optical images of flexible SSE prepared by cutting with a round-shaped cutter. (b) Specific capacity of the LFP//Li at 45 °C in the baseline A-1 and UB series with various PC:NMU amounts, with a ratio of NMU:PC from 0.67 to 1.5.
Figure 6
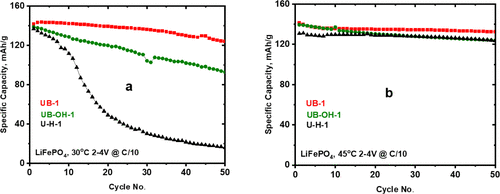
Figure 6. Specific capacity of the LFP//Li at (a) 30 and (b) 45 °C in baseline A-1 and UB-1.
Figure 7
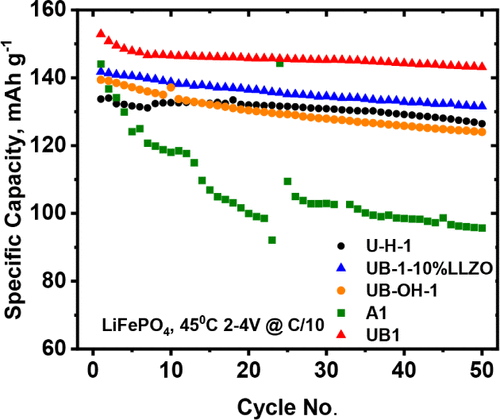
Figure 7. Cycling performance comparison of LFP//Li using SSE with different additives at 45 °C.
Figure 8

Figure 8. Nanoindentation process shows the maximum indentation load vs the depth at maximum load. UB-1 shows a 4-fold increase in strength than that of A-1.
Figure 9
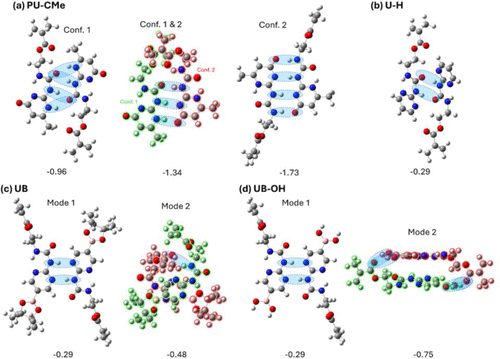
Figure 9. Molecular structures of two interacting monomers of (a) PU-CMe, (b) U–H, (c) UB, and (d) UB–OH, with corresponding interaction Gibbs free energies in eV. (a) For PU-CMe, both conformers (Conf. 1 and 2) in a side-by-side alignment are considered since they are isoenergetic (Figure 2). For (c) UB and (d) UB-OH, two modes of interactions, side-by-side (Mode 1) and π- π stacking (Mode 2), are presented..
Figure 10
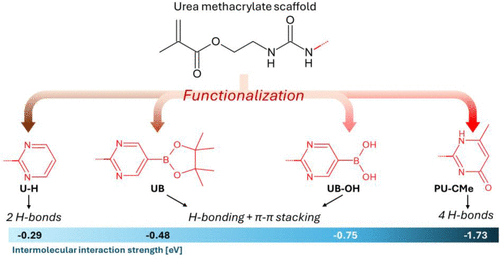
Figure 10. Summary of the role of functionalization in their intermolecular interaction modes and corresponding interaction Gibbs free energies from DFT calculations in eV.
Figure 11

Figure 11. Restricted ion diffusion within the polymer membranes revealed by PFG-NMR. Measured diffusion coefficients (a-c) and estimated diffusion lengths (d-f) of Li+ and TFSI– within the membranes as a function of diffusion time at 30 °C (a,d), 45 °C (b,e), and 60 °C (c,f). The insets in (b) and (c) show examples of 7Li and 19F NMR spectra during PFG measurements.
References
This article references 32 other publications.
- 1Johnshon, P. Toyota confirms 750 mi range solid-state EV battery plans to catch up to Tesla, but when?; Electrek, 2024.There is no corresponding record for this reference.
- 2Sahore, R.; Dogan, F.; Bloom, I. D. Identification of Electrolyte-Soluble Organic Cross-Talk Species in a Lithium-Ion Battery via a Two-Compartment Cell. Chem. Mater. 2019, 31 (8), 2884– 2891, DOI: 10.1021/acs.chemmater.9b00063There is no corresponding record for this reference.
- 3Murugan, R.; Thangadurai, V.; Weppner, W. Fast Lithium Ion Conduction in Garnet-Type Li7La3Zr2O12. Angew. Chem., Int. Ed. 2007, 46 (41), 7778– 7781, DOI: 10.1002/anie.200701144There is no corresponding record for this reference.
- 4Lee, Y.-G.; Fujiki, S.; Jung, C.; Suzuki, N.; Yashiro, N.; Omoda, R.; Ko, D.-S.; Shiratsuchi, T.; Sugimoto, T.; Ryu, S.; Ku, J. H.; Watanabe, T.; Park, Y.; Aihara, Y.; Im, D.; Han, I. T. High-energy long-cycling all-solid-state lithium metal batteries enabled by silver–carbon composite anodes. Nat. Energy 2020, 5 (4), 299– 308, DOI: 10.1038/s41560-020-0575-zThere is no corresponding record for this reference.
- 5Janek, J.; Zeier, W. G. Challenges in speeding up solid-state battery development. Nat. Energy 2023, 8 (3), 230– 240, DOI: 10.1038/s41560-023-01208-9There is no corresponding record for this reference.
- 6CATL, BYD, others unite in China for solid-state battery breakthrough; https://asia.nikkei.com/Business/Technology/CATL-BYD-others-unite-in-China-for-solid-state-battery-breakthrough. (Accessed 12 February 2024).There is no corresponding record for this reference.
- 7Jing, M.-X.; Yang, H.; Han, C.; Chen, F.; Yuan, W.-Y.; Ju, B.-W.; Tu, F.-Y.; Shen, X.-Q.; Qin, S.-B. Improving room-temperature electrochemical performance of solid-state lithium battery by using electrospun La2Zr2O7 fibers-filled composite solid electrolyte. Ceram. Int. 2019, 45 (15), 18614– 18622, DOI: 10.1016/j.ceramint.2019.06.085There is no corresponding record for this reference.
- 8Zhang, L.-K.; Jing, M.-X.; Yang, H.; Liu, Q.-Y.; Chen, F.; Yuan, W.-Y.; Liu, M.-Q.; Ji, Y.-S.; Shen, X.-Q. Highly Efficient Interface Modification between Poly(Propylene Carbonate)-Based Solid Electrolytes and a Lithium Anode by Facile Graphite Coating. ACS Sustainable Chem. Eng. 2020, 8 (46), 17106– 17115, DOI: 10.1021/acssuschemeng.0c05153There is no corresponding record for this reference.
- 9Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451 (7181), 977– 980, DOI: 10.1038/nature06669There is no corresponding record for this reference.
- 10Tee, B. C.-K.; Wang, C.; Allen, R.; Bao, Z. An electrically and mechanically self-healing composite with pressure- and flexion-sensitive properties for electronic skin applications. Nat. Nanotechnol. 2012, 7 (12), 825– 832, DOI: 10.1038/nnano.2012.192There is no corresponding record for this reference.
- 11Wang, C.; Wu, H.; Chen, Z.; McDowell, M. T.; Cui, Y.; Bao, Z. Self-healing chemistry enables the stable operation of silicon microparticle anodes for high-energy lithium-ion batteries. Nat. Chem. 2013, 5 (12), 1042– 1048, DOI: 10.1038/nchem.1802There is no corresponding record for this reference.
- 12Manthiram, A.; Yu, X.; Wang, S. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2017, 2 (4), 16103, DOI: 10.1038/natrevmats.2016.103There is no corresponding record for this reference.
- 13Gracia, I.; Armand, M.; Shanmukaraj, D. Li Metal Polymer Batteries. In Solid Electrolytes for Advanced Applications: Garnets and Competitors; Murugan, R.; Weppner, W., Eds.; Springer International Publishing: Cham, 2019, pp. 347– 373.There is no corresponding record for this reference.
- 14Wang, H.; Song, J.; Zhang, K.; Fang, Q.; Zuo, Y.; Yang, T.; Yang, Y.; Gao, C.; Wang, X.; Pang, Q.; Xia, D. A strongly complexed solid polymer electrolyte enables a stable solid state high-voltage lithium metal battery. Energy Environ. Sci. 2022, 15 (12), 5149– 5158, DOI: 10.1039/D2EE02904AThere is no corresponding record for this reference.
- 15Streipert, B.; Janßen, P.; Cao, X.; Kasnatscheew, J.; Wagner, R.; Cekic-Laskovic, I.; Winter, M.; Placke, T. Evaluation of Allylboronic Acid Pinacol Ester as Effective Shutdown Overcharge Additive for Lithium Ion Cells. J. Electro. Soc. 2017, 164 (2), A168, DOI: 10.1149/2.0711702jesThere is no corresponding record for this reference.
- 16Kelchtermans, A.-S.; De Sloovere, D.; Mercken, J.; Vranken, T.; Mangione, G.; Joos, B.; Vercruysse, W.; Vandamme, D.; Hamed, H.; Safari, M.; Derveaux, E.; Adriaensens, P.; Van Bael, M. K.; Hardy, A. Superconcentration Strategy Allows Sodium Metal Compatibility in Deep Eutectic Solvents for Sodium-Ion Batteries. ACS Omega 2024, 9 (41), 42343– 42352, DOI: 10.1021/acsomega.4c02896There is no corresponding record for this reference.
- 17Boisset, A.; Menne, S.; Jacquemin, J.; Balducci, A.; Anouti, M. Deep eutectic solvents based on N-methylacetamide and a lithium salt as suitable electrolytes for lithium-ion batteries. Phys. Chem. Chem. Phys. 2013, 15 (46), 20054– 20063, DOI: 10.1039/c3cp53406eThere is no corresponding record for this reference.
- 18Meles Neguse, S.; Yoon, S.; Lim, H.; Jang, J.; Baek, S.; Jöckel, D. M.; Widenmeyer, M.; Balke-Grünewald, B.; Weidenkaff, A. The Pitfalls of Deep Eutectic Solvents in the Recycling of Lithium-Ion Batteries. Energy Technol. 2024, 12 (4), 2301213, DOI: 10.1002/ente.202301213There is no corresponding record for this reference.
- 19Minkiewicz, J.; Jones, G. M.; Ghanizadeh, S.; Bostanchi, S.; Wasely, T. J.; Yamini, S. A.; Nekouie, V. Large-scale manufacturing of solid-state electrolytes: Challenges, progress, and prospects. Open Ceram. 2023, 16, 100497, DOI: 10.1016/j.oceram.2023.100497There is no corresponding record for this reference.
- 20Bae, J.; Zhu, Z.; Yan, J.; Kim, D.-M.; Ko, Y.; Jain, A.; Helms, B. A. Closed-loop cathode recycling in solid-state batteries enabled by supramolecular electrolytes. Sci. Adv. 2023, 9 (32), eadh9020 DOI: 10.1126/sciadv.adh9020There is no corresponding record for this reference.
- 21Stewart, J. J. P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19 (1), 1– 32, DOI: 10.1007/s00894-012-1667-xThere is no corresponding record for this reference.
- 22Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10 (44), 6615– 6620, DOI: 10.1039/b810189bThere is no corresponding record for this reference.
- 23Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Gaussian 16; Revision, C; Gaussian, Inc: Wallingford CT, 2016.There is no corresponding record for this reference.
- 24Liu, K.; Li, X.; Cai, J.; Yang, Z.; Chen, Z.; Key, B.; Zhang, Z.; Dzwiniel, T. L.; Liao, C. Design of High-Voltage Stable Hybrid Electrolyte with an Ultrahigh Li Transference Number. ACS Energy Lett. 2021, 6 (4), 1315– 1323, DOI: 10.1021/acsenergylett.0c02559There is no corresponding record for this reference.
- 25Matsumi, N.; Sugai, K.; Ohno, H. Selective Ion Transport in Organoboron Polymer Electrolytes Bearing a Mesitylboron Unit. Macromolecules 2002, 35 (15), 5731– 5733, DOI: 10.1021/ma0121666There is no corresponding record for this reference.
- 26Matsumi, N.; Sugai, K.; Ohno, H. Ion Conductive Characteristics of Alkylborane Type and Boric Ester Type Polymer Electrolytes Derived from Mesitylborane. Macromolecules 2003, 36 (7), 2321– 2326, DOI: 10.1021/ma021734uThere is no corresponding record for this reference.
- 27Nishihara, Y.; Miyazaki, M.; Tomita, Y.; Kadono, Y.; Takagi, K. Synthesis and ion conductive characteristics of inorganic–organic hybrid polymers bearing a tetraarylpentaborate unit. J. Polym. Sci., Part A: Polym. Chem. 2008, 46 (23), 7913– 7918, DOI: 10.1002/pola.23070There is no corresponding record for this reference.
- 28Yuan, C.; Xu, J. Quantitative regulation of electrochemical-mechanical performance of composite cathode in all-solid-state batteries. Nano Energy 2024, 121, 109193, DOI: 10.1016/j.nanoen.2023.109193There is no corresponding record for this reference.
- 29Yuan, C.; Lu, W.; Xu, J. Electrochemical-mechanical coupling failure mechanism of composite cathode in all-solid-state batteries. Energy Storage Mater. 2023, 60, 102834, DOI: 10.1016/j.ensm.2023.102834There is no corresponding record for this reference.
- 30Orisekeh, D.; Roh, B.-M.; Xiao, X. Solid-to-Solid Manufacturing Processes for High-Performance Li-Ion Solid-State Batteries. Polymer 2025, 17 (13), 1788, DOI: 10.3390/polym17131788There is no corresponding record for this reference.
- 31Monroe, C.; Newman, J. The Impact of Elastic Deformation on Deposition Kinetics at Lithium/Polymer Interfaces. J. Electro. Soc. 2005, 152 (2), A396, DOI: 10.1149/1.1850854There is no corresponding record for this reference.
- 32Xu, R.; Cheng, X.-B.; Yan, C.; Zhang, X.-Q.; Xiao, Y.; Zhao, C.-Z.; Huang, J.-Q.; Zhang, Q. Artificial Interphases for Highly Stable Lithium Metal Anode. Matter 2019, 1 (2), 317– 344, DOI: 10.1016/j.matt.2019.05.016There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsaem.5c02674.
Materials, syntheses, preparation of polymer electrolyte precursor solution, physical and electrochemical properties, and electrochemical characterization (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.