



This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Fentanyl-Rewired: A 2-Azaspiro[3.3]heptane Core Preserves μ-Opioid FunctionClick to copy article linkArticle link copied!
- Arran W. StewartArran W. StewartDepartment of Chemistry and Immunology, The Skaggs Institute for Chemical Biology, Worm Institute of Research and Medicine (WIRM), The Scripps Research Institute, La Jolla, California 92037, United StatesMore by Arran W. Stewart
- Lisa M. EubanksLisa M. EubanksDepartment of Chemistry and Immunology, The Skaggs Institute for Chemical Biology, Worm Institute of Research and Medicine (WIRM), The Scripps Research Institute, La Jolla, California 92037, United StatesMore by Lisa M. Eubanks
- Mingliang LinMingliang LinDepartment of Chemistry and Immunology, The Skaggs Institute for Chemical Biology, Worm Institute of Research and Medicine (WIRM), The Scripps Research Institute, La Jolla, California 92037, United StatesMore by Mingliang Lin
- Kim D. Janda*Kim D. Janda*[email protected]Department of Chemistry and Immunology, The Skaggs Institute for Chemical Biology, Worm Institute of Research and Medicine (WIRM), The Scripps Research Institute, La Jolla, California 92037, United StatesMore by Kim D. Janda
Abstract
Fentanyl is a benchmark μ-opioid analgesic but is constrained by respiratory risk. Searching for new entities with reduced respiratory liability, pharmacophore portability was probed by replacing the piperidine moiety with 2-azaspiro[3.3]heptane while preserving phenethyl/anilide geometry. This spiro analogue retained fentanyl-class behavior─μ-opioid receptor (MOR)-preferred binding (MOR > κ-opioid receptor (KOR) ≫ δ-opioid receptor (DOR)), absent β-arrestin-2 recruitment, and full hot-plate/tail-flick antinociception─despite ∼102-fold right-shift in potency versus fentanyl. In mice, it was stable and short-acting with a serum half-life of ∼27 min after an intravenous bolus dose. Whole-body plethysmography showed rapid, dose-dependent depression of respiration that was evident only at high doses. In sum, these studies present a topology-level core swap; preserving the fentanyl signature while decoupling potency from exposure, mapping the pharmacophore’s boundary conditions and providing an actionable, spiro-enabled blueprint to tune MOR signaling and disposition─and recover affinity via structure–activity relationship (SAR)─for next-generation opioid leads.
This publication is licensed for personal use by The American Chemical Society.
Opioid analgesics remain essential for severe pain, (1) yet their use is constrained by dose-limiting adverse effects and a public-health crisis driven by potent synthetics. (2) Fentanyl is an exceptionally efficacious MOR agonist that produces rapid, profound analgesia, (2) but also carries a well-documented risk of life-threatening respiratory depression (3) and has been a major driver of opioid overdose mortality when used nonmedically or encountered as illicitly manufactured fentanyl in the drug supply. (4) These pressures sharpen a central goal in opioid structure activity studies: preserve MOR-mediated analgesia while reshaping pharmacology to reduce risk.
One route to examine the phenylpiperidine class of synthetic opioid pharmacology is through scaffold hopping within the fentanyl chemotype to survey which elements encode its biological signature. Piperidine is the canonical core of fentanyl and a privileged motif across many drugs. (5) Compact spirocyclic amines─especially azaspiro[3.3]heptanes─have emerged as practical piperidine bioisosteres that maintain amine basicity and relative orientation of substituents while adding conformational restraint and three-dimensional shape. (6,7) Across matched pairs, azaspiro[3.3]heptanes typically lower lipophilicity, (8) increase intrinsic solubility, (7) and often reduce microsomal clearance versus six-membered monocyclic analogues (7)─advantages that have translated to comparable activity with improved metabolic stability in other series. (7)
Prior ring edits to the 4-anilidopiperidine scaffold─either conformationally restricting it to tropane frameworks or expanding it to perhydroazepines─showed potency to be highly stereochemistry-dependent, with several analogues losing efficacy or retaining morphine-like liability profiles. (9,10) Because these approaches primarily probed ring contraction/expansion within the 4-anilidopiperidine framework without clearly uncoupling analgesia from morphine-like dependence, we replaced the piperidine with a compact 2-azaspiro[3.3]heptane bioisostere within the fentanyl chemotype, preserving key vectors while introducing a compact 3D constraint and new physicochemical/pharmacokinetic tuning potential to test whether μ-opioid pharmacology can be retained yet signaling and disposition shifted.
In parallel, attempts to dissociate analgesia from respiratory depression have focused on MOR signaling, (11) particularly the balance between G-protein and β-arrestin-2 pathways. (12) Fentanyl engages both pathways at MOR. (13) By contrast, weak β-arrestin-2-recruiting ligands display distinct in vivo effects, (14,15) implying that the relative engagement of G-protein versus β-arrestin-2 pathways modulates the overall phenotype. (14,16) While the clinical generality remains debated, (17,18) deliberately tuning MOR signaling remains a compelling strategy for next-generation opioids.
Based upon this agenda, we probed portability by swapping the piperidine for 2-azaspiro[3.3]heptane while preserving phenethyl and anilide orientations (Figure 1). Across orthogonal assays─MOR/DOR/KOR binding, β-arrestin-2 readout, mouse-serum stability, pharmacokinetics, antinociception, acute tolerability, and whole-body plethysmography─the spiro core preserved a fentanyl-like profile yet shifted potency, exposure, and β-arrestin recruitment. The outcome establishes a practical template: spiro substitution can retain class-defining μ-opioid pharmacology while opening tractable levers in signaling and disposition to engineer wider therapeutic indices.
Figure 1
Figure 1. Structures of bioisostere analogue 1 and fentanyl, 2.
To enable evaluation, we developed a concise four-step synthesis of analogue 1 from commercial tert-butyl 6-oxo 2-azaspiro[3.3]heptane-2-carboxylate (Scheme 1). Synthesis started with the reductive amination of the C-6 ketone with aniline and NaBH(OAc)3, giving secondary amine 3 quantitatively, while the Boc group was retained to dampen basicity on the N(spiro) ring as well as suppress side reactions during the condensation/reduction. With 3 secure, acylation of the aniline with propionyl chloride furnished propionanilide 4 in 56% yield. It should be mentioned that Boc protection upon the N(spiro) functionality prevented competing acylation and ensured anilide selectivity. Boc deprotection with TFA was then employed to unmask the ring’s secondary amine, affording 5. Finally, N-alkylation with 2-phenethyl iodide afforded N-phenethyl tertiary amine 1 in 73% yield over two steps. It is worth noting that the iodide electrophile consistently outperformed the bromide in this SN2 step, giving higher conversion and fewer byproducts, which simplified purification. Finally, it should be understood that this route mirrors canonical fentanyl syntheses (19) from 4-piperidone while enabling the spirocyclic replacement of piperidine.
Scheme 1
To translate the chemical design into pharmacology, in vitro binding studies for 1 were performed off-site under a standardized protocol. The binding affinity of 1 for the opioid receptors was evaluated by using competitive radioligand displacement assays in a two-stage workflow. In the primary screen, 1 was tested at a concentration of 10 μM resulting in%-inhibition of 78.79 ± 2.14% at MOR, 65.94 ± 5.41% at KOR, and 28.60 ± 5.10% at DOR, thus exhibiting the rank order MOR > KOR ≫ DOR. Owing to the low DOR signal (<50% inhibition), secondary radioligand binding assays focused on MOR and KOR using [3H]DAMGO and [3H]U69593, respectively (Figure 2). Full concentration curves of 1 were generated across two independent assays per target receptor resulting in binding affinities of Ki = 1.17–1.68 μM (geometric mean 1.40 μM, pKi ≈ 5.85) at MOR and Ki = 3.70–5.03 μM (geometric mean 4.32 μM, pKi ≈ 5.36) at KOR, corresponding to ∼3-fold selectivity for MOR over KOR.
Figure 2
Figure 2. Competitive radioligand binding of 1 at (A) the μ-opioid receptor (MOR) and (B) the κ-opioid receptor (KOR), with morphine and salvinorin A included as positive control ligands, respectively.
The β-arrestin activity of 1 was assessed using a β-arrestin-2 recruitment assay in comparison to the reference MOR agonist DAMGO (Figure S1). While DAMGO produced a robust β-arrestin-2 recruitment response, 1 produced no detectable signal over the concentration range tested (up to ∼30 μM), indicating no measurable β-arrestin-2 recruitment under these conditions and implying that any β-arrestin-2 engagement, if present, lies below the assay’s detection threshold.
With serum degradation unlikely to limit exposure, we next characterized the in vivo disposition of 1. Analysis of the plasma concentration–time profile (Figure 3) shows rapid systemic exposure following administration of a intravenous (IV) bolus dose of 150 μg/kg, with a subsequent monoexponential decline. Noncompartmental modeling afforded an elimination half-life of approximately 27 min, a clearance of 13.3 mL·min–1·kg–1 and a volume of distribution of 0.52 L·kg–1. Extrapolation to time zero gave C0 ≈ 6.7 μM, and the area under the curve was AUC0–∞ ≈ 10 μM·min. Together, these observations are characteristic of a short-acting profile with low-moderate clearance and modest distribution
Figure 3
Figure 3. Plasma concentration–time profile of fentanyl bioisostere 1. Compound 1 displayed a rapid peak in plasma concentration at 5 min, followed by a monoexponential decline consistent with first-order elimination kinetics. The observed pharmacokinetic profile supports rapid systemic exposure and moderate clearance. Mice (n = 6) were administered 150 μg/kg drug (IV) and blood samples were collected at 5, 15, 30, 60, and 120 min post-injection, alternating three mice every other time point. Bars denote means ± SEM.
For context, previous acute mouse biodistribution studies comparing respiratory-depressant-relevant doses of fentanyl and morphine reported that fentanyl (0.3 mg/kg sc) reaches maximal concentrations in key tissues on a rapid time scale, with tmax ∼ 15 min in whole blood, brain, liver, and lung, and exhibits greater early redistribution from blood into brain, liver, lung, and heart than morphine. (20) Against this benchmark, the IV pharmacokinetics of 1─with rapid exposure evident by the earliest sampling point (5 min) and a short elimination half-life (∼27 min)─is consistent with an acute, short-acting profile appropriate for fentanyl-class comparison.
To evaluate potential toxicity, mice (n = 4 per dose) received increasing amounts of 1 (IV) and were monitored continuously for adverse effects and mortality. At 10 mg kg–1, mice appeared normal with no indication of distress or signs of acute toxicity except for slight piloerection. At 30 mg kg–1, mild and brief seizure activity was observed in all animals along with immediate and prominent piloerection. At 50 mg kg–1, generalized clonic-tonic seizures occurred rapidly and persisted in all mice; 2 of 4 mice died within 5–10 min at this dose. These data establish a dose-dependent neurotoxic threshold under acute IV bolus conditions and delineate a practical upper bound for subsequent efficacy studies.
MOR activation triggers intracellular signaling pathways thought to be associated with analgesia and respiratory depression, respectively. (11) To determine whether the spiro substitution preserves system-level analgesia, we evaluated 1 alongside fentanyl (reference control) in both hot plate and tail flick antinociception assays, which are often used to measure supraspinally and spinally mediated analgesic potencies of opioid-like drugs in rodents. In these assays, animals are cumulatively dosed with drug (IP) and their response to thermal stimuli measured until 100% maximal potential effect (MPE) is reached. Effective dose 50 (ED50) values calculated from the dose–response curves enables quantitative comparison of drug analgesic potencies. As anticipated, fentanyl produced strong analgesic effects in the μg/kg range, with ED50 values of 82 and 87 μg/kg for hot plate and tail flick tests, respectively (Figure 4). In contrast, a mg/kg dosing range of 1 was required to produce a measurable effect resulting in hot plate and tail flick ED50 values of 9 and 14 mg/kg. These ED50 values correspond to ∼115-fold (hot plate) and ∼165-fold (tail flick) lower potency for 1, yet full antinociceptive efficacy was still achieved. Notably, during dosing of 1, piloerection was observed at all concentrations and became more pronounced with increasing doses. However, the straub tail effect, a classic opioid-induced response caused by CNS activation, was only noticeable in fentanyl-dosed mice and not observed in mice dosed with 1.
Figure 4
Figure 4. Behavioral results for antinociception assays. Mice (n = 6) were cumulatively dosed (IP) with fentanyl or fentanyl bioisostere 1 and latency to nociception was measured in hot plate (A) and tail flick (B) tests. Calculated ED50 values were from dose–response curves (C). Bars denote means ± SEM ***P < 0.001, unpaired t-test.
Drug-induced respiratory depression is a serious and often fatal side effect of opioid medications caused by activation of MOR. (21,22) Thus, whole-body plethysmography was used to measure the potential respiratory depressive effects of 1 (Figure 5). Mice dosed with 1 (IP) generally showed a dose-dependent decrease in minute volume (MV) between 5 and 40 mg/kg with maximal respiratory depression observed within ∼10 min, consistent with other opioids. (23) However, MV returned to baseline within 25 min post-injection of all doses.
Figure 5
Figure 5. Effect of analogue 1 on mouse respiration. Minute volume (MV) was dose-dependently depressed by 1 reaching maximal respiratory depression at ∼10 min but returning to baseline within 25–30 min. Mice (n = 8 per group) were administered drug (5–40 mg/kg, IP), and respiration was measured by whole-body plethysmography. MV is plotted as the percent baseline with respect to time post-drug challenge. Statistical comparison between groups was made by two-way ANOVA [Fdose (4, 35) = 5.43; P = 0.0017] with Bonferroni’s comparison. Closed symbols indicate significant differences compared to the no drug control group at individual time points (p ≤ 0.05). Bars denote mean ± SEM.
Thus, 1 produces a rapid but transient ventilatory liability typical of opioids, normalizing by ∼25–30 min. Framed by this profile, we asked how far the fentanyl pharmacophore can be altered while preserving MOR preference and robust antinociception while retuning signaling and disposition. To probe this query, we replaced the central piperidine to a 2-azaspiro[3.3]heptane, with phenethyl/anilide geometry preserved, which delivered a recognizably fentanyl-class phenotype (MOR-preferred binding; full analgesic efficacy) but diverged from fentanyl in pathway engagement, showing abolished β-arrestin-2 recruitment activity despite fentanyl’s known engagement of this pathway. (24)
Receptor pharmacology quantified the portability of such a bioisosteric replacement approach. Thus, in primary affinity analysis the analogue favored MOR over KOR ≫ DOR, and secondary determinations placed MOR affinity in the low micromolar range (Ki ≈ 1.17–1.68 μM) with weaker KOR binding (Ki ≈ 3.70–5.03 μM). Although absolute potency is markedly reduced versus the typical low-nanomolar MOR Ki of fentanyl, the selectivity pattern is preserved using the spiro core, providing a clear baseline for structure–activity efforts to recover affinity while maintaining target preference.
Moreover, pharmacokinetics indicated a short-acting compound with moderate clearance and a modest volume of distribution. After an IV bolus dose, plasma concentrations declined monoexponentially; a parameter set that does not suggest extensive tissue retention. The molecule was stable in mouse serum for more than 24 h, indicating that the brief exposure reflects its pharmacokinetic properties rather than rapid degradation.
Assessing acute toxicity via bolus IV injections, we observed liabilities only at higher exposures. At 10 mg kg–1, animals displayed piloerection without seizure activity. At 30 mg kg–1, brief seizure activity occurred without mortality; at 50 mg kg–1, seizures were universal with 50% lethality within ∼5–10 min. During antinociception testing, seizure activity was noted in one mouse at a cumulative IP dose of 60 mg kg–1. However, these adverse effects arose at drug doses well above those producing maximal antinociceptive effects, indicating that significant efficacy is attainable below the threshold for neurobehavioral toxicity. These findings motivate exposure–response studies (brain/plasma Cmax and rate-of-rise), evaluation of route/formulation influences, receptor-level antagonism, and EEG/telemetry, as well as SAR around the spiro amine and anilide/phenethyl regions, to further separate efficacy from high-dose liabilities.
To contextualize the in vitro pharmacology, we docked fentanyl and 1 into the MOR. In our model, 1 adopts a pose that preserves the canonical ionic/hydrogen-bonding anchor to Asp3.32 (Asp149 in our system) and additionally engages His6.52 (His299) (25) via π–π stacking (Figure 6). By comparison, fentanyl retains the Asp3.32 interaction and exhibits a more extensive secondary stabilization network within the aromatic pocket, including H-bonding to Tyr3.33 (Tyr150) (26) and π–π/edge-to-face contacts with Trp6.48 (Trp295), Tyr7.43 (Tyr328), and Trp2.63 (Trp135) (Figure 6). Thus, relative to fentanyl, (25,27) the spiro analogue appears to attenuate aromatic cage engagement beyond the primary Asp3.32 anchor, with fewer cooperative contacts spanning TM2/3/6/7, consistent with a qualitatively shallower aromatic packing environment. In line with this interaction profile, fentanyl scored more favorably than 1 (docking scores: fentanyl – 7.445 vs 1 – 5.674; more negative indicates stronger predicted affinity). We note that docking provides a static, receptor-state-dependent snapshot; thus, while these interactions are consistent with known MOR pharmacophore elements (D3.32 salt bridge/H-bond and a multiresidue aromatic cage across TM2/3/6/7), they should be interpreted cautiously with respect to efficacy or bias. Complementing the binding-site interaction modeling, docking renderings of the MOR orthosteric site show the poses of bioisostere 1, fentanyl, and their overlay to illustrate conserved D3.32 anchoring and the reduced/shifted aromatic engagement of 1 relative to fentanyl (Figure 7).
Figure 6
Figure 6. Binding-site interactions for (A) fentanyl and (B) bioisostere 1 at the MOR, highlighting polar anchors and aromatic contacts. We note that docking provides a static, receptor-state-dependent snapshot; thus, while these interactions are consistent with known MOR pharmacophore elements (D3.32 salt bridge/H-bond and a multiresidue aromatic cage across TM2/3/6/7), they should be interpreted cautiously with respect to efficacy or bias.
Figure 7
Figure 7. Molecular docking at the MOR orthosteric site. (A) 2-azaspiro[3.3]heptane bioisostere 1 (red sticks), (B) fentanyl (green sticks), and (C) overlay of 1 (red) and fentanyl (green).
In summary, these results reframe what is possible for the fentanyl chemotype. A wholesale piperidine→2-azaspiro[3.3]heptane swap preserves a recognizably fentanyl-like phenotype─maintained MOR preference, full antinociception, and a transient, frequency-driven ventilatory effect─while decisively shifting both potency and exposure. Crucially, the analogue also separates from fentanyl along two liability-relevant axes: no apparent β-arrestin-2 engagement (fentanyl recruits this pathway (24)) and respiratory depression that emerges only at mg/kg doses and appears short-lived, rather than the μg/kg range typical for fentanyl. (28) Put simply, this is not just pharmacophore portability; it is a usable design lever.
The spiro core offers a concrete way to disentangle efficacy from features that often track with risk. It retains the essential Asp3.32 anchoring logic of the fentanyl scaffold, yet opens multiple, experimentally tractable knobs for optimization. First, anilide electronics can potentially be tuned with simple para-substitution (e.g., p-F, p-Cl, p-CF3, p-OMe) to rebalance aromatic-pocket stabilization without disrupting the conserved ionic/H-bonding anchor. Second, the phenethyl vector provides a high-yield opportunity to shape conformational preference and deepen engagement with the MOR aromatic cage; α-methyl or 3-methyl substitutions are obvious, fentanyl-validated moves to test in this new topological context. Third, the basicity and residence of the 2-azaspiro[3.3]heptane nitrogen can be systematically modulated via proximal electron-withdrawing substitution (e.g., α-fluoro or gem-difluoro patterns on the spiro framework), offering a clean way to interrogate potency–bias relationships while keeping the scaffold hop intact.
Taken together, this blueprint suggests a disciplined path forward: preserve the “fentanyl signature” at MOR, deliberately reprogram signaling and exposure through the spiro topology, and then recover affinity by iterating these specific SAR levers under exposure-matched functional readouts─including respiratory time course. The broader implication is simple and powerful: topological remodeling can convert a historically rigid chemotype into a modular platform for separating desirable opioid efficacy from key mechanistic liabilities.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.5c00672.
Experimental procedures, NMR spectra, HPLC traces, Supporting Figure 1, Supplementary tables, Supplementary references (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Kim D. Janda - Department of Chemistry and Immunology, The Skaggs Institute for Chemical Biology, Worm Institute of Research and Medicine (WIRM), The Scripps Research Institute, La Jolla, California 92037, United States;
 https://orcid.org/0000-0001-6759-4227;
https://orcid.org/0000-0001-6759-4227;
- Arran W. Stewart - Department of Chemistry and Immunology, The Skaggs Institute for Chemical Biology, Worm Institute of Research and Medicine (WIRM), The Scripps Research Institute, La Jolla, California 92037, United States;https://orcid.org/0009-0001-6276-3922
- Lisa M. Eubanks - Department of Chemistry and Immunology, The Skaggs Institute for Chemical Biology, Worm Institute of Research and Medicine (WIRM), The Scripps Research Institute, La Jolla, California 92037, United States;https://orcid.org/0000-0001-5288-6294
- Mingliang Lin - Department of Chemistry and Immunology, The Skaggs Institute for Chemical Biology, Worm Institute of Research and Medicine (WIRM), The Scripps Research Institute, La Jolla, California 92037, United States;https://orcid.org/0000-0002-3325-4539
Arran W. Stewart: Writing – review and editing, Writing – original draft, Visualization, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Lisa M. Eubanks: Writing – review and editing, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Mingliang Lin: Validation, Methodology, Investigation, Formal analysis, Data curation. Kim D. Janda: Writing – review and editing, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
Acknowledgments
We thank the NIMH Psychoactive Drug Screening Program (PDSP, UNC Chapel Hill) for conducting the MOR/β-arrestin assays. This work was supported by the Shadek Family Foundation.
| ANOVA | analysis of variance |
| AUC | Area under the curve |
| Boc | tert-butoxycarbonyl |
| Cmax | maximum concentration |
| CNS | central nervous system |
| DAMGO | [d-Ala2, N-MePhe4, Gly-ol]-enkephalin |
| DOR | δ-opioid receptor |
| ED50 | effective dose 50 |
| EEG | electroencephalography |
| Ki | inhibition constant |
| KOR | κ-opioid receptor |
| MOR | μ-opioid receptor |
| MPE | maximal possible effect |
| MV | minute volume |
| NaBH(OAc)3 | sodium triacetoxyborohydride |
| NIMH | National Institute of Mental Health |
| PDSP | Psychoactive Drug Screening Program |
| pKi | negative log10 of Ki value |
| SAR | structure–activity relationship |
| SN2 | bimolecular nucleophilic substitution |
| TFA | trifluoroacetic acid |
| t1/2 | elimination half-life |
References
This article references 28 other publications.
- 1Burns, S. M.; Cunningham, C. W.; Mercer, S. L. DARK Classics in Chemical Neuroscience: Fentanyl. ACS Chem. Neurosci. 2018, 9 (10), 2428– 2437, DOI: 10.1021/acschemneuro.8b00174Google ScholarThere is no corresponding record for this reference.
- 2Han, Y.; Yan, W.; Zheng, Y.; Khan, M. Z.; Yuan, K.; Lu, L. The rising crisis of illicit fentanyl use, overdose, and potential therapeutic strategies. Translational Psychiatry 2019, 9 (1), 282, DOI: 10.1038/s41398-019-0625-0Google ScholarThere is no corresponding record for this reference.
- 3Baird, A.; White, S. A.; Das, R.; Tatum, N.; Bisgaard, E. K. Whole body physiology model to simulate respiratory depression of fentanyl and associated naloxone reversal. Communications Medicine 2024, 4 (1), 114, DOI: 10.1038/s43856-024-00536-5Google ScholarThere is no corresponding record for this reference.
- 4Nelson, L.; Schwaner, R. Transdermal fentanyl: Pharmacology and toxicology. Journal of Medical Toxicology 2009, 5 (4), 230– 241, DOI: 10.1007/BF03178274Google ScholarThere is no corresponding record for this reference.
- 5Frolov, N. A.; Vereshchagin, A. N. Piperidine Derivatives: Recent Advances in Synthesis and Pharmacological Applications. International Journal of Molecular Sciences 2023, 24, 2937, DOI: 10.3390/ijms24032937Google ScholarThere is no corresponding record for this reference.
- 6Burkhard, J. A.; Wagner, B.; Fischer, H.; Schuler, F.; Müller, K.; Carreira, E. M. Synthesis of Azaspirocycles and their Evaluation in Drug Discovery. Angew. Chem., Int. Ed. 2010, 49 (20), 3524– 3527, DOI: 10.1002/anie.200907108Google ScholarThere is no corresponding record for this reference.
- 7Kirichok, A. A.; Tkachuk, H.; Kozyriev, Y.; Shablykin, O.; Datsenko, O.; Granat, D.; Yegorova, T.; Bas, Y. P.; Semirenko, V.; Pishel, I. 1-Azaspiro[3.3]heptane as a Bioisostere of Piperidine. Angew. Chem., Int. Ed. 2023, 62 (51), e202311583 DOI: 10.1002/anie.202311583Google ScholarThere is no corresponding record for this reference.
- 8Degorce, S. L.; Bodnarchuk, M. S.; Scott, J. S. Lowering Lipophilicity by Adding Carbon: AzaSpiroHeptanes, a logD Lowering Twist. ACS Med. Chem. Lett. 2019, 10 (8), 1198– 1204, DOI: 10.1021/acsmedchemlett.9b00248Google ScholarThere is no corresponding record for this reference.
- 9Finney, Z. G.; Riley, T. N. 4-Anilidopiperidine analgesics. 3. 1-Substituted 4-(propananilido)perhydroazepines as ring-expanded analogs. J. Med. Chem. 1980, 23 (8), 895– 899, DOI: 10.1021/jm00182a016Google ScholarThere is no corresponding record for this reference.
- 10Riley, T. N.; Bagley, J. R. 4-Anilidopiperidine analgesics. 2. A study of the conformational aspects of the analgesic activity of the 4-anilidopiperidines utilizing isomeric N-substituted 3-(propananilido)nortropane analogs. J. Med. Chem. 1979, 22 (10), 1167– 1171, DOI: 10.1021/jm00196a004Google ScholarThere is no corresponding record for this reference.
- 11Conibear, A.; Bailey, C. P.; Kelly, E. Biased signalling in analgesic research and development. Current Opinion in Pharmacology 2024, 76, 102465, DOI: 10.1016/j.coph.2024.102465Google ScholarThere is no corresponding record for this reference.
- 12Che, T.; Dwivedi-Agnihotri, H.; Shukla, A. K.; Roth, B. L. Biased ligands at opioid receptors: Current status and future directions. Science Signaling 2021, 14 (677), eaav0320 DOI: 10.1126/scisignal.aav0320Google ScholarThere is no corresponding record for this reference.
- 13Bateman, J. T.; Levitt, E. S. Evaluation of G protein bias and β-arrestin 2 signaling in opioid-induced respiratory depression. American Journal of Physiology-Cell Physiology 2021, 321 (4), C681– C683, DOI: 10.1152/ajpcell.00259.2021Google ScholarThere is no corresponding record for this reference.
- 14Manglik, A.; Lin, H.; Aryal, D. K.; McCorvy, J. D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R. C.; Bernat, V.; Hübner, H. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537 (7619), 185– 190, DOI: 10.1038/nature19112Google ScholarThere is no corresponding record for this reference.
- 15DeWire, S. M.; Yamashita, D. S.; Rominger, D. H.; Liu, G.; Cowan, C. L.; Graczyk, T. M.; Chen, X.-T.; Pitis, P. M.; Gotchev, D.; Yuan, C. A G Protein-Biased Ligand at the μ-Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphine. Journal of Pharmacology and Experimental Therapeutics 2013, 344 (3), 708– 717, DOI: 10.1124/jpet.112.201616Google ScholarThere is no corresponding record for this reference.
- 16Ramos-Gonzalez, N.; Paul, B.; Majumdar, S. IUPHAR themed review: Opioid efficacy, bias, and selectivity. Pharmacol. Res. 2023, 197, 106961, DOI: 10.1016/j.phrs.2023.106961Google ScholarThere is no corresponding record for this reference.
- 17Kelly, E.; Conibear, A.; Henderson, G. Biased Agonism: Lessons from Studies of Opioid Receptor Agonists. Annual Review of Pharmacology and Toxicology 2023, 63, 491– 515, DOI: 10.1146/annurev-pharmtox-052120-091058Google ScholarThere is no corresponding record for this reference.
- 18Haouzi, P.; McCann, M.; Tubbs, N. Respiratory effects of low and high doses of fentanyl in control and β-arrestin 2-deficient mice. Journal of Neurophysiology 2021, 125 (4), 1396– 1407, DOI: 10.1152/jn.00711.2020Google ScholarThere is no corresponding record for this reference.
- 19Valdez, C. A.; Leif, R. N.; Mayer, B. P. An Efficient, Optimized Synthesis of Fentanyl and Related Analogs. PLoS One 2014, 9 (9), e108250 DOI: 10.1371/journal.pone.0108250Google ScholarThere is no corresponding record for this reference.
- 20Goodson, R.; Poklis, J.; Elder, H. J.; Walentiny, D. M.; Dewey, W.; Halquist, M. Acute Biodistribution Comparison of Fentanyl and Morphine. Psychoactives 2024, 3, 437– 460, DOI: 10.3390/psychoactives3040027Google ScholarThere is no corresponding record for this reference.
- 21Stein, C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433– 451, DOI: 10.1146/annurev-med-062613-093100Google ScholarThere is no corresponding record for this reference.
- 22Kelly, E.; Sutcliffe, K.; Cavallo, D.; Ramos-Gonzalez, N.; Alhosan, N.; Henderson, G. The anomalous pharmacology of fentanyl. Br. J. Pharmacol. 2023, 180 (7), 797– 812, DOI: 10.1111/bph.15573Google ScholarThere is no corresponding record for this reference.
- 23Hill, R. A.-O.; Santhakumar, R.; Dewey, W.; Kelly, E. A.-O.; Henderson, G. A.-O. Fentanyl depression of respiration: Comparison with heroin and morphine. Br. J. Pharmacol. 2020, 177, 254, DOI: 10.1111/bph.14860Google ScholarThere is no corresponding record for this reference.
- 24de Waal, P. W.; Shi, J.; You, E.; Wang, X.; Melcher, K.; Jiang, Y.; Xu, H. E.; Dickson, B. M. Molecular mechanisms of fentanyl mediated β-arrestin biased signaling. PLOS Computational Biology 2020, 16 (4), e1007394 DOI: 10.1371/journal.pcbi.1007394Google ScholarThere is no corresponding record for this reference.
- 25Vo, Q. N.; Mahinthichaichan, P.; Shen, J.; Ellis, C. R. How μ-opioid receptor recognizes fentanyl. Nat. Commun. 2021, 12 (1), 984, DOI: 10.1038/s41467-021-21262-9Google ScholarThere is no corresponding record for this reference.
- 26Zádor, F.; Király, K.; Essmat, N.; Al-Khrasani, M. Recent Molecular Insights into Agonist-specific Binding to the Mu-Opioid Receptor. Frontiers in Molecular Biosciences 2022, 9, DOI: 10.3389/fmolb.2022.900547Google ScholarThere is no corresponding record for this reference.
- 27Lipiński, P. F. J.; Kosson, P.; Matalińska, J.; Roszkowski, P.; Czarnocki, Z.; Jarończyk, M.; Misicka, A.; Dobrowolski, J. C.; Sadlej, J. Fentanyl Family at the Mu-Opioid Receptor: Uniform Assessment of Binding and Computational Analysis. Molecules 2019, 24, 740, DOI: 10.3390/molecules24040740Google ScholarThere is no corresponding record for this reference.
- 28Williamson, J.; Kermanizadeh, A. A Review of Toxicological Profile of Fentanyl─A 2024 Update. Toxics 2024, 12, 690, DOI: 10.3390/toxics12100690Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract

Figure 1

Figure 1. Structures of bioisostere analogue 1 and fentanyl, 2.
Scheme 1
 Scheme 1. Synthesis of the 2-Azaspiro[3.3]heptane Fentanyl Analogue 1
Scheme 1. Synthesis of the 2-Azaspiro[3.3]heptane Fentanyl Analogue 1Figure 2
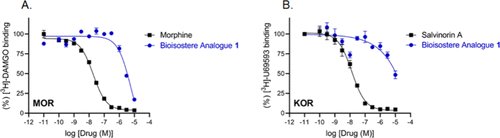
Figure 2. Competitive radioligand binding of 1 at (A) the μ-opioid receptor (MOR) and (B) the κ-opioid receptor (KOR), with morphine and salvinorin A included as positive control ligands, respectively.
Figure 3
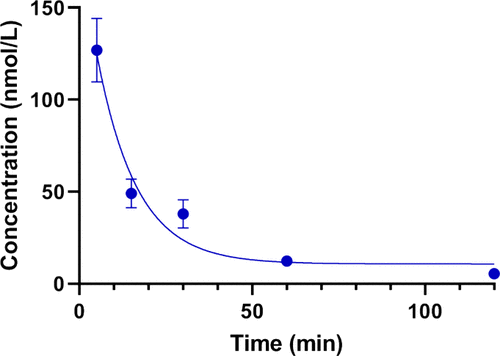
Figure 3. Plasma concentration–time profile of fentanyl bioisostere 1. Compound 1 displayed a rapid peak in plasma concentration at 5 min, followed by a monoexponential decline consistent with first-order elimination kinetics. The observed pharmacokinetic profile supports rapid systemic exposure and moderate clearance. Mice (n = 6) were administered 150 μg/kg drug (IV) and blood samples were collected at 5, 15, 30, 60, and 120 min post-injection, alternating three mice every other time point. Bars denote means ± SEM.
Figure 4

Figure 4. Behavioral results for antinociception assays. Mice (n = 6) were cumulatively dosed (IP) with fentanyl or fentanyl bioisostere 1 and latency to nociception was measured in hot plate (A) and tail flick (B) tests. Calculated ED50 values were from dose–response curves (C). Bars denote means ± SEM ***P < 0.001, unpaired t-test.
Figure 5

Figure 5. Effect of analogue 1 on mouse respiration. Minute volume (MV) was dose-dependently depressed by 1 reaching maximal respiratory depression at ∼10 min but returning to baseline within 25–30 min. Mice (n = 8 per group) were administered drug (5–40 mg/kg, IP), and respiration was measured by whole-body plethysmography. MV is plotted as the percent baseline with respect to time post-drug challenge. Statistical comparison between groups was made by two-way ANOVA [Fdose (4, 35) = 5.43; P = 0.0017] with Bonferroni’s comparison. Closed symbols indicate significant differences compared to the no drug control group at individual time points (p ≤ 0.05). Bars denote mean ± SEM.
Figure 6

Figure 6. Binding-site interactions for (A) fentanyl and (B) bioisostere 1 at the MOR, highlighting polar anchors and aromatic contacts. We note that docking provides a static, receptor-state-dependent snapshot; thus, while these interactions are consistent with known MOR pharmacophore elements (D3.32 salt bridge/H-bond and a multiresidue aromatic cage across TM2/3/6/7), they should be interpreted cautiously with respect to efficacy or bias.
Figure 7

Figure 7. Molecular docking at the MOR orthosteric site. (A) 2-azaspiro[3.3]heptane bioisostere 1 (red sticks), (B) fentanyl (green sticks), and (C) overlay of 1 (red) and fentanyl (green).
References
This article references 28 other publications.
- 1Burns, S. M.; Cunningham, C. W.; Mercer, S. L. DARK Classics in Chemical Neuroscience: Fentanyl. ACS Chem. Neurosci. 2018, 9 (10), 2428– 2437, DOI: 10.1021/acschemneuro.8b00174There is no corresponding record for this reference.
- 2Han, Y.; Yan, W.; Zheng, Y.; Khan, M. Z.; Yuan, K.; Lu, L. The rising crisis of illicit fentanyl use, overdose, and potential therapeutic strategies. Translational Psychiatry 2019, 9 (1), 282, DOI: 10.1038/s41398-019-0625-0There is no corresponding record for this reference.
- 3Baird, A.; White, S. A.; Das, R.; Tatum, N.; Bisgaard, E. K. Whole body physiology model to simulate respiratory depression of fentanyl and associated naloxone reversal. Communications Medicine 2024, 4 (1), 114, DOI: 10.1038/s43856-024-00536-5There is no corresponding record for this reference.
- 4Nelson, L.; Schwaner, R. Transdermal fentanyl: Pharmacology and toxicology. Journal of Medical Toxicology 2009, 5 (4), 230– 241, DOI: 10.1007/BF03178274There is no corresponding record for this reference.
- 5Frolov, N. A.; Vereshchagin, A. N. Piperidine Derivatives: Recent Advances in Synthesis and Pharmacological Applications. International Journal of Molecular Sciences 2023, 24, 2937, DOI: 10.3390/ijms24032937There is no corresponding record for this reference.
- 6Burkhard, J. A.; Wagner, B.; Fischer, H.; Schuler, F.; Müller, K.; Carreira, E. M. Synthesis of Azaspirocycles and their Evaluation in Drug Discovery. Angew. Chem., Int. Ed. 2010, 49 (20), 3524– 3527, DOI: 10.1002/anie.200907108There is no corresponding record for this reference.
- 7Kirichok, A. A.; Tkachuk, H.; Kozyriev, Y.; Shablykin, O.; Datsenko, O.; Granat, D.; Yegorova, T.; Bas, Y. P.; Semirenko, V.; Pishel, I. 1-Azaspiro[3.3]heptane as a Bioisostere of Piperidine. Angew. Chem., Int. Ed. 2023, 62 (51), e202311583 DOI: 10.1002/anie.202311583There is no corresponding record for this reference.
- 8Degorce, S. L.; Bodnarchuk, M. S.; Scott, J. S. Lowering Lipophilicity by Adding Carbon: AzaSpiroHeptanes, a logD Lowering Twist. ACS Med. Chem. Lett. 2019, 10 (8), 1198– 1204, DOI: 10.1021/acsmedchemlett.9b00248There is no corresponding record for this reference.
- 9Finney, Z. G.; Riley, T. N. 4-Anilidopiperidine analgesics. 3. 1-Substituted 4-(propananilido)perhydroazepines as ring-expanded analogs. J. Med. Chem. 1980, 23 (8), 895– 899, DOI: 10.1021/jm00182a016There is no corresponding record for this reference.
- 10Riley, T. N.; Bagley, J. R. 4-Anilidopiperidine analgesics. 2. A study of the conformational aspects of the analgesic activity of the 4-anilidopiperidines utilizing isomeric N-substituted 3-(propananilido)nortropane analogs. J. Med. Chem. 1979, 22 (10), 1167– 1171, DOI: 10.1021/jm00196a004There is no corresponding record for this reference.
- 11Conibear, A.; Bailey, C. P.; Kelly, E. Biased signalling in analgesic research and development. Current Opinion in Pharmacology 2024, 76, 102465, DOI: 10.1016/j.coph.2024.102465There is no corresponding record for this reference.
- 12Che, T.; Dwivedi-Agnihotri, H.; Shukla, A. K.; Roth, B. L. Biased ligands at opioid receptors: Current status and future directions. Science Signaling 2021, 14 (677), eaav0320 DOI: 10.1126/scisignal.aav0320There is no corresponding record for this reference.
- 13Bateman, J. T.; Levitt, E. S. Evaluation of G protein bias and β-arrestin 2 signaling in opioid-induced respiratory depression. American Journal of Physiology-Cell Physiology 2021, 321 (4), C681– C683, DOI: 10.1152/ajpcell.00259.2021There is no corresponding record for this reference.
- 14Manglik, A.; Lin, H.; Aryal, D. K.; McCorvy, J. D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R. C.; Bernat, V.; Hübner, H. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537 (7619), 185– 190, DOI: 10.1038/nature19112There is no corresponding record for this reference.
- 15DeWire, S. M.; Yamashita, D. S.; Rominger, D. H.; Liu, G.; Cowan, C. L.; Graczyk, T. M.; Chen, X.-T.; Pitis, P. M.; Gotchev, D.; Yuan, C. A G Protein-Biased Ligand at the μ-Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphine. Journal of Pharmacology and Experimental Therapeutics 2013, 344 (3), 708– 717, DOI: 10.1124/jpet.112.201616There is no corresponding record for this reference.
- 16Ramos-Gonzalez, N.; Paul, B.; Majumdar, S. IUPHAR themed review: Opioid efficacy, bias, and selectivity. Pharmacol. Res. 2023, 197, 106961, DOI: 10.1016/j.phrs.2023.106961There is no corresponding record for this reference.
- 17Kelly, E.; Conibear, A.; Henderson, G. Biased Agonism: Lessons from Studies of Opioid Receptor Agonists. Annual Review of Pharmacology and Toxicology 2023, 63, 491– 515, DOI: 10.1146/annurev-pharmtox-052120-091058There is no corresponding record for this reference.
- 18Haouzi, P.; McCann, M.; Tubbs, N. Respiratory effects of low and high doses of fentanyl in control and β-arrestin 2-deficient mice. Journal of Neurophysiology 2021, 125 (4), 1396– 1407, DOI: 10.1152/jn.00711.2020There is no corresponding record for this reference.
- 19Valdez, C. A.; Leif, R. N.; Mayer, B. P. An Efficient, Optimized Synthesis of Fentanyl and Related Analogs. PLoS One 2014, 9 (9), e108250 DOI: 10.1371/journal.pone.0108250There is no corresponding record for this reference.
- 20Goodson, R.; Poklis, J.; Elder, H. J.; Walentiny, D. M.; Dewey, W.; Halquist, M. Acute Biodistribution Comparison of Fentanyl and Morphine. Psychoactives 2024, 3, 437– 460, DOI: 10.3390/psychoactives3040027There is no corresponding record for this reference.
- 21Stein, C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433– 451, DOI: 10.1146/annurev-med-062613-093100There is no corresponding record for this reference.
- 22Kelly, E.; Sutcliffe, K.; Cavallo, D.; Ramos-Gonzalez, N.; Alhosan, N.; Henderson, G. The anomalous pharmacology of fentanyl. Br. J. Pharmacol. 2023, 180 (7), 797– 812, DOI: 10.1111/bph.15573There is no corresponding record for this reference.
- 23Hill, R. A.-O.; Santhakumar, R.; Dewey, W.; Kelly, E. A.-O.; Henderson, G. A.-O. Fentanyl depression of respiration: Comparison with heroin and morphine. Br. J. Pharmacol. 2020, 177, 254, DOI: 10.1111/bph.14860There is no corresponding record for this reference.
- 24de Waal, P. W.; Shi, J.; You, E.; Wang, X.; Melcher, K.; Jiang, Y.; Xu, H. E.; Dickson, B. M. Molecular mechanisms of fentanyl mediated β-arrestin biased signaling. PLOS Computational Biology 2020, 16 (4), e1007394 DOI: 10.1371/journal.pcbi.1007394There is no corresponding record for this reference.
- 25Vo, Q. N.; Mahinthichaichan, P.; Shen, J.; Ellis, C. R. How μ-opioid receptor recognizes fentanyl. Nat. Commun. 2021, 12 (1), 984, DOI: 10.1038/s41467-021-21262-9There is no corresponding record for this reference.
- 26Zádor, F.; Király, K.; Essmat, N.; Al-Khrasani, M. Recent Molecular Insights into Agonist-specific Binding to the Mu-Opioid Receptor. Frontiers in Molecular Biosciences 2022, 9, DOI: 10.3389/fmolb.2022.900547There is no corresponding record for this reference.
- 27Lipiński, P. F. J.; Kosson, P.; Matalińska, J.; Roszkowski, P.; Czarnocki, Z.; Jarończyk, M.; Misicka, A.; Dobrowolski, J. C.; Sadlej, J. Fentanyl Family at the Mu-Opioid Receptor: Uniform Assessment of Binding and Computational Analysis. Molecules 2019, 24, 740, DOI: 10.3390/molecules24040740There is no corresponding record for this reference.
- 28Williamson, J.; Kermanizadeh, A. A Review of Toxicological Profile of Fentanyl─A 2024 Update. Toxics 2024, 12, 690, DOI: 10.3390/toxics12100690There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.5c00672.
Experimental procedures, NMR spectra, HPLC traces, Supporting Figure 1, Supplementary tables, Supplementary references (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.