



This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Write and Read: Harnessing Synthetic DNA Modifications for Nanopore SequencingClick to copy article linkArticle link copied!
- Uri BertocchiUri BertocchiSchool of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelSagol School of Neuroscience, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelMore by Uri Bertocchi
- Assaf GrunwaldAssaf GrunwaldSchool of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelMore by Assaf Grunwald
- Gal GoldnerGal GoldnerSchool of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelMore by Gal Goldner
- Eliran EitanEliran EitanSchool of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelMore by Eliran Eitan
- Sigal AvrahamSigal AvrahamSchool of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelMore by Sigal Avraham
- Shani Dvir
- Jasline DeekJasline DeekSchool of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelMore by Jasline Deek
- Yael MichaeliYael MichaeliSchool of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelMore by Yael Michaeli
- Brian YaoBrian YaoDepartment of Electrical Engineering & Computer Sciences, University of California, Berkeley, California 94720, United StatesMore by Brian Yao
- Jennifer ListgartenJennifer ListgartenDepartment of Electrical Engineering & Computer Sciences, University of California, Berkeley, California 94720, United StatesMore by Jennifer Listgarten
- Jared T. SimpsonJared T. SimpsonOntario Institute for Cancer Research, Toronto, Ontario M5G 0A3, CanadaDepartment of Molecular Genetics, University of Toronto, Toronto, Ontario M5S 1A8, CanadaMore by Jared T. Simpson
- Winston TimpWinston TimpDepartment of Biomedical Engineering, Johns Hopkins University, Baltimore, Maryland 21218, United StatesMore by Winston Timp
- Yuval Ebenstein*Yuval Ebenstein*Email: [email protected]School of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelSchool of Biomedical Engineering, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelSagol School of Neuroscience, Tel Aviv University, Tel Aviv-Yafo 6997801, IsraelMore by Yuval Ebenstein
ACS Nano
Copyright © 2025 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
An exciting feature of nanopore sequencing is its ability to record multiomic information on the same sequenced DNA molecule. Well-trained models allow the detection of nucleotide-specific molecular signatures through changes in ionic current as DNA molecules translocate through the nanopore. Thus, naturally occurring DNA modifications, such as DNA methylation and hydroxymethylation, may be recorded simultaneously with the genetic sequence. Additional genomic information, such as chromatin state or the locations of bound transcription factors, may also be recorded if their locations are chemically encoded into the DNA. Here, we present a versatile “write-and-read” framework, where chemo-enzymatic DNA labeling with unnatural synthetic tags results in predictable electrical fingerprints in nanopore sequencing. As a proof-of-concept, we explore a DNA glucosylation approach that selectively modifies 5-hydroxymethylcytosine (5hmC) with glucose or glucose-azide adducts. We demonstrate that these modifications generate distinct and reproducible electrical shifts, enabling the direct detection of chemically altered nucleotides. We further demonstrate that enzymatic alkylation, such as the enzymatic transfer of azide residues to the N6 position of adenines, also produces characteristic nanopore signal shifts relative to the native adenine and 6-methyladenine. Beyond direct nucleotide detection, this approach enables bio-orthogonal DNA labeling, enabling an extended alphabet of sequence-specific detectable moieties. The future use of programmable chemical modifications for simultaneous analysis of multiple omics features on individual molecules can significantly advance genetic research and discovery.
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
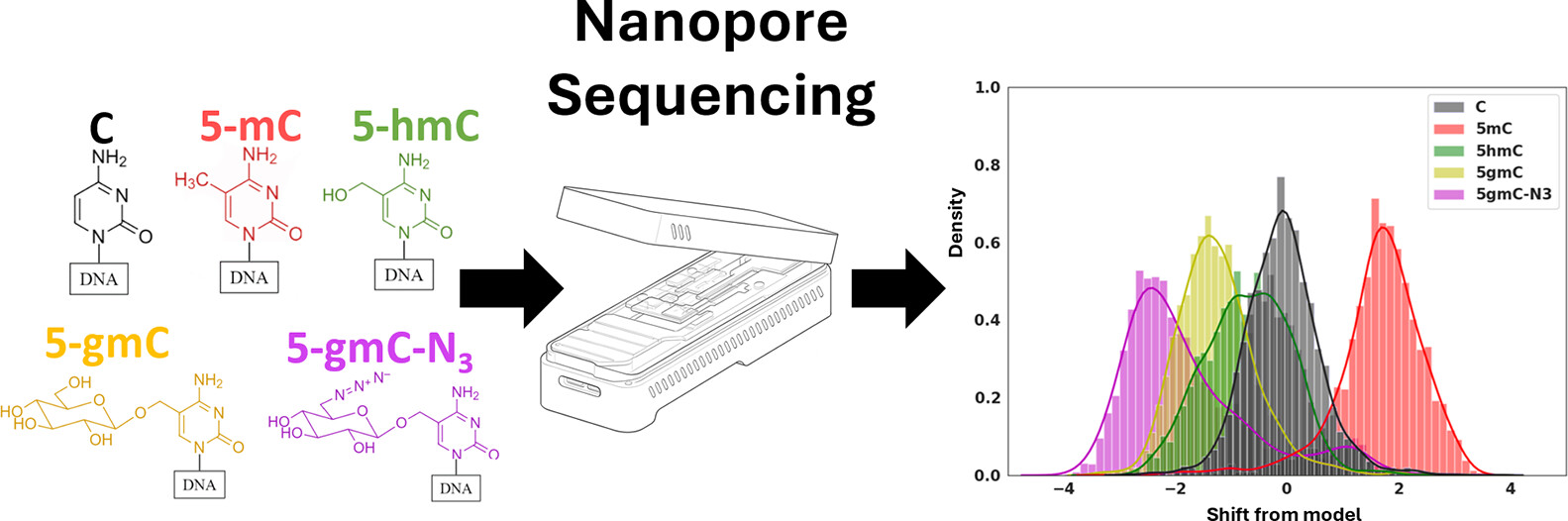
Figure 1
Figure 1. This plot shows an illustration of our write-and-read paradigm: (A) write: chemoenzymatic conversion of designated genomic bases with synthetic epigenetic modifications. (B) Nanopore sequencing of the modified strands. (C) Read: detection of modified signal using modified-basecalling algorithms.
Results
DNA Glucosylation Detection
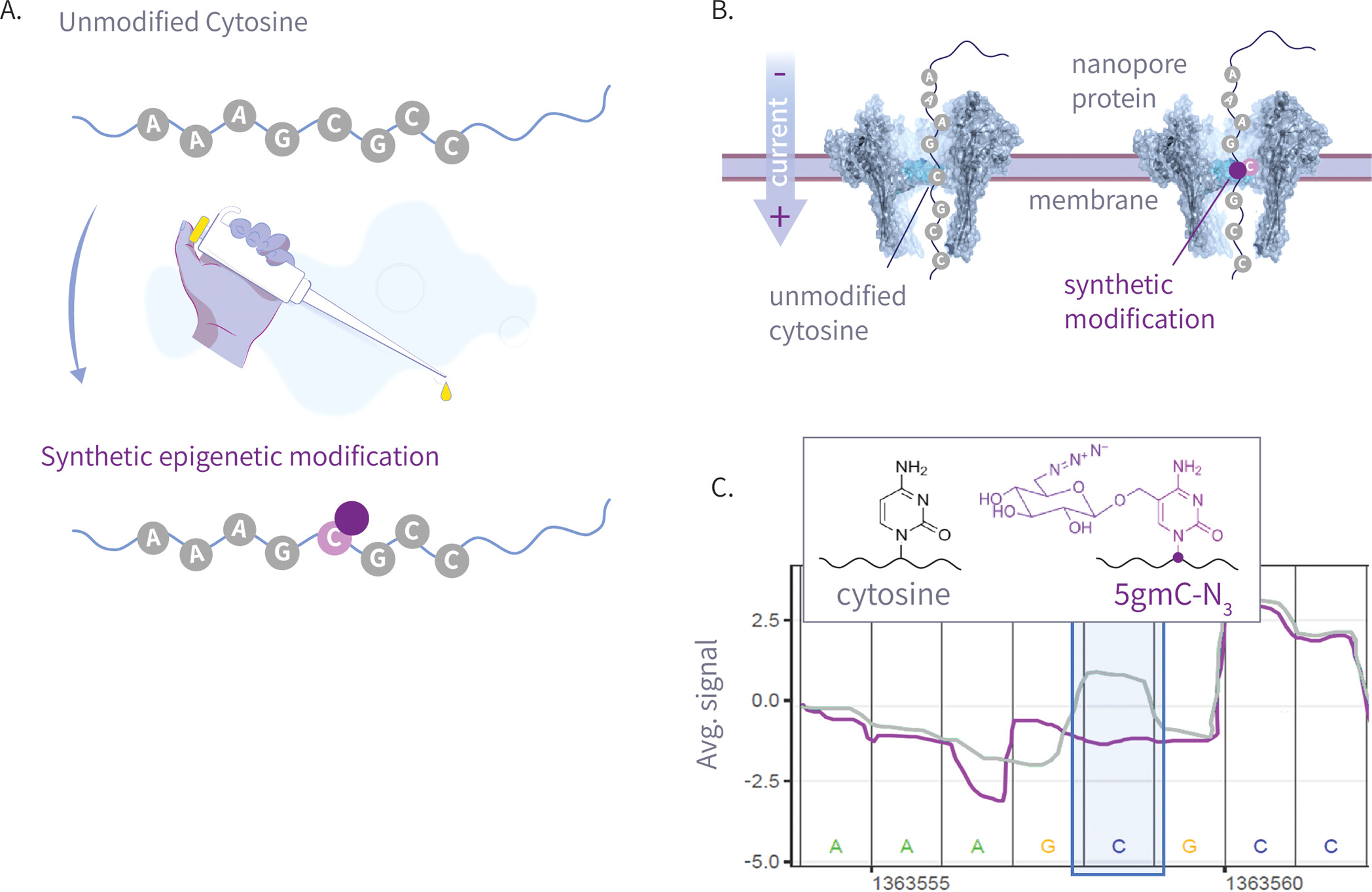
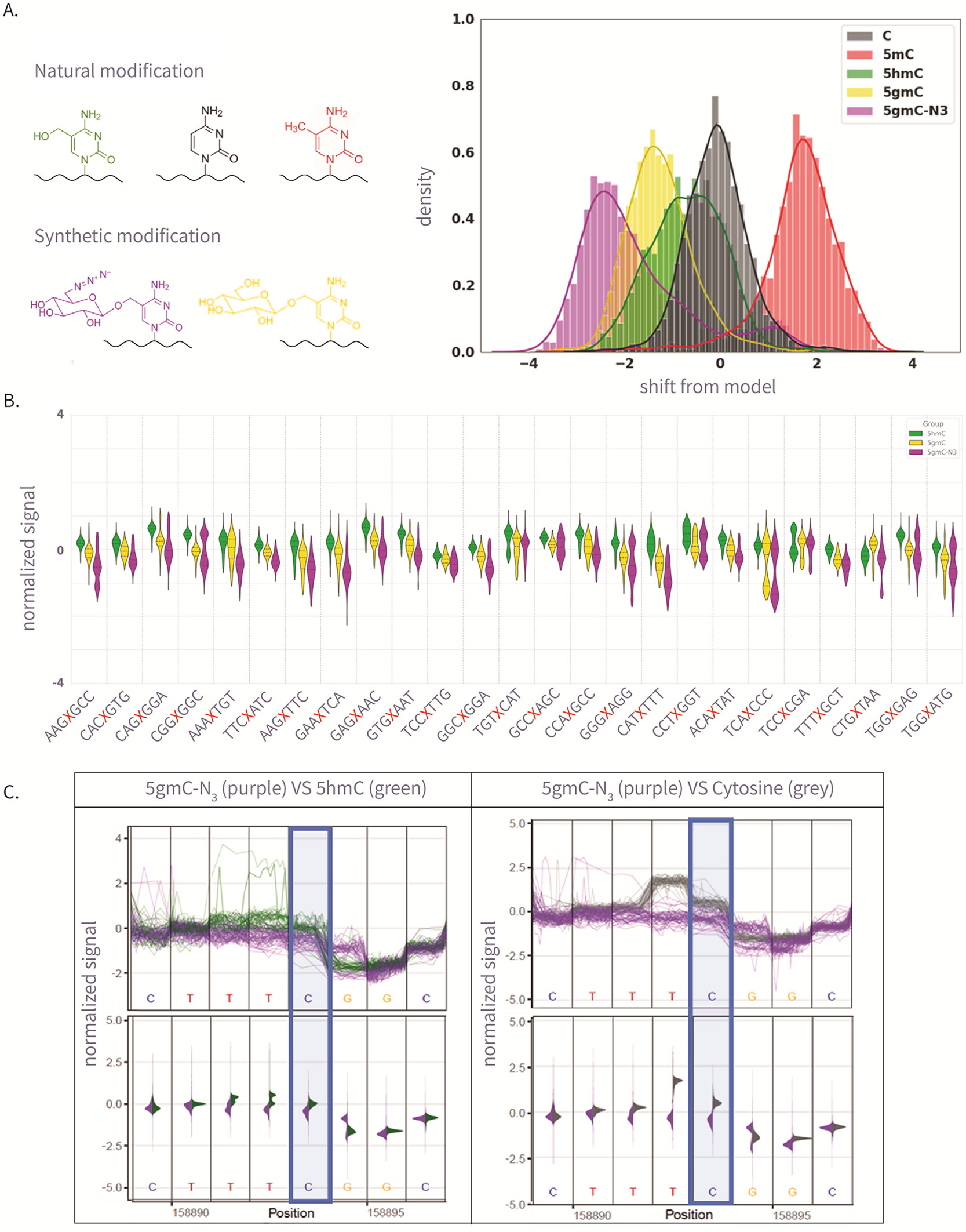
Figure 2
Figure 2. (A) The left panel is the molecular structure of various natural and synthetic cytosine derivatives: unmodified C, 5mC, 5hmC, 5gmC, and 5gmC-N3; The right panel is the signal shift distributions of a single k-mer (GATGCG) from ONT’s trained model expected signal, which demonstrates clear differentiation between the natural and synthetic modifications. (B) The 25 Amplicon sequences showing normalized signal for 5hmC, 5gmC, 5gmC-N3. (C) Plots comparing ionic current signals for 5gmC-N3 (purple) versus 5hmC (green) on the left, and unmodified cytosine (gray) on the right. The y-axis represents normalized signal, and the x-axis denotes genomic position. Upper panels show raw signal traces, while lower density plots display signal distributions. The modified cytosine position of the CG context is highlighted in blue.
DNA Alkylation Detection
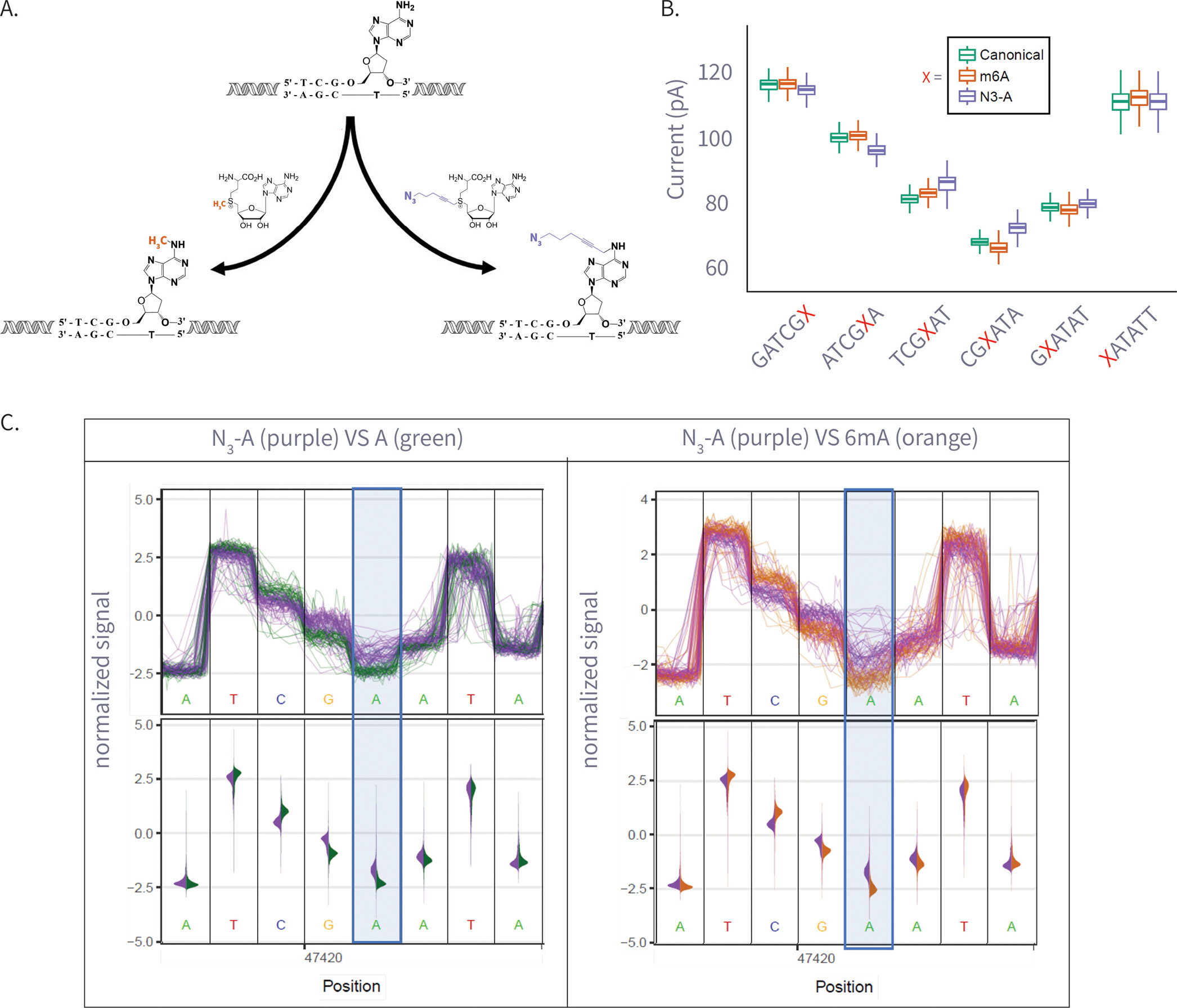
Figure 3
Figure 3. (A) Enzymatic transfer of the azide-methyl group to adenine by M.Taql methyltransferase using S-adenosylmethionine (AdoMet) cofactor and its azide-modified analog (Azide-AdoMet). (B) Box plots of ionic current (pA) for canonical adenine (N = 6569), N6-methyladenine (6 mA, N = 7224), and azide-modified adenine (N3-A, N = 5191) across selected k-mers, showing distinct current shifts; A or modified-A position in the 5-mer is colored in red. (C) Plots comparing ionic current signals for azide-adenine (N3-A, purple) versus unmodified adenine (A, green; left) and N6-methyladenine (6 mA, gray; right). The y-axis represents normalized ionic current (pA), and the x-axis denotes genomic position. Upper panels show raw signal traces, while lower violin plots display signal distributions. The modified adenine position is highlighted in blue.
Discussion
Figure 4
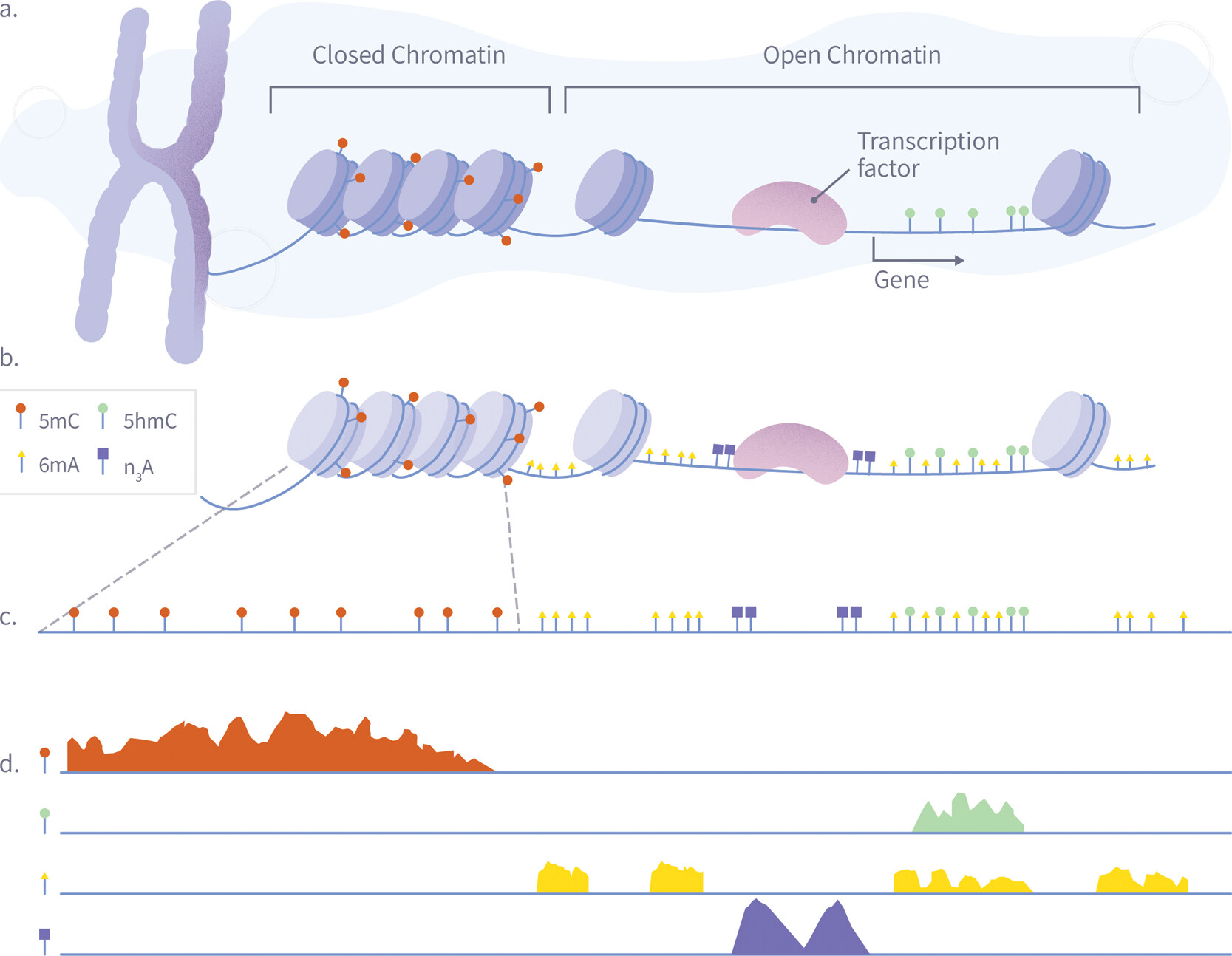
Figure 4. A schematic illustration of a single-pass multiomic experiment demonstrating how nanopore sequencing can simultaneously detect multiple epigenetic and chromatin features. The diagram shows the process in four stages: (A) Nuclear chromatin is composed of genomic DNA, carrying native 5mC (red) and 5hmC (green) modifications, and associated with a plethora of DNA-binding proteins such as nucleosomes and transcription factors. (B) Exposed genomic DNA is labeled within permeabilized nuclei to mark protein footprints by utilizing the high frequency of Adenine, which provides high-resolution mapping. Transcription factor binding sites are marked with N3-A tags (purple) via antibody-mediated proximity labeling, while chromatin accessibility is marked by 6 mA tags (yellow). (C) The DNA is extracted for nanopore sequencing, during which it is stripped from histones, transcription factors, and other bound proteins, maintaining both synthetic and natural modifications. (D) After sequencing and basecalling, the resulting data enable the simultaneous analysis of 5mC, 5hmC, chromatin accessibility, and transcription factor binding sites using standard bioinformatics tools.
Methods
Samples Preparation and Sequencing
Data Analysis
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.5c12530.
Supplementary Figures (S1–S10) Tables (S1–S8) additional methods statistical details raw nanopore current traces for cytosine and adenine modifications analysis of nonassigned events PCR and labeling conditions and sequencing yield summaries (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Yuval Ebenstein - School of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, Israel; School of Biomedical Engineering, Tel Aviv University, Tel Aviv-Yafo 6997801, Israel; Sagol School of Neuroscience, Tel Aviv University, Tel Aviv-Yafo 6997801, Israel;
 https://orcid.org/0000-0002-7107-7529;
https://orcid.org/0000-0002-7107-7529;
- Gal Goldner - School of Chemistry, Tel Aviv University, Tel Aviv-Yafo 6997801, Israel;https://orcid.org/0000-0003-1385-6011
WT, JTS and YE are supported by the National Human Genome Research Institute (5R01HG009190). YE acknowledges the European Research Council consolidator grant [817,811], the Israel Science Foundation [771/21], and the Koret-UC Berkeley-Tel Aviv University Initiative in Computational Biology and Bioinformatics. The Ontario Institute for Cancer Research supports JTS through funds provided by the Government of Ontario, the Government of Canada through Genome Canada and Ontario Genomics (OGI-136 and OGI-201).
| DNA | deoxyribonucleic acid |
| RNA | ribonucleic acid |
| PCR | polymerase chain reaction |
| HPLC | high-performance liquid chromatography |
| ONT | oxford nanopore technologies |
| BGT | β-glucosyltransferase |
| Mtase | methyltransferase |
| AdoMet | S-adenosylmethionine |
| SAM | S-adenosylmethionine |
| K-mer | oligonucleotide of length k |
| 5hmC | 5-hydroxymethylcytosine |
| 5mC | 5-methylcytosine |
| 5gmC | 5-glucosyl-methylcytosine |
| 5gmC-N3 | 5-azide-glucosyl-methylcytosine |
| 6 mA | N6-methyladenine |
| N3-A | azide-modified adenine |
References
This article references 60 other publications.
- 1Lee, I.; Razaghi, R.; Gilpatrick, T.; Molnar, M.; Gershman, A.; Sadowski, N.; Sedlazeck, F. J.; Hansen, K. D.; Simpson, J. T.; Timp, W. Simultaneous Profiling of Chromatin Accessibility and Methylation on Human Cell Lines with Nanopore Sequencing. Nat. Methods 2020, 17 (12), 1191– 1199, DOI: 10.1038/s41592-020-01000-7Google ScholarThere is no corresponding record for this reference.
- 2Simpson, J. T.; Workman, R. E.; Zuzarte, P. C.; David, M.; Dursi, L. J.; Timp, W. Detecting DNA Cytosine Methylation Using Nanopore Sequencing. Nat. Methods 2017, 14 (4), 407– 410, DOI: 10.1038/nmeth.4184Google ScholarThere is no corresponding record for this reference.
- 3Kovaka, S.; Fan, Y.; Ni, B.; Timp, W.; Schatz, M. C. Targeted Nanopore Sequencing by Real-Time Mapping of Raw Electrical Signal with UNCALLED. Nat. Biotechnol. 2021, 39 (4), 431– 441, DOI: 10.1038/s41587-020-0731-9Google ScholarThere is no corresponding record for this reference.
- 4Jain, M.; Koren, S.; Miga, K. H.; Quick, J.; Rand, A. C.; Sasani, T. A.; Tyson, J. R.; Beggs, A. D.; Dilthey, A. T.; Fiddes, I. T.; Malla, S.; Marriott, H.; Nieto, T.; O’Grady, J.; Olsen, H. E.; Pedersen, B. S.; Rhie, A.; Richardson, H.; Quinlan, A. R.; Snutch, T. P.; Tee, L.; Paten, B.; Phillippy, A. M.; Simpson, J. T.; Loman, N. J.; Loose, M. Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36 (4), 338– 345, DOI: 10.1038/nbt.4060Google ScholarThere is no corresponding record for this reference.
- 5Howorka, S.; Siwy, Z. Nanopore Analytics: Sensing of Single Molecules. Chem. Soc. Rev. 2009, 38 (8), 2360– 2384, DOI: 10.1039/b813796jGoogle ScholarThere is no corresponding record for this reference.
- 6Manrao, E. A.; Derrington, I. M.; Laszlo, A. H.; Langford, K. W.; Hopper, M. K.; Gillgren, N.; Pavlenok, M.; Niederweis, M.; Gundlach, J. H. Reading DNA at Single-Nucleotide Resolution with a Mutant MspA Nanopore and Phi29 DNA Polymerase. Nat. Biotechnol. 2012, 30 (4), 349– 353, DOI: 10.1038/nbt.2171Google ScholarThere is no corresponding record for this reference.
- 7Howorka, S.; Cheley, S.; Bayley, H. Sequence-Specific Detection of Individual DNA Strands Using Engineered Nanopores. Nat. Biotechnol. 2001, 19 (7), 636– 639, DOI: 10.1038/90236Google ScholarThere is no corresponding record for this reference.
- 8Jain, M.; Abu-Shumays, R.; Olsen, H. E.; Akeson, M. Advances in Nanopore Direct RNA Sequencing. Nat. Methods 2022, 19 (10), 1160– 1164, DOI: 10.1038/s41592-022-01633-wGoogle ScholarThere is no corresponding record for this reference.
- 9Motone, K.; Kontogiorgos-Heintz, D.; Wee, J.; Kurihara, K.; Yang, S.; Roote, G.; Fox, O. E.; Fang, Y.; Queen, M.; Tolhurst, M.; Cardozo, N.; Jain, M.; Nivala, J. Multi-Pass, Single-Molecule Nanopore Reading of Long Protein Strands. Nature 2024, 633 (8030), 662– 669, DOI: 10.1038/s41586-024-07935-7Google ScholarThere is no corresponding record for this reference.
- 10Stephenson, W.; Razaghi, R.; Busan, S.; Weeks, K. M.; Timp, W.; Smibert, P. Direct Detection of RNA Modifications and Structure Using Single-Molecule Nanopore Sequencing. Cell Genomics 2022, 2 (2), 100097, DOI: 10.1016/j.xgen.2022.100097Google ScholarThere is no corresponding record for this reference.
- 11Gabrieli, T.; Sharim, H.; Nifker, G.; Jeffet, J.; Shahal, T.; Arielly, R.; Levi-Sakin, M.; Hoch, L.; Arbib, N.; Michaeli, Y.; Ebenstein, Y. Epigenetic Optical Mapping of 5-Hydroxymethylcytosine in Nanochannel Arrays. ACS Nano 2018, 12 (7), 7148– 7158, DOI: 10.1021/acsnano.8b03023Google ScholarThere is no corresponding record for this reference.
- 12Fu, Y.; Timp, W.; Sedlazeck, F. J. Computational Analysis of DNA Methylation from Long-Read Sequencing. Nat. Rev. Genet. 2025, 26 (9), 620– 634, DOI: 10.1038/s41576-025-00822-5Google ScholarThere is no corresponding record for this reference.
- 13Zirkin, S.; Fishman, S.; Sharim, H.; Michaeli, Y.; Don, J.; Ebenstein, Y. Lighting Up Individual DNA Damage Sites by In Vitro Repair Synthesis. J. Am. Chem. Soc. 2014, 136 (21), 7771– 7776, DOI: 10.1021/ja503677nGoogle ScholarThere is no corresponding record for this reference.
- 14Michaeli, Y.; Shahal, T.; Torchinsky, D.; Grunwald, A.; Hoch, R.; Ebenstein, Y. Optical Detection of Epigenetic Marks: Sensitive Quantification and Direct Imaging of Individual Hydroxymethylcytosine Bases. Chem. Commun. 2013, 49 (77), 8599– 8601, DOI: 10.1039/c3cc42543fGoogle ScholarThere is no corresponding record for this reference.
- 15Nifker, G.; Levy-Sakin, M.; Berkov-Zrihen, Y.; Shahal, T.; Gabrieli, T.; Fridman, M.; Ebenstein, Y. One-Pot Chemoenzymatic Cascade for Labeling of the Epigenetic Marker 5-Hydroxymethylcytosine. ChemBioChem 2015, 16 (13), 1857– 1860, DOI: 10.1002/cbic.201500329Google ScholarThere is no corresponding record for this reference.
- 16Avraham, S.; Schütz, L.; Käver, L.; Dankers, A.; Margalit, S.; Michaeli, Y.; Zirkin, S.; Torchinsky, D.; Gilat, N.; Bahr, O.; Nifker, G.; Koren-Michowitz, M.; Weinhold, E.; Ebenstein, Y. Chemo-Enzymatic Fluorescence Labeling Of Genomic DNA For Simultaneous Detection Of Global 5-Methylcytosine And 5-Hydroxymethylcytosine. ChemBioChem 2023, 24 (20), e202300400 DOI: 10.1002/cbic.202300400Google ScholarThere is no corresponding record for this reference.
- 17Neely, R. K.; Dedecker, P.; Hotta, J.-i.; Urbanavičiu̅tė, G.; Klimašauskas, S.; Hofkens, J. DNA Fluorocode: A Single Molecule, Optical Map of DNA with Nanometre Resolution. Chem. Sci. 2010, 1 (4), 453– 460, DOI: 10.1039/C0SC00277AGoogle ScholarThere is no corresponding record for this reference.
- 18Jin, X.; Hapsari, N. D.; Lee, S.; Jo, K. DNA Binding Fluorescent Proteins as Single-Molecule Probes. Analyst 2020, 145 (12), 4079– 4095, DOI: 10.1039/D0AN00218FGoogle ScholarThere is no corresponding record for this reference.
- 19McCaffrey, J.; Sibert, J.; Zhang, B.; Zhang, Y.; Hu, W.; Riethman, H.; Xiao, M. CRISPR-CAS9 D10A Nickase Target-Specific Fluorescent Labeling of Double Strand DNA for Whole Genome Mapping and Structural Variation Analysis. Nucleic Acids Res. 2016, 44 (2), e11 DOI: 10.1093/nar/gkv878Google ScholarThere is no corresponding record for this reference.
- 20Grunwald, A.; Dahan, M.; Giesbertz, A.; Nilsson, A.; Nyberg, L. K.; Weinhold, E.; Ambjörnsson, T.; Westerlund, F.; Ebenstein, Y. Bacteriophage Strain Typing by Rapid Single Molecule Analysis. Nucleic Acids Res. 2015, 43 (18), e117 DOI: 10.1093/nar/gkv563Google ScholarThere is no corresponding record for this reference.
- 21Gabrieli, T.; Michaeli, Y.; Avraham, S.; Torchinsky, D.; Margalit, S.; Schütz, L.; Juhasz, M.; Coruh, C.; Arbib, N.; Zhou, Z. S.; Law, J. A.; Weinhold, E.; Ebenstein, Y. Chemoenzymatic Labeling of DNA Methylation Patterns for Single-Molecule Epigenetic Mapping. Nucleic Acids Res. 2022, 50 (16), e92 DOI: 10.1093/nar/gkac460Google ScholarThere is no corresponding record for this reference.
- 22Nifker, G.; Grunwald, A.; Margalit, S.; Tulpova, Z.; Michaeli, Y.; Har-Gil, H.; Maimon, N.; Roichman, E.; Schütz, L.; Weinhold, E.; Ebenstein, Y. Dam Assisted Fluorescent Tagging of Chromatin Accessibility (DAFCA) for Optical Genome Mapping in Nanochannel Arrays. ACS Nano 2023, 17 (10), 9178– 9187, DOI: 10.1021/acsnano.2c12755Google ScholarThere is no corresponding record for this reference.
- 23Detinis Zur, T.; Margalit, S.; Jeffet, J.; Grunwald, A.; Fishman, S.; Tulpová, Z.; Michaeli, Y.; Deek, J.; Ebenstein, Y. Single-Molecule Toxicogenomics: Optical Genome Mapping of DNA-Damage in Nanochannel Arrays. DNA Repair 2025, 146, 103808, DOI: 10.1016/j.dnarep.2025.103808Google ScholarThere is no corresponding record for this reference.
- 24Margalit, S.; Tulpová, Z.; Detinis Zur, T.; Michaeli, Y.; Deek, J.; Nifker, G.; Haldar, R.; Gnatek, Y.; Omer, D.; Dekel, B.; Baris Feldman, H.; Grunwald, A.; Ebenstein, Y. Long-Read Structural and Epigenetic Profiling of a Kidney Tumor-Matched Sample with Nanopore Sequencing and Optical Genome Mapping. NAR:Genomics Bioinf. 2025, 7 (1), lqae190, DOI: 10.1093/nargab/lqae190Google ScholarThere is no corresponding record for this reference.
- 25Raseley, K.; Jinwala, Z.; Zhang, D.; Xiao, M. Single-Molecule Telomere Assay via Optical Mapping (SMTA-OM) Can Potentially Define the ALT Positivity of Cancer. Genes 2023, 14 (6), 1278, DOI: 10.3390/genes14061278Google ScholarThere is no corresponding record for this reference.
- 26Müller, V.; Dvirnas, A.; Andersson, J.; Singh, V.; Kk, S.; Johansson, P.; Ebenstein, Y.; Ambjörnsson, T.; Westerlund, F. Enzyme-Free Optical DNA Mapping of the Human Genome Using Competitive Binding. Nucleic Acids Res. 2019, 47 (15), e89 DOI: 10.1093/nar/gkz489Google ScholarThere is no corresponding record for this reference.
- 27Margalit, S.; Abramson, Y.; Sharim, H.; Manber, Z.; Bhattacharya, S.; Chen, Y.-W.; Vilain, E.; Barseghyan, H.; Elkon, R.; Sharan, R.; Ebenstein, Y. Long Reads Capture Simultaneous Enhancer–Promoter Methylation Status for Cell-Type Deconvolution. Bioinformatics 2021, 37 (Supplement_1), i327– i333, DOI: 10.1093/bioinformatics/btab306Google ScholarThere is no corresponding record for this reference.
- 28Sharim, H.; Grunwald, A.; Gabrieli, T.; Michaeli, Y.; Margalit, S.; Torchinsky, D.; Arielly, R.; Nifker, G.; Juhasz, M.; Gularek, F.; Almalvez, M.; Dufault, B.; Chandra, S. S.; Liu, A.; Bhattacharya, S.; Chen, Y.-W.; Vilain, E.; Wagner, K. R.; Pevsner, J.; Reifenberger, J.; Lam, E. T.; Hastie, A. R.; Cao, H.; Barseghyan, H.; Weinhold, E.; Ebenstein, Y. Long-Read Single-Molecule Maps of the Functional Methylome. Genome Res. 2019, 29 (4), 646– 656, DOI: 10.1101/gr.240739.118Google ScholarThere is no corresponding record for this reference.
- 29Kinney, S. M.; Chin, H. G.; Vaisvila, R.; Bitinaite, J.; Zheng, Y.; Estève, P.-O.; Feng, S.; Stroud, H.; Jacobsen, S. E.; Pradhan, S. Tissue-Specific Distribution and Dynamic Changes of 5-Hydroxymethylcytosine in Mammalian Genomes. J. Biol. Chem. 2011, 286 (28), 24685– 24693, DOI: 10.1074/jbc.M110.217083Google ScholarThere is no corresponding record for this reference.
- 30Klimašauskas, S.; Weinhold, E. A New Tool for Biotechnology: AdoMet-Dependent Methyltransferases. Trends Biotechnol. 2007, 25 (3), 99– 104, DOI: 10.1016/j.tibtech.2007.01.006Google ScholarThere is no corresponding record for this reference.
- 31Lukinavičius, G.; Tomkuvienė, M.; Masevičius, V.; Klimašauskas, S. Enhanced Chemical Stability of Adomet Analogues for Improved Methyltransferase-Directed Labeling of DNA. ACS Chem. Biol. 2013, 8 (6), 1134– 1139, DOI: 10.1021/cb300669xGoogle ScholarThere is no corresponding record for this reference.
- 32Al Temimi, A. H. K.; Martin, M.; Meng, Q.; Lenstra, D. C.; Qian, P.; Guo, H.; Weinhold, E.; Mecinović, J. Lysine Ethylation by Histone Lysine Methyltransferases. ChemBioChem 2020, 21 (3), 392– 400, DOI: 10.1002/cbic.201900359Google ScholarThere is no corresponding record for this reference.
- 33Willnow, S.; Martin, M.; Lüscher, B.; Weinhold, E. A Selenium-Based Click AdoMet Analogue for Versatile Substrate Labeling with Wild-Type Protein Methyltransferases. ChemBioChem 2012, 13 (8), 1167– 1173, DOI: 10.1002/cbic.201100781Google ScholarThere is no corresponding record for this reference.
- 34Holstein, J. M.; Stummer, D.; Rentmeister, A. Enzymatic Modification of 5′-Capped RNA with a 4-Vinylbenzyl Group Provides a Platform for Photoclick and Inverse Electron-Demand Diels–Alder Reaction. Chem. Sci. 2015, 6 (2), 1362– 1369, DOI: 10.1039/C4SC03182BGoogle ScholarThere is no corresponding record for this reference.
- 35Muttach, F.; Mäsing, F.; Studer, A.; Rentmeister, A. New AdoMet Analogues as Tools for Enzymatic Transfer of Photo-Cross-Linkers and Capturing RNA–Protein Interactions. Chem. - Eur. J. 2017, 23 (25), 5988– 5993, DOI: 10.1002/chem.201605663Google ScholarThere is no corresponding record for this reference.
- 36Song, C.-X.; Szulwach, K. E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.-H.; Zhang, W.; Jian, X.; Wang, J.; Zhang, L.; Looney, T. J.; Zhang, B.; Godley, L. A.; Hicks, L. M.; Lahn, B. T.; Jin, P.; He, C. Selective Chemical Labeling Reveals the Genome-Wide Distribution of 5-Hydroxymethylcytosine. Nat. Biotechnol. 2011, 29 (1), 68– 72, DOI: 10.1038/nbt.1732Google ScholarThere is no corresponding record for this reference.
- 37Dalhoff, C.; Lukinavicius, G.; Klimasăuskas, S.; Weinhold, E. Direct Transfer of Extended Groups from Synthetic Cofactors by DNA Methyltransferases. Nat. Chem. Biol. 2006, 2 (1), 31– 32, DOI: 10.1038/nchembio754Google ScholarThere is no corresponding record for this reference.
- 38Josse, J.; Kornberg, A. Glucosylation of Deoxyribonucleic Acid: III. α- AND β-GLUCOSYL TRANSFERASES FROM T4-INFECTED ESCHERICHIA COLI. J. Biol. Chem. 1962, 237 (6), 1968– 1976, DOI: 10.1016/S0021-9258(19)73968-4Google ScholarThere is no corresponding record for this reference.
- 39Pastor, W. A.; Huang, Y.; Henderson, H. R.; Agarwal, S.; Rao, A. The GLIB Technique for Genome-Wide Mapping of 5-Hydroxymethylcytosine. Nat. Protoc. 2012, 7 (10), 1909– 1917, DOI: 10.1038/nprot.2012.104Google ScholarThere is no corresponding record for this reference.
- 40Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K. F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39 (11), 1348– 1365, DOI: 10.1038/s41587-021-01108-xGoogle ScholarThere is no corresponding record for this reference.
- 41Sanger, F.; Coulson, A. R.; Hong, G. F.; Hill, D. F.; Petersen, G. B. Nucleotide Sequence of Bacteriophage λ DNA. J. Mol. Biol. 1982, 162 (4), 729– 773, DOI: 10.1016/0022-2836(82)90546-0Google ScholarThere is no corresponding record for this reference.
- 42Enterobacteria Phage Lambda, Complete Genome, 2023. http://www.ncbi.nlm.nih.gov/nuccore/NC_001416.1 (accessed June 24, 2025).Google ScholarThere is no corresponding record for this reference.
- 43Laszlo, A. H.; Derrington, I. M.; Brinkerhoff, H.; Langford, K. W.; Nova, I. C.; Samson, J. M.; Bartlett, J. J.; Pavlenok, M.; Gundlach, J. H. Detection and Mapping of 5-Methylcytosine and 5-Hydroxymethylcytosine with Nanopore MspA. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (47), 18904– 18909, DOI: 10.1073/pnas.1310240110Google ScholarThere is no corresponding record for this reference.
- 44Yao, B.; Hsu, C.; Goldner, G.; Michaeli, Y.; Ebenstein, Y.; Listgarten, J. Effective Training of Nanopore Callers for Epigenetic Marks with Limited Labelled Data. Open Biol. 2024, 14 (6), 230449, DOI: 10.1098/rsob.230449Google ScholarThere is no corresponding record for this reference.
- 45Kriukienė, E.; Labrie, V.; Khare, T.; Urbanavičiu̅tė, G.; Lapinaitė, A.; Koncevičius, K.; Li, D.; Wang, T.; Pai, S.; Ptak, C.; Gordevičius, J.; Wang, S.-C.; Petronis, A.; Klimašauskas, S. DNA Unmethylome Profiling by Covalent Capture of CpG Sites. Nat. Commun. 2013, 4 (1), 2190, DOI: 10.1038/ncomms3190Google ScholarThere is no corresponding record for this reference.
- 46Klimasauskas, S.; Weinhold, E. A New Tool for Biotechnology: AdoMet-Dependent Methyltransferases. Trends Biotechnol. 2007, 25 (3), 99– 104, DOI: 10.1016/j.tibtech.2007.01.006Google ScholarThere is no corresponding record for this reference.
- 47Hanz, G. M.; Jung, B.; Giesbertz, A.; Juhasz, M.; Weinhold, E. Sequence-Specific Labeling of Nucleic Acids and Proteins with Methyltransferases and Cofactor Analogues. J. Visualized Exp. 2014, 93, e52014 DOI: 10.3791/52014Google ScholarThere is no corresponding record for this reference.
- 48Weng, Z.; Ruan, F.; Chen, W.; Chen, Z.; Xie, Y.; Luo, M.; Xie, Z.; Zhang, C.; Wang, J.; Sun, Y.; Fang, Y.; Guo, M.; Tan, C.; Chen, W.; Tong, Y.; Li, Y.; Wang, H.; Tang, C. BIND&MODIFY: A Long-Range Method for Single-Molecule Mapping of Chromatin Modifications in Eukaryotes. Genome Biol. 2023, 24 (1), 61, DOI: 10.1186/s13059-023-02896-yGoogle ScholarThere is no corresponding record for this reference.
- 49Thatavarty, A.; Sagy, N.; Erdos, M. R.; Lee, I.; Simpson, J. T.; Timp, W.; Collins, F. S.; Bar, D. Z. Detecting Protein-DNA Binding in Single Molecules Using Antibody Guided Methylation. Epigenet. Chromatin 2025, 18 (1), 39, DOI: 10.1186/s13072-025-00602-9Google ScholarThere is no corresponding record for this reference.
- 50Goedecke, K.; Pignot, M.; Goody, R. S.; Scheidig, A. J.; Weinhold, E. Structure of the N6-Adenine DNA Methyltransferase M•TaqI in Complex with DNA and a Cofactor Analog. Nat. Struct. Biol. 2001, 8 (2), 121– 125, DOI: 10.1038/84104Google ScholarThere is no corresponding record for this reference.
- 51Shahal, T.; Gilat, N.; Michaeli, Y.; Redy-Keisar, O.; Shabat, D.; Ebenstein, Y. Spectroscopic Quantification of 5-Hydroxymethylcytosine in Genomic DNA. Anal. Chem. 2014, 86 (16), 8231– 8237, DOI: 10.1021/ac501609dGoogle ScholarThere is no corresponding record for this reference.
- 52Products Scheduled to Be Discontinued; Oxford Nanopore Technologies. https://nanoporetech.com/products/prepare/discontinued-kits (accessed 2025–02–06).Google ScholarThere is no corresponding record for this reference.
- 53Sanderson, N. D.; Kapel, N.; Rodger, G.; Webster, H.; Lipworth, S.; Street, T. L.; Peto, T.; Crook, D.; Stoesser, N. Comparison of R9.4.1/Kit10 and R10/Kit12 Oxford Nanopore Flowcells and Chemistries in Bacterial Genome Reconstruction. Microb. Genomics 2023, 9 (1), 000910, DOI: 10.1099/mgen.0.000910Google ScholarThere is no corresponding record for this reference.
- 54Ni, Y.; Liu, X.; Simeneh, Z. M.; Yang, M.; Li, R. Benchmarking of Nanopore R10.4 and R9.4.1 Flow Cells in Single-Cell Whole-Genome Amplification and Whole-Genome Shotgun Sequencing. Comput. Struct. Biotechnol. J. 2023, 21, 2352– 2364, DOI: 10.1016/j.csbj.2023.03.038Google ScholarThere is no corresponding record for this reference.
- 55Jung, C.; Hawkins, J. A.; Jones, S. K.; Xiao, Y.; Rybarski, J. R.; Dillard, K. E.; Hussmann, J.; Saifuddin, F. A.; Savran, C. A.; Ellington, A. D.; Ke, A.; Press, W. H.; Finkelstein, I. J. Massively Parallel Biophysical Analysis of CRISPR-Cas Complexes on Next Generation Sequencing Chips. Cell 2017, 170 (1), 35– 47, DOI: 10.1016/j.cell.2017.05.044Google ScholarThere is no corresponding record for this reference.
- 56Escherichia Coli Str. K-12 Substr. MG1655, Complete Genome, 2022. http://www.ncbi.nlm.nih.gov/nuccore/NC_000913.3 (accessed 2025–03–10).Google ScholarThere is no corresponding record for this reference.
- 57ONT Fast5 API. https://github.com/nanoporetech/ont_fast5_api (accessed 2024–11–14).Google ScholarThere is no corresponding record for this reference.
- 58Tombo, 2024. https://github.com/nanoporetech/tombo (accessed 2024–11–14).Google ScholarThere is no corresponding record for this reference.
- 59Loman, N. J.; Quick, J.; Simpson, J. T. A Complete Bacterial Genome Assembled de Novo Using Only Nanopore Sequencing Data. Nat. Methods 2015, 12 (8), 733– 735, DOI: 10.1038/nmeth.3444Google ScholarThere is no corresponding record for this reference.
- 60Bertocchi, U.; Grunwald, A.; Goldner, G.; Eitan, E.; Avraham, S.; Deek, J.; Michaeli, Y.; Yao, B.; Listgarten, J.; Simpson, J. T.; Timp, W.; Ebenstein, Y. Write and Read: Harnessing Synthetic DNA Modifications for Nanopore Sequencing. bioRxiv 2025, 2025.04.14.648727, DOI: 10.1101/2025.04.14.648727Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACS Nano
Copyright © 2025 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
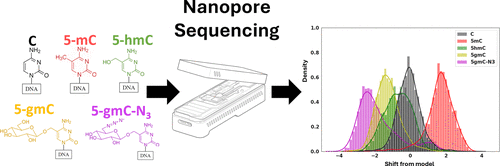
Figure 1
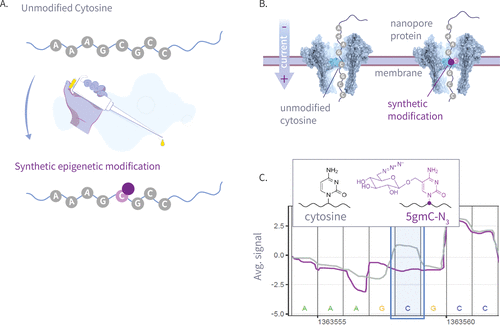
Figure 1. This plot shows an illustration of our write-and-read paradigm: (A) write: chemoenzymatic conversion of designated genomic bases with synthetic epigenetic modifications. (B) Nanopore sequencing of the modified strands. (C) Read: detection of modified signal using modified-basecalling algorithms.
Figure 2
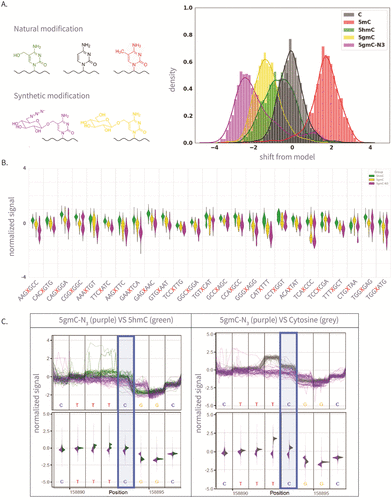
Figure 2. (A) The left panel is the molecular structure of various natural and synthetic cytosine derivatives: unmodified C, 5mC, 5hmC, 5gmC, and 5gmC-N3; The right panel is the signal shift distributions of a single k-mer (GATGCG) from ONT’s trained model expected signal, which demonstrates clear differentiation between the natural and synthetic modifications. (B) The 25 Amplicon sequences showing normalized signal for 5hmC, 5gmC, 5gmC-N3. (C) Plots comparing ionic current signals for 5gmC-N3 (purple) versus 5hmC (green) on the left, and unmodified cytosine (gray) on the right. The y-axis represents normalized signal, and the x-axis denotes genomic position. Upper panels show raw signal traces, while lower density plots display signal distributions. The modified cytosine position of the CG context is highlighted in blue.
Figure 3
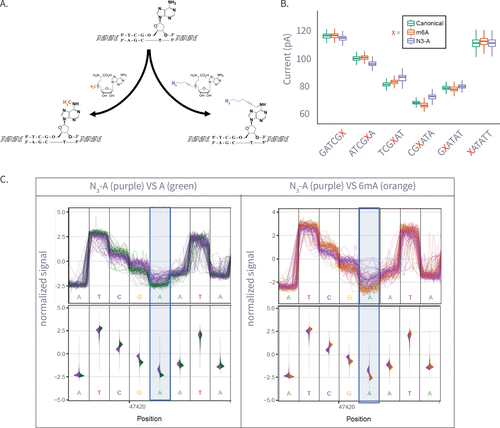
Figure 3. (A) Enzymatic transfer of the azide-methyl group to adenine by M.Taql methyltransferase using S-adenosylmethionine (AdoMet) cofactor and its azide-modified analog (Azide-AdoMet). (B) Box plots of ionic current (pA) for canonical adenine (N = 6569), N6-methyladenine (6 mA, N = 7224), and azide-modified adenine (N3-A, N = 5191) across selected k-mers, showing distinct current shifts; A or modified-A position in the 5-mer is colored in red. (C) Plots comparing ionic current signals for azide-adenine (N3-A, purple) versus unmodified adenine (A, green; left) and N6-methyladenine (6 mA, gray; right). The y-axis represents normalized ionic current (pA), and the x-axis denotes genomic position. Upper panels show raw signal traces, while lower violin plots display signal distributions. The modified adenine position is highlighted in blue.
Figure 4
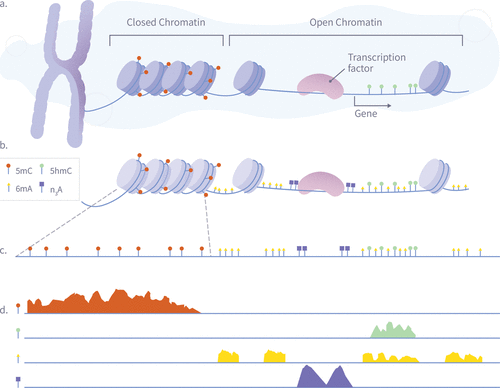
Figure 4. A schematic illustration of a single-pass multiomic experiment demonstrating how nanopore sequencing can simultaneously detect multiple epigenetic and chromatin features. The diagram shows the process in four stages: (A) Nuclear chromatin is composed of genomic DNA, carrying native 5mC (red) and 5hmC (green) modifications, and associated with a plethora of DNA-binding proteins such as nucleosomes and transcription factors. (B) Exposed genomic DNA is labeled within permeabilized nuclei to mark protein footprints by utilizing the high frequency of Adenine, which provides high-resolution mapping. Transcription factor binding sites are marked with N3-A tags (purple) via antibody-mediated proximity labeling, while chromatin accessibility is marked by 6 mA tags (yellow). (C) The DNA is extracted for nanopore sequencing, during which it is stripped from histones, transcription factors, and other bound proteins, maintaining both synthetic and natural modifications. (D) After sequencing and basecalling, the resulting data enable the simultaneous analysis of 5mC, 5hmC, chromatin accessibility, and transcription factor binding sites using standard bioinformatics tools.
References
This article references 60 other publications.
- 1Lee, I.; Razaghi, R.; Gilpatrick, T.; Molnar, M.; Gershman, A.; Sadowski, N.; Sedlazeck, F. J.; Hansen, K. D.; Simpson, J. T.; Timp, W. Simultaneous Profiling of Chromatin Accessibility and Methylation on Human Cell Lines with Nanopore Sequencing. Nat. Methods 2020, 17 (12), 1191– 1199, DOI: 10.1038/s41592-020-01000-7There is no corresponding record for this reference.
- 2Simpson, J. T.; Workman, R. E.; Zuzarte, P. C.; David, M.; Dursi, L. J.; Timp, W. Detecting DNA Cytosine Methylation Using Nanopore Sequencing. Nat. Methods 2017, 14 (4), 407– 410, DOI: 10.1038/nmeth.4184There is no corresponding record for this reference.
- 3Kovaka, S.; Fan, Y.; Ni, B.; Timp, W.; Schatz, M. C. Targeted Nanopore Sequencing by Real-Time Mapping of Raw Electrical Signal with UNCALLED. Nat. Biotechnol. 2021, 39 (4), 431– 441, DOI: 10.1038/s41587-020-0731-9There is no corresponding record for this reference.
- 4Jain, M.; Koren, S.; Miga, K. H.; Quick, J.; Rand, A. C.; Sasani, T. A.; Tyson, J. R.; Beggs, A. D.; Dilthey, A. T.; Fiddes, I. T.; Malla, S.; Marriott, H.; Nieto, T.; O’Grady, J.; Olsen, H. E.; Pedersen, B. S.; Rhie, A.; Richardson, H.; Quinlan, A. R.; Snutch, T. P.; Tee, L.; Paten, B.; Phillippy, A. M.; Simpson, J. T.; Loman, N. J.; Loose, M. Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36 (4), 338– 345, DOI: 10.1038/nbt.4060There is no corresponding record for this reference.
- 5Howorka, S.; Siwy, Z. Nanopore Analytics: Sensing of Single Molecules. Chem. Soc. Rev. 2009, 38 (8), 2360– 2384, DOI: 10.1039/b813796jThere is no corresponding record for this reference.
- 6Manrao, E. A.; Derrington, I. M.; Laszlo, A. H.; Langford, K. W.; Hopper, M. K.; Gillgren, N.; Pavlenok, M.; Niederweis, M.; Gundlach, J. H. Reading DNA at Single-Nucleotide Resolution with a Mutant MspA Nanopore and Phi29 DNA Polymerase. Nat. Biotechnol. 2012, 30 (4), 349– 353, DOI: 10.1038/nbt.2171There is no corresponding record for this reference.
- 7Howorka, S.; Cheley, S.; Bayley, H. Sequence-Specific Detection of Individual DNA Strands Using Engineered Nanopores. Nat. Biotechnol. 2001, 19 (7), 636– 639, DOI: 10.1038/90236There is no corresponding record for this reference.
- 8Jain, M.; Abu-Shumays, R.; Olsen, H. E.; Akeson, M. Advances in Nanopore Direct RNA Sequencing. Nat. Methods 2022, 19 (10), 1160– 1164, DOI: 10.1038/s41592-022-01633-wThere is no corresponding record for this reference.
- 9Motone, K.; Kontogiorgos-Heintz, D.; Wee, J.; Kurihara, K.; Yang, S.; Roote, G.; Fox, O. E.; Fang, Y.; Queen, M.; Tolhurst, M.; Cardozo, N.; Jain, M.; Nivala, J. Multi-Pass, Single-Molecule Nanopore Reading of Long Protein Strands. Nature 2024, 633 (8030), 662– 669, DOI: 10.1038/s41586-024-07935-7There is no corresponding record for this reference.
- 10Stephenson, W.; Razaghi, R.; Busan, S.; Weeks, K. M.; Timp, W.; Smibert, P. Direct Detection of RNA Modifications and Structure Using Single-Molecule Nanopore Sequencing. Cell Genomics 2022, 2 (2), 100097, DOI: 10.1016/j.xgen.2022.100097There is no corresponding record for this reference.
- 11Gabrieli, T.; Sharim, H.; Nifker, G.; Jeffet, J.; Shahal, T.; Arielly, R.; Levi-Sakin, M.; Hoch, L.; Arbib, N.; Michaeli, Y.; Ebenstein, Y. Epigenetic Optical Mapping of 5-Hydroxymethylcytosine in Nanochannel Arrays. ACS Nano 2018, 12 (7), 7148– 7158, DOI: 10.1021/acsnano.8b03023There is no corresponding record for this reference.
- 12Fu, Y.; Timp, W.; Sedlazeck, F. J. Computational Analysis of DNA Methylation from Long-Read Sequencing. Nat. Rev. Genet. 2025, 26 (9), 620– 634, DOI: 10.1038/s41576-025-00822-5There is no corresponding record for this reference.
- 13Zirkin, S.; Fishman, S.; Sharim, H.; Michaeli, Y.; Don, J.; Ebenstein, Y. Lighting Up Individual DNA Damage Sites by In Vitro Repair Synthesis. J. Am. Chem. Soc. 2014, 136 (21), 7771– 7776, DOI: 10.1021/ja503677nThere is no corresponding record for this reference.
- 14Michaeli, Y.; Shahal, T.; Torchinsky, D.; Grunwald, A.; Hoch, R.; Ebenstein, Y. Optical Detection of Epigenetic Marks: Sensitive Quantification and Direct Imaging of Individual Hydroxymethylcytosine Bases. Chem. Commun. 2013, 49 (77), 8599– 8601, DOI: 10.1039/c3cc42543fThere is no corresponding record for this reference.
- 15Nifker, G.; Levy-Sakin, M.; Berkov-Zrihen, Y.; Shahal, T.; Gabrieli, T.; Fridman, M.; Ebenstein, Y. One-Pot Chemoenzymatic Cascade for Labeling of the Epigenetic Marker 5-Hydroxymethylcytosine. ChemBioChem 2015, 16 (13), 1857– 1860, DOI: 10.1002/cbic.201500329There is no corresponding record for this reference.
- 16Avraham, S.; Schütz, L.; Käver, L.; Dankers, A.; Margalit, S.; Michaeli, Y.; Zirkin, S.; Torchinsky, D.; Gilat, N.; Bahr, O.; Nifker, G.; Koren-Michowitz, M.; Weinhold, E.; Ebenstein, Y. Chemo-Enzymatic Fluorescence Labeling Of Genomic DNA For Simultaneous Detection Of Global 5-Methylcytosine And 5-Hydroxymethylcytosine. ChemBioChem 2023, 24 (20), e202300400 DOI: 10.1002/cbic.202300400There is no corresponding record for this reference.
- 17Neely, R. K.; Dedecker, P.; Hotta, J.-i.; Urbanavičiu̅tė, G.; Klimašauskas, S.; Hofkens, J. DNA Fluorocode: A Single Molecule, Optical Map of DNA with Nanometre Resolution. Chem. Sci. 2010, 1 (4), 453– 460, DOI: 10.1039/C0SC00277AThere is no corresponding record for this reference.
- 18Jin, X.; Hapsari, N. D.; Lee, S.; Jo, K. DNA Binding Fluorescent Proteins as Single-Molecule Probes. Analyst 2020, 145 (12), 4079– 4095, DOI: 10.1039/D0AN00218FThere is no corresponding record for this reference.
- 19McCaffrey, J.; Sibert, J.; Zhang, B.; Zhang, Y.; Hu, W.; Riethman, H.; Xiao, M. CRISPR-CAS9 D10A Nickase Target-Specific Fluorescent Labeling of Double Strand DNA for Whole Genome Mapping and Structural Variation Analysis. Nucleic Acids Res. 2016, 44 (2), e11 DOI: 10.1093/nar/gkv878There is no corresponding record for this reference.
- 20Grunwald, A.; Dahan, M.; Giesbertz, A.; Nilsson, A.; Nyberg, L. K.; Weinhold, E.; Ambjörnsson, T.; Westerlund, F.; Ebenstein, Y. Bacteriophage Strain Typing by Rapid Single Molecule Analysis. Nucleic Acids Res. 2015, 43 (18), e117 DOI: 10.1093/nar/gkv563There is no corresponding record for this reference.
- 21Gabrieli, T.; Michaeli, Y.; Avraham, S.; Torchinsky, D.; Margalit, S.; Schütz, L.; Juhasz, M.; Coruh, C.; Arbib, N.; Zhou, Z. S.; Law, J. A.; Weinhold, E.; Ebenstein, Y. Chemoenzymatic Labeling of DNA Methylation Patterns for Single-Molecule Epigenetic Mapping. Nucleic Acids Res. 2022, 50 (16), e92 DOI: 10.1093/nar/gkac460There is no corresponding record for this reference.
- 22Nifker, G.; Grunwald, A.; Margalit, S.; Tulpova, Z.; Michaeli, Y.; Har-Gil, H.; Maimon, N.; Roichman, E.; Schütz, L.; Weinhold, E.; Ebenstein, Y. Dam Assisted Fluorescent Tagging of Chromatin Accessibility (DAFCA) for Optical Genome Mapping in Nanochannel Arrays. ACS Nano 2023, 17 (10), 9178– 9187, DOI: 10.1021/acsnano.2c12755There is no corresponding record for this reference.
- 23Detinis Zur, T.; Margalit, S.; Jeffet, J.; Grunwald, A.; Fishman, S.; Tulpová, Z.; Michaeli, Y.; Deek, J.; Ebenstein, Y. Single-Molecule Toxicogenomics: Optical Genome Mapping of DNA-Damage in Nanochannel Arrays. DNA Repair 2025, 146, 103808, DOI: 10.1016/j.dnarep.2025.103808There is no corresponding record for this reference.
- 24Margalit, S.; Tulpová, Z.; Detinis Zur, T.; Michaeli, Y.; Deek, J.; Nifker, G.; Haldar, R.; Gnatek, Y.; Omer, D.; Dekel, B.; Baris Feldman, H.; Grunwald, A.; Ebenstein, Y. Long-Read Structural and Epigenetic Profiling of a Kidney Tumor-Matched Sample with Nanopore Sequencing and Optical Genome Mapping. NAR:Genomics Bioinf. 2025, 7 (1), lqae190, DOI: 10.1093/nargab/lqae190There is no corresponding record for this reference.
- 25Raseley, K.; Jinwala, Z.; Zhang, D.; Xiao, M. Single-Molecule Telomere Assay via Optical Mapping (SMTA-OM) Can Potentially Define the ALT Positivity of Cancer. Genes 2023, 14 (6), 1278, DOI: 10.3390/genes14061278There is no corresponding record for this reference.
- 26Müller, V.; Dvirnas, A.; Andersson, J.; Singh, V.; Kk, S.; Johansson, P.; Ebenstein, Y.; Ambjörnsson, T.; Westerlund, F. Enzyme-Free Optical DNA Mapping of the Human Genome Using Competitive Binding. Nucleic Acids Res. 2019, 47 (15), e89 DOI: 10.1093/nar/gkz489There is no corresponding record for this reference.
- 27Margalit, S.; Abramson, Y.; Sharim, H.; Manber, Z.; Bhattacharya, S.; Chen, Y.-W.; Vilain, E.; Barseghyan, H.; Elkon, R.; Sharan, R.; Ebenstein, Y. Long Reads Capture Simultaneous Enhancer–Promoter Methylation Status for Cell-Type Deconvolution. Bioinformatics 2021, 37 (Supplement_1), i327– i333, DOI: 10.1093/bioinformatics/btab306There is no corresponding record for this reference.
- 28Sharim, H.; Grunwald, A.; Gabrieli, T.; Michaeli, Y.; Margalit, S.; Torchinsky, D.; Arielly, R.; Nifker, G.; Juhasz, M.; Gularek, F.; Almalvez, M.; Dufault, B.; Chandra, S. S.; Liu, A.; Bhattacharya, S.; Chen, Y.-W.; Vilain, E.; Wagner, K. R.; Pevsner, J.; Reifenberger, J.; Lam, E. T.; Hastie, A. R.; Cao, H.; Barseghyan, H.; Weinhold, E.; Ebenstein, Y. Long-Read Single-Molecule Maps of the Functional Methylome. Genome Res. 2019, 29 (4), 646– 656, DOI: 10.1101/gr.240739.118There is no corresponding record for this reference.
- 29Kinney, S. M.; Chin, H. G.; Vaisvila, R.; Bitinaite, J.; Zheng, Y.; Estève, P.-O.; Feng, S.; Stroud, H.; Jacobsen, S. E.; Pradhan, S. Tissue-Specific Distribution and Dynamic Changes of 5-Hydroxymethylcytosine in Mammalian Genomes. J. Biol. Chem. 2011, 286 (28), 24685– 24693, DOI: 10.1074/jbc.M110.217083There is no corresponding record for this reference.
- 30Klimašauskas, S.; Weinhold, E. A New Tool for Biotechnology: AdoMet-Dependent Methyltransferases. Trends Biotechnol. 2007, 25 (3), 99– 104, DOI: 10.1016/j.tibtech.2007.01.006There is no corresponding record for this reference.
- 31Lukinavičius, G.; Tomkuvienė, M.; Masevičius, V.; Klimašauskas, S. Enhanced Chemical Stability of Adomet Analogues for Improved Methyltransferase-Directed Labeling of DNA. ACS Chem. Biol. 2013, 8 (6), 1134– 1139, DOI: 10.1021/cb300669xThere is no corresponding record for this reference.
- 32Al Temimi, A. H. K.; Martin, M.; Meng, Q.; Lenstra, D. C.; Qian, P.; Guo, H.; Weinhold, E.; Mecinović, J. Lysine Ethylation by Histone Lysine Methyltransferases. ChemBioChem 2020, 21 (3), 392– 400, DOI: 10.1002/cbic.201900359There is no corresponding record for this reference.
- 33Willnow, S.; Martin, M.; Lüscher, B.; Weinhold, E. A Selenium-Based Click AdoMet Analogue for Versatile Substrate Labeling with Wild-Type Protein Methyltransferases. ChemBioChem 2012, 13 (8), 1167– 1173, DOI: 10.1002/cbic.201100781There is no corresponding record for this reference.
- 34Holstein, J. M.; Stummer, D.; Rentmeister, A. Enzymatic Modification of 5′-Capped RNA with a 4-Vinylbenzyl Group Provides a Platform for Photoclick and Inverse Electron-Demand Diels–Alder Reaction. Chem. Sci. 2015, 6 (2), 1362– 1369, DOI: 10.1039/C4SC03182BThere is no corresponding record for this reference.
- 35Muttach, F.; Mäsing, F.; Studer, A.; Rentmeister, A. New AdoMet Analogues as Tools for Enzymatic Transfer of Photo-Cross-Linkers and Capturing RNA–Protein Interactions. Chem. - Eur. J. 2017, 23 (25), 5988– 5993, DOI: 10.1002/chem.201605663There is no corresponding record for this reference.
- 36Song, C.-X.; Szulwach, K. E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.-H.; Zhang, W.; Jian, X.; Wang, J.; Zhang, L.; Looney, T. J.; Zhang, B.; Godley, L. A.; Hicks, L. M.; Lahn, B. T.; Jin, P.; He, C. Selective Chemical Labeling Reveals the Genome-Wide Distribution of 5-Hydroxymethylcytosine. Nat. Biotechnol. 2011, 29 (1), 68– 72, DOI: 10.1038/nbt.1732There is no corresponding record for this reference.
- 37Dalhoff, C.; Lukinavicius, G.; Klimasăuskas, S.; Weinhold, E. Direct Transfer of Extended Groups from Synthetic Cofactors by DNA Methyltransferases. Nat. Chem. Biol. 2006, 2 (1), 31– 32, DOI: 10.1038/nchembio754There is no corresponding record for this reference.
- 38Josse, J.; Kornberg, A. Glucosylation of Deoxyribonucleic Acid: III. α- AND β-GLUCOSYL TRANSFERASES FROM T4-INFECTED ESCHERICHIA COLI. J. Biol. Chem. 1962, 237 (6), 1968– 1976, DOI: 10.1016/S0021-9258(19)73968-4There is no corresponding record for this reference.
- 39Pastor, W. A.; Huang, Y.; Henderson, H. R.; Agarwal, S.; Rao, A. The GLIB Technique for Genome-Wide Mapping of 5-Hydroxymethylcytosine. Nat. Protoc. 2012, 7 (10), 1909– 1917, DOI: 10.1038/nprot.2012.104There is no corresponding record for this reference.
- 40Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K. F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39 (11), 1348– 1365, DOI: 10.1038/s41587-021-01108-xThere is no corresponding record for this reference.
- 41Sanger, F.; Coulson, A. R.; Hong, G. F.; Hill, D. F.; Petersen, G. B. Nucleotide Sequence of Bacteriophage λ DNA. J. Mol. Biol. 1982, 162 (4), 729– 773, DOI: 10.1016/0022-2836(82)90546-0There is no corresponding record for this reference.
- 42Enterobacteria Phage Lambda, Complete Genome, 2023. http://www.ncbi.nlm.nih.gov/nuccore/NC_001416.1 (accessed June 24, 2025).There is no corresponding record for this reference.
- 43Laszlo, A. H.; Derrington, I. M.; Brinkerhoff, H.; Langford, K. W.; Nova, I. C.; Samson, J. M.; Bartlett, J. J.; Pavlenok, M.; Gundlach, J. H. Detection and Mapping of 5-Methylcytosine and 5-Hydroxymethylcytosine with Nanopore MspA. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (47), 18904– 18909, DOI: 10.1073/pnas.1310240110There is no corresponding record for this reference.
- 44Yao, B.; Hsu, C.; Goldner, G.; Michaeli, Y.; Ebenstein, Y.; Listgarten, J. Effective Training of Nanopore Callers for Epigenetic Marks with Limited Labelled Data. Open Biol. 2024, 14 (6), 230449, DOI: 10.1098/rsob.230449There is no corresponding record for this reference.
- 45Kriukienė, E.; Labrie, V.; Khare, T.; Urbanavičiu̅tė, G.; Lapinaitė, A.; Koncevičius, K.; Li, D.; Wang, T.; Pai, S.; Ptak, C.; Gordevičius, J.; Wang, S.-C.; Petronis, A.; Klimašauskas, S. DNA Unmethylome Profiling by Covalent Capture of CpG Sites. Nat. Commun. 2013, 4 (1), 2190, DOI: 10.1038/ncomms3190There is no corresponding record for this reference.
- 46Klimasauskas, S.; Weinhold, E. A New Tool for Biotechnology: AdoMet-Dependent Methyltransferases. Trends Biotechnol. 2007, 25 (3), 99– 104, DOI: 10.1016/j.tibtech.2007.01.006There is no corresponding record for this reference.
- 47Hanz, G. M.; Jung, B.; Giesbertz, A.; Juhasz, M.; Weinhold, E. Sequence-Specific Labeling of Nucleic Acids and Proteins with Methyltransferases and Cofactor Analogues. J. Visualized Exp. 2014, 93, e52014 DOI: 10.3791/52014There is no corresponding record for this reference.
- 48Weng, Z.; Ruan, F.; Chen, W.; Chen, Z.; Xie, Y.; Luo, M.; Xie, Z.; Zhang, C.; Wang, J.; Sun, Y.; Fang, Y.; Guo, M.; Tan, C.; Chen, W.; Tong, Y.; Li, Y.; Wang, H.; Tang, C. BIND&MODIFY: A Long-Range Method for Single-Molecule Mapping of Chromatin Modifications in Eukaryotes. Genome Biol. 2023, 24 (1), 61, DOI: 10.1186/s13059-023-02896-yThere is no corresponding record for this reference.
- 49Thatavarty, A.; Sagy, N.; Erdos, M. R.; Lee, I.; Simpson, J. T.; Timp, W.; Collins, F. S.; Bar, D. Z. Detecting Protein-DNA Binding in Single Molecules Using Antibody Guided Methylation. Epigenet. Chromatin 2025, 18 (1), 39, DOI: 10.1186/s13072-025-00602-9There is no corresponding record for this reference.
- 50Goedecke, K.; Pignot, M.; Goody, R. S.; Scheidig, A. J.; Weinhold, E. Structure of the N6-Adenine DNA Methyltransferase M•TaqI in Complex with DNA and a Cofactor Analog. Nat. Struct. Biol. 2001, 8 (2), 121– 125, DOI: 10.1038/84104There is no corresponding record for this reference.
- 51Shahal, T.; Gilat, N.; Michaeli, Y.; Redy-Keisar, O.; Shabat, D.; Ebenstein, Y. Spectroscopic Quantification of 5-Hydroxymethylcytosine in Genomic DNA. Anal. Chem. 2014, 86 (16), 8231– 8237, DOI: 10.1021/ac501609dThere is no corresponding record for this reference.
- 52Products Scheduled to Be Discontinued; Oxford Nanopore Technologies. https://nanoporetech.com/products/prepare/discontinued-kits (accessed 2025–02–06).There is no corresponding record for this reference.
- 53Sanderson, N. D.; Kapel, N.; Rodger, G.; Webster, H.; Lipworth, S.; Street, T. L.; Peto, T.; Crook, D.; Stoesser, N. Comparison of R9.4.1/Kit10 and R10/Kit12 Oxford Nanopore Flowcells and Chemistries in Bacterial Genome Reconstruction. Microb. Genomics 2023, 9 (1), 000910, DOI: 10.1099/mgen.0.000910There is no corresponding record for this reference.
- 54Ni, Y.; Liu, X.; Simeneh, Z. M.; Yang, M.; Li, R. Benchmarking of Nanopore R10.4 and R9.4.1 Flow Cells in Single-Cell Whole-Genome Amplification and Whole-Genome Shotgun Sequencing. Comput. Struct. Biotechnol. J. 2023, 21, 2352– 2364, DOI: 10.1016/j.csbj.2023.03.038There is no corresponding record for this reference.
- 55Jung, C.; Hawkins, J. A.; Jones, S. K.; Xiao, Y.; Rybarski, J. R.; Dillard, K. E.; Hussmann, J.; Saifuddin, F. A.; Savran, C. A.; Ellington, A. D.; Ke, A.; Press, W. H.; Finkelstein, I. J. Massively Parallel Biophysical Analysis of CRISPR-Cas Complexes on Next Generation Sequencing Chips. Cell 2017, 170 (1), 35– 47, DOI: 10.1016/j.cell.2017.05.044There is no corresponding record for this reference.
- 56Escherichia Coli Str. K-12 Substr. MG1655, Complete Genome, 2022. http://www.ncbi.nlm.nih.gov/nuccore/NC_000913.3 (accessed 2025–03–10).There is no corresponding record for this reference.
- 57ONT Fast5 API. https://github.com/nanoporetech/ont_fast5_api (accessed 2024–11–14).There is no corresponding record for this reference.
- 58Tombo, 2024. https://github.com/nanoporetech/tombo (accessed 2024–11–14).There is no corresponding record for this reference.
- 59Loman, N. J.; Quick, J.; Simpson, J. T. A Complete Bacterial Genome Assembled de Novo Using Only Nanopore Sequencing Data. Nat. Methods 2015, 12 (8), 733– 735, DOI: 10.1038/nmeth.3444There is no corresponding record for this reference.
- 60Bertocchi, U.; Grunwald, A.; Goldner, G.; Eitan, E.; Avraham, S.; Deek, J.; Michaeli, Y.; Yao, B.; Listgarten, J.; Simpson, J. T.; Timp, W.; Ebenstein, Y. Write and Read: Harnessing Synthetic DNA Modifications for Nanopore Sequencing. bioRxiv 2025, 2025.04.14.648727, DOI: 10.1101/2025.04.14.648727There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.5c12530.
Supplementary Figures (S1–S10) Tables (S1–S8) additional methods statistical details raw nanopore current traces for cytosine and adenine modifications analysis of nonassigned events PCR and labeling conditions and sequencing yield summaries (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.