



This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
C–C Bond Formation via Direct Functionalization of Indolizines with a Bichromophoric Ruthenium PhotocatalystClick to copy article linkArticle link copied!
- Kevin Klaus StefanoniKevin Klaus StefanoniInstitute of Organic Chemistry, Clausthal University of Technology, Leibnizstr. 6, Clausthal-Zellerfeld 38678, GermanyMore by Kevin Klaus Stefanoni
- René Wilhelm*René Wilhelm*E-mail: [email protected]Institute of Organic Chemistry, Clausthal University of Technology, Leibnizstr. 6, Clausthal-Zellerfeld 38678, GermanyMore by René Wilhelm
ACS Organic & Inorganic Au
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
An unprecedented photocatalyzed radical C(sp2)–C(sp3) alkylation protocol to prepare a range of substituted 3-alkylated indolizine derivatives, mediated by 2-mercaptothiazolidinium salts as radical sources and a new dyad-like Ruthenium complex as a photoredox catalyst, under green light irradiation, resulted in yields of up to 99%. The mild, robust, and chemoselective procedure employs inexpensive, air-insensitive, and readily accessible reagents, enabling convenient synthesis of the substituted indolizines. Moreover, different N-heteroarenes, such as 1H-indoles and 2H-indazoles, were successfully alkylated under the optimized conditions. The resulting alkylated products are scaffolds with significance for drug design in medicinal chemistry.
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Introduction
Figure 1
Figure 1. Selected known carbon-centered radical precursors for photoredox SET processes.
Scheme 1
Results and Discussion

| Entry | Photoredox Catalyst | Radical Source eq | Time | LEDs | Yielda (2a: 2a′: 1a) |
|---|---|---|---|---|---|
| 1 | PC1 | 1.5 | 3 h | green (15 W, 515 nm) | 67%: 18%: 0% |
| 2 | PC2 | 1.5 | 3 h | green (15 W, 515 nm) | 70%: 16%: 0% |
| 3 | PC3 | 1.5 | 3 h | green (15 W, 515 nm) | 68%: 17%: 0% |
| 4 | PC4 | 1.5 | 3 h | green (15 W, 515 nm) | 60%: 20%: 0% |
| 5 | [Ru(bpy)3](PF6)2 | 1.5 | 3 h | green (15 W, 515 nm) | 47%: 0%: 50% |
| 6 | [Ru(phen)3](PF6)2 | 1.5 | 3 h | green (15 W, 515 nm) | 43%: 0%: 57% |
| 7 | [Ru(dpp)3](PF6)2 | 1.5 | 3 h | green (15 W, 515 nm) | 52%: 19%: 0% |
| 8 | Ir(ppy)3 | 1.5 | 3 h | green (15 W, 515 nm) | 59%: 23%: 0% |
| 9 | 4-CzIPN | 1.5 | 3 h | green (15 W, 515 nm) | 30%: 0%: 70% |
| 10 | – | 1.5 | 3 h | green (15 W, 515 nm) | 0%: 0%: 100% |
| 11 | PC2 | 1.1 | 3 h | green (15 W, 515 nm) | 69%: 8%: 13% |
| 12 | PC2 | 1.25 | 3 h | green (15 W, 515 nm) | 70%: 9%: 5% |
| 13 | PC2 | 1.5 | 1 h | green (15 W, 515 nm) | 67%: 9%: 9% |
| 14 | PC2 | 1.5 | 2 h | green (15 W, 515 nm) | 70%: 12%: 0% (isolated) |
| 15 | PC2 | 1.5 | 16 h | green (3 W, 515 nm) | 19%: 0%: 81% |
Yields were determined by 1H NMR using dibromomethane as the internal standard, unless otherwise stated.
Values for the excited-state redox potentials in V Vs SCE were obtained according to the literature. (32)
Scheme 2
Scheme 3
Scheme 4
Scheme 5
Scheme 6
Scheme 7
Scheme 8
Scheme 9
Scheme 10
Conclusion
Experimental Section
General Experimental
General Procedure for the Photoredox Experiments
General Procedure A
General Procedure B
General Procedure C
General Procedure D
Data Availability
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.5c00110.
Full experimental description, spectral data of the compounds, and additional spectroscopy data (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- René Wilhelm - Institute of Organic Chemistry, Clausthal University of Technology, Leibnizstr. 6, Clausthal-Zellerfeld 38678, Germany;
 https://orcid.org/0000-0003-3856-2757;
https://orcid.org/0000-0003-3856-2757;
- Kevin Klaus Stefanoni - Institute of Organic Chemistry, Clausthal University of Technology, Leibnizstr. 6, Clausthal-Zellerfeld 38678, Germany;https://orcid.org/0009-0002-3212-4019
K.K.S. performed the radical C(sp2)–C(sp3) alkylation of indolizines and synthesized and characterized the Ru(II) complexes (i.e., PC1–4). R.W. supervised each part of the project and wrote the manuscript with contributions from all the authors. All authors have given approval of the final version of the manuscript.
References
This article references 51 other publications.
- 1Sharma, V.; Kumar, V. Indolizine: A Biologically Active Moiety. Med. Chem. Res. 2014, 23 (8), 3593– 3606, DOI: 10.1007/s00044-014-0940-1Google ScholarThere is no corresponding record for this reference.
- 2Singh, G. S.; Mmatli, E. E. Recent Progress in Synthesis and Bioactivity Studies of Indolizines. Eur. J. Med. Chem. 2011, 46 (11), 5237– 5257, DOI: 10.1016/j.ejmech.2011.08.042Google ScholarThere is no corresponding record for this reference.
- 3Harrell, W. B.; Doerge, R. F. Mannich Bases from 2-Phenylindolizines I: 3-Alkyl-1-Dialkylaminomethyl Derivatives. J. Pharm. Sci. 1967, 56 (2), 225– 228, DOI: 10.1002/jps.2600560215Google ScholarThere is no corresponding record for this reference.
- 4Badaro, J. S. A.; Godlewski, B.; Gryko, D. T. Advances in the Synthesis of Indolizines and Their π-Expanded Analogues: Update 2016–2024. Org. Chem. Front. 2025, 12 (8), 2860– 2907, DOI: 10.1039/D4QO02082KGoogle ScholarThere is no corresponding record for this reference.
- 5Priyanka; Rani, P.; Kiran; Sindhu, J. Indolizine: A Promising Framework for Developing a Diverse Array of C–H Functionalized Hybrids. ChemistrySelect 2023, 8 (1), e202203531 DOI: 10.1002/slct.202203531Google ScholarThere is no corresponding record for this reference.
- 6Nevskaya, A. A.; Zinoveva, A. D.; Van der Eycken, E. V.; Voskressensky, L. G. Synthetic Strategies for the Construction of Indolizines and Indolizine-Fused Compounds: Thienoindolizines and Indolizinoindoles. Asian J. Org. Chem. 2023, 12 (10), e202300359 DOI: 10.1002/ajoc.202300359Google ScholarThere is no corresponding record for this reference.
- 7Hui, J.; Ma, Y.; Zhao, J.; Cao, H. Recent Advances in the Synthesis of Indolizine and Its Derivatives by Radical Cyclization/Cross-Coupling. Org. Biomol. Chem. 2021, 19 (47), 10245– 10258, DOI: 10.1039/D1OB01431EGoogle ScholarThere is no corresponding record for this reference.
- 8Sadowski, B.; Klajn, J.; Gryko, D. T. Recent Advances in the Synthesis of Indolizines and Their π-Expanded Analogues. Org. Biomol. Chem. 2016, 14 (33), 7804– 7828, DOI: 10.1039/C6OB00985AGoogle ScholarThere is no corresponding record for this reference.
- 9Botezatu, A. V.; Furdui, B.; Busuioc, A.; Dinică, R. M. Recent Developments in the Synthesis of Indolizines and Their Derivatives as Compounds of Interest in Medicinal Chemistry: A Review. Eur. J. Med. Chem. 2025, 297, 117908, DOI: 10.1016/j.ejmech.2025.117908Google ScholarThere is no corresponding record for this reference.
- 10Cho, S. H.; Kim, J. Y.; Kwak, J.; Chang, S. Recent Advances in the Transition Metal-Catalyzed Twofold Oxidative C–H Bond Activation Strategy for C–C and C–N Bond Formation. Chem. Soc. Rev. 2011, 40 (10), 5068– 5083, DOI: 10.1039/c1cs15082kGoogle ScholarThere is no corresponding record for this reference.
- 11Dalton, T.; Faber, T.; Glorius, F. C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7 (2), 245– 261, DOI: 10.1021/acscentsci.0c01413Google ScholarThere is no corresponding record for this reference.
- 12Wencel-Delord, J.; Glorius, F. C–H Bond Activation Enables the Rapid Construction and Late-Stage Diversification of Functional Molecules. Nat. Chem. 2013, 5 (5), 369– 375, DOI: 10.1038/nchem.1607Google ScholarThere is no corresponding record for this reference.
- 13Bellotti, P.; Huang, H.-M.; Faber, T.; Glorius, F. Photocatalytic Late-Stage C–H Functionalization. Chem. Rev. 2023, 123 (8), 4237– 4352, DOI: 10.1021/acs.chemrev.2c00478Google ScholarThere is no corresponding record for this reference.
- 14Holmberg-Douglas, N.; Nicewicz, D. A. Photoredox-Catalyzed C–H Functionalization Reactions. Chem. Rev. 2022, 122 (2), 1925– 2016, DOI: 10.1021/acs.chemrev.1c00311Google ScholarThere is no corresponding record for this reference.
- 15Wang, C.-S.; Dixneuf, P. H.; Soulé, J.-F. Photoredox Catalysis for Building C–C Bonds from C(Sp2)–H Bonds. Chem. Rev. 2018, 118 (16), 7532– 7585, DOI: 10.1021/acs.chemrev.8b00077Google ScholarThere is no corresponding record for this reference.
- 16Xie, J.; Jin, H.; Xu, P.; Zhu, C. When C–H Bond Functionalization Meets Visible-Light Photoredox Catalysis. Tetrahedron Lett. 2014, 55 (1), 36– 48, DOI: 10.1016/j.tetlet.2013.10.090Google ScholarThere is no corresponding record for this reference.
- 17Correia, J. T. M.; Fernandes, V. A.; Matsuo, B. T.; Delgado, J. A. C.; Souza, W. C. D.; Paixão, M. W. Photoinduced Deaminative Strategies: Katritzky Salts as Alkyl Radical Precursors. Chem. Commun. 2020, 56 (4), 503– 514, DOI: 10.1039/C9CC08348KGoogle ScholarThere is no corresponding record for this reference.
- 18Gutiérrez-Bonet, Á.; Tellis, J. C.; Matsui, J. K.; Vara, B. A.; Molander, G. A. 1,4-Dihydropyridines as Alkyl Radical Precursors: Introducing the Aldehyde Feedstock to Nickel/Photoredox Dual Catalysis. ACS Catal. 2016, 6 (12), 8004– 8008, DOI: 10.1021/acscatal.6b02786Google ScholarThere is no corresponding record for this reference.
- 19Liu, Y.; Zhou, J.; Sun, Z. Direct Synthesis of Unnatural Amino Acids and Modifications of Peptides via LADA Strategy. Chin. Chem. Lett. 2024, 35 (1), 108553, DOI: 10.1016/j.cclet.2023.108553Google ScholarThere is no corresponding record for this reference.
- 20Reed, M. A.; Noden, M. Using Bench-Stable 2-Mercaptothiazolinium Salts for the Photoredox Alkylation of N-Heteroarenes. Synfacts 2024, 20, 1020, DOI: 10.1055/s-0043-1775358Google ScholarThere is no corresponding record for this reference.
- 21Geniller, L.; Taillefer, M.; Jaroschik, F.; Prieto, A. Photo-Induced Desulfurative Processes for Carbon Radical Generation. ChemCatchem 2023, 15 (20), e202300808 DOI: 10.1002/cctc.202300808Google ScholarThere is no corresponding record for this reference.
- 22Zemtsov, A. A.; Ashirbaev, S. S.; Levin, V. V.; Kokorekin, V. A.; Korlyukov, A. A.; Dilman, A. D. Photoredox Reaction of 2-Mercaptothiazolinium Salts with Silyl Enol Ethers. J. Org. Chem. 2019, 84 (23), 15745– 15753, DOI: 10.1021/acs.joc.9b02478Google ScholarThere is no corresponding record for this reference.
- 23Tian, Y.; Fu, X.-X.; Cheng, R.-M.; Feng, J.; Shen, M.-H.; Zhu, C.-F.; Xu, H.-D. Visible-Light-Mediated Alkylation of N-Heteroarenes Using 2-Mercaptothiazolinium Salts as Alkyl Radical Source. Eur. J. Org. Chem. 2024, 27, e202400344 DOI: 10.1002/ejoc.202400344Google ScholarThere is no corresponding record for this reference.
- 24Zhu, C.-F.; Li, F.; Mai, J.-J.; Li, X.-J.; Dong, X.; Shi, M.; Shen, M.-H.; Xu, H.-D. Visible-light-promoted synthesis of alkylated Indolo[2,1-α]isoquinolines using 2-Mercaptothiazolinium salts as alkyl radical source. Tetrahedron Lett. 2025, 154, 155397, DOI: 10.1016/j.tetlet.2024.155397Google ScholarThere is no corresponding record for this reference.
- 25Neuba, A.; Ortmeyer, J.; Konieczna, D. D.; Weigel, G.; Flörke, U.; Henkel, G.; Wilhelm, R. Synthesis of New Copper(i) Based Linear 1-D-Coordination Polymers with Neutral Imidazolinium-Dithiocarboxylate Ligands. RSC Adv. 2015, 5 (12), 9217– 9220, DOI: 10.1039/C4RA09033KGoogle ScholarThere is no corresponding record for this reference.
- 26Rosenthal, M.; Lindner, J. K. N.; Gerstmann, U.; Meier, A.; Schmidt, W. G.; Wilhelm, R. A Photoredox Catalysed Heck Reaction via Hole Transfer from a Ru(ii)-Bis(Terpyridine) Complex to Graphene Oxide. RSC Adv. 2020, 10 (70), 42930– 42937, DOI: 10.1039/D0RA08749AGoogle ScholarThere is no corresponding record for this reference.
- 27Rosenthal, M.; Biktagirov, T.; Schmidt, W. G.; Wilhelm, R. Synthesis of New Graphene Oxide/TiO2 and TiO2/SiO2 Nanocomposites and Their Evaluation as Photocatalysts. Catal. Sci. Technol. 2023, 13 (15), 4367– 4377, DOI: 10.1039/D3CY00461AGoogle ScholarThere is no corresponding record for this reference.
- 28Konieczna, D. D.; Biller, H.; Witte, M.; Schmidt, W. G.; Neuba, A.; Wilhelm, R. New Pyridinium Based Ionic Dyes for the Hydrogen Evolution Reaction. Tetrahedron 2018, 74 (1), 142– 149, DOI: 10.1016/j.tet.2017.11.053Google ScholarThere is no corresponding record for this reference.
- 29Stefanoni, K. K.; Wilhelm, R. Access to SCF3-Substituted Indolizines via a Photocatalytic Late-Stage Functionalization Protocol. Org. Lett. 2025, 27 (31), 8389– 8393, DOI: 10.1021/acs.orglett.5c02079Google ScholarThere is no corresponding record for this reference.
- 30Stefanoni, K. K.; Schmitz, M.; Treuheit, J.; Kerzig, C.; Wilhelm, R. Bichromophoric Ruthenium Complexes for Photocatalyzed Late-Stage Synthesis of Trifluoromethylated Indolizines. J. Org. Chem. 2025, 90 (19), 6491– 6503, DOI: 10.1021/acs.joc.5c00319Google ScholarThere is no corresponding record for this reference.
- 31Meier, A.; Badalov, S. V.; Biktagirov, T.; Schmidt, W. G.; Wilhelm, R. Diquat Based Dyes: A New Class of Photoredox Catalysts and Their Use in Aerobic Thiocyanation. Chem. – Eur. J. 2023, 29 (22), e202203541 DOI: 10.1002/chem.202203541Google ScholarThere is no corresponding record for this reference.
- 32Buzzetti, L.; Crisenza, G. E. M.; Melchiorre, P. Mechanistic Studies in Photocatalysis. Angew. Chem., Int. Ed. 2019, 58 (12), 3730– 3747, DOI: 10.1002/anie.201809984Google ScholarThere is no corresponding record for this reference.
- 33Baggott, M. J. Indolizine Compounds for the Treatment of Mental Disorders or Inflammation. WO 2,023,183,613 A2, 2023.Google ScholarThere is no corresponding record for this reference.
- 34Antonini, I.; Claudi, F.; Gulini, U.; Micossi, L.; Venturi, F. Indolizine Derivatives with Biological Activity IV: 3-(2-Aminoethyl)-2-Methylindolizine, 3-(2-Aminoethyl)-2-Methyl-5,6,7,8-Tetrahydroindolizine, and Their N-Alkyl Derivatives. J. Pharm. Sci. 1979, 68 (3), 321– 324, DOI: 10.1002/jps.2600680317Google ScholarThere is no corresponding record for this reference.
- 35Hansch, C.; Leo, A.; Taft, R. W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91 (2), 165– 195, DOI: 10.1021/cr00002a004Google ScholarThere is no corresponding record for this reference.
- 36Almirante, L.; Polo, L.; Mugnaini, A.; Provinciali, E.; Rugarli, P.; Biancotti, A.; Gamba, A.; Murmann, W. Derivatives of Imidazole. I. Synthesis and Reactions of Imidazo[1,2-α]Pyridines with Analgesic, Antiinflammatory, Antipyretic, and Anticonvulsant Activity. J. Med. Chem. 1965, 8 (3), 305– 312, DOI: 10.1021/jm00327a007Google ScholarThere is no corresponding record for this reference.
- 37Singh, D. K.; Kim, S.; Lee, J. H.; Lee, N. K.; Kim, J.; Lee, J.; Kim, I. 6-(Hetero)Arylindolizino[1,2-]Quinolines as Highly Fluorescent Chemical Space: Synthesis and Photophysical Properties. J. Heterocycl. Chem. 2020, 57 (8), 3018– 3028, DOI: 10.1002/jhet.4000Google ScholarThere is no corresponding record for this reference.
- 38Haddadin, M. J.; Conrad, W. E.; Kurth, M. J. The Davis-Beirut Reaction: A Novel Entry into 2H-Indazoles and Indazolones. Recent Biological Activity of Indazoles. Mini-Rev. Med. Chem. 2012, 12 (12), 1293– 1300, DOI: 10.2174/138955712802762059Google ScholarThere is no corresponding record for this reference.
- 39Lee, J. C.; Cuthbertson, J. D.; Mitchell, N. J. Chemoselective Late-Stage Functionalization of Peptides via Photocatalytic C2-Alkylation of Tryptophan. Org. Lett. 2023, 25 (29), 5459– 5464, DOI: 10.1021/acs.orglett.3c01795Google ScholarThere is no corresponding record for this reference.
- 40Enguehard-Gueiffier, C.; Gueiffier, A. Recent Progress in the Pharmacology of Imidazo[1,2-a]Pyridines. Mini-Rev. Med. Chem. 2007, 7 (9), 888– 899, DOI: 10.2174/138955707781662645Google ScholarThere is no corresponding record for this reference.
- 41Sumalatha, Y.; Reddy, T. R.; Reddy, P. P.; Satyanarayana, B. A. Simple Efficient and Scalable Synthesis of Hypnotic Agent, Zolpidem; ARKIVOC, 2009.Google ScholarThere is no corresponding record for this reference.
- 42Shrivastava, S. K.; Srivastava, P.; Bandresh, R.; Tripathi, P. N.; Tripathi, A. Design Synthesis, and Biological Evaluation of Some Novel Indolizine Derivatives as Dual Cyclooxygenase and Lipoxygenase Inhibitor for Anti-Inflammatory Activity. Bioorg. Med. Chem. 2017, 25 (16), 4424– 4432, DOI: 10.1016/j.bmc.2017.06.027Google ScholarThere is no corresponding record for this reference.
- 43Wu, Z.; Lu, Y.; Li, L.; Zhao, R.; Wang, B.; Lv, K.; Liu, M.; You, X. Identification of N-(2-Phenoxyethyl)Imidazo[1,2-a]Pyridine-3-Carboxamides as New Antituberculosis Agents. ACS Med. Chem. Lett. 2016, 7 (12), 1130– 1133, DOI: 10.1021/acsmedchemlett.6b00330Google ScholarThere is no corresponding record for this reference.
- 44Jose, G.; Suresha Kumara, T. H.; Nagendrappa, G.; Sowmya, H. B. V.; Sriram, D.; Yogeeswari, P.; Sridevi, J. P.; Guru Row, T. N.; Hosamani, A. A.; Sujan Ganapathy, P. S.; Chandrika, N.; Narendra, L. V. Synthesis Molecular Docking and Anti-Mycobacterial Evaluation of New Imidazo[1,2-a]Pyridine-2-Carboxamide Derivatives. Eur. J. Med. Chem. 2015, 89, 616– 627, DOI: 10.1016/j.ejmech.2014.10.079Google ScholarThere is no corresponding record for this reference.
- 45Khetmalis, Y. M.; Chitti, S.; Wunnava, A. U.; Kumar, B. K.; Kumar, M. M. K.; Murugesan, S.; Sekhar, K. V. G. C. Design Synthesis and Anti-Mycobacterial Evaluation of Imidazo[1,2-a]Pyridine Analogues. RSC Med. Chem. 2022, 13 (3), 327– 342, DOI: 10.1039/D1MD00367DGoogle ScholarThere is no corresponding record for this reference.
- 46Nahm, S.; Weinreb, S. M. N-Methoxy-n-Methylamides as Effective Acylating Agents. Tetrahedron Lett. 1981, 22 (39), 3815– 3818, DOI: 10.1016/S0040-4039(01)91316-4Google ScholarThere is no corresponding record for this reference.
- 47Kaur, H.; Karabulut, S.; Gauld, J. W.; Fagot, S. A.; Holloway, K. N.; Shaw, H. E.; Fantegrossi, W. E. Balancing Therapeutic Efficacy and Safety of MDMA and Novel MDXX Analogues as Novel Treatments for Autism Spectrum Disorder. Psychedelic Med. 2023, 1 (3), 166– 185, DOI: 10.1089/psymed.2023.0023Google ScholarThere is no corresponding record for this reference.
- 48Oeri, H. E. Beyond Ecstasy: Alternative Entactogens to 3,4-Methylenedioxymethamphetamine with Potential Applications in Psychotherapy. J. Psychopharmacol. 2021, 35 (5), 512– 536, DOI: 10.1177/0269881120920420Google ScholarThere is no corresponding record for this reference.
- 49He, X.; Chen, Z.; Zhu, X.; Liu, H.; Chen, Y.; Sun, Z.; Chu, W. Photoredox-Catalyzed Trifluoromethylation of 2H-Indazoles Using TT-CF3+OTf– in Ionic Liquids. Org. Biomol. Chem. 2023, 21 (8), 1814– 1820, DOI: 10.1039/D3OB00096FGoogle ScholarThere is no corresponding record for this reference.
- 50Jia, H.; Häring, A. P.; Berger, F.; Zhang, L.; Ritter, T. Trifluoromethyl Thianthrenium Triflate: A Readily Available Trifluoromethylating Reagent with Formal CF3+, CF3•, and CF3– Reactivity. J. Am. Chem. Soc. 2021, 143 (20), 7623– 7628, DOI: 10.1021/jacs.1c02606Google ScholarThere is no corresponding record for this reference.
- 51Malpani, Y. R.; Biswas, B. K.; Han, H. S.; Jung, Y.-S.; Han, S. B. Multicomponent Oxidative Trifluoromethylation of Alkynes with Photoredox Catalysis: Synthesis of α-Trifluoromethyl Ketones. Org. Lett. 2018, 20 (7), 1693– 1697, DOI: 10.1021/acs.orglett.8b00410Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACS Organic & Inorganic Au
Copyright © 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract

Figure 1

Figure 1. Selected known carbon-centered radical precursors for photoredox SET processes.
Scheme 1
 Scheme 1. 2-Mercaptothiazolidinium Salts as Alkyl Radical Sources
Scheme 1. 2-Mercaptothiazolidinium Salts as Alkyl Radical SourcesScheme 2
 Scheme 2. Radical Sources Substrate Scope for the Direct Radical Alkylation
Scheme 2. Radical Sources Substrate Scope for the Direct Radical AlkylationScheme 3
 Scheme 3. Indolizines Substrate Scope for the Direct Radical Alkylation
Scheme 3. Indolizines Substrate Scope for the Direct Radical AlkylationScheme 4
 Scheme 4. Substrate Scope of N-Heteroarenes
Scheme 4. Substrate Scope of N-HeteroarenesScheme 5
 Scheme 5. Reaction Scale-Up
Scheme 5. Reaction Scale-UpScheme 6
 Scheme 6. One-Step Synthesis of Zolpidem Bioisostere
Scheme 6. One-Step Synthesis of Zolpidem BioisostereScheme 7
 Scheme 7. Synthesis of a Precursor for an Anti-TB Agents Bioisostere from 2o
Scheme 7. Synthesis of a Precursor for an Anti-TB Agents Bioisostere from 2oScheme 8
 Scheme 8. Derivatization of Indolizine 2a
Scheme 8. Derivatization of Indolizine 2aScheme 9
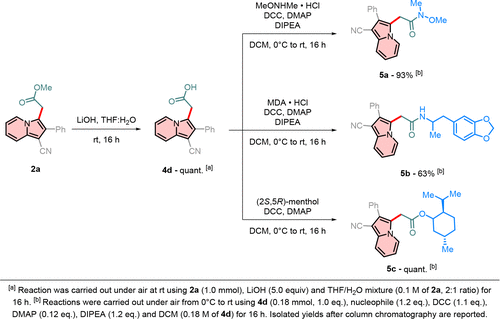 Scheme 9. Derivatization of the Carboxylate Moiety
Scheme 9. Derivatization of the Carboxylate MoietyScheme 10
 Scheme 10. Proposed Reaction Mechanism
Scheme 10. Proposed Reaction MechanismReferences
This article references 51 other publications.
- 1Sharma, V.; Kumar, V. Indolizine: A Biologically Active Moiety. Med. Chem. Res. 2014, 23 (8), 3593– 3606, DOI: 10.1007/s00044-014-0940-1There is no corresponding record for this reference.
- 2Singh, G. S.; Mmatli, E. E. Recent Progress in Synthesis and Bioactivity Studies of Indolizines. Eur. J. Med. Chem. 2011, 46 (11), 5237– 5257, DOI: 10.1016/j.ejmech.2011.08.042There is no corresponding record for this reference.
- 3Harrell, W. B.; Doerge, R. F. Mannich Bases from 2-Phenylindolizines I: 3-Alkyl-1-Dialkylaminomethyl Derivatives. J. Pharm. Sci. 1967, 56 (2), 225– 228, DOI: 10.1002/jps.2600560215There is no corresponding record for this reference.
- 4Badaro, J. S. A.; Godlewski, B.; Gryko, D. T. Advances in the Synthesis of Indolizines and Their π-Expanded Analogues: Update 2016–2024. Org. Chem. Front. 2025, 12 (8), 2860– 2907, DOI: 10.1039/D4QO02082KThere is no corresponding record for this reference.
- 5Priyanka; Rani, P.; Kiran; Sindhu, J. Indolizine: A Promising Framework for Developing a Diverse Array of C–H Functionalized Hybrids. ChemistrySelect 2023, 8 (1), e202203531 DOI: 10.1002/slct.202203531There is no corresponding record for this reference.
- 6Nevskaya, A. A.; Zinoveva, A. D.; Van der Eycken, E. V.; Voskressensky, L. G. Synthetic Strategies for the Construction of Indolizines and Indolizine-Fused Compounds: Thienoindolizines and Indolizinoindoles. Asian J. Org. Chem. 2023, 12 (10), e202300359 DOI: 10.1002/ajoc.202300359There is no corresponding record for this reference.
- 7Hui, J.; Ma, Y.; Zhao, J.; Cao, H. Recent Advances in the Synthesis of Indolizine and Its Derivatives by Radical Cyclization/Cross-Coupling. Org. Biomol. Chem. 2021, 19 (47), 10245– 10258, DOI: 10.1039/D1OB01431EThere is no corresponding record for this reference.
- 8Sadowski, B.; Klajn, J.; Gryko, D. T. Recent Advances in the Synthesis of Indolizines and Their π-Expanded Analogues. Org. Biomol. Chem. 2016, 14 (33), 7804– 7828, DOI: 10.1039/C6OB00985AThere is no corresponding record for this reference.
- 9Botezatu, A. V.; Furdui, B.; Busuioc, A.; Dinică, R. M. Recent Developments in the Synthesis of Indolizines and Their Derivatives as Compounds of Interest in Medicinal Chemistry: A Review. Eur. J. Med. Chem. 2025, 297, 117908, DOI: 10.1016/j.ejmech.2025.117908There is no corresponding record for this reference.
- 10Cho, S. H.; Kim, J. Y.; Kwak, J.; Chang, S. Recent Advances in the Transition Metal-Catalyzed Twofold Oxidative C–H Bond Activation Strategy for C–C and C–N Bond Formation. Chem. Soc. Rev. 2011, 40 (10), 5068– 5083, DOI: 10.1039/c1cs15082kThere is no corresponding record for this reference.
- 11Dalton, T.; Faber, T.; Glorius, F. C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7 (2), 245– 261, DOI: 10.1021/acscentsci.0c01413There is no corresponding record for this reference.
- 12Wencel-Delord, J.; Glorius, F. C–H Bond Activation Enables the Rapid Construction and Late-Stage Diversification of Functional Molecules. Nat. Chem. 2013, 5 (5), 369– 375, DOI: 10.1038/nchem.1607There is no corresponding record for this reference.
- 13Bellotti, P.; Huang, H.-M.; Faber, T.; Glorius, F. Photocatalytic Late-Stage C–H Functionalization. Chem. Rev. 2023, 123 (8), 4237– 4352, DOI: 10.1021/acs.chemrev.2c00478There is no corresponding record for this reference.
- 14Holmberg-Douglas, N.; Nicewicz, D. A. Photoredox-Catalyzed C–H Functionalization Reactions. Chem. Rev. 2022, 122 (2), 1925– 2016, DOI: 10.1021/acs.chemrev.1c00311There is no corresponding record for this reference.
- 15Wang, C.-S.; Dixneuf, P. H.; Soulé, J.-F. Photoredox Catalysis for Building C–C Bonds from C(Sp2)–H Bonds. Chem. Rev. 2018, 118 (16), 7532– 7585, DOI: 10.1021/acs.chemrev.8b00077There is no corresponding record for this reference.
- 16Xie, J.; Jin, H.; Xu, P.; Zhu, C. When C–H Bond Functionalization Meets Visible-Light Photoredox Catalysis. Tetrahedron Lett. 2014, 55 (1), 36– 48, DOI: 10.1016/j.tetlet.2013.10.090There is no corresponding record for this reference.
- 17Correia, J. T. M.; Fernandes, V. A.; Matsuo, B. T.; Delgado, J. A. C.; Souza, W. C. D.; Paixão, M. W. Photoinduced Deaminative Strategies: Katritzky Salts as Alkyl Radical Precursors. Chem. Commun. 2020, 56 (4), 503– 514, DOI: 10.1039/C9CC08348KThere is no corresponding record for this reference.
- 18Gutiérrez-Bonet, Á.; Tellis, J. C.; Matsui, J. K.; Vara, B. A.; Molander, G. A. 1,4-Dihydropyridines as Alkyl Radical Precursors: Introducing the Aldehyde Feedstock to Nickel/Photoredox Dual Catalysis. ACS Catal. 2016, 6 (12), 8004– 8008, DOI: 10.1021/acscatal.6b02786There is no corresponding record for this reference.
- 19Liu, Y.; Zhou, J.; Sun, Z. Direct Synthesis of Unnatural Amino Acids and Modifications of Peptides via LADA Strategy. Chin. Chem. Lett. 2024, 35 (1), 108553, DOI: 10.1016/j.cclet.2023.108553There is no corresponding record for this reference.
- 20Reed, M. A.; Noden, M. Using Bench-Stable 2-Mercaptothiazolinium Salts for the Photoredox Alkylation of N-Heteroarenes. Synfacts 2024, 20, 1020, DOI: 10.1055/s-0043-1775358There is no corresponding record for this reference.
- 21Geniller, L.; Taillefer, M.; Jaroschik, F.; Prieto, A. Photo-Induced Desulfurative Processes for Carbon Radical Generation. ChemCatchem 2023, 15 (20), e202300808 DOI: 10.1002/cctc.202300808There is no corresponding record for this reference.
- 22Zemtsov, A. A.; Ashirbaev, S. S.; Levin, V. V.; Kokorekin, V. A.; Korlyukov, A. A.; Dilman, A. D. Photoredox Reaction of 2-Mercaptothiazolinium Salts with Silyl Enol Ethers. J. Org. Chem. 2019, 84 (23), 15745– 15753, DOI: 10.1021/acs.joc.9b02478There is no corresponding record for this reference.
- 23Tian, Y.; Fu, X.-X.; Cheng, R.-M.; Feng, J.; Shen, M.-H.; Zhu, C.-F.; Xu, H.-D. Visible-Light-Mediated Alkylation of N-Heteroarenes Using 2-Mercaptothiazolinium Salts as Alkyl Radical Source. Eur. J. Org. Chem. 2024, 27, e202400344 DOI: 10.1002/ejoc.202400344There is no corresponding record for this reference.
- 24Zhu, C.-F.; Li, F.; Mai, J.-J.; Li, X.-J.; Dong, X.; Shi, M.; Shen, M.-H.; Xu, H.-D. Visible-light-promoted synthesis of alkylated Indolo[2,1-α]isoquinolines using 2-Mercaptothiazolinium salts as alkyl radical source. Tetrahedron Lett. 2025, 154, 155397, DOI: 10.1016/j.tetlet.2024.155397There is no corresponding record for this reference.
- 25Neuba, A.; Ortmeyer, J.; Konieczna, D. D.; Weigel, G.; Flörke, U.; Henkel, G.; Wilhelm, R. Synthesis of New Copper(i) Based Linear 1-D-Coordination Polymers with Neutral Imidazolinium-Dithiocarboxylate Ligands. RSC Adv. 2015, 5 (12), 9217– 9220, DOI: 10.1039/C4RA09033KThere is no corresponding record for this reference.
- 26Rosenthal, M.; Lindner, J. K. N.; Gerstmann, U.; Meier, A.; Schmidt, W. G.; Wilhelm, R. A Photoredox Catalysed Heck Reaction via Hole Transfer from a Ru(ii)-Bis(Terpyridine) Complex to Graphene Oxide. RSC Adv. 2020, 10 (70), 42930– 42937, DOI: 10.1039/D0RA08749AThere is no corresponding record for this reference.
- 27Rosenthal, M.; Biktagirov, T.; Schmidt, W. G.; Wilhelm, R. Synthesis of New Graphene Oxide/TiO2 and TiO2/SiO2 Nanocomposites and Their Evaluation as Photocatalysts. Catal. Sci. Technol. 2023, 13 (15), 4367– 4377, DOI: 10.1039/D3CY00461AThere is no corresponding record for this reference.
- 28Konieczna, D. D.; Biller, H.; Witte, M.; Schmidt, W. G.; Neuba, A.; Wilhelm, R. New Pyridinium Based Ionic Dyes for the Hydrogen Evolution Reaction. Tetrahedron 2018, 74 (1), 142– 149, DOI: 10.1016/j.tet.2017.11.053There is no corresponding record for this reference.
- 29Stefanoni, K. K.; Wilhelm, R. Access to SCF3-Substituted Indolizines via a Photocatalytic Late-Stage Functionalization Protocol. Org. Lett. 2025, 27 (31), 8389– 8393, DOI: 10.1021/acs.orglett.5c02079There is no corresponding record for this reference.
- 30Stefanoni, K. K.; Schmitz, M.; Treuheit, J.; Kerzig, C.; Wilhelm, R. Bichromophoric Ruthenium Complexes for Photocatalyzed Late-Stage Synthesis of Trifluoromethylated Indolizines. J. Org. Chem. 2025, 90 (19), 6491– 6503, DOI: 10.1021/acs.joc.5c00319There is no corresponding record for this reference.
- 31Meier, A.; Badalov, S. V.; Biktagirov, T.; Schmidt, W. G.; Wilhelm, R. Diquat Based Dyes: A New Class of Photoredox Catalysts and Their Use in Aerobic Thiocyanation. Chem. – Eur. J. 2023, 29 (22), e202203541 DOI: 10.1002/chem.202203541There is no corresponding record for this reference.
- 32Buzzetti, L.; Crisenza, G. E. M.; Melchiorre, P. Mechanistic Studies in Photocatalysis. Angew. Chem., Int. Ed. 2019, 58 (12), 3730– 3747, DOI: 10.1002/anie.201809984There is no corresponding record for this reference.
- 33Baggott, M. J. Indolizine Compounds for the Treatment of Mental Disorders or Inflammation. WO 2,023,183,613 A2, 2023.There is no corresponding record for this reference.
- 34Antonini, I.; Claudi, F.; Gulini, U.; Micossi, L.; Venturi, F. Indolizine Derivatives with Biological Activity IV: 3-(2-Aminoethyl)-2-Methylindolizine, 3-(2-Aminoethyl)-2-Methyl-5,6,7,8-Tetrahydroindolizine, and Their N-Alkyl Derivatives. J. Pharm. Sci. 1979, 68 (3), 321– 324, DOI: 10.1002/jps.2600680317There is no corresponding record for this reference.
- 35Hansch, C.; Leo, A.; Taft, R. W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91 (2), 165– 195, DOI: 10.1021/cr00002a004There is no corresponding record for this reference.
- 36Almirante, L.; Polo, L.; Mugnaini, A.; Provinciali, E.; Rugarli, P.; Biancotti, A.; Gamba, A.; Murmann, W. Derivatives of Imidazole. I. Synthesis and Reactions of Imidazo[1,2-α]Pyridines with Analgesic, Antiinflammatory, Antipyretic, and Anticonvulsant Activity. J. Med. Chem. 1965, 8 (3), 305– 312, DOI: 10.1021/jm00327a007There is no corresponding record for this reference.
- 37Singh, D. K.; Kim, S.; Lee, J. H.; Lee, N. K.; Kim, J.; Lee, J.; Kim, I. 6-(Hetero)Arylindolizino[1,2-]Quinolines as Highly Fluorescent Chemical Space: Synthesis and Photophysical Properties. J. Heterocycl. Chem. 2020, 57 (8), 3018– 3028, DOI: 10.1002/jhet.4000There is no corresponding record for this reference.
- 38Haddadin, M. J.; Conrad, W. E.; Kurth, M. J. The Davis-Beirut Reaction: A Novel Entry into 2H-Indazoles and Indazolones. Recent Biological Activity of Indazoles. Mini-Rev. Med. Chem. 2012, 12 (12), 1293– 1300, DOI: 10.2174/138955712802762059There is no corresponding record for this reference.
- 39Lee, J. C.; Cuthbertson, J. D.; Mitchell, N. J. Chemoselective Late-Stage Functionalization of Peptides via Photocatalytic C2-Alkylation of Tryptophan. Org. Lett. 2023, 25 (29), 5459– 5464, DOI: 10.1021/acs.orglett.3c01795There is no corresponding record for this reference.
- 40Enguehard-Gueiffier, C.; Gueiffier, A. Recent Progress in the Pharmacology of Imidazo[1,2-a]Pyridines. Mini-Rev. Med. Chem. 2007, 7 (9), 888– 899, DOI: 10.2174/138955707781662645There is no corresponding record for this reference.
- 41Sumalatha, Y.; Reddy, T. R.; Reddy, P. P.; Satyanarayana, B. A. Simple Efficient and Scalable Synthesis of Hypnotic Agent, Zolpidem; ARKIVOC, 2009.There is no corresponding record for this reference.
- 42Shrivastava, S. K.; Srivastava, P.; Bandresh, R.; Tripathi, P. N.; Tripathi, A. Design Synthesis, and Biological Evaluation of Some Novel Indolizine Derivatives as Dual Cyclooxygenase and Lipoxygenase Inhibitor for Anti-Inflammatory Activity. Bioorg. Med. Chem. 2017, 25 (16), 4424– 4432, DOI: 10.1016/j.bmc.2017.06.027There is no corresponding record for this reference.
- 43Wu, Z.; Lu, Y.; Li, L.; Zhao, R.; Wang, B.; Lv, K.; Liu, M.; You, X. Identification of N-(2-Phenoxyethyl)Imidazo[1,2-a]Pyridine-3-Carboxamides as New Antituberculosis Agents. ACS Med. Chem. Lett. 2016, 7 (12), 1130– 1133, DOI: 10.1021/acsmedchemlett.6b00330There is no corresponding record for this reference.
- 44Jose, G.; Suresha Kumara, T. H.; Nagendrappa, G.; Sowmya, H. B. V.; Sriram, D.; Yogeeswari, P.; Sridevi, J. P.; Guru Row, T. N.; Hosamani, A. A.; Sujan Ganapathy, P. S.; Chandrika, N.; Narendra, L. V. Synthesis Molecular Docking and Anti-Mycobacterial Evaluation of New Imidazo[1,2-a]Pyridine-2-Carboxamide Derivatives. Eur. J. Med. Chem. 2015, 89, 616– 627, DOI: 10.1016/j.ejmech.2014.10.079There is no corresponding record for this reference.
- 45Khetmalis, Y. M.; Chitti, S.; Wunnava, A. U.; Kumar, B. K.; Kumar, M. M. K.; Murugesan, S.; Sekhar, K. V. G. C. Design Synthesis and Anti-Mycobacterial Evaluation of Imidazo[1,2-a]Pyridine Analogues. RSC Med. Chem. 2022, 13 (3), 327– 342, DOI: 10.1039/D1MD00367DThere is no corresponding record for this reference.
- 46Nahm, S.; Weinreb, S. M. N-Methoxy-n-Methylamides as Effective Acylating Agents. Tetrahedron Lett. 1981, 22 (39), 3815– 3818, DOI: 10.1016/S0040-4039(01)91316-4There is no corresponding record for this reference.
- 47Kaur, H.; Karabulut, S.; Gauld, J. W.; Fagot, S. A.; Holloway, K. N.; Shaw, H. E.; Fantegrossi, W. E. Balancing Therapeutic Efficacy and Safety of MDMA and Novel MDXX Analogues as Novel Treatments for Autism Spectrum Disorder. Psychedelic Med. 2023, 1 (3), 166– 185, DOI: 10.1089/psymed.2023.0023There is no corresponding record for this reference.
- 48Oeri, H. E. Beyond Ecstasy: Alternative Entactogens to 3,4-Methylenedioxymethamphetamine with Potential Applications in Psychotherapy. J. Psychopharmacol. 2021, 35 (5), 512– 536, DOI: 10.1177/0269881120920420There is no corresponding record for this reference.
- 49He, X.; Chen, Z.; Zhu, X.; Liu, H.; Chen, Y.; Sun, Z.; Chu, W. Photoredox-Catalyzed Trifluoromethylation of 2H-Indazoles Using TT-CF3+OTf– in Ionic Liquids. Org. Biomol. Chem. 2023, 21 (8), 1814– 1820, DOI: 10.1039/D3OB00096FThere is no corresponding record for this reference.
- 50Jia, H.; Häring, A. P.; Berger, F.; Zhang, L.; Ritter, T. Trifluoromethyl Thianthrenium Triflate: A Readily Available Trifluoromethylating Reagent with Formal CF3+, CF3•, and CF3– Reactivity. J. Am. Chem. Soc. 2021, 143 (20), 7623– 7628, DOI: 10.1021/jacs.1c02606There is no corresponding record for this reference.
- 51Malpani, Y. R.; Biswas, B. K.; Han, H. S.; Jung, Y.-S.; Han, S. B. Multicomponent Oxidative Trifluoromethylation of Alkynes with Photoredox Catalysis: Synthesis of α-Trifluoromethyl Ketones. Org. Lett. 2018, 20 (7), 1693– 1697, DOI: 10.1021/acs.orglett.8b00410There is no corresponding record for this reference.
Supporting Information
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.5c00110.
Full experimental description, spectral data of the compounds, and additional spectroscopy data (PDF)
Terms & Conditions
Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html.