



This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Degradable Vinyl-Based Polymers by Radical Ring-Opening Polymerization: A User GuideClick to copy article linkArticle link copied!
- Bastien LuzelBastien LuzelAix-Marseille Université, CNRS, Institut de Chimie Radicalaire, UMR 7273, F-13397 Marseille, FranceMore by Bastien Luzel
- Sophia KouiderSophia KouiderAix-Marseille Université, CNRS, Institut de Chimie Radicalaire, UMR 7273, F-13397 Marseille, FranceMore by Sophia Kouider
- Franck D’AgostoFranck D’AgostoUniversite Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5128, Catalysis, Polymerization, Processes and Materials, 69616 Villeurbanne, FranceMore by Franck D’Agosto
- Didier GigmesDidier GigmesAix-Marseille Université, CNRS, Institut de Chimie Radicalaire, UMR 7273, F-13397 Marseille, FranceMore by Didier Gigmes
- Muriel LansalotMuriel LansalotUniversite Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5128, Catalysis, Polymerization, Processes and Materials, 69616 Villeurbanne, FranceMore by Muriel Lansalot
- Christopher M. BatesChristopher M. BatesMaterials Research Laboratory, University of California, Santa Barbara, California 93106, United StatesMaterials Department, University of California, Santa Barbara, California 93106, United StatesMore by Christopher M. Bates
- Elise AckermanElise AckermanDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United StatesMore by Elise Ackerman
- Steven LabalmeSteven LabalmeDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United StatesMore by Steven Labalme
- Jeremiah A. JohnsonJeremiah A. JohnsonDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United StatesMore by Jeremiah A. Johnson
- Jia NiuJia NiuDepartment of Chemistry, Boston College, Chestnut Hill, Massachusetts 02467, United StatesMore by Jia Niu
- Julien NicolasJulien NicolasUniversité Paris-Saclay, CNRS, Institut Galien Paris-Saclay, F-91400 Orsay, FranceMore by Julien Nicolas
- Yohann Guillaneuf*Yohann Guillaneuf*Email: [email protected]Aix-Marseille Université, CNRS, Institut de Chimie Radicalaire, UMR 7273, F-13397 Marseille, FranceMore by Yohann Guillaneuf
- Catherine Lefay*Catherine Lefay*Email: [email protected]Aix-Marseille Université, CNRS, Institut de Chimie Radicalaire, UMR 7273, F-13397 Marseille, FranceMore by Catherine Lefay
ACS Polymers Au
© 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:

Creative Commons (CC): This is a Creative Commons license.

Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Abstract
Low weight, low price, and excellent long-term stability are the main advantages of vinyl-based polymers. Such polymers are obtained by chain-growth processes leading to all-carbon backbones, which are non(bio)degradable and nonchemically recyclable. Unfortunately, this chemical stability manifests as postuse persistence; coupled with poor waste management practices, polymers including vinyl derivatives pose major environmental problems today. Given that it is very difficult and costly to design entirely new materials that have both desired properties (mechanical, thermal, solvent resistance, etc.) and recyclability and/or biodegradability at the end of their life cycle, it seems worthwhile to transform already known materials into (bio)degradable/chemically recyclable equivalents. One approach is based on the introduction of cleavable bonds into the polymer backbone, so that degradation (by hydrolysis, for example) produces oligomers which can then be further recycled and/or bioassimilated by micro-organisms. An effective method for incorporating weak bonds randomly into the C–C backbone of a vinyl polymer is the copolymerization of vinyl monomers with cyclic monomers by radical ring-opening polymerization (rROP). This method combines the advantages of ring-opening and radical polymerization, i.e., the production of polymers with heteroatoms and/or functional groups in the main chain, with the robustness, ease of use, and mild polymerization conditions of a radical process. The aim of this tutorial review is to provide polymer chemists with guidelines to use rROP to prepare vinyl-based materials with predictable degradation. This review thus presents the rROP principle, the main families of cyclic monomers copolymerizable with vinyl monomers, and the main applications of the resulting (bio)degradable/chemically recyclable materials (polymers for packaging, latexes and degradable surfaces, 3D printing, biomaterials and water-soluble polymers).
This publication is licensed under
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
License Summary*
You are free to share(copy and redistribute) this article in any medium or format and to adapt(remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
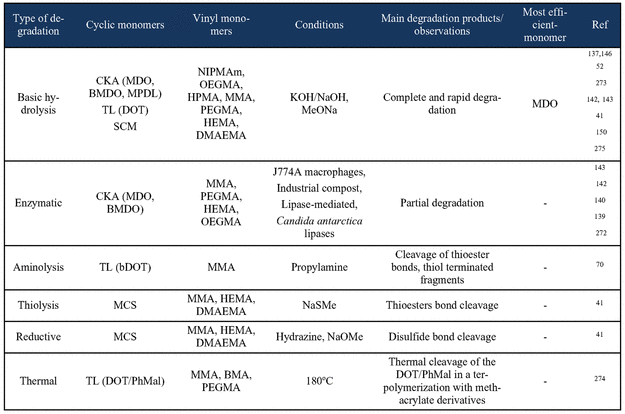
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Introduction
Figure 1
Figure 1. Radical copolymerization of cyclic and vinyl monomers aimed at developing degradable materials.
Figure 2
Figure 2. Structures of the two main categories of cyclic monomers that can be polymerized by radical ring-opening polymerization (rROP).
Figure 3
Figure 3. Competition between radical ring-opening (β-scission) and ring retention (1,2-addition).
History of rROP
Figure 4
Figure 4. Timeline of the history of rROP with the different families of monomers used.
Main Monomer Families
Monomer Syntheses
Cyclic Ketene Acetals (CKA)
Figure 5
Figure 5. Structures of the most efficient CKAs in rROP.
Figure 6
Figure 6. Synthesis of CKA via the transacetalization and dehydrochloration reaction.
Figure 7
Figure 7. Two other synthesis pathways: a new acetal pathway and carbonate pathway.
Figure 8
Figure 8. Synthesis pathway of Glu-CKA.
Sulfide Cyclic Methacrylate (SCM)
Figure 9
Figure 9. Structures of sulfide cyclic methacrylate monomers.
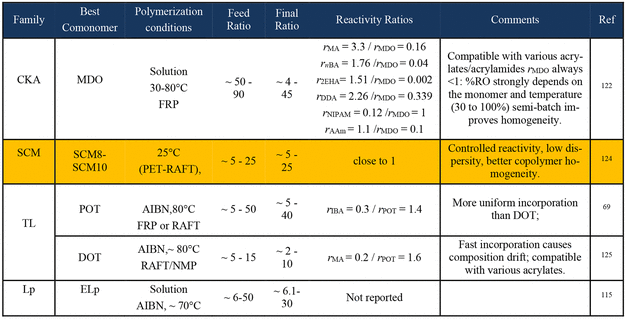
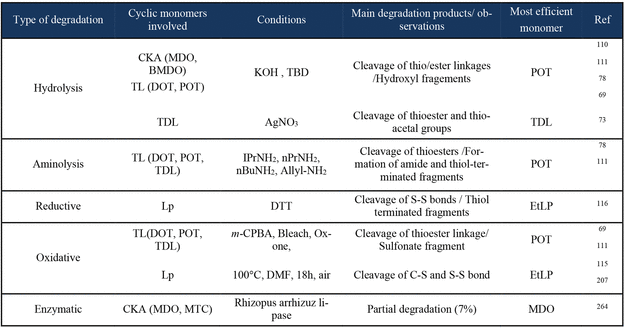
Figure 10
Figure 11
Figure 11. Synthesis of sulfide cyclic methacrylate-type monomers (SCM). a) First generation, b) second generation, and c) cyclic sulfide diene (CSD).
Thionolactone (TL)
Figure 12
Figure 12. Synthesis of dibenzo[c,e]-oxepine-5(7H)-thione (DOT).
Figure 13
Figure 13. Synthesis of 7-phenyloxepane-2-thione (POT).
Figure 14
Figure 14. Synthesis of 10-fluoro-7-(4-(trifluoromethyl) phenyl) DOT (F-p-CF3PhDOT).
Lipoates
Figure 15
Figure 15. Synthesis of ethyl lipoate and structures of monomers previously reported in the literature.
Homopolymerization via rROP
Figure 16
Figure 16. (A) Kinetic competition between vinyl propagation and ring opening. (B) Percentage of ring opening for 5-, 6-, and 7-membered CKA monomers (filled points: experimental data; empty points: theoretical data). Reproduced from ref (82) with permission. Copyright 2020 Wiley-VCH.
SCM Homopolymerization
Figure 17
Figure 17. Mechanism of radical polymerization via ring-opening of sulfide cyclic methacrylates (SCM1–SCM7).
Figure 18
Figure 18. a) Mechanism of radical polymerization via ring-opening of sulfide cyclic methacrylates (SCM8–SCM10). b) Mechanism of radical polymerization via ring opening of CSD monomers.
Thionolactone Homopolymerization
Figure 19
Figure 19. Homopolymerization of (A) DBT and (B) TIC.
Figure 20
Figure 20. Homopolymerization of POT.
Lipoate Homopolymerization
Reactivity of Monomers in rROP
Figure 21
Figure 21. Copolymerization kinetics and associated reactivity ratios.
Figure 22
Figure 22. Simulation of individual chain degradation, obtained from kinetic Monte Carlo simulations: Calculated size exclusion chromatography (SEC) traces for polymer chains and degradation products. SEC traces before and after copolymer hydrolysis under various conditions: (top) RDRP (orange) versus uncontrolled (green) radical polymerization. (Bottom) Comparison between the most heterogeneous (black) and most homogeneous (orange) RDRP degradation products. In each panel, the degraded product appears to the left of the corresponding initial polymer (same color). Reproduced from ref (103) with permission. Copyright 2018 Wiley-VCH.
Figure 23
Figure 23. (a) Schematic of the cleavable comonomer additive (CCA) approach for deconstructable copolymers. CCAs copolymerize with standard monomers (“M1”), introducing cleavable sites along the backbone. (b) Relative decrease in molecular weight (Mw,deg/Mw,poly) as a function of reactivity ratio pairs, r1 and rCCA, for M1 and CCA, respectively. For all simulations presented, a degree of polymerization of 1000 was targeted with a CCA loading of 2.5 mol %. (c) Fractional decrease in number-average molecular weight (Mn,deg/Mn,poly) as a function of reactivity ratio pairs. (d) Dispersity (Đ) of the deconstructed fragments as a function of reactivity ratio pairs. Reproduced from ref (105) with permission. Copyright 2024 American Chemical Society.
Figure 24
Figure 24. (a) Elementary steps involved in the ring-opening polymerization (ROP) of thionolactones with vinyl monomers, along with the corresponding rate constants: kadd: rate constant for addition, k–add rate constant for reverse addition, kβ: rate constant for fragmentation, kp: rate constant for propagation. (b) Definition of the transfer constant ktr. (c) Determination of the kp/ktr ratio to estimate copolymerization behavior. Adapted from ref (69) with permission. Copyright 2023 American Chemical Society. To analyze this process, the transfer rate constant (ktr) was used. This encompasses the three previously mentioned steps and allows for modeling of the addition–fragmentation mechanism. It was then compared to the propagation constant of the vinyl monomer (kp), providing a relevant criterion to assess the reactivity of the comonomer pair by determining the reactivity ratio rv. In these systems, the cyclic monomer is typically introduced as an additive in low concentration (less than 10 mol %), meaning that the majority of the growing macroradicals are polyvinyl macroradical. This approach simplified the calculations by avoiding the determination of reactivity ratios specific to thionolactones.
Synthesis of Copolymers in rROP
Poly(Styrene)
Cyclic Ketene Acetal (CKA)
Figure 25
Figure 25. (a, b) Important protons used for the 1H NMR (CDCl3) analysis of P(CKA-co-S) copolymers and degraded styrenic oligomers in P(MDO-co-S) copolymers and P(BMDO-co-S) copolymers. Reprinted from ref (110) with permission. Copyright 2020 MDPI.
Sulfide Cyclic Methacrylates (SCM)
Thionolactone (TL)
Figure 26
Figure 26. Analysis of substituent effects on the copolymerization of styrene with DOT-based thionolactone. Reproduced from ref (78) with permission. Copyright 2022 American Chemical Society.
Figure 27
Figure 27. Experimental and theoretical composition of the POT monomer as a function of overall molar conversion during solution copolymerization at 80 °C initiated with 0.5 mol % AIBN from a mixture of POT (5 mol %) and styrene in anisole: Cumulative average molar thioester content in the copolymers. Adapted from ref (69) with permission. Copyright 2023 American Chemical Society.
Lipoates (Lp)
Summary

Polyacrylates/Acrylamides
Cyclic Ketene Acetal (CKA)
Sulfide Cyclic Methacrylates (SCM)
Thionolactone (TL)
Figure 28
Figure 28. Preparation of a polyacrylate-based P(nBA-b-tBA) diblock copolymer containing 5 mol % of DOT into the two blocks. Reproduced from ref (37) with permission. Copyright 2019 American Chemical Society.
Figure 29
Figure 29. Experimental and theoretical composition of the POT monomer as a function of overall molar conversion during solution copolymerization at 80 °C, initiated with 0.5 mol % AIBN from a mixture of POT (5 mol %) with isobornyl acrylate: Cumulative average molar thioester composition of the copolymers. Adapted from ref (69) with permission. Copyright 2023 American Chemical Society.
Lipoate (Lp)
Figure 30
Figure 30. Tunable degradation of poly(acrylate) copolymers by controlling the concentration and temperature of polymerization. (a) The degradability of lipoic-acid–acrylate copolymers can be synthetically tuned through polymerization conditions that control the average number of disulfide bonds per polymer chain. (b, c) As evidenced by size-exclusion chromatography, (b) higher monomer concentrations ([M]), and (c) lower polymerization temperatures (T) improve degradability. Reproduced from ref (130) with permission. Copyright 2023 American Chemical Society.
Summary
Polymethacrylates
Cyclic Ketene Acetal (CKA)
Figure 31
Figure 31. Real-time 1H NMR monitoring of copolymerization. (A) Reaction scheme; (B–D) real-time 1H NMR tracking of conversion versus reaction time: (B) Glu-CKA/MMA = 1:1; (C) Glu-CKA/MI/MMA = 1:1:1; and (D) Glu-CKA/MI/MMA = 1:2:5. Reproduced from ref (57) with permission. Copyright 2024 American Chemical Society.
Sulfide Cyclic Methacrylate (SCM)
Thionolactone (TL)
Figure 32
Figure 32. (A) Relative Gibbs free energy profile for an MMA radical reacting either with MMA or with DOT, calculated to model homopropagation and cross-propagation of a chain terminating in MMA. (B) The effect of benzylic substituent(s) on the energy profile was evaluated using DFT calculations. Calculations were performed at the wB97X-D3/def2-SVP level of theory; electronic energies of all optimized structures were re-evaluated using wB97X-D3/def2-TZVP/CPCM (toluene). (C) A Monte Carlo simulation evaluates the efficiency of aromatic bDOTs as cleavable comonomers. The heat map generated by the simulation visualizes the ratio of degraded fragment size to copolymer size as a function of reactivity ratios, for a 2.5% molar loading of CC in copolymers with DP 1000. (D) A series of bDOTs was synthesized for optimization of copolymerization reactivity. Reproduced from ref (70) with permission. Copyright 2024 American Chemical Society.
Figure 33
Figure 33. (A) Preparation of degradable PMMA derivatives via terpolymerization of MMA, DOT, and N-phenylmaleimide (PhMal; in red). (B) Simulated monomer sequences for modeling-assisted copolymerization with [MMA]0:[PhMal]0:[DOT]0 = 90:18:28 (30% solvent). Monomer sequences follow the color code from panel (C). On the right: selection of chains from the left panel, showing isolated MMA-PhMal units (red box) and MMA-DOT-PhMal triads (green box). Reproduced from ref (125) with permission. Copyright 2025 Springer Nature.
Lipoate (Lp)
Summary

Nonstabilized Monomers
Cyclic Ketene Acetal (CKA)
Thionolactone (TL)
Figure 34
Figure 34. Radical ring-opening copolymerization of cyclic thionocarbamates with N-vinylpyrrolidone. Reproduced from ref (172) with permission. Copyright 2024 American Chemical Society.
Summary

Maleic Anhydride and Maleimides
Cyclic Ketene Acetal (CKA)
Thionolactone
Figure 35
Figure 35. Molar DOT content in maleimide copolymer vs molar DOT fraction in the monomer feed with nonlinear least-squares fitted curves for (A) N-methylmaleimide, (B) N-phenylmaleimide, and (C) N-2,3,4,5,6-pentafluorophenylmaleimide. Adapted from ref (97) with permission. Copyright 2020 American Chemical Society.
Applications
Degradable Latexes
Cyclic Ketene Acetals
Figure 36
Figure 36. (A) Synthesis of SDS-stabilized P(MMA-co-BMDO) latexes by aqueous emulsion polymerization. (B) Photos of the latexes obtained with various BMDO contents. (C) SEC traces of the dry extracts of P(MMA-co-BMDO) latexes (plain lines) and their degradation products (dashed lines) as a function of incorporated BMDO content. Adapted from ref (187) with permission. Copyright 2023 Royal Society of Chemistry.
Figure 37
Figure 37. Synthesis of block copolymer nanoparticles with degradable cores via self-assembly induced by radical ring-opening copolymerization (rROPISA) mediated by RAFT from cyclic ketene acetals (CKAs). Reproduced from ref (196) with permission. Copyright 2019 American Chemical Society.
Thionolactones (TL)
Figure 38
Figure 38. (A) Synthesis of SDS-stabilized latexes of P(BA-co-DOT), P(S-co-DOT), and P(BA-co-S-co-DOT) by aqueous emulsion polymerization. (B) Molar mass distribution of the dry extracts of P(S-co-DOT) latexes (plain lines) and their degradation products with TBD (dashed lines) as a function of incorporated DOT content (up to 4.7 mol %). (C) Evolution of the Tg depending on the average molar fraction BA/styrene in the monomer mixture for emulsion polymerization with 2 mol % of DOT. Adapted from ref (201) with permission. Copyright 2022 Wiley-VCH.
Figure 39
Figure 39. (A) Synthesis of PDMAC43-P(MEA100-co-DOTm) (m = 2 or 4) spheres and PDMAC43-P(MEA300-co-DOT6) and PDMAC43-P(MEA400-co-DOTn) (n = 4, 8, or 16) vesicles via aqueous rROPISA with 20% w/w solids. The MEA/DOT mixture was added either all at once or gradually using a syringe pump (0.2 mL·h–1 over 2 h). (B) Scheme showing the Nile Red probe (red spheres) loaded in the membrane of PDMAC43-P(MEA400-co-DOT8) vesicles. Degradation of these vesicles in the presence of 10 mM l-cysteine and 10 mM glutathione leads to precipitation of insoluble probes. (C) Fluorescence micrographs (λex = 550 nm, λem = 605 nm) were recorded for 1% w/w dispersions at two time points (0 and 96 h) during hydrolytic degradation. Reproduced from ref (203) with permission. Copyright 2025 American Chemical Society.
Figure 40
Figure 40. (A) Synthesis of PAA-b-P(nBA-co-DOT) and PAA-b-P(S-co-DOT) copolymers by rROPISA in water. (B) SEC traces of the dry extracts and NPs composed of a PAA-b-P(nBA-co-DOT) copolymers with 1.3 mol % DOT before and after degradation in the presence of TBD or isopropylamine. Reproduced from ref (204) with permission. Copyright 2022 American Chemical Society.
α-Lipoic Acid and Lipoates
Figure 41
Figure 41. (A) Miniemulsion polymerization of α-lipoic acid with n-butyl acrylate. (B) Degradation for different amounts of ethyl lipoate with TCEP in an H2O/THF mixture. Reproduced from ref (129) with permission. Copyright 2024 American Chemical Society.
Sulfide Cyclic Methacrylate (SCM)
Degradable Surface Coatings
Figure 42
Figure 42. (A) Preparation of surface coatings in the form of polymer brushes grafted onto silica surfaces, obtained by copolymerizing poly(ethylene glycol) methacrylate (PEGMA) with the cyclic monomer BMDO. (B) 3D AFM images and 2D cross-sectional profiles of P(PEGMA) brushes with and without BMDO, taken at different time intervals during exposure to a pH 3 solution at 25 °C. Reproduced from ref (208) with permission. Copyright 2009 American Chemical Society.
Figure 43
Figure 43. (A) Antifouling mechanism of degradable and hydrolyzable polymers. (B) Structures of degradable and hydrolyzable polymers. Reproduced from ref (213) with permission. Copyright 2022 American Chemical Society.
Degradable Thermoset
Figure 44
Figure 44. (a) Scheme for producing T-REX thermosets from polyplexes, reversible encapsulation, and subsequent characterization. (b) A comparative analysis of error rates in 210-bp dsDNA segments encoding digital data between samples stored in a frozen state without encapsulation and DNA recovered from both T-REX and silica-based encapsulated samples. (c) Comparison of error rates of T-REX-encapsulated samples containing 210-bp dsDNA encoding an image file subjected to real-time and accelerated weathering conditions. Reproduced from ref (221) with permission. Copyright 2024 American Chemical Society.
Figure 45
Figure 45. (a) During the DLW process, aliphatic polyester units are incorporated into the cross-linked network; after treatment with a nucleophile (Nu–), these units break down, degrading the microstructure. Cleavage of the ester bond by the nucleophile occurs between the carbonyl carbon and the oxygen (not shown for clarity). (b) Partial degradation (SEM images) of microdonut structures fabricated with MDO:PETA 90:10 (scale bar 20 μm). Only the final part degrades, leaving a bite mark. Reproduced from ref (222) with permission. Copyright 2022 Wiley-VCH.
Figure 46
Figure 46. (A) Preparation of degradable 3D objects by VAT photopolymerization and chain cleavage. (B) Example of a 3D product with and without cleavable copolymer additive. Reproduced from ref (223) with permission. Copyright 2022 Wiley-VCH. (C) Concept of self-destruct materials via the combination of thermolatent base and cleavable comonomers.
Figure 47
Figure 47. (A) Chemical structures of a 3D printing resin derived from α-lipoic acid building blocks. The resin components include n-butyl acrylate, a mixture of cross-linkers DIS-Lp2/TEG-Lp2, and the photoinitiator (BAPO). (B) Diagram illustrating self-healing, degradation, and recycling of the printed material using DIS-Lp2 as the cross-linker. Reproduced from ref (225) with permission. Copyright 2024 American Chemical Society.
Figure 48
Figure 48. Method enabling polymerization–depolymerization cycles of dynamic disulfide bonds, allowing for the formulation of 3D-printing resins from renewable sources that are suitable for closed-loop chemical recycling. (a) Chemical composition of the formulated resin. (b) An example of a complex 3D-printed part. (c) Photograph of 3D-printed parts in powder form. (d) Photograph of the resin recovered in 98% yield after depolymerizing a 3D-printed part. e) SEC of initial resin compared to recovered resin. Adapted from ref (81) with permission. Copyright 2024 Springer Nature.
Degradable Biomaterials
Figure 49
Figure 49. a) Synthesis strategy for the design of gemcitabine-based degradable polymeric prodrugs via nitroxide-mediated polymerization initiated by a Gem-alkoxyamine initiator. Reproduced from ref (230) with permission. Copyright 2018 Royal Society of Chemistry. b) Design and preclinical development of (degradable) polyacrylamide (PAAm)-based prodrugs for the SC administration of the anticancer drug gemcitabine (Gem). Reproduced from ref (231) with permission. Copyright 2025 Royal Society of Chemistry.
Figure 50
Figure 50. (A) Synthesis of poly(N,N-dimethylaminoethyl methacrylate-co-methacrylic acid-co-5,6-benzo-2-methylene-1,3-dioxepane) poly(DMAEMA-co-MAA-co-BMDO) terpolymers by RAFT terpolymerization of DMAEMA, TBDMSMA, and BMDO with CPADB as a RAFT agent, followed by deprotection of TBDMSMA units. (234) (B) Schematic of the monolayer cryopreservation and post-thaw process. (C) Cell recovery 24 h post-thaw, relative to prefreezing, determined using Trypan blue exclusion test (left) and cell viability 24 h post thaw determined using Trypan blue exclusion test (right). One-way ANOVA with Tukey’s posthoc test. * = p < 0.05, ** = p < 0.001 considered as statistically significant different using a 95% confidence level, ns = not significant. Reproduced from ref (234) with permission. Copyright 2022 American Chemical Society.
Degradable Water-Soluble Polymers
Figure 51
Figure 51. (A) rROP of MDO and tBA yielding poly(MDO-co-tBA), followed by acid-mediated tert-butyl deprotection to obtain degradable poly(MDO-co-AA). (B) Overview of biodegradability, through initial hydrolysis of the main-chain esters into short oligomers, followed by complete biodegradation. Reproduced from ref (237) with permission. Copyright 2022 Elsevier.
Figure 52
Figure 52. (A) Synthesis of P(AAm-co-BMDO). (B) Evolution of the Mn with time during hydrolytic degradation in physiological conditions (PBS, pH 7.4, T = 37 °C) of (1) PAAm (P9), P(AAm-co-BMDO) copolymers with different BMDO contents (P10–P13 and P17) and (2) PLA and PLGA. (C) Evolution of the Mn with time during enzymatic degradation with lipases (Candida antartica, PBS, pH 7.4, T = 37 °C) of (1) PAAm (P9), P(AAm-co-BMDO) copolymers P13 and P17 and (2) PLA and PLGA. Reproduced from ref (239) with permission. Copyright 2022 Springer Nature.
Figure 53
Figure 53. Proposed reaction pathway for a 17O-labeled experiment for the hydrolysis of MDO with H217O at pH 2. Reproduced from ref (50) with permission. Copyright 2023 Wiley-VCH.
Degradable Adhesive
Figure 54
Figure 54. (a) Synthesis of UV cross-linkable degradable thioester-functional PSA. (c) Photos of dye-labeled photo-cross-linked copolymer films on glass substrates of the nondegradable control BA-ABP0.05-NBDA0.25 (left) and degradable BA-ABP0.05-DOT0.25-NBDA0.25 (right) A) before immersion and B) after immersion in 2 M n-propylamine in THF for 120 s, confirming visually the presence of insoluble residue for the control sample only. Reproduced from ref (244) with permission. Copyright 2023 Wiley-VCH.
Figure 55
Figure 55. (a) In situ rROP of CKA and comonomers to form a degradable and functional macromolecular chain by redox initiation benzoyl peroxide/N,N-dimethyl-p-toluidine (BPO/DMPT). (b) Tunable preparation of the adhesive called backbone-degradable robust adhesives (BDRAs) that achieve strong adhesion by forming a covalent interpenetrating network by in situ rROP and the synergy of intermolecular and chemical bonds. (c) Adhesion strength and setting time of BDRAs and the existing tissue adhesives for hard and soft tissues. (d) Bearing capacity of bonded fractured bovine bone using BDRAs. (e) Adhesion strength of BDRAs and commercial medical adhesives on different biological tissues, represented by flexural strength for bone and shear strength for pigskin. Data are presented as the means ± SDs, n = 3 independent samples per group. (f) Shear adhesion strength for low-surface-energy polymers adhered by a BDRA and commercial engineering adhesives. PP polypropylene, PE polyethylene, PTFE polytetrafluoroethylene. Data are presented as the means ± SDs, n = 3 independent samples. Reproduced from ref (251) with permission. Copyright 2023 Springer Nature.
Degradable Polyethylene
Figure 56
Figure 56. (A) rROP of ethylene, vinyl acetate and thionolactone for the production of chemically degradable PE and EVA. (B) Thermogravimetric analyses of P(E-co-TCL). (C) SEC analyses of pristine and degraded (dashed lines) P(E-co-VAc-co-TCL). Adapted from ref (178) with permission. Copyright 2024 American Chemical Society.
Miscellaneous
Figure 57
Figure 57. a) Degradation of a PMMA-based copolymer via a specific TBAF triggering. b) SEC of pristine and degraded copolymer ([MMA]:[BMDO]:[SiOMMA] = 25:18:57, Mn = 31,100 g·mol–1, D = 1.54). c) Evolution of Mp with an increasing of amount of MMA in the copolymer. Reproduced from ref (257) with permission. Copyright 2024 American Chemical Society.
Figure 58
Figure 58. (a) Triblock copolymer synthesis and its stepwise photodegradation: a diblock copolymer consisting of a PDMA nondegradable block and photodegradable copolymer of the coumarin cycloadduct and DMA block is prepared by green light-initiated RAFT polymerization. Chain extension of this polymer with RAFT copolymerization of DMA and the cyclic monomer resulted from intramolecular [2 + 2] cycloaddition of styrylpyrene under blue light yields a triblock polymer with the copolymer of DMA and styrylpyrene cycloadduct as the third block. Under UVA, the styrylpyrene cycloadduct experiences [2 + 2] cycloreversion, leading to the fragmentation of the third block. Subsequent UVB irradiation initiates the degradation of the second block as the coumarin dimer in the polymer backbone undergoes [2 + 2] cycloreversion. Reproduced from ref (259) with permission. Copyright 2023 Wiley-VCH. (b) Synthesis of a polypeptide mimic and its SEC traces before (P1-tBu) and after (P1) deprotection. DOSY NMR of P1 at a 1 mg/mL concentration and CD spectra of P1 (0.2 mg/mL) at basic (pink) and acidic (purple) pH. The CD traces have not been smoothed. Reproduced from ref (260) with permission. Copyright 2024 Wiley-VCH.
(BIO)Degradation
Homopolymerization
Figure 59
Figure 59. a) Degree of hydrolysis of PCL, PMDOs, and PMe-MDO as a function of time. b) Biodegradation test results of PCL, PMDO-DB 10%, PMDO-DB 18% and PMe-MDO in river water. Mean and standard deviations (SDs) are calculated based on the biodegradation achieved in three replicate bottles for two biological replicate per inoculum. Reproduced from ref (263) with permission. Copyright 2023 Royal Society of Chemistry.
Figure 60
Figure 60. Proposed degradation pathways of a pDOT chain, where X = H and Me, and Y = OH and OTf for BF3·Et2O-initiated and MeOTf-initiated polymers, respectively. Reproduced from ref (265) with permission. Copyright 2024 Elsevier.
Copolymerization
Polystyrene
Figure 61
Figure 61. Evolution of the molar mass distribution of a P(S-co-POT) prepared at 80 °C and 5% POT before and after various degradation conditions. Reproduced from ref (69) with permission. Copyright 2023 American Chemical Society.
Polyacrylates/Acrylamides
Figure 62
Figure 62. Temperature influence on the enzymatic degradation by proteinase K of P(NIPAM-co-MDO) hydrogels. Reproduced from ref (119) with permission. Copyright 2003 Wiley-VCH.
Figure 63
Figure 63. OECD 301 D Closed Bottle Test biodegradability (%) results of degradable P(MDO-co-AA) and nondegradable poly(AA) using secondary effluent from domestic wastewater treatment plant, as inoculum. Results shown are the average of the triplicates, Following the OECD Guideline for Ready Biodegradability, the test results were valid since reference compound achieves more than 60% biodegradability on Day 14. Reproduced from ref (237) with permission. Copyright 2022 Elsevier.
Figure 64
Figure 64. Various ecmhanisms of degradation for polyacrylate and polyacrylamide-based copolymers containing DOT units into the backbone. Reproduced from ref (267) with permission. Copyright 2022 American Chemical Society.

Polymethacrylates/Methacrylamides
Figure 65
Figure 65. Evolution of the number-average molar mass, Mn, with time of different P(OEGA-co-MPDL) copolymers, PLA and PCL during the hydrolytic degradation in PBS (0.1 M, pH 7.4, 37 °C). Reproduced from ref (272) with permission. Copyright 2018 American Chemical Society.
Figure 66
Figure 66. a) Various degradation conditions for polymethacrylate derivatives containing SCM5–7 monomers. b) SEC traces of the PMMA-based copolymer containing 0.3% of SCM7 and 0.8% of SCM5 and its products after stepwise degradation. Adapted from ref (41) with permission. Copyright 2009 American Chemical Society.
Figure 67
Figure 67. Change of SEC-measured molar mass (normalized to intact species) versus time during the hydrolysis (in 8 mM NaOH in water–methanol) of three p(R-SCM-co-HPMAm) copolymers. Values are the fitted rates of degradation for each copolymer. Adapted from ref (275) with permission. Copyright 2025 American Chemical Society.
Nonstabilized Monomers
Figure 68
Figure 68. OECD 301 D Closed Bottle Test biodegradability (%) results of solid sample of nondegradable PVA (Mn = 5,100 g·mol–1), degradable P(VA-co-MDO) (88% VA units, Mn = 1,700 g·mol–1) and nanoparticles of degradable P(VA-b-MDO) (35% VA units, Mn = 7,300 g·mol–1) and degradable P(VA-b-MDO) (64% VA units, Mn = 12,000 g·mol–1) using secondary effluent from domestic wastewater treatment plant, as inoculum. The results shown are the average of the triplicates. Adapted from refs (199) and (200) with permission. Copyright 2023 Elsevier.
Figure 69
Figure 69. Enzymatic hydrolysis test results for CKA-NVP polymers. Adapted from ref (163) with permission. Copyright 2024 Elsevier.
Figure 70
Figure 70. a) Biodegradation curves of CKA-based polymers following OECD 301F (solid lines) and modified OECD 302B (dashed line) protocols. b) SEC analysis of poly(MTC-co-NVP) before and after modified OECD 302B biodegradation test. The assigned mass peak sets for species comprising one MTC unit from the spectrum (B) are listed in spectra (C–F), corresponding to oligomer structures (2)–(5). (G) Proposed biodegradation pathways, based on mass spectrometry analysis. Adapted from ref (163) with permission. Copyright 2024 Elsevier.
Figure 71
Figure 71. Evolution of the weight-average molar mass (Mw) during the hydrolytic degradation under accelerated conditions (0.05 wt % NaOH MeOH:THF 1:1 v/v) (a, b) or under physiological conditions (PBS, pH 7.4, 37 °C) (c, d) of P(MDO-co-BVE) and P(MTC-co-BVE) copolymers, and P(MDO-co-TEGVE) and P(MTC-co-TEGVE) copolymers. Adapted from ref (168) with permission. Copyright 2023 American Chemical Society.
Figure 72
Figure 72. Schematic representation of the two-step (peroxidation/aminolysis) and one-step (bleach) degradation processes for P (vinyl pivalate-co-thionocaprolactone). Experimental SEC chromatograms before and after degradation: P(TCL0.68-co-VP0.32) with benzyl peroxide (BPO) and subsequent addition of N-isopropylamine (IPA); IPA and subsequent addition of BPO; and bleach in comparison to the two-step degradation. Adapted from ref (276) with permission. Copyright 2023 American Chemical Society.

Recycling
Figure 73
Figure 73. a) Synthetic scheme illustrating the deconstruction and recycling of high-molar-mass PS. SEC traces for the deconstruction/recycling cycles of P(S-co-DOT). b) Application of the deconstruction/reconstruction strategy to an acrylic copolymer; SEC traces for the deconstruction/recycling cycles of P(nBA-co-DOT). Reproduced from ref (78) with permission. Copyright 2022 American Chemical Society.
Figure 74
Figure 74. A) Removal of label adhesive (αLA-ELp-nBA and nBA-AA) attached to recyclable plastic bottles. B) Model adhesive (ELp-nBA) with functional chain-ends produced after degradation can undergo repeated oxidative repolymerization and reductive degradation for closed-loop recycling, as evidenced by c) size-exclusion chromatography analysis. Reproduced from ref (130) with permission. Copyright 2023 American Chemical Society.
Figure 75
Figure 75. Closed-loop recycling of PS-PnBA multiblock copolymers containing disulfide linkages. (i) Mild oxidation (I2/pyridine) reconnects α,ω-dithiol PS and PnBA blocks into high-molar-mass copolymers; (ii) reductive treatment (TCEP) cleaves the disulfide bonds. Reproduced from ref (278) with permission. Copyright 2025 Wiley-VCH.
Figure 76
Figure 76. Degradation and regelation scheme for PBA-DOT networks prepared by RAFT polymerization. Reproduced from ref (217) with permission. Copyright 2023 Royal Society of Chemistry.
Conclusion
Author Information
- Yohann Guillaneuf - Aix-Marseille Université, CNRS, Institut de Chimie Radicalaire, UMR 7273, F-13397 Marseille, France;
 https://orcid.org/0000-0002-5390-1553;
https://orcid.org/0000-0002-5390-1553;
- Catherine Lefay - Aix-Marseille Université, CNRS, Institut de Chimie Radicalaire, UMR 7273, F-13397 Marseille, France;https://orcid.org/0000-0001-8989-2962;
- Franck D’Agosto - Universite Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5128, Catalysis, Polymerization, Processes and Materials, 69616 Villeurbanne, France;https://orcid.org/0000-0003-2730-869X
- Muriel Lansalot - Universite Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5128, Catalysis, Polymerization, Processes and Materials, 69616 Villeurbanne, France;https://orcid.org/0000-0001-9010-6746
- Christopher M. Bates - Materials Research Laboratory, University of California, Santa Barbara, California 93106, United States; Materials Department, University of California, Santa Barbara, California 93106, United States;https://orcid.org/0000-0002-1598-794X
- Steven Labalme - Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States;https://orcid.org/0009-0009-7163-168X
- Jeremiah A. Johnson - Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States;https://orcid.org/0000-0001-9157-6491
- Jia Niu - Department of Chemistry, Boston College, Chestnut Hill, Massachusetts 02467, United States;https://orcid.org/0000-0002-5622-6362
- Julien Nicolas - Université Paris-Saclay, CNRS, Institut Galien Paris-Saclay, F-91400 Orsay, France;https://orcid.org/0000-0002-1597-0873
Acknowledgments
We thank the French National Research Agency (ANR-22-CE06-0017 and ANR-23-CE06-0009) for the PhD funding of Bastien Luzel and Sophia Kouider. The Centre National de la Recherche Scientifique and Aix-Marseille Université are acknowledged for financial support. C.M.B. acknowledges support from the National Science Foundation Award No. DMR-2348679. Some results described in this review article have received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement No. 771829) and from the Agence Nationale de la Recherche (Grant No. ANR-18-CE06-0014 CKAPART).
References
This article references 278 other publications.
- 1Braunecker, W.; Matyjaszewski, K. Controlled/Living Radical Polymerization: Features, Developments, and Perspectives. Prog. Polym. Sci. 2007, 32 (1), 93– 146, DOI: 10.1016/j.progpolymsci.2006.11.002Google ScholarThere is no corresponding record for this reference.
- 2Corrigan, N.; Jung, K.; Moad, G.; Hawker, C.; Matyjaszewski, K.; Boyer, C. Reversible-Deactivation Radical Polymerization (Controlled/Living Radical Polymerization): From Discovery to Materials Design and Applications. Prog. Polym. Sci. 2020, 111, 101311, DOI: 10.1016/j.progpolymsci.2020.101311Google ScholarThere is no corresponding record for this reference.
- 3Perrier, S. 50th Anniversary Perspective: RAFT Polymerization-A User Guide. Macromolecules 2017, 50 (19), 7433– 7447, DOI: 10.1021/acs.macromol.7b00767Google ScholarThere is no corresponding record for this reference.
- 4Moad, G.; Rizzardo, E.; Thang, S. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58 (6), 379– 410, DOI: 10.1071/CH05072Google ScholarThere is no corresponding record for this reference.
- 5Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-Mediated Polymerization. Prog. Polym. Sci. 2013, 38 (1), 63– 235, DOI: 10.1016/j.progpolymsci.2012.06.002Google ScholarThere is no corresponding record for this reference.
- 6Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101 (9), 2921– 2990, DOI: 10.1021/cr940534gGoogle ScholarThere is no corresponding record for this reference.
- 7Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45 (10), 4015– 4039, DOI: 10.1021/ma3001719Google ScholarThere is no corresponding record for this reference.
- 8Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-Catalyzed Living Radical Polymerization. Chem. Rev. 2001, 101 (12), 3689– 3745, DOI: 10.1021/cr9901182Google ScholarThere is no corresponding record for this reference.
- 9Pan, X.; Tasdelen, M.; Laun, J.; Junkers, T.; Yagci, Y.; Matyjaszewski, K. Photomediated Controlled Radical Polymerization. Prog. Polym. Sci. 2016, 62, 73– 125, DOI: 10.1016/j.progpolymsci.2016.06.005Google ScholarThere is no corresponding record for this reference.
- 10Chen, M.; Zhong, M.; Johnson, J. Light-Controlled Radical Polymerization: Mechanisms, Methods, and Applications. Chem. Rev. 2016, 116 (17), 10167– 10211, DOI: 10.1021/acs.chemrev.5b00671Google ScholarThere is no corresponding record for this reference.
- 11Blasco, E.; Sims, M.; Goldmann, A.; Sumerlin, B.; Barner-Kowollik, C. 50th Anniversary Perspective: Polymer Functionalization. Macromolecules 2017, 50 (14), 5215– 5252, DOI: 10.1021/acs.macromol.7b00465Google ScholarThere is no corresponding record for this reference.
- 12Iha, R.; Wooley, K.; Nyström, A.; Burke, D.; Kade, M.; Hawker, C. Applications of Orthogonal “Click” Chemistries in the Synthesis of Functional Soft Materials. Chem. Rev. 2009, 109 (11), 5620– 5686, DOI: 10.1021/cr900138tGoogle ScholarThere is no corresponding record for this reference.
- 13Günay, K.; Theato, P.; Klok, H. Standing on the Shoulders of Hermann Staudinger: Post-Polymerization Modification from Past to Present. J. Polym. Sci., Part A: Polym. Chem. 2013, 51 (1), 1– 28, DOI: 10.1002/pola.26333Google ScholarThere is no corresponding record for this reference.
- 14Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and Chemical Recycling of Solid Plastic Waste. Waste Management 2017, 69, 24– 58, DOI: 10.1016/j.wasman.2017.07.044Google ScholarThere is no corresponding record for this reference.
- 15Al-Salem, S.; Lettieri, P.; Baeyens, J. Recycling and Recovery Routes of Plastic Solid Waste (PSW): A Review. Waste Management 2009, 29 (10), 2625– 2643, DOI: 10.1016/j.wasman.2009.06.004Google ScholarThere is no corresponding record for this reference.
- 16Geyer, R.; Jambeck, J. R.; Law, K. L. Production, Use, and Fate of All Plastics Ever Made. Sci. Adv. 2017, 3, e1700782 DOI: 10.1126/sciadv.1700782Google ScholarThere is no corresponding record for this reference.
- 17Lefay, C.; Guillaneuf, Y. Recyclable/Degradable Materials via the Insertion of Labile/Cleavable Bonds Using a Comonomer Approach. Prog. Polym. Sci. 2023, 147, 101764, DOI: 10.1016/j.progpolymsci.2023.101764Google ScholarThere is no corresponding record for this reference.
- 18Tardy, A.; Nicolas, J.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. Radical Ring-Opening Polymerization: Scope, Limitations, and Application to (Bio)Degradable Materials. Chem. Rev. 2017, 117 (3), 1319– 1406, DOI: 10.1021/acs.chemrev.6b00319Google ScholarThere is no corresponding record for this reference.
- 19Colombani, D. Driving Forces in Free Radical Addition-Fragmentation Processes. Prog. Polym. Sci. 1999, 24 (3), 425– 480, DOI: 10.1016/S0079-6700(99)00005-2Google ScholarThere is no corresponding record for this reference.
- 20Errede, L. A. The Chemistry of Xylylenes. X. Some Polymers and Telomers of Spiro-Di-o-Xylylene. J. Polym. Sci. 1961, 49, 253, DOI: 10.1002/pol.1961.1204915209Google ScholarThere is no corresponding record for this reference.
- 21Van Volkenburgh, R.; Greenlee, K. W.; Derfer, J. M.; Boord, C. E. A Synthesis of Vinylcyclopropane. J. Am. Chem. Soc. 1949, 71 (11), 3595– 3597, DOI: 10.1021/ja01179a004Google ScholarThere is no corresponding record for this reference.
- 22Takahashi, T.; Yamashita, I. 1,5-Polymerization of Vinylcyclopropane. Journal of Polymer Science Part B: Polymer Letters 1965, 3 (4), 251– 255, DOI: 10.1002/pol.1965.110030403Google ScholarThere is no corresponding record for this reference.
- 23Endo, T.; Bailey, W. J. Radical Ring-Opening Polymerization of 3,9-Dimethylene-1,5,7,11-Tetraoxaspiro-[5,5] Undecane. Journal of Polymer Science: Polymer Letters Edition 1975, 13 (4), 193– 195, DOI: 10.1002/pol.1975.130130401Google ScholarThere is no corresponding record for this reference.
- 24Endo, T.; Bailey, W. J. Synthesis and Radical Ring-Opening Polymerization of 2-Methylene-1,4,6-Trioxaspiro[4,4] Nonane. Journal of Polymer Science: Polymer Letters Edition 1980, 18 (1), 25– 27, DOI: 10.1002/pol.1980.130180106Google ScholarThere is no corresponding record for this reference.
- 25Lalande, R.; Maillard, B.; Cazaux, M. Addition radicalaire du dioxolanne-1,3 a l’octene-1. Tetrahedron Lett. 1969, 10 (9), 745– 748, DOI: 10.1016/S0040-4039(01)87798-4Google ScholarThere is no corresponding record for this reference.
- 26Bailey, W. J.; Ni, Z.; Wu, S.-R. Synthesis of Poly-r-Caprolactone via a Free Radical Mechanism. Free Radical Ring-Opening Polymerization of 2-Methylene- 1,3-Dioxepane. Journal of Polymer Science: Polymer Chemistry Edition 1982, 20, 3021– 3030, DOI: 10.1002/pol.1982.170201101Google ScholarThere is no corresponding record for this reference.
- 27Cho, I.; Kim, J.-B. Exploratory Ring-Opening Polymerization. VIII. Radical Ring-Opening Polymerization of 2-Phenyl-3-Vinyloxirane: A C–C Bond Scission Polymerization of the Epoxide Ring. Journal of Polymer Science: Polymer Letters Edition 1983, 21 (6), 433– 436, DOI: 10.1002/pol.1983.130210605Google ScholarThere is no corresponding record for this reference.
- 28Bailey, W. J. Free Radical Ring-Opening Polymerization. Polym. J. 1985, 17, 85, DOI: 10.1295/polymj.17.85Google ScholarThere is no corresponding record for this reference.
- 29Cho, I.; Kim, S.-K.; Lee, M.-H. Exploratory Ring-Opening Polymerization. XIIL Ring-Opening Polymerization of 2-Vinyl Cyclic Sulfones. Journal of Polymer Science: Polymer Symposia 1986, 74 (1), 219– 226, DOI: 10.1002/polc.5070740119Google ScholarThere is no corresponding record for this reference.
- 30Bailey, W. J.; Chou, J. L.; Feng, P.-Z.; Issari, B.; Kuruganti, V.; Zhou, L.-L. Recent Advances in Free-Radical Ring-Opening Polymerization. Journal of Macromolecular Science: Part A - Chemistry 1988, 25 (5–7), 781– 798, DOI: 10.1080/00222338808053398Google ScholarThere is no corresponding record for this reference.
- 31Evans, R. A.; Moad, G.; Rizzardo, E.; Thang, S. H. New Free-Radical Ring-Opening Acrylate Monomers. Macromolecules 1994, 27 (26), 7935– 7937, DOI: 10.1021/ma00104a062Google ScholarThere is no corresponding record for this reference.
- 32Evans, R. A.; Rizzardo, E. Free-Radical Ring-Opening Polymerization of Cyclic Allylic Sulfides. Macromolecules 1996, 29 (22), 6983– 6989, DOI: 10.1021/ma960573pGoogle ScholarThere is no corresponding record for this reference.
- 33Tang, H.; Tsarevsky, N. V. Lipoates as Building Blocks of Sulfur-Containing Branched Macromolecules. Polym. Chem. 2015, 6 (39), 6936– 6945, DOI: 10.1039/C5PY01005EGoogle ScholarThere is no corresponding record for this reference.
- 34Albanese, K. R.; Morris, P. T.; Read De Alaniz, J.; Bates, C. M.; Hawker, C. J. Controlled-Radical Polymerization of α-Lipoic Acid: A General Route to Degradable Vinyl Copolymers. J. Am. Chem. Soc. 2023, 145 (41), 22728– 22734, DOI: 10.1021/jacs.3c08248Google ScholarThere is no corresponding record for this reference.
- 35Huang, H.; Sun, B.; Huang, Y.; Niu, J. Radical Cascade-Triggered Controlled Ring-Opening Polymerization of Macrocyclic Monomers. J. Am. Chem. Soc. 2018, 140 (33), 10402– 10406, DOI: 10.1021/jacs.8b05365Google ScholarThere is no corresponding record for this reference.
- 36Bingham, N. M.; Roth, P. J. Degradable Vinyl Copolymers through Thiocarbonyl Addition-Ring-Opening (TARO) Polymerization. Chem. Commun. 2019, 55 (1), 55– 58, DOI: 10.1039/C8CC08287AGoogle ScholarThere is no corresponding record for this reference.
- 37Smith, R. A.; Fu, G. Y.; McAteer, O.; Xu, M. Z.; Gutekunst, W. R. Radical Approach to Thioester-Containing Polymers. J. Am. Chem. Soc. 2019, 141 (4), 1446– 1451, DOI: 10.1021/jacs.8b12154Google ScholarThere is no corresponding record for this reference.
- 38Jackson, A. W. Reversible-Deactivation Radical Polymerization of Cyclic Ketene Acetals. Polym. Chem. 2020, 11 (21), 3525– 3545, DOI: 10.1039/D0PY00446DGoogle ScholarThere is no corresponding record for this reference.
- 39Agarwal, S. Chemistry, Chances and Limitations of the Radical Ring-Opening Polymerization of Cyclic Ketene Acetals for the Synthesis of Degradable Polyesters. Polym. Chem. 2010, 1, 953– 964, DOI: 10.1039/c0py00040jGoogle ScholarThere is no corresponding record for this reference.
- 40Deng, Y.; Mehner, F.; Gaitzsch, J. Current Standing on Radical Ring-Opening Polymerizations of Cyclic Ketene Acetals as Homopolymers and Copolymers with One Another. Macromol. Rapid Commun. 2023, 44, e2200941 DOI: 10.1002/marc.202200941Google ScholarThere is no corresponding record for this reference.
- 41Paulusse, J. M. J.; Amir, R. J.; Evans, R. A.; Hawker, C. J. Free Radical Polymers with Tunable and Selective Bio- and Chemical Degradability. J. Am. Chem. Soc. 2009, 131 (28), 9805– 9812, DOI: 10.1021/ja903245pGoogle ScholarThere is no corresponding record for this reference.
- 42Zhang, S.; Wang, Y.; Huang, H.; Cao, D. A Strategy for Controlling the Polymerizations of Thiyl Radical Propagation by RAFT Agents. Angew. Chem. Int. Ed 2023, 62 (37), e202308524 DOI: 10.1002/anie.202308524Google ScholarThere is no corresponding record for this reference.
- 43Hu, H.; Yi, P.; He, J.; Cao, D.; Huang, H. Controlling Thiyl Radical Polymerization via in Situ Desulfurization. Polym. Chem. 2025, 16 (2), 174– 180, DOI: 10.1039/D4PY01140FGoogle ScholarThere is no corresponding record for this reference.
- 44Wang, Y.; Du, J.; Huang, H. Reversible Thiyl Radical Addition-Fragmentation Chain Transfer Polymerization. Angew. Chem. Int. Ed. 2024, 63 (12). DOI: 10.1002/anie.202318898 .Google ScholarThere is no corresponding record for this reference.
- 45Stockmayer, W. H.; Howard, R. O.; Clarke, J. T. Copolymerization of Vinyl Acetate with a Cyclic Disulfide. J. Am. Chem. Soc. 1953, 75 (7), 1756– 1757, DOI: 10.1021/ja01103a528Google ScholarThere is no corresponding record for this reference.
- 46Tobolsky, A. V.; Baysal, B. The Reaction between Styrene and Ring Disulfides: Copolymerization Effected by the Chain Transfer Reaction. J. Am. Chem. Soc. 1953, 75 (7), 1757– 1757, DOI: 10.1021/ja01103a529Google ScholarThere is no corresponding record for this reference.
- 47Nambu, Y.; Acar, M. H.; Suzuki, T.; Endo, T. Thermal and Photoinitiated Copolymerization of the Cyclic Disulfide Lipoamide with Styrene. Makromol. Chem. 1988, 189 (3), 495– 500, DOI: 10.1002/macp.1988.021890302Google ScholarThere is no corresponding record for this reference.
- 48Suzuki, T.; Nambu, Y.; Endo, T. Radical Copolymerization of Lipoamide with Vinyl Monomers. Macromolecules 1990, 23 (6), 1579– 1582, DOI: 10.1021/ma00208a004Google ScholarThere is no corresponding record for this reference.
- 49Albanese, K. R.; Read De Alaniz, J.; Hawker, C. J.; Bates, C. M. From Health Supplement to Versatile Monomer: Radical Ring-Opening Polymerization and Depolymerization of α-Lipoic Acid. Polymer 2024, 304, 127167, DOI: 10.1016/j.polymer.2024.127167Google ScholarThere is no corresponding record for this reference.
- 50Mothe, S. R.; Chennamaneni, L. R.; Tan, J.; Lim, F. C. H.; Zhao, W.; Thoniyot, P. A Mechanistic Study on the Hydrolysis of Cyclic Ketene Acetal (CKA) and Proof of Concept of Polymerization in Water. Macro Chemistry & Physics 2023, 224 (19), 2300221, DOI: 10.1002/macp.202300221Google ScholarThere is no corresponding record for this reference.
- 51McElvain, S. M.; Curry, M. J. Ketene Acetals. XIX. 2-Methylene-1,3-Dioxolanes and 1,3-Dioxanes. J. Am. Chem. Soc. 1948, 70 (11), 3781– 3786, DOI: 10.1021/ja01191a071Google ScholarThere is no corresponding record for this reference.
- 52Tran, J.; Guégain, E.; Ibrahim, N.; Harrisson, S.; Nicolas, J. Efficient Synthesis of 2-Methylene-4-Phenyl-1,3-Dioxolane, a Cyclic Ketene Acetal for Controlling the NMP of Methyl Methacrylate and Conferring Tunable Degradability. Polym. Chem. 2016, 7 (26), 4427– 4435, DOI: 10.1039/C6PY00778CGoogle ScholarThere is no corresponding record for this reference.
- 53Folini, J.; Murad, W.; Mehner, F.; Meier, W.; Gaitzsch, J. Updating Radical Ring-Opening Polymerisation of Cyclic Ketene Acetals from Synthesis to Degradation. Eur. Polym. J. 2020, 134, 109851, DOI: 10.1016/j.eurpolymj.2020.109851Google ScholarThere is no corresponding record for this reference.
- 54Folini, J.; Huang, C.-H.; Anderson, J. C.; Meier, W. P.; Gaitzsch, J. Novel Monomers in Radical Ring-Opening Polymerisation for Biodegradable and pH Responsive Nanoparticles. Polym. Chem. 2019, 10 (39), 5285– 5288, DOI: 10.1039/C9PY01103JGoogle ScholarThere is no corresponding record for this reference.
- 55Petasis, N. A.; Lu, S.-P. Methylenations of Heteroatom-Substituted Carbonyls with Dimethyl Titanocene. Tetrahedron Lett. 1995, 36 (14), 2393– 2396, DOI: 10.1016/0040-4039(95)00320-CGoogle ScholarThere is no corresponding record for this reference.
- 56Deng, Y.; Frezel, A.; Mehner, F.; Friedel, P.; Gaitzsch, J. Amine-Bearing Cyclic Ketene Acetals for pH-Responsive and Degradable Polyesters through Radical Ring-Opening Polymerisation. Polym. Chem. 2023, 14 (37), 4275– 4281, DOI: 10.1039/D3PY00929GGoogle ScholarThere is no corresponding record for this reference.
- 57Jiang, N.-C.; Zhou, Z.; Niu, J. Quantitative, Regiospecific, and Stereoselective Radical Ring-Opening Polymerization of Monosaccharide Cyclic Ketene Acetals. J. Am. Chem. Soc. 2024, 146 (8), 5056– 5062, DOI: 10.1021/jacs.3c14244Google ScholarThere is no corresponding record for this reference.
- 58Ko, Y.-J.; Shim, S.-B.; Shin, J.-H. Facile Synthesis of 2- O -Iodoacetyl Protected Glycosyl Iodides: Useful Precursors of 1→2-Linked 1,2- Trans -Glycosides. Org. Lett. 2009, 11 (3), 609– 612, DOI: 10.1021/ol8026472Google ScholarThere is no corresponding record for this reference.
- 59Sznaidman, M. L.; Johnson, S. C.; Crasto, C.; Hecht, S. M. Carbohydrate Cyclic Ketene Acetals. J. Org. Chem. 1995, 60 (13), 3942– 3943, DOI: 10.1021/jo00118a006Google ScholarThere is no corresponding record for this reference.
- 60Hardy, C.; Levere, M. E.; Kociok-Köhn, G.; Buchard, A. Radical Ring Opening Polymerization of Cyclic Ketene Acetals Derived From D-Glucal. ACS Macro Lett. 2023, 12 (11), 1443– 1449, DOI: 10.1021/acsmacrolett.3c00397Google ScholarThere is no corresponding record for this reference.
- 61Evans, R. A.; Rizzardo, E. Free Radical Ring-Opening Polymerization of Cyclic Allylic Sulfides: Liquid Monomers with Low Polymerization Volume Shrinkage. J. Polym. Sci. A Polym. Chem. 2001, 39 (1), 202– 215, DOI: 10.1002/1099-0518(20010101)39:1<202::AID-POLA230>3.0.CO;2-6Google ScholarThere is no corresponding record for this reference.
- 62Chaumont, P.; Asgarzadeh, F.; Colombani, D.; Arotcarena, M.; Baudouin, A. Synthesis, Characterization and Hydrolysis of Poly[Styrene-Co-(6-Methylene-1,4-Oxathiepane-7-One)] and Poly[Styrene-Co-(6-Methylene-5-Methyl-1,4-Oxathiepane-7-One)]. Macromol. Chem. Phys. 1998, 199 (11), 2577– 2582, DOI: 10.1002/macp.1998.021991128Google ScholarThere is no corresponding record for this reference.
- 63Phelan, M.; Aldabbagh, F.; Zetterlund, P. B.; Yamada, B. Mechanism and Kinetics of the Free Radical Ring-Opening Polymerization of Eight-Membered Cyclic Allylic Disulfide Monomers. Macromolecules 2005, 38 (6), 2143– 2147, DOI: 10.1021/ma047894iGoogle ScholarThere is no corresponding record for this reference.
- 64Sbordone, F.; Veskova, J.; Richardson, B.; Do, P.; Micallef, A.; Frisch, H. Embedding Peptides into Synthetic Polymers: Radical Ring-Opening Copolymerization of Cyclic Peptides. J. Am. Chem. Soc. 2023, 145 (11), 6221– 6229, DOI: 10.1021/jacs.2c12517Google ScholarThere is no corresponding record for this reference.
- 65Do, P.; Sbordone, F.; Kalmer, H.; Sokolova, A.; Zhang, C.; Thai, L.; Golberg, D.; Chapman, R.; Poad, B.; Frisch, H. Main Chain Selective Polymer Degradation: Controlled by the Wavelength and Assembly. Chemical Science 2024, 15 (31), 12410– 12419, DOI: 10.1039/D4SC02172JGoogle ScholarThere is no corresponding record for this reference.
- 66Huang, H.; Wang, W.; Zhou, Z.; Sun, B.; An, M.; Haeffner, F.; Niu, J. Radical Ring-Closing/Ring-Opening Cascade Polymerization. J. Am. Chem. Soc. 2019, 141 (32), 12493– 12497, DOI: 10.1021/jacs.9b05568Google ScholarThere is no corresponding record for this reference.
- 67Zhang, S.; Cao, C.; Jiang, S.; Huang, H. A General Strategy for Radical Ring-Opening Polymerization of Macrocyclic Allylic Sulfides. Macromolecules 2022, 55 (21), 9411– 9419, DOI: 10.1021/acs.macromol.2c01636Google ScholarThere is no corresponding record for this reference.
- 68Meijs, G. F.; Rizzardo, E.; Le, T. P. T.; Chen, Y.-C. Influence of Thionoesters on the Degree of Polymerization of Styrene, Meth. Makromol. Chem. 1992, 193 (2), 369– 378, DOI: 10.1002/macp.1992.021930209Google ScholarThere is no corresponding record for this reference.
- 69Luzel, B.; Gil, N.; Désirée, P.; Monot, J.; Bourissou, D.; Siri, D.; Gigmes, D.; Martin-Vaca, B.; Lefay, C.; Guillaneuf, Y. Development of an Efficient Thionolactone for Radical Ring-Opening Polymerization by a Combined Theoretical/Experimental Approach. J. Am. Chem. Soc. 2023, 145 (50), 27437– 27449, DOI: 10.1021/jacs.3c08610Google ScholarThere is no corresponding record for this reference.
- 70Ko, K.; Lundberg, D.; Johnson, A.; Johnson, J. Mechanism-Guided Discovery of Cleavable Comonomers for Backbone Deconstructable Poly(Methyl Methacrylate). J. Am. Chem. Soc. 2024, 146 (13), 9142– 9154, DOI: 10.1021/jacs.3c14554Google ScholarThere is no corresponding record for this reference.
- 71Plummer, C. M.; Gil, N.; Dufils, P. E.; Wilson, D. J.; Lefay, C.; Gigmes, D.; Guillaneuf, Y. Mechanistic Investigation of Epsilon-Thiono-Caprolactone Radical Polymerization: An Interesting Tool to Insert Weak Bonds into Poly(Vinyl Esters). Acs Applied Polymer Materials 2021, 3 (6), 3264– 3271, DOI: 10.1021/acsapm.1c00569Google ScholarThere is no corresponding record for this reference.
- 72Ivanchenko, O.; Authesserre, U.; Coste, G.; Mazières, S.; Destarac, M.; Harrisson, S. ε-Thionocaprolactone: An Accessible Monomer for Preparation of Degradable Poly(Vinyl Esters) by Radical Ring-Opening Polymerization. Polym. Chem. 2021, 12 (13), 1931– 1938, DOI: 10.1039/D1PY00080BGoogle ScholarThere is no corresponding record for this reference.
- 73Kamiki, R.; Kubo, T.; Satoh, K. Addition-Fragmentation Ring-Opening Polymerization of Bio-Based Thiocarbonyl L-Lactide for Dual Degradable Vinyl Copolymers. Macromol. Rapid Commun. 2023, 44 (2), 2200537, DOI: 10.1002/marc.202200537Google ScholarThere is no corresponding record for this reference.
- 74Ivanchenko, O.; Mazières, S.; Harrisson, S.; Destarac, M. Lactide-Derived Monomers for Radical Thiocarbonyl Addition Ring-Opening Copolymerisation. Polym. Chem. 2022, 13 (39), 5525– 5529, DOI: 10.1039/D2PY00893AGoogle ScholarThere is no corresponding record for this reference.
- 75Ivanchenko, O.; Mazières, S.; Mallet-Ladeira, S.; Harrisson, S.; Destarac, M. Radical Copolymerization of Thionoglycolide and Derivatives for Preparation of Degradable Vinyl Polymers. Macromolecules 2024, 57 (16), 8059– 8066, DOI: 10.1021/acs.macromol.4c01259Google ScholarThere is no corresponding record for this reference.
- 76Brendel, J.; Schmidt, W.; Below, P. 2’-Substituted 1,1’-Biphenyl-2- Carboxamides, Processes for Their Preparation, Their Use as Medicaments, and Pharmaceutical Preparations Comprising Them. US 6,531,495 B1, March 11, 2003.Google ScholarThere is no corresponding record for this reference.
- 77Johnson, J. A.; Cardoso Da Costa, L. C.; Prince, E. Cleavable Comonomer Strategy for Accelerating Removal of Gel Nail Polish. WO2024119140A1, June 6, 2024.Google ScholarThere is no corresponding record for this reference.
- 78Kiel, G. R.; Lundberg, D. J.; Prince, E.; Husted, K. E. L.; Johnson, A. M.; Lensch, V.; Li, S.; Shieh, P.; Johnson, J. A. Cleavable Comonomers for Chemically Recyclable Polystyrene: A General Approach to Vinyl Polymer Circularity. J. Am. Chem. Soc. 2022, 144 (28), 12979– 12988, DOI: 10.1021/jacs.2c05374Google ScholarThere is no corresponding record for this reference.
- 79Luzel, B.; Raggio, L.; Benharrous, E.; Monot, J.; Bourissou, D.; Siri, D.; Gigmes, D.; Lefay, C.; Martin-Vaca, B.; Guillaneuf, Y. Closer Look at the Substituent Effects on the Copolymerization of Thionolactones by Radical Ring-Opening Polymerization. Macromolecules 2025, 58 (9), 4627– 4635, DOI: 10.1021/acs.macromol.5c00448Google ScholarThere is no corresponding record for this reference.
- 80Schomaker, J. M.; Travis, B. R.; Borhan, B. Direct Lactonization of Alkenols via Osmium Tetroxide-Mediated Oxidative Cleavage. Org. Lett. 2003, 5 (17), 3089– 3092, DOI: 10.1021/ol035057fGoogle ScholarThere is no corresponding record for this reference.
- 81Machado, T. O.; Stubbs, C. J.; Chiaradia, V.; Alraddadi, M. A.; Brandolese, A.; Worch, J. C.; Dove, A. P. A Renewably Sourced, Circular Photopolymer Resin for Additive Manufacturing. Nature 2024, 629 (8014), 1069– 1074, DOI: 10.1038/s41586-024-07399-9Google ScholarThere is no corresponding record for this reference.
- 82Tardy, A.; Gil, N.; Plummer, C. M.; Siri, D.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. Polyesters by a Radical Pathway: Rationalization of the Cyclic Ketene Acetal Efficiency. Angew. Chem. Int. Ed 2020, 59 (34), 14517– 14526, DOI: 10.1002/anie.202005114Google ScholarThere is no corresponding record for this reference.
- 83Ochiai, B.; Endo, T. Computational Evaluation of Radical Ring-Opening Polymerization. J. Pol. Sci.: Part A: Polym. Chem. 2007, 45, 2827– 2834, DOI: 10.1002/pola.22039Google ScholarThere is no corresponding record for this reference.
- 84Reddy Mothe, S.; Tan, J. S. J.; Chennamaneni, L. R.; Aidil, F.; Su, Y.; Kang, H. C.; Lim, F. C. H.; Thoniyot, P. A Systematic Investigation of the Ring Size Effects on the Free Radical Ring-Opening Polymerization (rROP) of Cyclic Ketene Acetal (CKA) Using Both Experimental and Theoretical Approach. J. Polym. Sci. 2020, 58 (12), 1728– 1738, DOI: 10.1002/pol.20200210Google ScholarThere is no corresponding record for this reference.
- 85Jin, S.; Gonsalves, K. E. A Study of the Mechanism of the Free-Radical Ring-Opening Polymerization of 2-Methylene-1,3-Dioxepane. Macromolecules 1997, 30 (10), 3104– 3106, DOI: 10.1021/ma961590hGoogle ScholarThere is no corresponding record for this reference.
- 86Agarwal, S.; Speyerer, C. Degradable Blends of Semi-Crystalline and Amorphous Branched Poly(Caprolactone): Effect of Microstructure on Blend Properties. Polymer 2010, 51 (5), 1024– 1032, DOI: 10.1016/j.polymer.2010.01.020Google ScholarThere is no corresponding record for this reference.
- 87Tardy, A.; Honoré, J.-C.; Siri, D.; Nicolas, J.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. A Comprehensive Kinetic Study of the Conventional Free-Radical Polymerization of Seven-Membered Cyclic Ketene Acetals. Polym. Chem. 2017, 8 (34), 5139– 5147, DOI: 10.1039/C7PY00337DGoogle ScholarThere is no corresponding record for this reference.
- 88Tardy, A.; Delplace, V.; Siri, D.; Lefay, C.; Harrisson, S.; De Fatima Albergaria Pereira, B.; Charles, L.; Gigmes, D.; Nicolas, J.; Guillaneuf, Y. Scope and Limitations of the Nitroxide-Mediated Radical Ring-Opening Polymerization of Cyclic Ketene Acetals. Polym. Chem. 2013, 4 (17), 4776, DOI: 10.1039/c3py00719gGoogle ScholarThere is no corresponding record for this reference.
- 89Delplace, V.; Tardy, A.; Harrisson, S.; Mura, S.; Gigmes, D.; Guillaneuf, Y.; Nicolas, J. Degradable and Comb-Like PEG-Based Copolymers by Nitroxide-Mediated Radical Ring-Opening Polymerization. Biomacromolecules 2013, 14 (10), 3769– 3779, DOI: 10.1021/bm401157gGoogle ScholarThere is no corresponding record for this reference.
- 90Delplace, V.; Harrisson, S.; Tardy, A.; Gigmes, D.; Guillaneuf, Y.; Nicolas, J. Nitroxide-Mediated Radical Ring-Opening Copolymerization: Chain-End Investigation and Block Copolymer Synthesis. Macromol. Rapid Commun. 2014, 35 (4), 484– 491, DOI: 10.1002/marc.201300809Google ScholarThere is no corresponding record for this reference.
- 91Delplace, V.; Guégain, E.; Harrisson, S.; Gigmes, D.; Guillaneuf, Y.; Nicolas, J. A Ring to Rule Them All: A Cyclic Ketene Acetal Comonomer Controls the Nitroxide-Mediated Polymerization of Methacrylates and Confers Tunable Degradability. Chem. Commun. 2015, 51 (64), 12847– 12850, DOI: 10.1039/C5CC04610FGoogle ScholarThere is no corresponding record for this reference.
- 92Albergaria Pereira, B. de F.; Tardy, A.; Monnier, V.; Guillaneuf, Y.; Gigmes, D.; Charles, L. Elucidation of a Side Reaction Occurring during Nitroxide-Mediated Polymerization of Cyclic Ketene Acetals by Tandem Mass Spectrometric End-Group Analysis of Aliphatic Polyesters. Rapid Commun. Mass Spectrom. 2015, 29 (23), 2302– 2308, DOI: 10.1002/rcm.7397Google ScholarThere is no corresponding record for this reference.
- 93Jackson, A. W.; Chennamaneni, L. R.; Thoniyot, P. Main-Chain Degradable, pH-Responsive and Covalently Cross-Linked Nanoparticles via a One-Step RAFT-Based Radical Ring-Opening Terpolymerization. Eur. Polym. J. 2020, 122, 109391, DOI: 10.1016/j.eurpolymj.2019.109391Google ScholarThere is no corresponding record for this reference.
- 94Jackson, A. W.; Chennamaneni, L. R.; Mothe, S. R.; Thoniyot, P. A General Strategy for Degradable Single-Chain Nanoparticles via Cross-Linker Mediated Chain Collapse of Radical Copolymers. Chem. Commun. 2020, 56 (68), 9838– 9841, DOI: 10.1039/D0CC03792CGoogle ScholarThere is no corresponding record for this reference.
- 95Wais, U.; Chennamaneni, L. R.; Thoniyot, P.; Zhang, H.; Jackson, A. W. Main-Chain Degradable Star Polymers Comprised of pH-Responsive Hyperbranched Cores and Thermoresponsive Polyethylene Glycol-Based Coronas. Polym. Chem. 2018, 9 (39), 4824– 4839, DOI: 10.1039/C8PY01113CGoogle ScholarThere is no corresponding record for this reference.
- 96Jiang, S.; Huang, H. Oxygen-Initiated Radical Ring-Opening Polymerization of Macrocyclic Allylic Sulfides under Ambient Conditions. Polymer 2024, 303, 127106, DOI: 10.1016/j.polymer.2024.127106Google ScholarThere is no corresponding record for this reference.
- 97Spick, M. P.; Bingham, N. M.; Li, Y.; de Jesus, J.; Costa, C.; Bailey, M. J.; Roth, P. J. Fully Degradable Thioester-Functional Homo- and Alternating Copolymers Prepared through Thiocarbonyl Addition-Ring-Opening RAFT Radical Polymerization. Macromolecules 2020, 53 (2), 539– 547, DOI: 10.1021/acs.macromol.9b02497Google ScholarThere is no corresponding record for this reference.
- 98Rix, M.; Higgs, S.; Dodd, E.; Coles, S.; Bingham, N. M.; Roth, P. J. Insertion of Degradable Thioester Linkages into Styrene and Methacrylate Polymers. ChemRxiv , 2022, DOI: 10.26434/chemrxiv-2022-cdt52 .Google ScholarThere is no corresponding record for this reference.
- 99Kazmi, T.; Hepburn, K. S.; Nisa, Q.; Neogi, S.; Bingham, N. M.; Roth, P. J. Single-Unit Comonomer Insertion Initiates Radical Polymerization of Thionolactone to Give Chemically and Thermally Degradable Polythioesters. Macromolecules 2025, 58 (18), 9617– 9628, DOI: 10.1021/acs.macromol.5c01418Google ScholarThere is no corresponding record for this reference.
- 100Prebihalo, E. A.; Luke, A. M.; Reddi, Y.; LaSalle, C. J.; Shah, V. M.; Cramer, C. J.; Reineke, T. M. Radical Ring-Opening Polymerization of Sustainably-Derived Thionoisochromanone. Chem. Sci. 2023, 14 (21), 5689– 5698, DOI: 10.1039/D2SC06040JGoogle ScholarThere is no corresponding record for this reference.
- 101Raeisi, M.; Tsarevsky, N. V. Radical Ring-opening Polymerization of Lipoates: Kinetic and Thermodynamic Aspects. J. Polym. Sci. 2021, 59 (8), 675– 684, DOI: 10.1002/pol.20200765Google ScholarThere is no corresponding record for this reference.
- 102Lee, K.; Han, S.; Morris, P. T.; Bates, C. M.; Hawker, C. J.; Boyer, C. Controlled Synthesis of Lipoate Homopolymers via Reversible Addition-Fragmentation Chain Transfer Polymerization. J. Am. Chem. Soc. 2025, 147 (42), 38796– 38806, DOI: 10.1021/jacs.5c14377Google ScholarThere is no corresponding record for this reference.
- 103Gigmes, D.; Van Steenberge, P. H. M.; Siri, D.; D’Hooge, D. R.; Guillaneuf, Y.; Lefay, C. Simulation of the Degradation of Cyclic Ketene Acetal and Vinyl-Based Copolymers Synthesized via a Radical Process: Influence of the Reactivity Ratios on the Degradability Properties. Macromol. Rapid Commun. 2018, 39 (19), 1800193, DOI: 10.1002/marc.201800193Google ScholarThere is no corresponding record for this reference.
- 104Liausvia, F.; Rusli, W.; van Herk, A. Prediction of the Oligomer Distribution after Degradation of (Co)Polymers with Inserted Break Points. Macromol. Theory Simul. 2021, 30 (6), 2100038, DOI: 10.1002/mats.202100038Google ScholarThere is no corresponding record for this reference.
- 105Lundberg, D. J.; Ko, K.; Kilgallon, L. J.; Johnson, J. A. Defining Reactivity-Deconstructability Relationships for Copolymerizations Involving Cleavable Comonomer Additives. ACS Macro Lett. 2024, 13 (5), 521– 527, DOI: 10.1021/acsmacrolett.4c00106Google ScholarThere is no corresponding record for this reference.
- 106Callan, D. H.; Bates, C. M. Efficient Deterministic Modeling of Reversible Copolymerizations. Macromolecules 2025, 58 (19), 10289– 10300, DOI: 10.1021/acs.macromol.5c01421Google ScholarThere is no corresponding record for this reference.
- 107Wen, M.; Wong, W.; Junkers, T. Synthesis of Biodegradable Vinyl Copolymers via Enforced Regular Sequence Distribution from Automated Radical Ring Opening Polymerisation. Chem. Sci. 2025, 16 (42), 19624– 19631, DOI: 10.1039/D5SC03738GGoogle ScholarThere is no corresponding record for this reference.
- 108Bailey, W. J.; Wu, S.-R.; Ni, Z. Free Radical Ring-Opening Polymerization of 4-n-Hexyl- and 4-n-Decyl-2-Methylene-1,3-Dioxolanes. Journal of Macromolecular Science: Part A - Chemistry 1982, 18 (6), 973– 986, DOI: 10.1080/00222338208077212Google ScholarThere is no corresponding record for this reference.
- 109Bailey, W. J.; Endo, T.; Gapud, B.; Lin, Y.-N.; Ni, Z.; Pan, C.-Y.; Shaffer, S. E.; Wu, S.-R.; Yamazaki, N.; Yonezawa, K. Synthesis of Functionally-Terminated Oligomers by Free Radical Ring-Opening Polymerization. Journal of Macromolecular Science: Part A - Chemistry 1984, 21 (8–9), 979– 995, DOI: 10.1080/00222338408056586Google ScholarThere is no corresponding record for this reference.
- 110Jackson, A. W.; Mothe, S. R.; Chennamaneni, L. R.; van Herk, A.; Thoniyot, P. Unraveling the History and Revisiting the Synthesis of Degradable Polystyrene Analogues via Radical Ring-Opening Copolymerization with Cyclic Ketene Acetals. Materials 2020, 13 (10), 2325, DOI: 10.3390/ma13102325Google ScholarThere is no corresponding record for this reference.
- 111Gil, N.; Caron, B.; Siri, D.; Roche, J.; Hadiouch, S.; Khedaioui, D.; Ranque, S.; Cassagne, C.; Montarnal, D.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. Degradable Polystyrene via the Cleavable Comonomer Approach. Macromolecules 2022, 55 (15), 6680– 6694, DOI: 10.1021/acs.macromol.2c00651Google ScholarThere is no corresponding record for this reference.
- 112Wickel, H.; Agarwal, S. Synthesis and Characterization of Copolymers of 5,6-Benzo-2-Methylene-1,3-Dioxepane and Styrene. Macromolecules 2003, 36 (16), 6152– 6159, DOI: 10.1021/ma034551wGoogle ScholarThere is no corresponding record for this reference.
- 113Hiracuri, Y.; Tokiwa, Y. Synthesis of Copolymers Composed of 2-Methylene-1,3,6-Trioxocane and Vinyl Monomers and Their Enzymatic Degradation. J. Polym. Sci., Part A: Polym. Chem. 1993, 31 (12), 3159– 3163, DOI: 10.1002/pola.1993.080311233Google ScholarThere is no corresponding record for this reference.
- 114Rix, M. F. I.; Collins, K.; Higgs, S. J.; Dodd, E. M.; Coles, S. J.; Bingham, N. M.; Roth, P. J. Insertion of Degradable Thioester Linkages into Styrene and Methacrylate Polymers: Insights into the Reactivity of Thionolactones. Macromolecules 2023, 56 (23), 9787– 9795, DOI: 10.1021/acs.macromol.3c01811Google ScholarThere is no corresponding record for this reference.
- 115Okayama, Y.; Morris, P.; Albanese, K.; Olsen, S.; Mori, A.; de Alaniz, J.; Bates, C.; Hawker, C. Enhanced Degradation of Vinyl Copolymers Based on Lipoic Acid. J. Polym. Sci. 2025, 63 (6), 1345– 1351, DOI: 10.1002/pol.20241085Google ScholarThere is no corresponding record for this reference.
- 116Li, J.; Yang, H.; Jiang, Q.; Li, J.; Jiang, B.; Xue, X.; Komarneni, S.; Huang, W. A New Strategy to Synthesize Degradable Polystyrene of Ultra-High Molecular Weight. Chem. Commun. 2025, 61 (18), 3692– 3695, DOI: 10.1039/D4CC05166AGoogle ScholarThere is no corresponding record for this reference.
- 117Sun, L. F.; Zhuo, R. X.; Liu, Z. L. Synthesis and Enzymatic Degradation of 2-Methylene-1,3-Dioxepane and Methyl Acrylate Copolymers. J. Polym. Sci., Part A: Polym. Chem. 2003, 41 (18), 2898– 2904, DOI: 10.1002/pola.10868Google ScholarThere is no corresponding record for this reference.
- 118Maji, S.; Zheng, M.; Agarwal, S. Functional Degradable Polymers via Radical Ring-Opening Polymerization and Click Chemistry. Macromol. Chem. Phys. 2011, 212 (23), 2573– 2582, DOI: 10.1002/macp.201100375Google ScholarThere is no corresponding record for this reference.
- 119Sun, L.-F.; Zhuo, R.-X.; Liu, Z.-L. Studies on the Synthesis and Properties of Temperature Responsive and Biodegradable Hydrogels. Macromol. Biosci. 2003, 3 (12), 725– 728, DOI: 10.1002/mabi.200300012Google ScholarThere is no corresponding record for this reference.
- 120Huang, J.; Gil, R.; Matyjaszewski, K. Synthesis and Characterization of Copolymers of 5,6-Benzo-2-Methylene-1,3-Dioxepane and n-Butyl Acrylate. Polymer 2005, 46 (25), 11698– 11706, DOI: 10.1016/j.polymer.2005.09.048Google ScholarThere is no corresponding record for this reference.
- 121Yuan, J.-Y.; Pan, C.-Y. “Living” Free Radical Ring-Opening Copolymerization of 4,7-Dimethyl-2-Methylene-1,3-Dioxepane and Conventional Vinyl Monomers. Eur. Polym. J. 2002, 38 (10), 2069– 2076, DOI: 10.1016/S0014-3057(02)00085-XGoogle ScholarThere is no corresponding record for this reference.
- 122Lena, J.-B.; Van Herk, A. M. Toward Biodegradable Chain-Growth Polymers and Polymer Particles: Re-Evaluation of Reactivity Ratios in Copolymerization of Vinyl Monomers with Cyclic Ketene Acetal Using Nonlinear Regression with Proper Error Analysis. Ind. Eng. Chem. Res. 2019, 58 (46), 20923– 20931, DOI: 10.1021/acs.iecr.9b02375Google ScholarThere is no corresponding record for this reference.
- 123Lena, J.-B.; Jackson, A. W.; Chennamaneni, L. R.; Wong, C. T.; Lim, F.; Andriani, Y.; Thoniyot, P.; Van Herk, A. M. Degradable Poly(Alkyl Acrylates) with Uniform Insertion of Ester Bonds, Comparing Batch and Semibatch Copolymerizations. Macromolecules 2020, 53 (10), 3994– 4011, DOI: 10.1021/acs.macromol.0c00207Google ScholarThere is no corresponding record for this reference.
- 124Wang, W.; Zhou, Z.; Sathe, D.; Tang, X.; Moran, S.; Jin, J.; Haeffner, F.; Wang, J.; Niu, J. Degradable Vinyl Random Copolymers via Photocontrolled Radical Ring-Opening Cascade Copolymerization. Angew. Chem., Int. Ed. 2022, 61 (8), e202113302 DOI: 10.1002/anie.202113302Google ScholarThere is no corresponding record for this reference.
- 125Luzel, B.; Efimov, I.; Edeleva, M.; Kouider, S.; Gil, N.; Montarnal, D.; Lefay, C.; Gigmes, D.; Van Steenberge, P. H. M.; D’hooge, D. R.; Guillaneuf, Y. The successful impossible radical ring-opening copolymerization of thionolactones and methacrylates via an auxiliary third comonomer. Nature Communications 2025, 1, 9468, DOI: 10.1038/s41467-025-64502-yGoogle ScholarThere is no corresponding record for this reference.
- 126Bingham, N. M.; Nisa, Q. un; Chua, S. H. L.; Fontugne, L.; Spick, M. P.; Roth, P. J. Thioester-Functional Polyacrylamides: Rapid Selective Backbone Degradation Triggers Solubility Switch Based on Aqueous Lower Critical Solution Temperature/Upper Critical Solution Temperature. ACS Appl. Polym. Mater. 2020, 2 (8), 3440– 3449, DOI: 10.1021/acsapm.0c00503Google ScholarThere is no corresponding record for this reference.
- 127Un Nisa, Q.; Theobald, W.; Hepburn, K. S.; Riddlestone, I.; Bingham, N. M.; Kopeć, M.; Roth, P. J. Degradable Linear and Bottlebrush Thioester-Functional Copolymers through Atom-Transfer Radical Ring-Opening Copolymerization of a Thionolactone. Macromolecules 2022, 55 (17), 7392– 7400, DOI: 10.1021/acs.macromol.2c01317Google ScholarThere is no corresponding record for this reference.
- 128Jazani, A. M.; Bernat, R.; Matyjaszewski, K. Visible Light-Induced Photo-Radical Ring-Opening Copolymerization of Thionolactone and Acrylates. Polymer 2024, 302, 127032, DOI: 10.1016/j.polymer.2024.127032Google ScholarThere is no corresponding record for this reference.
- 129Morris, P. T.; Watanabe, K.; Albanese, K. R.; Kent, G. T.; Gupta, R.; Gerst, M.; Read De Alaniz, J.; Hawker, C. J.; Bates, C. M. Scalable Synthesis of Degradable Copolymers Containing α-Lipoic Acid via Miniemulsion Polymerization. J. Am. Chem. Soc. 2024, 146 (44), 30662– 30667, DOI: 10.1021/jacs.4c12438Google ScholarThere is no corresponding record for this reference.
- 130Albanese, K. R.; Okayama, Y.; Morris, P. T.; Gerst, M.; Gupta, R.; Speros, J. C.; Hawker, C. J.; Choi, C.; De Alaniz, J. R.; Bates, C. M. Building Tunable Degradation into High-Performance Poly(Acrylate) Pressure-Sensitive Adhesives. ACS Macro Lett. 2023, 12 (6), 787– 793, DOI: 10.1021/acsmacrolett.3c00204Google ScholarThere is no corresponding record for this reference.
- 131Albanese, K. R.; Morris, P. T.; Roehrich, B.; Read de Alaniz, J.; Hawker, C. J.; Bates, C. M. Selective Electrochemical Degradation of Bottlebrush Elastomers. J. Polym. Sci. 2024, 62 (18), 4326– 4331, DOI: 10.1002/pol.20240393Google ScholarThere is no corresponding record for this reference.
- 132Giri, A.; Aquib, M.; Choudhury, A.; Kannaujiya, V. K.; Lim, J. L.; Gu, Z.; Lenardon, M. D.; Boyer, C. Lipoic Acid Based Redox-Responsive Degradable Antimicrobial Polymers. Macromol. Rapid Commun. 2025, 46 (17), e00224 DOI: 10.1002/marc.202500224Google ScholarThere is no corresponding record for this reference.
- 133Choi, C.; Okayama, Y.; Morris, P. T.; Robinson, L. L.; Gerst, M.; Speros, J. C.; Hawker, C. J.; Read de Alaniz, J.; Bates, C. M. Digital Light Processing of Dynamic Bottlebrush Materials. Adv. Funct. Mater. 2022, 32 (25), 2200883, DOI: 10.1002/adfm.202200883Google ScholarThere is no corresponding record for this reference.
- 134Lai, H.; Le Dot, M.; Chen, J.; Zhang, J.; Xiao, P. Biomass-Derived Photoresins for Digital Light Processing 3D Printing of Degradable Objects. ACS Sustainable Resour. Manage. 2025, 2 (5), 833– 841, DOI: 10.1021/acssusresmgt.5c00094Google ScholarThere is no corresponding record for this reference.
- 135Pesenti, T.; Nicolas, J. 100th Anniversary of Macromolecular Science Viewpoint: Degradable Polymers from Radical Ring-Opening Polymerization: Latest Advances, New Directions, and Ongoing Challenges. ACS Macro Lett. 2020, 9 (12), 1812– 1835, DOI: 10.1021/acsmacrolett.0c00676Google ScholarThere is no corresponding record for this reference.
- 136Roberts, G. E.; Coote, M. L.; Heuts, J. P. A.; Morris, L. M.; Davis, T. P. Radical Ring-Opening Copolymerization of 2-Methylene 1,3-Dioxepane and Methyl Methacrylate: Experiments Originally Designed To Probe the Origin of the Penultimate Unit Effect. Macromolecules 1999, 32 (5), 1332– 1340, DOI: 10.1021/ma9813587Google ScholarThere is no corresponding record for this reference.
- 137Agarwal, S. Microstructural Characterisation and Properties Evaluation of Poly (Methyl Methacrylate-Co-Ester)s. Polym. J. 2007, 39 (2), 163– 174, DOI: 10.1295/polymj.PJ2006137Google ScholarThere is no corresponding record for this reference.
- 138Zhang, Y.; Zheng, M.; Kissel, T.; Agarwal, S. Design and Biophysical Characterization of Bioresponsive Degradable Poly(Dimethylaminoethyl Methacrylate) Based Polymers for In Vitro DNA Transfection. Biomacromolecules 2012, 13 (2), 313– 322, DOI: 10.1021/bm2015174Google ScholarThere is no corresponding record for this reference.
- 139Lutz, J.-F.; Andrieu, J.; Üzgün, S.; Rudolph, C.; Agarwal, S. Biocompatible, Thermoresponsive, and Biodegradable: Simple Preparation of “All-in-One” Biorelevant Polymers. Macromolecules 2007, 40 (24), 8540– 8543, DOI: 10.1021/ma7021474Google ScholarThere is no corresponding record for this reference.
- 140Agarwal, S.; Ren, L.. Polycaprolactone-Based Novel Degradable Ionomers by Radical Ring-Opening Polymerization of 2-Methylene-1,3-Dioxepane. Macromolecules 2009, 42 (5), 1574– 1579, DOI: 10.1021/ma802615fGoogle ScholarThere is no corresponding record for this reference.
- 141Zhang, Y.; Aigner, A.; Agarwal, S. Degradable and Biocompatible Poly(N,N-Dimethylaminoethyl Methacrylate-Co-Caprolactone)s as DNA Transfection Agents. Macromol. Biosci. 2013, 13 (9), 1267– 1275, DOI: 10.1002/mabi.201300043Google ScholarThere is no corresponding record for this reference.
- 142Jin, Q.; Maji, S.; Agarwal, S. Novel Amphiphilic, Biodegradable, Biocompatible, Cross-Linkable Copolymers: Synthesis, Characterization and Drug Delivery Applications. Polym. Chem. 2012, 3 (10), 2785, DOI: 10.1039/c2py20364bGoogle ScholarThere is no corresponding record for this reference.
- 143Zhang, Y.; Chu, D.; Zheng, M.; Kissel, T.; Agarwal, S. Biocompatible and Degradable Poly(2-Hydroxyethyl Methacrylate) Based Polymers for Biomedical Applications. Polym. Chem. 2012, 3 (10), 2752, DOI: 10.1039/c2py20403gGoogle ScholarThere is no corresponding record for this reference.
- 144Ko, J. H.; Terashima, T.; Sawamoto, M.; Maynard, H. D. Fluorous Comonomer Modulates the Reactivity of Cyclic Ketene Acetal and Degradation of Vinyl Polymers. Macromolecules 2017, 50 (23), 9222– 9232, DOI: 10.1021/acs.macromol.7b01973Google ScholarThere is no corresponding record for this reference.
- 145Lai, H.; Ouchi, M. Backbone-Degradable Polymers via Radical Copolymerizations of Pentafluorophenyl Methacrylate with Cyclic Ketene Acetal: Pendant Modification and Efficient Degradation by Alternating-Rich Sequence. ACS Macro Lett. 2021, 10 (10), 1223– 1228, DOI: 10.1021/acsmacrolett.1c00513Google ScholarThere is no corresponding record for this reference.
- 146Grabe, N.; Zhang, Y.; Agarwal, S. Degradable Elastomeric Block Copolymers Based on Polycaprolactone by Free-Radical Chemistry. Macromol. Chem. Phys. 2011, 212 (13), 1327– 1334, DOI: 10.1002/macp.201100031Google ScholarThere is no corresponding record for this reference.
- 147Gardoni, G.; Zanoni, A.; González De San Román, E.; Moscatelli, D.; Gabirondo, E.; Leiza, J. R.; Barquero, A.; Sardon, H. Enhancing the Incorporation of 2-Methylen-1,3-Dioxepane (MDO) into Industrial Monomers by the Addition of Crotonate Comonomers. Polymer 2024, 307, 127298, DOI: 10.1016/j.polymer.2024.127298Google ScholarThere is no corresponding record for this reference.
- 148Gao, Y.; Böhmer, V. I.; Zhou, D.; Zhao, T.; Wang, W.; Paulusse, J. M. J. Main-Chain Degradable Single-Chain Cyclized Polymers as Gene Delivery Vectors. J. Controlled Release 2016, 244, 375– 383, DOI: 10.1016/j.jconrel.2016.07.046Google ScholarThere is no corresponding record for this reference.
- 149Ratcliffe, L. P. D.; Couchon, C.; Armes, S. P.; Paulusse, J. M. J. Inducing an Order-Order Morphological Transition via Chemical Degradation of Amphiphilic Diblock Copolymer Nano-Objects. Biomacromolecules 2016, 17 (6), 2277– 2283, DOI: 10.1021/acs.biomac.6b00540Google ScholarThere is no corresponding record for this reference.
- 150Wang, W.; Rondon, B.; Wang, Z.; Wang, J.; Niu, J. Macrocyclic Allylic Sulfone as a Universal Comonomer in Organocatalyzed Photocontrolled Radical Copolymerization with Vinyl Monomers. Macromolecules 2023, 56 (5), 2052– 2061, DOI: 10.1021/acs.macromol.2c02025Google ScholarThere is no corresponding record for this reference.
- 151Keyser, S. P.; Fairbanks, B. D.; Bahns, T.; Bowman, C. N. Dithiolane-Ene Copolymerization: Enabling Tunable, Dynamic, Dual-Cure Networks via Real-Time UV-Vis/FTIR Kinetics and Compositional Analysis. Macromolecules 2025, 58 (15), 8468– 8478, DOI: 10.1021/acs.macromol.5c00977Google ScholarThere is no corresponding record for this reference.
- 152Agarwal, S.; Kumar, R.; Kissel, T.; Reul, R. Synthesis of Degradable Materials Based on Caprolactone and Vinyl Acetate Units Using Radical Chemistry. Polym. J. 2009, 41 (8), 650– 660, DOI: 10.1295/polymj.PJ2009091Google ScholarThere is no corresponding record for this reference.
- 153Undin, J.; Illanes, T.; Finne-Wistrand, A.; Albertsson, A.-C. Random Introduction of Degradable Linkages into Functional Vinyl Polymers by Radical Ring-Opening Polymerization, Tailored for Soft Tissue Engineering. Polym. Chem. 2012, 3 (5), 1260, DOI: 10.1039/c2py20034aGoogle ScholarThere is no corresponding record for this reference.
- 154Bell, C. A.; Hedir, G. G.; O’Reilly, R. K.; Dove, A. P. Controlling the Synthesis of Degradable Vinyl Polymers by Xanthate-Mediated Polymerization. Polym. Chem. 2015, 6 (42), 7447– 7454, DOI: 10.1039/C5PY01156FGoogle ScholarThere is no corresponding record for this reference.
- 155Hedir, G. G.; Bell, C. A.; Ieong, N. S.; Chapman, E.; Collins, I. R.; O’Reilly, R. K.; Dove, A. P. Functional Degradable Polymers by Xanthate-Mediated Polymerization. Macromolecules 2014, 47 (9), 2847– 2852, DOI: 10.1021/ma500428eGoogle ScholarThere is no corresponding record for this reference.
- 156Bailey, W. J.; Wu, S.-R.; Ni, Z. Synthesis and Free Radical Ring-Opening Polymerization of 2-Methylene-4-Phenyl-1,3-Dioxolane. Makromol. Chem. 1982, 183 (8), 1913– 1920, DOI: 10.1002/macp.1982.021830811Google ScholarThere is no corresponding record for this reference.
- 157Tardy, A.; Honoré, J.-C.; Tran, J.; Siri, D.; Delplace, V.; Bataille, I.; Letourneur, D.; Perrier, J.; Nicoletti, C.; Maresca, M.; Lefay, C.; Gigmes, D.; Nicolas, J.; Guillaneuf, Y. Radical Copolymerization of Vinyl Ethers and Cyclic Ketene Acetals as a Versatile Platform to Design Functional Polyesters. Angew. Chem., Int. Ed. 2017, 56 (52), 16515– 16520, DOI: 10.1002/anie.201707043Google ScholarThere is no corresponding record for this reference.
- 158Hedir, G.; Stubbs, C.; Aston, P.; Dove, A. P.; Gibson, M. I. Synthesis of Degradable Poly(Vinyl Alcohol) by Radical Ring-Opening Copolymerization and Ice Recrystallization Inhibition Activity. ACS Macro Lett. 2017, 6 (12), 1404– 1408, DOI: 10.1021/acsmacrolett.7b00905Google ScholarThere is no corresponding record for this reference.
- 159Hedir, G. G.; Bell, C. A.; O’Reilly, R. K.; Dove, A. P. Functional Degradable Polymers by Radical Ring-Opening Copolymerization of MDO and Vinyl Bromobutanoate: Synthesis, Degradability and Post-Polymerization Modification. Biomacromolecules 2015, 16 (7), 2049– 2058, DOI: 10.1021/acs.biomac.5b00476Google ScholarThere is no corresponding record for this reference.
- 160Ding, D.; Pan, X.; Zhang, Z.; Li, N.; Zhu, J.; Zhu, X. A Degradable Copolymer of 2-Methylene-1,3-Dioxepane and Vinyl Acetate by Photo-Induced Cobalt-Mediated Radical Polymerization. Polym. Chem. 2016, 7 (33), 5258– 5264, DOI: 10.1039/C6PY01061JGoogle ScholarThere is no corresponding record for this reference.
- 161Odian, G. Principles of Polymerization; John Wiley & Sons, 2004.Google ScholarThere is no corresponding record for this reference.
- 162Jin, S.; Gonsalves, K. E. Synthesis and Characterization of Functionalized Poly(ε-Caprolactone) Copolymers by Free-Radical Polymerization. Macromolecules 1998, 31 (4), 1010– 1015, DOI: 10.1021/ma9707289Google ScholarThere is no corresponding record for this reference.
- 163Du, Y.; Ren, L.; Sloan, J.; Chong, S.; Lamprou, A.; Du, Y.; Coughlin, E. B. Comparative Study of N-Vinyl Pyrrolidone and Cyclic Ketene Acetal Copolymer Degradation under Alkaline, Enzymatic, or Wastewater Conditions. Polymer 2024, 309, 127444, DOI: 10.1016/j.polymer.2024.127444Google ScholarThere is no corresponding record for this reference.
- 164Du, Y.; Du, Y.; Lazzari, S.; Reimers, T.; Konradi, R.; Holcombe, T. W.; Coughlin, E. B. Mechanistic Investigation of Cyclic Ketene Acetal Radical Ring-Opening Homo- and Co-Polymerization and Preparation of PEO Graft Copolymers with Tunable Composition. Polym. Chem. 2022, 13 (41), 5829– 5840, DOI: 10.1039/D2PY00986BGoogle ScholarThere is no corresponding record for this reference.
- 165Hiraguri, Y.; Tokiwa, Y. Synthesis of a Novel Water-Soluble Copolymer Composed of 2-Methylene-1,3,6-Trioxocane and N-Vinyl 2-Pyrrolidone and Its Enzymatic Degradation. Clean Prod Processes 2001, 3 (3), 303– 306, DOI: 10.1007/s100980100120Google ScholarThere is no corresponding record for this reference.
- 166Choi, S.; Lee, K.; Kwon, S.; Kim, H. Preparation of Fine Particles of Poly(N-Vinyl-2-Pyrrolidone-Co-2-Methylene-1,3-Dioxepane) Using Supercritical Antisolvent. Journal of Supercritical Fluids 2006, 37 (3), 287– 291, DOI: 10.1016/j.supflu.2005.11.023Google ScholarThere is no corresponding record for this reference.
- 167Pamungkas, M. S.; Ando, T.; Ajiro, H. Copolymer Synthesis of N -Vinylacetamide and 2-Methylene-1,3-Dioxepane Provides a New Degradable Polymer. J. Polym. Sci. 2025, 63 (6), 1306– 1318, DOI: 10.1002/pol.20240935Google ScholarThere is no corresponding record for this reference.
- 168Pesenti, T.; Gillon, E.; Ishii, S.; Messaoudi, S.; Guillaneuf, Y.; Imberty, A.; Nicolas, J. Increasing the Hydrophilicity of Cyclic Ketene Acetals Improves the Hydrolytic Degradation of Vinyl Copolymers and the Interaction of Glycopolymer Nanoparticles with Lectins. Biomacromolecules 2023, 24 (2), 991– 1002, DOI: 10.1021/acs.biomac.2c01419Google ScholarThere is no corresponding record for this reference.
- 169Pesenti, T.; Domingo-Lopez, D.; Gillon, E.; Ibrahim, N.; Messaoudi, S.; Imberty, A.; Nicolas, J. Degradable Glycopolyester-like Nanoparticles by Radical Ring-Opening Polymerization. Biomacromolecules 2022, 23 (9), 4015– 4028, DOI: 10.1021/acs.biomac.2c00851Google ScholarThere is no corresponding record for this reference.
- 170Tran, J.; Pesenti, T.; Cressonnier, J.; Lefay, C.; Gigmes, D.; Guillaneuf, Y.; Nicolas, J. Degradable Copolymer Nanoparticles from Radical Ring-Opening Copolymerization between Cyclic Ketene Acetals and Vinyl Ethers. Biomacromolecules 2019, 20 (1), 305– 317, DOI: 10.1021/acs.biomac.8b01500Google ScholarThere is no corresponding record for this reference.
- 171Ivanchenko, O.; Mazières, S.; Poli, R.; Harrisson, S.; Destarac, M. Ring Size-Reactivity Relationship in Radical Ring-Opening Copolymerisation of Thionolactones with Vinyl Pivalate. Polym. Chem. 2022, 13 (45), 6284– 6292, DOI: 10.1039/D2PY01153KGoogle ScholarThere is no corresponding record for this reference.
- 172Calderón-Díaz, A.; Boggiano, A. C.; Xiong, W.; Kaiser, N.; Gutekunst, W. R. Degradable N -Vinyl Copolymers through Radical Ring-Opening Polymerization of Cyclic Thionocarbamates. ACS Macro Lett. 2024, 13 (11), 1390– 1395, DOI: 10.1021/acsmacrolett.4c00550Google ScholarThere is no corresponding record for this reference.
- 173Hepburn, K. S.; Zulkifli, N. S.; Delorme, C.; Kazmi, T.; Abu Bakar, R.; Roth, P. J. Thioester-Rich Degradable Copolymers from a Thionolactone and S-Vinyl and P-Vinyl Monomers. Polymer 2024, 311, 127485, DOI: 10.1016/j.polymer.2024.127485Google ScholarThere is no corresponding record for this reference.
- 174Shi, Y.; Agarwal, S. Thermally Stable Optically Transparent Copolymers of 2-Methylene-1,3-Dioxepane and N-Phenyl Maleimide with Degradable Ester Linkages. e-Polym. 2015, 15 (4), 217– 226, DOI: 10.1515/epoly-2015-0096Google ScholarThere is no corresponding record for this reference.
- 175Hill, M. R.; Guégain, E.; Tran, J.; Figg, C. A.; Turner, A. C.; Nicolas, J.; Sumerlin, B. S. Radical Ring-Opening Copolymerization of Cyclic Ketene Acetals and Maleimides Affords Homogeneous Incorporation of Degradable Units. ACS Macro Lett. 2017, 6 (10), 1071– 1077, DOI: 10.1021/acsmacrolett.7b00572Google ScholarThere is no corresponding record for this reference.
- 176Hill, M. R.; Kubo, T.; Goodrich, S. L.; Figg, C. A.; Sumerlin, B. S. Alternating Radical Ring-Opening Polymerization of Cyclic Ketene Acetals: Access to Tunable and Functional Polyester Copolymers. Macromolecules 2018, 51 (14), 5079– 5084, DOI: 10.1021/acs.macromol.8b00889Google ScholarThere is no corresponding record for this reference.
- 177Hall, H. K.; Padias, A. B. Bond Forming Initiation of “Charge-Transfer” Polymerizations and the Accompanying Cycloadditions. Acc. Chem. Res. 1997, 30 (8), 322– 329, DOI: 10.1021/ar950222fGoogle ScholarThere is no corresponding record for this reference.
- 178Vu, N. D.; Ivanchenko, O.; Baffie, F.; Guillaneuf, Y.; Gigmes, D.; Harrisson, S.; Monteil, V.; Destarac, M.; D’Agosto, F. Synthesis of Degradable Polyethylene and Poly(Ethylene- Co -Vinyl Acetate) by Radical Co- and Terpolymerization of Ethylene, ε-Thionocaprolactone, and Vinyl Acetate. Macromolecules 2024, 57 (9), 4516– 4523, DOI: 10.1021/acs.macromol.4c00085Google ScholarThere is no corresponding record for this reference.
- 179Hiraguri, Y.; Katase, K.; Tokiwa, Y. Synthesis of Biodegradable Detergent Builder by Alternating Copolymerization of 2-Methylene-1,3,6-trioxocane and Maleic Anhydride. Journal of Macromolecular Science, Part A 2007, 44 (8), 893– 897, DOI: 10.1080/10601320701407979Google ScholarThere is no corresponding record for this reference.
- 180Du, Y.; Yang, Y.; Deglmann, P.; Holcombe, T. W.; Du, Y.; Coughlin, E. B. The Competition between Radical Copolymerization and Charge Separation of Cyclic Ketene Acetals and Electron-deficient Olefins. J. Polym. Sci. 2024, 62 (16), 3716– 3729, DOI: 10.1002/pol.20230480Google ScholarThere is no corresponding record for this reference.
- 181Lovell, P. A.; Schork, F. J. Fundamentals of Emulsion Polymerization. Biomacromolecules 2020, 21 (11), 4396– 4441, DOI: 10.1021/acs.biomac.0c00769Google ScholarThere is no corresponding record for this reference.
- 182Schork, F. J. In Support of Commercial Applications of Miniemulsion Polymerization. Ind. Eng. Chem. Res. 2023, 62 (5), 2329– 2335, DOI: 10.1021/acs.iecr.2c01746Google ScholarThere is no corresponding record for this reference.
- 183Asua, J. M. Miniemulsion Polymerization. In Encyclopedia of Polymeric Nanomaterials; Kobayashi, S., Müllen, K., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2015; pp 1267– 1275. DOI: 10.1007/978-3-642-29648-2_263 .Google ScholarThere is no corresponding record for this reference.
- 184Siebert, J. M.; Baumann, D.; Zeller, A.; Mailänder, V.; Landfester, K. Synthesis of Polyester Nanoparticles in Miniemulsion Obtained by Radical Ring-Opening of BMDO and Their Potential as Biodegradable Drug Carriers. Macromol. Biosci. 2012, 12 (2), 165– 175, DOI: 10.1002/mabi.201100236Google ScholarThere is no corresponding record for this reference.
- 185d’Ayala, G. G.; Malinconico, M.; Laurienzo, P.; Tardy, A.; Guillaneuf, Y.; Lansalot, M.; D’Agosto, F.; Charleux, B. RAFT/MADIX Copolymerization of Vinyl Acetate and 5,6-benzo-2-methylene-1,3-dioxepane. J. Polym. Sci., Part A: Polym. Chem. 2014, 52 (1), 104– 111, DOI: 10.1002/pola.26976Google ScholarThere is no corresponding record for this reference.
- 186Wickel, H.; Agarwal, S.; Greiner, A. Homopolymers and Random Copolymers of 5,6-Benzo-2-Methylene-1,3-Dioxepane and Methyl Methacrylate: Structural Characterization Using 1D and 2D NMR. Macromolecules 2003, 36 (7), 2397– 2403, DOI: 10.1021/ma025983uGoogle ScholarThere is no corresponding record for this reference.
- 187Galanopoulo, P.; Sinniger, L.; Gil, N.; Gigmes, D.; Lefay, C.; Guillaneuf, Y.; Lages, M.; Nicolas, J.; D’Agosto, F.; Lansalot, M. Degradable Vinyl Polymer Particles by Radical Aqueous Emulsion Copolymerization of Methyl Methacrylate and 5,6-Benzo-2-Methylene-1,3-Dioxepane. Polym. Chem. 2023, 14 (11), 1224– 1231, DOI: 10.1039/D3PY00040KGoogle ScholarThere is no corresponding record for this reference.
- 188Carter, M. C. D.; Hejl, A.; Woodfin, S.; Einsla, B.; Janco, M.; DeFelippis, J.; Cooper, R. J.; Even, R. C. Backbone-Degradable Vinyl Acetate Latex: Coatings for Single-Use Paper Products. ACS Macro Lett. 2021, 10 (5), 591– 597, DOI: 10.1021/acsmacrolett.1c00172Google ScholarThere is no corresponding record for this reference.
- 189Carter, M. C. D.; DeFelippis, J.; Even, R. C.; Hejl, A. Aqueous Dispersion of Copolymer Particles of Vinyl Acetate and a Cyclic Ketene Acetal Monomer. US 11,111,328 B2, July 9, 2021.Google ScholarThere is no corresponding record for this reference.
- 190Kordes, B. R.; Ascherl, L.; Rüdinger, C.; Melchin, T.; Agarwal, S. Competition between Hydrolysis and Radical Ring-Opening Polymerization of MDO in Water. Who Makes the Race?. Macromolecules 2023, 56 (3), 1033– 1044, DOI: 10.1021/acs.macromol.2c01653Google ScholarThere is no corresponding record for this reference.
- 191Carter, M. C. D.; Hejl, A.; Janco, M.; DeFelippis, J.; Yang, P.; Gallagher, M.; Liang, Y. Emulsion Polymerization of 2-Methylene-1,3-Dioxepane and Vinyl Acetate: Process Analysis and Characterization. Macromolecules 2023, 56 (15), 5718– 5729, DOI: 10.1021/acs.macromol.3c00571Google ScholarThere is no corresponding record for this reference.
- 192Wenzel, F.; Aguirre, M.; Leiza, J. R. Emulsion Copolymerization of 2-Methylene-1,3-Dioxepane (MDO) and Acrylate Monomers: Incorporation vs Hydrolysis. Polymer 2024, 307, 127285, DOI: 10.1016/j.polymer.2024.127285Google ScholarThere is no corresponding record for this reference.
- 193Movafagh, M.; Meek, K. M.; Dubé, M. A. Butyl Acrylate/2-Methylene-1,3-Dioxepane/Vinyl Acetate Emulsion Terpolymerization: Incorporating Backbone Degradable Linkages into Adhesive Applications. ChemSusChem 2025, 18 (9), e202402478 DOI: 10.1002/cssc.202402478Google ScholarThere is no corresponding record for this reference.
- 194Mothe, S. R.; Zhao, W.; Van Herk, A. M.; Thoniyot, P. Backbone-Degradable Acrylate Latex: Toward Overcoming Hydrolysis Limitations of Cyclic Ketene Acetal Monomers. Macromolecules 2024, 57 (6), 2937– 2949, DOI: 10.1021/acs.macromol.3c02474Google ScholarThere is no corresponding record for this reference.
- 195Zhu, C.; Nicolas, J. Towards Nanoparticles with Site-Specific Degradability by Ring-Opening Copolymerization Induced Self-Assembly in Organic Medium. Polym. Chem. 2021, 12 (4), 594– 607, DOI: 10.1039/D0PY01425GGoogle ScholarThere is no corresponding record for this reference.
- 196Guégain, E.; Zhu, C.; Giovanardi, E.; Nicolas, J. Radical Ring-Opening Copolymerization-Induced Self-Assembly (rROPISA). Macromolecules 2019, 52 (10), 3612– 3624, DOI: 10.1021/acs.macromol.9b00161Google ScholarThere is no corresponding record for this reference.
- 197D’Agosto, F.; Rieger, J.; Lansalot, M. RAFT-Mediated Polymerization-Induced Self-Assembly. Angew. Chem. Int. Ed 2020, 59 (22), 8368– 8392, DOI: 10.1002/anie.201911758Google ScholarThere is no corresponding record for this reference.
- 198Zhu, C.; Denis, S.; Nicolas, J. A Simple Route to Aqueous Suspensions of Degradable Copolymer Nanoparticles Based on Radical Ring-Opening Polymerization-Induced Self-Assembly (rROPISA). Chem. Mater. 2022, 34 (4), 1875– 1888, DOI: 10.1021/acs.chemmater.1c04151Google ScholarThere is no corresponding record for this reference.
- 199Mothe, S. R.; Kanaujia, P.; Oh, A. B. Y.; Ang, P.; Thoniyot, P. PVA Free-Radical Diblock Copolymer for Nanoencapsulation with Enhanced Biodegradability in the Environment. Eur. Polym. J. 2023, 196, 112287, DOI: 10.1016/j.eurpolymj.2023.112287Google ScholarThere is no corresponding record for this reference.
- 200Mothe, S. R.; Ang, P.; Lau, H. H.; Oh, A. B. Y.; Thoniyot, P. Investigating the Potential of Degradable Poly(Vinyl Acetate) Copolymer Microparticles for Encapsulation and in Vitro Release Studies. Next Materials 2023, 1 (4), 100055, DOI: 10.1016/j.nxmate.2023.100055Google ScholarThere is no corresponding record for this reference.
- 201Galanopoulo, P.; Gil, N.; Gigmes, D.; Lefay, C.; Guillaneuf, Y.; Lages, M.; Nicolas, J.; Lansalot, M.; D’Agosto, F. One-Step Synthesis of Degradable Vinylic Polymer-Based Latexes via Aqueous Radical Emulsion Polymerization. Angew. Chem., Int. Ed. 2022, 61 (15), e202117498 DOI: 10.1002/anie.202117498Google ScholarThere is no corresponding record for this reference.
- 202Galanopoulo, P.; Gil, N.; Gigmes, D.; Lefay, C.; Guillaneuf, Y.; Lages, M.; Nicolas, J.; d’Agosto, F.; Lansalot, M. RAFT-Mediated Emulsion Polymerization-Induced Self-Assembly for the Synthesis of Core-Degradable Waterborne Particles. Angew. Chem., Int. Ed. 2023, 62 (16), e202302093 DOI: 10.1002/anie.202302093Google ScholarThere is no corresponding record for this reference.
- 203Georgiou, P. G.; Neal, T. J.; Newell, M. A.; Hepburn, K. S.; Tyler, J. J. S.; Farmer, M. A. H.; Chohan, P.; Roth, P. J.; Nicolas, J.; Armes, S. P. Degradable Diblock Copolymer Vesicles via Radical Ring-Opening Polymerization-Induced Self-Assembly in Aqueous Media. J. Am. Chem. Soc. 2025, 147 (23), 19817– 19828, DOI: 10.1021/jacs.5c03744Google ScholarThere is no corresponding record for this reference.
- 204Lages, M.; Gil, N.; Galanopoulo, P.; Mougin, J.; Lefay, C.; Guillaneuf, Y.; Lansalot, M.; D’Agosto, F.; Nicolas, J. Degradable Vinyl Copolymer Nanoparticles/Latexes by Aqueous Nitroxide-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2022, 55 (21), 9790– 9801, DOI: 10.1021/acs.macromol.2c01734Google ScholarThere is no corresponding record for this reference.
- 205Lages, M.; Gil, N.; Galanopoulo, P.; Mougin, J.; Lefay, C.; Guillaneuf, Y.; Lansalot, M.; D’Agosto, F.; Nicolas, J. Degradable Latexes by Nitroxide-Mediated Aqueous Seeded Emulsion Copolymerization Using a Thionolactone. Macromolecules 2023, 56 (19), 7973– 7983, DOI: 10.1021/acs.macromol.3c01478Google ScholarThere is no corresponding record for this reference.
- 206Fuji, K.; Kitayama, Y.; Harada, A. Miniemulsion Ring-Opening Radical Polymerization with Dibenzo[c, e]Oxepan-5-Thione for Degradable Polymer Particles. ACS Appl. Polym. Mater. 2025, 7 (5), 3349– 3357, DOI: 10.1021/acsapm.5c00088Google ScholarThere is no corresponding record for this reference.
- 207Fadil, Y.; Fan, D.; Thompson, S. W.; Zetterlund, P. B. Synthesis of Degradable Vinyl Copolymers Based on Lipoic Acid via Ab Initio Emulsion Polymerization. ACS Sustainable Chem. Eng. 2025, 13 (38), 16117– 16126, DOI: 10.1021/acssuschemeng.5c06894Google ScholarThere is no corresponding record for this reference.
- 208Riachi, C.; Schüwer, N.; Klok, H.-A. Degradable Polymer Brushes Prepared via Surface-Initiated Controlled Radical Polymerization. Macromolecules 2009, 42 (21), 8076– 8081, DOI: 10.1021/ma901537xGoogle ScholarThere is no corresponding record for this reference.
- 209Xie, F.; Deng, X.; Kratzer, D.; Cheng, K. C. K.; Friedmann, C.; Qi, S.; Solorio, L.; Lahann, J. Backbone-Degradable Polymers Prepared by Chemical Vapor Deposition. Angew. Chem. 2017, 129 (1), 209– 213, DOI: 10.1002/ange.201609307Google ScholarThere is no corresponding record for this reference.
- 210Dai, G.; Xie, Q.; Chen, S.; Ma, C.; Zhang, G. Biodegradable Poly(Ester)-Poly(Methyl Methacrylate) Copolymer for Marine Anti-Biofouling. Prog. Org. Coat. 2018, 124, 55– 60, DOI: 10.1016/j.porgcoat.2018.08.003Google ScholarThere is no corresponding record for this reference.
- 211Xie, Q.; Ma, C.; Zhang, G.; Bressy, C. Poly(Ester)-Poly(Silyl Methacrylate) Copolymers: Synthesis and Hydrolytic Degradation Kinetics. Polym. Chem. 2018, 9 (12), 1448– 1454, DOI: 10.1039/C8PY00052BGoogle ScholarThere is no corresponding record for this reference.
- 212Dai, G.; Xie, Q.; Ai, X.; Ma, C.; Zhang, G. Self-Generating and Self-Renewing Zwitterionic Polymer Surfaces for Marine Anti-Biofouling. ACS Appl. Mater. Interfaces 2019, 11 (44), 41750– 41757, DOI: 10.1021/acsami.9b16775Google ScholarThere is no corresponding record for this reference.
- 213Pan, J.; Ai, X.; Ma, C.; Zhang, G. Degradable Vinyl Polymers for Combating Marine Biofouling. Acc. Chem. Res. 2022, 55 (11), 1586– 1598, DOI: 10.1021/acs.accounts.2c00187Google ScholarThere is no corresponding record for this reference.
- 214Ai, X.; Pan, J.; Xie, Q.; Ma, C.; Zhang, G. UV-Curable Hyperbranched Poly(Ester- Co -Vinyl) by Radical Ring-Opening Copolymerization for Antifouling Coatings. Polym. Chem. 2021, 12 (31), 4524– 4531, DOI: 10.1039/D1PY00810BGoogle ScholarThere is no corresponding record for this reference.
- 215Irvine, G.; Dawson, F.; George, A.; Kopec, M. Are Polymer Gels Synthesized by Free Radical Polymerization with Cleavable Crosslinkers Really Degradable?. Eur. Polym. J. 2024, 213, 113089, DOI: 10.1016/j.eurpolymj.2024.113089Google ScholarThere is no corresponding record for this reference.
- 216Elliss, H.; Dawson, F.; Nisa, Q. un; Bingham, N. M.; Roth, P. J.; Kopeć, M. Fully Degradable Polyacrylate Networks from Conventional Radical Polymerization Enabled by Thionolactone Addition. Macromolecules 2022, 55 (15), 6695– 6702, DOI: 10.1021/acs.macromol.2c01140Google ScholarThere is no corresponding record for this reference.
- 217Dawson, F.; Kazmi, T.; Roth, P.; Kopec, M. Strands vs. Crosslinks: Topology-Dependent Degradation and Regelation of Polyacrylate Networks Synthesised by RAFT Polymerisation. Polym. Chem. 2023, 14 (47), 5166– 5177, DOI: 10.1039/D3PY01008BGoogle ScholarThere is no corresponding record for this reference.
- 218Dawson, F.; Irvine, G.; Kopeć, M. Lipoic Acid/Ethyl Lipoate as Cleavable Comonomers for Synthesis of Degradable Polymer Networks. Polym. Chem. 2025, 16 (22), 2659– 2669, DOI: 10.1039/D5PY00379BGoogle ScholarThere is no corresponding record for this reference.
- 219Choi, C.; Self, J. L.; Okayama, Y.; Levi, A. E.; Gerst, M.; Speros, J. C.; Hawker, C. J.; Read De Alaniz, J.; Bates, C. M. Light-Mediated Synthesis and Reprocessing of Dynamic Bottlebrush Elastomers under Ambient Conditions. J. Am. Chem. Soc. 2021, 143 (26), 9866– 9871, DOI: 10.1021/jacs.1c03686Google ScholarThere is no corresponding record for this reference.
- 220Maw, M.; Dashtimoghadam, E.; Keith, A. N.; Morgan, B. J.; Tanas, A. K.; Nikitina, E.; Ivanov, D. A.; Vatankhah-Varnosfaderani, M.; Dobrynin, A. V.; Sheiko, S. S. Sticky Architecture: Encoding Pressure Sensitive Adhesion in Polymer Networks. ACS Cent. Sci. 2023, 9 (2), 197– 205, DOI: 10.1021/acscentsci.2c01407Google ScholarThere is no corresponding record for this reference.
- 221Prince, E.; Cheng, H.; Banal, J.; Johnson, J. Reversible Nucleic Acid Storage in Deconstructable Glassy Polymer Networks. J. Am. Chem. Soc. 2024, 146 (25), 17066– 17074, DOI: 10.1021/jacs.4c01925Google ScholarThere is no corresponding record for this reference.
- 222Carlotti, M.; Tricinci, O.; Mattoli, V. Novel, High-Resolution, Subtractive Photoresist Formulations for 3D Direct Laser Writing Based on Cyclic Ketene Acetals. Advanced Materials Technologies 2022, 7 (9), 2101590, DOI: 10.1002/admt.202101590Google ScholarThere is no corresponding record for this reference.
- 223Gil, N.; Thomas, C.; Mhanna, R.; Mauriello, J.; Maury, R.; Leuschel, B.; Malval, J.-P.; Clément, J.-L.; Gigmes, D.; Lefay, C.; Soppera, O.; Guillaneuf, Y. Thionolactone as a Resin Additive to Prepare (Bio)Degradable 3D Objects via VAT Photopolymerization. Angew. Chem., Int. Ed. 2022, 61 (18), e202117700 DOI: 10.1002/anie.202117700Google ScholarThere is no corresponding record for this reference.
- 224Kouider, S.; Buchon, L.; Choulot, L.; Bratasanu, J.; Désirée, P.; Mauriello, J.; Maarek, E.; Clément, J.-L.; Montarnal, D.; Soppera, O.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. Self-Destruct Thermosets: A Combination of the Comonomer Approach and Thermolatent-Base Compounds. ChemRxiv , 2025, DOI: 10.26434/chemrxiv-2025-z051f .Google ScholarThere is no corresponding record for this reference.
- 225Han, S.; Bobrin, V. A.; Michelas, M.; Hawker, C. J.; Boyer, C. Sustainable and Recyclable Acrylate Resins for Liquid-Crystal Display 3D Printing Based on Lipoic Acid. ACS Macro Lett. 2024, 13 (11), 1495– 1502, DOI: 10.1021/acsmacrolett.4c00600Google ScholarThere is no corresponding record for this reference.
- 226Biying, A. O.; Mothe, S. R.; Jackson, A. W.; Kanaujia, P.; Thoniyot, P. Effects of Polymer Microstructure Introduced by Radical Ring-Opening Polymerization on Nanoencapsulation and Controlled Release. Polymer 2024, 307, 127284, DOI: 10.1016/j.polymer.2024.127284Google ScholarThere is no corresponding record for this reference.
- 227Balaji, A.; Prior, A. R.; O’Reilly, R. K.; Dove, A. P.; Thurecht, K. J.; Bell, C. A. Biodegradable mPEG- b -Poly(MDO- Co -Vinyl Esters) Block Copolymers as a Viable Nanocarrier Platform with Tuneable Disassembly. Polym. Chem. 2024, 15 (12), 1152– 1165, DOI: 10.1039/D4PY00049HGoogle ScholarThere is no corresponding record for this reference.
- 228Ahmed, S. E.; Fletcher, N. L.; Prior, A. R.; Huda, P.; Bell, C. A.; Thurecht, K. J. Development of Targeted Micelles and Polymersomes Prepared from Degradable RAFT-Based Diblock Copolymers and Their Potential Role as Nanocarriers for Chemotherapeutics. Polym. Chem. 2022, 13 (27), 4004– 4017, DOI: 10.1039/D2PY00257DGoogle ScholarThere is no corresponding record for this reference.
- 229Deng, Y.; Debbeler, J.; Traeger, A.; Gaitzsch, J. Degradable Nanocarriers Capable of Gene Delivery Derived from pH-Responsive Polyester: RROP Copolymerization Between Cyclic Ketene Acetals. Macromol. Rapid Commun. 2025, e00722 DOI: 10.1002/marc.202500722Google ScholarThere is no corresponding record for this reference.
- 230Guégain, E.; Tran, J.; Deguettes, Q.; Nicolas, J. Degradable Polymer Prodrugs with Adjustable Activity from Drug-Initiated Radical Ring-Opening Copolymerization. Chem. Sci. 2018, 9 (43), 8291– 8306, DOI: 10.1039/C8SC02256AGoogle ScholarThere is no corresponding record for this reference.
- 231Guerassimoff, L.; Cao, J.; Auguste, M.; Bossion, A.; Zhu, C.; Le, D.; Cailleau, C.; Ismail, S. M.; Mercier-Nomé, F.; Nicolas, J. Degradable Water-Soluble Polymer Prodrugs for Subcutaneous Delivery of Irritant Anticancer Drugs. Chem. Sci. 2025, 16 (31), 14323– 14341, DOI: 10.1039/D5SC02967HGoogle ScholarThere is no corresponding record for this reference.
- 232Zhu, C.; Beauseroy, H.; Mougin, J.; Lages, M.; Nicolas, J. In Situ Synthesis of Degradable Polymer Prodrug Nanoparticles. Chem. Sci. 2025, 16 (6), 2619– 2633, DOI: 10.1039/D4SC07746FGoogle ScholarThere is no corresponding record for this reference.
- 233Lau, U. Y.; Pelegri-O’Day, E. M.; Maynard, H. D. Synthesis and Biological Evaluation of a Degradable Trehalose Glycopolymer Prepared by RAFT Polymerization. Macromol. Rapid Commun. 2018, 39 (5), 1700652, DOI: 10.1002/marc.201700652Google ScholarThere is no corresponding record for this reference.
- 234Pesenti, T.; Zhu, C.; Gonzalez-Martinez, N.; Tomás, R. M. F.; Gibson, M. I.; Nicolas, J. Degradable Polyampholytes from Radical Ring-Opening Copolymerization Enhance Cellular Cryopreservation. ACS Macro Lett. 2022, 11 (7), 889– 894, DOI: 10.1021/acsmacrolett.2c00298Google ScholarThere is no corresponding record for this reference.
- 235Barber Nunez, B.; Guillaneuf, Y.; Grande, D.; Carbonnier, B.; Le Droumaguet, B. Degradable Biporous Polymeric Networks Based on 2-Methylene-1,3-Dioxepane: Towards Hierarchically Structured Materials Meant for Biomedical Applications. Polymer 2024, 308, 127332, DOI: 10.1016/j.polymer.2024.127332Google ScholarThere is no corresponding record for this reference.
- 236Barber Nunez, B.; Carbonnier, B.; Grande, D.; Le Droumaguet, B. Biporous Polycaprolactone-like Networks from 2-Methylene-1,3-Dioxepane: A Versatile Platform towards Functional and Reactive Polyesters. React. Funct. Polym. 2025, 214, 106284, DOI: 10.1016/j.reactfunctpolym.2025.106284Google ScholarThere is no corresponding record for this reference.
- 237Jackson, A. W.; Mothe, S. R.; Ang, P.; Chennamaneni, L. R.; Herk, A. M. V.; Thoniyot, P. Backbone Degradable Poly(Acrylic Acid) Analogue via Radical Ring-Opening Copolymerization and Enhanced Biodegradability. Chemosphere 2022, 293, 133487, DOI: 10.1016/j.chemosphere.2021.133487Google ScholarThere is no corresponding record for this reference.
- 238Däbritz, S. B.; Neubauer, K.; Kropf, C.; Agarwal, S. Evaluating Polymerization Methods and Deprotection Strategies for Making Water Soluble Poly(Acrylic Acid) with Hydrolyzable Breaking Points. Macro Chemistry & Physics 2025, 226 (11), 2500080, DOI: 10.1002/macp.202500080Google ScholarThere is no corresponding record for this reference.
- 239Bossion, A.; Zhu, C.; Guerassimoff, L.; Mougin, J.; Nicolas, J. Vinyl Copolymers with Faster Hydrolytic Degradation than Aliphatic Polyesters and Tunable Upper Critical Solution Temperatures. Nat. Commun. 2022, 13 (1), 2873, DOI: 10.1038/s41467-022-30220-yGoogle ScholarThere is no corresponding record for this reference.
- 240Kertsomboon, T.; Agarwal, S.; Chirachanchai, S. UCST-Type Copolymer through the Combination of Water-Soluble Polyacrylamide and Polycaprolactone-Like Polyester. Macromol. Rapid Commun. 2020, 41 (21), 2000243, DOI: 10.1002/marc.202000243Google ScholarThere is no corresponding record for this reference.
- 241Sugihara, S.; Endo, A.; Raje, K.; Matsumoto, A.; Fujita, S.; Maeda, Y. Design and Synthesis of Thermoresponsive Degradable Copolymers: Integrating Hydroxy-Functional Vinyl Ethers with Cyclic Ketene Acetals. Polym. Chem. 2025, 16 (26), 3059– 3069, DOI: 10.1039/D5PY00398AGoogle ScholarThere is no corresponding record for this reference.
- 242Neal, T. J.; Nicolas, J. Aqueous Degradability of Water-Soluble, Thioester-Containing Polyacrylamides with UCST-Type Behaviour in Salt Solutions Obtained by rROP. Chem. Commun. 2024, 60 (96), 14260– 14263, DOI: 10.1039/D4CC04718DGoogle ScholarThere is no corresponding record for this reference.
- 243Levkovsky, I. O.; Trachsel, L.; Murata, H.; Matyjaszewski, K. Versatile and Controlled Synthesis of Degradable, Water-Soluble Bottlebrush Polymers with Poly(Disulfide) Backbones Derived from α-Lipoic Acid. ACS Macro Lett. 2025, 14 (2), 207– 213, DOI: 10.1021/acsmacrolett.4c00839Google ScholarThere is no corresponding record for this reference.
- 244Abu Bakar, R.; Hepburn, K. S.; Keddie, J. L.; Roth, P. J. Degradable, Ultraviolet-Crosslinked Pressure-Sensitive Adhesives Made from Thioester-Functional Acrylate Copolymers. Angew. Chem. Int. Ed 2023, 62 (34), e202307009 DOI: 10.1002/anie.202307009Google ScholarThere is no corresponding record for this reference.
- 245Kohsaka, Y.; Naganuma, K. Main-Chain Scission of Acrylic Polymers via Intrachain Transesterification and Their Application in UV-Curable Dismountable Adhesives. ACS Polym. Au 2025, 5 (5), 481– 487, DOI: 10.1021/acspolymersau.5c00060Google ScholarThere is no corresponding record for this reference.
- 246Pal, S.; Shin, J.; DeFrates, K.; Arslan, M.; Dale, K.; Chen, H.; Ramirez, D.; Messersmith, P. B. Recyclable Surgical, Consumer, and Industrial Adhesives of Poly(α-Lipoic Acid). Science 2024, 385 (6711), 877– 883, DOI: 10.1126/science.ado6292Google ScholarThere is no corresponding record for this reference.
- 247Pal, S.; Salzman, E. E.; Ramirez, D.; Chen, H.; Perez, C. A.; Dale, K.; Ghosh, S. K.; Lin, L.; Messersmith, P. B. Versatile Solid-State Medical Superglue Precursors of α-Lipoic Acid. J. Am. Chem. Soc. 2025, 147 (16), 13377– 13384, DOI: 10.1021/jacs.4c18448Google ScholarThere is no corresponding record for this reference.
- 248Shi, Y.; Zhou, P.; Jérôme, V.; Freitag, R.; Agarwal, S. Enzymatically Degradable Polyester-Based Adhesives. ACS Biomater. Sci. Eng. 2015, 1 (10), 971– 977, DOI: 10.1021/acsbiomaterials.5b00217Google ScholarThere is no corresponding record for this reference.
- 249Yang, Y.; Shi, K.; Yu, K.; Xing, F.; Lai, H.; Zhou, Y.; Xiao, P. Degradable Hydrogel Adhesives with Enhanced Tissue Adhesion, Superior Self-Healing, Cytocompatibility, and Antibacterial Property. Adv. Healthcare Mater. 2022, 11 (4), 2101504, DOI: 10.1002/adhm.202101504Google ScholarThere is no corresponding record for this reference.
- 250Ding, J.; Han, Y.; Wu, J.; Chen, D.; Xiong, C.; Huang, D.; Xiong, Z. Degradable Underwater Adhesive Utilizing Free Radical Ring-Opening Polymerization. ACS Appl. Polym. Mater. 2024, 6 (10), 5980– 5987, DOI: 10.1021/acsapm.4c00666Google ScholarThere is no corresponding record for this reference.
- 251Yang, R.; Zhang, X.; Chen, B.; Yan, Q.; Yin, J.; Luan, S. Tunable Backbone-Degradable Robust Tissue Adhesives via in Situ Radical Ring-Opening Polymerization. Nat. Commun. 2023, 14 (1), 6063, DOI: 10.1038/s41467-023-41610-1Google ScholarThere is no corresponding record for this reference.
- 252Bailey, W. J.; Gapud, B. Synthesis of Biodegradable Polyethylene. In Polymer Stabilization and Degradation; ACS Symposium Series; American Chemical Society: Washington, D.C., 1985; Vol. 280, pp 423– 431. DOI: 10.1021/bk-1985-0280 .Google ScholarThere is no corresponding record for this reference.
- 253Wu, B.; Lenz, R. W. Synthesis, Characterization, and Hydrolytic Degradation of Copolymers of 2-Methylene-1,3-Dioxepane with Ethylene and with Styrene. Journal of Polymers and the Environment 1998, 6 (1), 23– 29, DOI: 10.1023/A:1022874411834Google ScholarThere is no corresponding record for this reference.
- 254Zeng, T.; You, W.; Chen, G.; Nie, X.; Zhang, Z.; Xia, L.; Hong, C.; Chen, C.; You, Y. Degradable PE-Based Copolymer with Controlled Ester Structure Incorporation by Cobalt-Mediated Radical Copolymerization under Mild Condition. iScience 2020, 23 (3), 100904, DOI: 10.1016/j.isci.2020.100904Google ScholarThere is no corresponding record for this reference.
- 255Wolpers, A.; Bergerbit, C.; Ebeling, B.; D’Agosto, F.; Monteil, V. Aromatic Xanthates and Dithiocarbamates for the Polymerization of Ethylene through Reversible Addition-Fragmentation Chain Transfer (RAFT). Angew. Chem. Int. Ed 2019, 58 (40), 14295– 14302, DOI: 10.1002/anie.201905629Google ScholarThere is no corresponding record for this reference.
- 256Zeng, T.-Y.; Xia, L.; Zhang, Z.; Hong, C.-Y.; You, Y.-Z. Dithiocarbamate-Mediated Controlled Copolymerization of Ethylene with Cyclic Ketene Acetals towards Polyethylene-Based Degradable Copolymers. Polym. Chem. 2021, 12 (2), 165– 171, DOI: 10.1039/D0PY00200CGoogle ScholarThere is no corresponding record for this reference.
- 257Kohsaka, Y.; Toyama, K.; Kawauchi, M.; Naganuma, K. Fast and Selective Main-Chain Scission of Vinyl Polymers Using the Domino Reaction in the Alternating Sequence for Transesterification. ACS Macro Lett. 2024, 13 (8), 1016– 1021, DOI: 10.1021/acsmacrolett.4c00295Google ScholarThere is no corresponding record for this reference.
- 258Yang, Y.; Xing, F.; Zhou, Y.; Xiao, P. Hydrolysis/Photolysis Dual-Stimuli-Responsive Backbone-Degradable Copolymers Featuring Cyclic Ketene Acetal and Ortho-Nitrobenzyl Pendants. Eur. Polym. J. 2022, 181, 111659, DOI: 10.1016/j.eurpolymj.2022.111659Google ScholarThere is no corresponding record for this reference.
- 259Do, P. T.; Poad, B. L. J.; Frisch, H. Programming Photodegradability into Vinylic Polymers via Radical Ring-Opening Polymerization. Angew. Chem. Int. Ed 2023, 62 (6), e202213511 DOI: 10.1002/anie.202213511Google ScholarThere is no corresponding record for this reference.
- 260Sbordone, F.; Micallef, A.; Frisch, H. pH-Controlled Reversible Folding of Copolymers via Formation of Β-sheet Secondary Structures. Angew. Chem. Int. Ed 2024, 63 (10), e202319839 DOI: 10.1002/anie.202319839Google ScholarThere is no corresponding record for this reference.
- 261Woodruff, M. A.; Hutmacher, D. W. The Return of a Forgotten Polymer─Polycaprolactone in the 21st Century. Prog. Polym. Sci. 2010, 35 (10), 1217– 1256, DOI: 10.1016/j.progpolymsci.2010.04.002Google ScholarThere is no corresponding record for this reference.
- 262Ikada, Y.; Tsuji, H. Biodegradable Polyesters for Medical and Ecological Applications. Macromol. Rapid Commun. 2000, 21 (3), 117– 132, DOI: 10.1002/(SICI)1521-3927(20000201)21:3<117::AID-MARC117>3.0.CO;2-XGoogle ScholarThere is no corresponding record for this reference.
- 263Mousa, M.; Jonsson, M.; Wilson, O.; Geerts, R.; Bergenudd, H.; Bengtsson, C.; Larsson Kron, A.; Malmström, E. Branched Polyesters from Radical Ring-Opening Polymerization of Cyclic Ketene Acetals: Synthesis, Chemical Hydrolysis and Biodegradation. Polym. Chem. 2023, 14 (47), 5154– 5165, DOI: 10.1039/D3PY00630AGoogle ScholarThere is no corresponding record for this reference.
- 264Hiracuri, Y.; Tokiwa, Y. Synthesis of Copolymers Composed of 2-methylene-1,3,6-trioxocane and Vinyl Monomers and Their Enzymatic Degradation. J. Polym. Sci. A Polym. Chem. 1993, 31 (12), 3159– 3163, DOI: 10.1002/pola.1993.080311233Google ScholarThere is no corresponding record for this reference.
- 265Neogi, S.; un Nisa, Q.; Abu Bakar, R.; Bingham, N. M.; Roth, P. J. Ambient Cationic Ring-Opening Polymerization of Dibenzo[c,e]Oxepine-5 (7H)-Thione (DOT): Thermal and Nucleophile-Initiated Depolymerization. Eur. Polym. J. 2024, 219, 113390, DOI: 10.1016/j.eurpolymj.2024.113390Google ScholarThere is no corresponding record for this reference.
- 266Truong, T. H. A.; Mothe, S. R.; Min, J. L.; Tan, H. M.; Jackson, A. W.; Nguyen, D. T.; Ye, D. K. J.; Kanaujia, P.; Thoniyot, P.; Dang, T. T. Immuno-Modulatory Effects of Microparticles Formulated from Degradable Polystyrene Analogue. Macromol. Biosci. 2022, 22 (7), 2100472, DOI: 10.1002/mabi.202100472Google ScholarThere is no corresponding record for this reference.
- 267Bingham, N. M.; Nisa, Q. U.; Gupta, P.; Young, N. P.; Velliou, E.; Roth, P. J. Biocompatibility and Physiological Thiolytic Degradability of Radically Made Thioester-Functional Copolymers: Opportunities for Drug Release. Biomacromolecules 2022, 23 (5), 2031– 2039, DOI: 10.1021/acs.biomac.2c00039Google ScholarThere is no corresponding record for this reference.
- 268Hiraguri, Y.; Tokiwa, Y. Synthesis of Thermosensitive Polymers Having Enzymatic Degradability. Clean Techn Environ. Policy 2002, 4 (2), 122– 124, DOI: 10.1007/s10098-002-0140-4Google ScholarThere is no corresponding record for this reference.
- 269Ren, L.; Agarwal, S. Synthesis, Characterization, and Properties Evaluation of Poly[(N-isopropylacrylamide)-co-ester]s. Macromol. Chem. Phys. 2007, 208 (3), 245– 253, DOI: 10.1002/macp.200600484Google ScholarThere is no corresponding record for this reference.
- 270Siegwart, D. J.; Bencherif, S. A.; Srinivasan, A.; Hollinger, J. O.; Matyjaszewski, K. Synthesis, Characterization, and in Vitro Cell Culture Viability of Degradable Poly(N -isopropylacrylamide- Co −5,6-benzo-2-methylene-1,3-dioxepane)-based Polymers and Crosslinked Gels. J. Biomedical Materials Res. 2008, 87A (2), 345– 358, DOI: 10.1002/jbm.a.31708Google ScholarThere is no corresponding record for this reference.
- 271Barbon, S. M.; Carter, M. C. D.; Yin, L.; Whaley, C. M.; Albright, V. C.; Tecklenburg, R. E. Synthesis and Biodegradation Studies of Low-Dispersity Poly(Acrylic Acid). Macromol. Rapid Commun. 2022, 43 (13), 2100773, DOI: 10.1002/marc.202100773Google ScholarThere is no corresponding record for this reference.
- 272Guégain, E.; Michel, J.-P.; Boissenot, T.; Nicolas, J. Tunable Degradation of Copolymers Prepared by Nitroxide-Mediated Radical Ring-Opening Polymerization and Point-by-Point Comparison with Traditional Polyesters. Macromolecules 2018, 51 (3), 724– 736, DOI: 10.1021/acs.macromol.7b02655Google ScholarThere is no corresponding record for this reference.
- 273Volz, H. K.-H.; Agarwal, S. High Tg Poly(Ester- Co -Vinyl)s with Compositional Homogeneity by Radical Ring-Opening Polymerization. ACS Appl. Polym. Mater. 2023, 5 (10), 8754– 8763, DOI: 10.1021/acsapm.3c01970Google ScholarThere is no corresponding record for this reference.
- 274Luzel, B.; Efimov, I.; Edeleva, M.; Kouider, S.; Gil, N.; Montarnal, D.; Lefay, C.; Gigmes, D.; Van Steenberge, P. H. M.; D’hooge, D. R.; Guillaneuf, Y. The Successful Impossible Radical Ring-Opening Copolymerization of Thionolactones and Methacrylates via an Auxiliary Third Comonomer. Nat. Commun. 2025, 16 (1), 9468, DOI: 10.1038/s41467-025-64502-yGoogle ScholarThere is no corresponding record for this reference.
- 275Bingham, N. M.; Collins, K. E.; Walsh, J.; Williams, A.; Roth, P. J. Bespoke Degradable Polymers: Modifying Cyclic Allyl Sulfide Lactones to Tune Polymer Degradation Rates and Lower Critical Solution Temperatures. Macromolecules 2025, 58 (1), 516– 525, DOI: 10.1021/acs.macromol.4c02681Google ScholarThere is no corresponding record for this reference.
- 276Ivanchenko, O.; Mazières, S.; Harrisson, S.; Destarac, M. On-Demand Degradation of Thioester/Thioketal Functions in Vinyl Pivalate-Derived Copolymers with Thionolactones. Macromolecules 2023, 56 (11), 4163– 4171, DOI: 10.1021/acs.macromol.3c00312Google ScholarThere is no corresponding record for this reference.
- 277Rahimi, A.; García, J. M. Chemical Recycling of Waste Plastics for New Materials Production. Nat. Rev. Chem. 2017, 1 (6), 0046, DOI: 10.1038/s41570-017-0046Google ScholarThere is no corresponding record for this reference.
- 278Czuczola, M.; Hossain, M. S.; Shannon, D. P.; Morris, P. T.; Getty, P. T.; Bates, C. M.; Read de Alaniz, J.; Hawker, C. J. Telechelic Dithiol Copolymers as Tunable Building Blocks for Synthesizing Multiblock Materials. J. Polym. Sci. 2025, 63 (3), 759– 765, DOI: 10.1002/pol.20240876Google ScholarThere is no corresponding record for this reference.
Cited By
This article has not yet been cited by other publications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACS Polymers Au
© 2026 The Authors. Published by American Chemical Society. This publication is licensed under
License Summary*
You are free to share (copy and redistribute) this article in any medium or format and to adapt (remix, transform, and build upon) the material for any purpose, even commercially within the parameters below:
Creative Commons (CC): This is a Creative Commons license.
Attribution (BY): Credit must be given to the creator.
*Disclaimer
This summary highlights only some of the key features and terms of the actual license. It is not a license and has no legal value. Carefully review the actual license before using these materials.
Article Views
Altmetric
Citations
Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.
Recommended Articles
Abstract
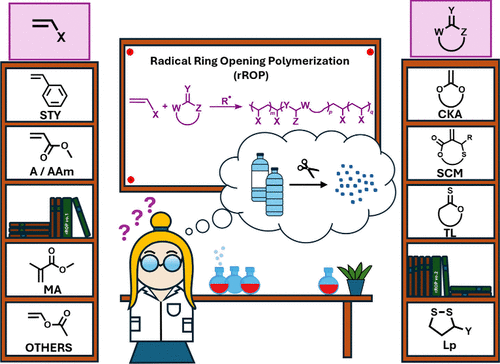
Figure 1

Figure 1. Radical copolymerization of cyclic and vinyl monomers aimed at developing degradable materials.
Figure 2

Figure 2. Structures of the two main categories of cyclic monomers that can be polymerized by radical ring-opening polymerization (rROP).
Figure 3

Figure 3. Competition between radical ring-opening (β-scission) and ring retention (1,2-addition).
Figure 4
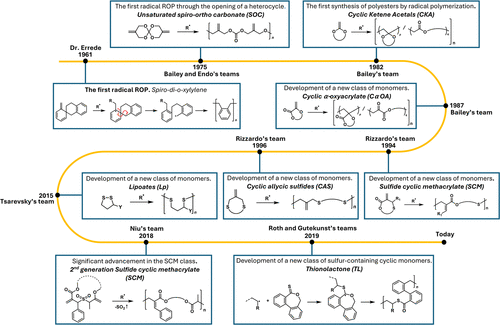
Figure 4. Timeline of the history of rROP with the different families of monomers used.
Figure 5
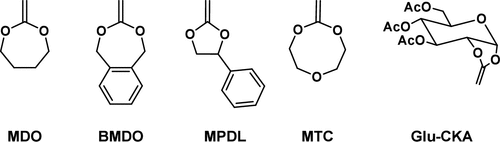
Figure 5. Structures of the most efficient CKAs in rROP.
Figure 6
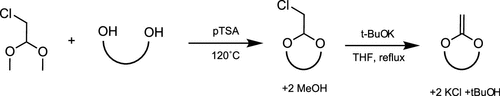
Figure 6. Synthesis of CKA via the transacetalization and dehydrochloration reaction.
Figure 7

Figure 7. Two other synthesis pathways: a new acetal pathway and carbonate pathway.
Figure 8
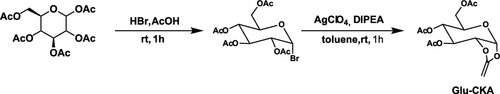
Figure 8. Synthesis pathway of Glu-CKA.
Figure 9
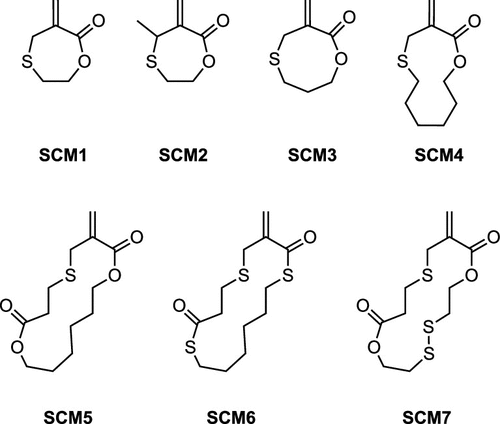
Figure 9. Structures of sulfide cyclic methacrylate monomers.
Figure 10
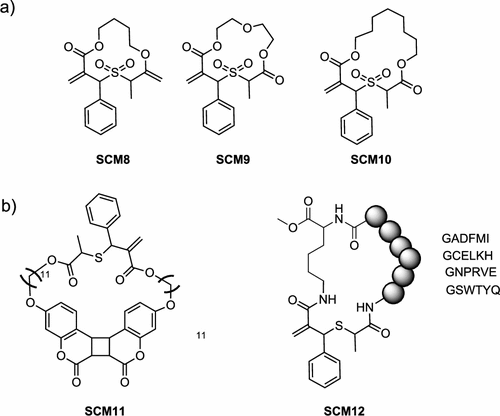
Figure 11

Figure 11. Synthesis of sulfide cyclic methacrylate-type monomers (SCM). a) First generation, b) second generation, and c) cyclic sulfide diene (CSD).
Figure 12
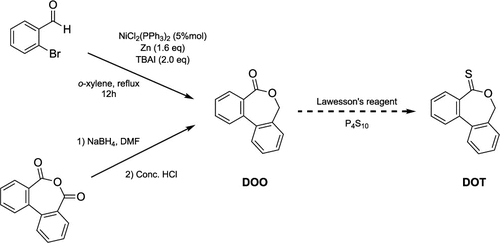
Figure 12. Synthesis of dibenzo[c,e]-oxepine-5(7H)-thione (DOT).
Figure 13

Figure 13. Synthesis of 7-phenyloxepane-2-thione (POT).
Figure 14

Figure 14. Synthesis of 10-fluoro-7-(4-(trifluoromethyl) phenyl) DOT (F-p-CF3PhDOT).
Figure 15
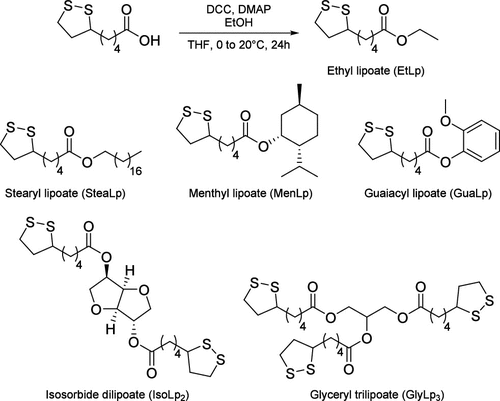
Figure 15. Synthesis of ethyl lipoate and structures of monomers previously reported in the literature.
Figure 16

Figure 16. (A) Kinetic competition between vinyl propagation and ring opening. (B) Percentage of ring opening for 5-, 6-, and 7-membered CKA monomers (filled points: experimental data; empty points: theoretical data). Reproduced from ref (82) with permission. Copyright 2020 Wiley-VCH.
Figure 17
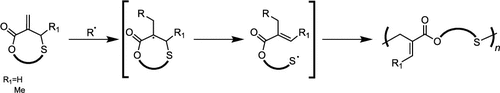
Figure 17. Mechanism of radical polymerization via ring-opening of sulfide cyclic methacrylates (SCM1–SCM7).
Figure 18

Figure 18. a) Mechanism of radical polymerization via ring-opening of sulfide cyclic methacrylates (SCM8–SCM10). b) Mechanism of radical polymerization via ring opening of CSD monomers.
Figure 19

Figure 19. Homopolymerization of (A) DBT and (B) TIC.
Figure 20

Figure 20. Homopolymerization of POT.
Figure 21

Figure 21. Copolymerization kinetics and associated reactivity ratios.
Figure 22

Figure 22. Simulation of individual chain degradation, obtained from kinetic Monte Carlo simulations: Calculated size exclusion chromatography (SEC) traces for polymer chains and degradation products. SEC traces before and after copolymer hydrolysis under various conditions: (top) RDRP (orange) versus uncontrolled (green) radical polymerization. (Bottom) Comparison between the most heterogeneous (black) and most homogeneous (orange) RDRP degradation products. In each panel, the degraded product appears to the left of the corresponding initial polymer (same color). Reproduced from ref (103) with permission. Copyright 2018 Wiley-VCH.
Figure 23

Figure 23. (a) Schematic of the cleavable comonomer additive (CCA) approach for deconstructable copolymers. CCAs copolymerize with standard monomers (“M1”), introducing cleavable sites along the backbone. (b) Relative decrease in molecular weight (Mw,deg/Mw,poly) as a function of reactivity ratio pairs, r1 and rCCA, for M1 and CCA, respectively. For all simulations presented, a degree of polymerization of 1000 was targeted with a CCA loading of 2.5 mol %. (c) Fractional decrease in number-average molecular weight (Mn,deg/Mn,poly) as a function of reactivity ratio pairs. (d) Dispersity (Đ) of the deconstructed fragments as a function of reactivity ratio pairs. Reproduced from ref (105) with permission. Copyright 2024 American Chemical Society.
Figure 24

Figure 24. (a) Elementary steps involved in the ring-opening polymerization (ROP) of thionolactones with vinyl monomers, along with the corresponding rate constants: kadd: rate constant for addition, k–add rate constant for reverse addition, kβ: rate constant for fragmentation, kp: rate constant for propagation. (b) Definition of the transfer constant ktr. (c) Determination of the kp/ktr ratio to estimate copolymerization behavior. Adapted from ref (69) with permission. Copyright 2023 American Chemical Society. To analyze this process, the transfer rate constant (ktr) was used. This encompasses the three previously mentioned steps and allows for modeling of the addition–fragmentation mechanism. It was then compared to the propagation constant of the vinyl monomer (kp), providing a relevant criterion to assess the reactivity of the comonomer pair by determining the reactivity ratio rv. In these systems, the cyclic monomer is typically introduced as an additive in low concentration (less than 10 mol %), meaning that the majority of the growing macroradicals are polyvinyl macroradical. This approach simplified the calculations by avoiding the determination of reactivity ratios specific to thionolactones.
Figure 25

Figure 25. (a, b) Important protons used for the 1H NMR (CDCl3) analysis of P(CKA-co-S) copolymers and degraded styrenic oligomers in P(MDO-co-S) copolymers and P(BMDO-co-S) copolymers. Reprinted from ref (110) with permission. Copyright 2020 MDPI.
Figure 26

Figure 26. Analysis of substituent effects on the copolymerization of styrene with DOT-based thionolactone. Reproduced from ref (78) with permission. Copyright 2022 American Chemical Society.
Figure 27

Figure 27. Experimental and theoretical composition of the POT monomer as a function of overall molar conversion during solution copolymerization at 80 °C initiated with 0.5 mol % AIBN from a mixture of POT (5 mol %) and styrene in anisole: Cumulative average molar thioester content in the copolymers. Adapted from ref (69) with permission. Copyright 2023 American Chemical Society.
Figure 28

Figure 28. Preparation of a polyacrylate-based P(nBA-b-tBA) diblock copolymer containing 5 mol % of DOT into the two blocks. Reproduced from ref (37) with permission. Copyright 2019 American Chemical Society.
Figure 29

Figure 29. Experimental and theoretical composition of the POT monomer as a function of overall molar conversion during solution copolymerization at 80 °C, initiated with 0.5 mol % AIBN from a mixture of POT (5 mol %) with isobornyl acrylate: Cumulative average molar thioester composition of the copolymers. Adapted from ref (69) with permission. Copyright 2023 American Chemical Society.
Figure 30

Figure 30. Tunable degradation of poly(acrylate) copolymers by controlling the concentration and temperature of polymerization. (a) The degradability of lipoic-acid–acrylate copolymers can be synthetically tuned through polymerization conditions that control the average number of disulfide bonds per polymer chain. (b, c) As evidenced by size-exclusion chromatography, (b) higher monomer concentrations ([M]), and (c) lower polymerization temperatures (T) improve degradability. Reproduced from ref (130) with permission. Copyright 2023 American Chemical Society.
Figure 31

Figure 31. Real-time 1H NMR monitoring of copolymerization. (A) Reaction scheme; (B–D) real-time 1H NMR tracking of conversion versus reaction time: (B) Glu-CKA/MMA = 1:1; (C) Glu-CKA/MI/MMA = 1:1:1; and (D) Glu-CKA/MI/MMA = 1:2:5. Reproduced from ref (57) with permission. Copyright 2024 American Chemical Society.
Figure 32

Figure 32. (A) Relative Gibbs free energy profile for an MMA radical reacting either with MMA or with DOT, calculated to model homopropagation and cross-propagation of a chain terminating in MMA. (B) The effect of benzylic substituent(s) on the energy profile was evaluated using DFT calculations. Calculations were performed at the wB97X-D3/def2-SVP level of theory; electronic energies of all optimized structures were re-evaluated using wB97X-D3/def2-TZVP/CPCM (toluene). (C) A Monte Carlo simulation evaluates the efficiency of aromatic bDOTs as cleavable comonomers. The heat map generated by the simulation visualizes the ratio of degraded fragment size to copolymer size as a function of reactivity ratios, for a 2.5% molar loading of CC in copolymers with DP 1000. (D) A series of bDOTs was synthesized for optimization of copolymerization reactivity. Reproduced from ref (70) with permission. Copyright 2024 American Chemical Society.
Figure 33

Figure 33. (A) Preparation of degradable PMMA derivatives via terpolymerization of MMA, DOT, and N-phenylmaleimide (PhMal; in red). (B) Simulated monomer sequences for modeling-assisted copolymerization with [MMA]0:[PhMal]0:[DOT]0 = 90:18:28 (30% solvent). Monomer sequences follow the color code from panel (C). On the right: selection of chains from the left panel, showing isolated MMA-PhMal units (red box) and MMA-DOT-PhMal triads (green box). Reproduced from ref (125) with permission. Copyright 2025 Springer Nature.
Figure 34

Figure 34. Radical ring-opening copolymerization of cyclic thionocarbamates with N-vinylpyrrolidone. Reproduced from ref (172) with permission. Copyright 2024 American Chemical Society.
Figure 35

Figure 35. Molar DOT content in maleimide copolymer vs molar DOT fraction in the monomer feed with nonlinear least-squares fitted curves for (A) N-methylmaleimide, (B) N-phenylmaleimide, and (C) N-2,3,4,5,6-pentafluorophenylmaleimide. Adapted from ref (97) with permission. Copyright 2020 American Chemical Society.
Figure 36

Figure 36. (A) Synthesis of SDS-stabilized P(MMA-co-BMDO) latexes by aqueous emulsion polymerization. (B) Photos of the latexes obtained with various BMDO contents. (C) SEC traces of the dry extracts of P(MMA-co-BMDO) latexes (plain lines) and their degradation products (dashed lines) as a function of incorporated BMDO content. Adapted from ref (187) with permission. Copyright 2023 Royal Society of Chemistry.
Figure 37

Figure 37. Synthesis of block copolymer nanoparticles with degradable cores via self-assembly induced by radical ring-opening copolymerization (rROPISA) mediated by RAFT from cyclic ketene acetals (CKAs). Reproduced from ref (196) with permission. Copyright 2019 American Chemical Society.
Figure 38
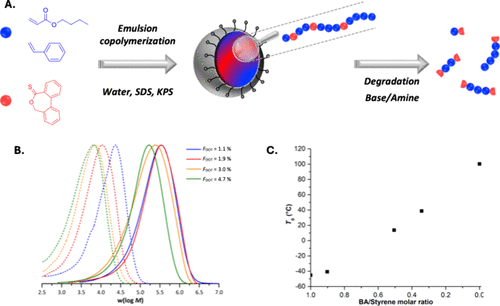
Figure 38. (A) Synthesis of SDS-stabilized latexes of P(BA-co-DOT), P(S-co-DOT), and P(BA-co-S-co-DOT) by aqueous emulsion polymerization. (B) Molar mass distribution of the dry extracts of P(S-co-DOT) latexes (plain lines) and their degradation products with TBD (dashed lines) as a function of incorporated DOT content (up to 4.7 mol %). (C) Evolution of the Tg depending on the average molar fraction BA/styrene in the monomer mixture for emulsion polymerization with 2 mol % of DOT. Adapted from ref (201) with permission. Copyright 2022 Wiley-VCH.
Figure 39
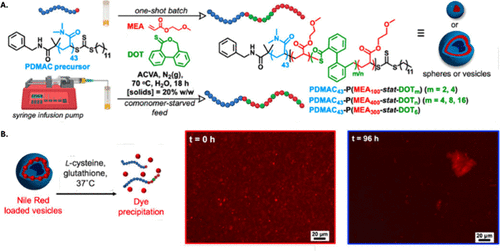
Figure 39. (A) Synthesis of PDMAC43-P(MEA100-co-DOTm) (m = 2 or 4) spheres and PDMAC43-P(MEA300-co-DOT6) and PDMAC43-P(MEA400-co-DOTn) (n = 4, 8, or 16) vesicles via aqueous rROPISA with 20% w/w solids. The MEA/DOT mixture was added either all at once or gradually using a syringe pump (0.2 mL·h–1 over 2 h). (B) Scheme showing the Nile Red probe (red spheres) loaded in the membrane of PDMAC43-P(MEA400-co-DOT8) vesicles. Degradation of these vesicles in the presence of 10 mM l-cysteine and 10 mM glutathione leads to precipitation of insoluble probes. (C) Fluorescence micrographs (λex = 550 nm, λem = 605 nm) were recorded for 1% w/w dispersions at two time points (0 and 96 h) during hydrolytic degradation. Reproduced from ref (203) with permission. Copyright 2025 American Chemical Society.
Figure 40

Figure 40. (A) Synthesis of PAA-b-P(nBA-co-DOT) and PAA-b-P(S-co-DOT) copolymers by rROPISA in water. (B) SEC traces of the dry extracts and NPs composed of a PAA-b-P(nBA-co-DOT) copolymers with 1.3 mol % DOT before and after degradation in the presence of TBD or isopropylamine. Reproduced from ref (204) with permission. Copyright 2022 American Chemical Society.
Figure 41

Figure 41. (A) Miniemulsion polymerization of α-lipoic acid with n-butyl acrylate. (B) Degradation for different amounts of ethyl lipoate with TCEP in an H2O/THF mixture. Reproduced from ref (129) with permission. Copyright 2024 American Chemical Society.
Figure 42

Figure 42. (A) Preparation of surface coatings in the form of polymer brushes grafted onto silica surfaces, obtained by copolymerizing poly(ethylene glycol) methacrylate (PEGMA) with the cyclic monomer BMDO. (B) 3D AFM images and 2D cross-sectional profiles of P(PEGMA) brushes with and without BMDO, taken at different time intervals during exposure to a pH 3 solution at 25 °C. Reproduced from ref (208) with permission. Copyright 2009 American Chemical Society.
Figure 43

Figure 43. (A) Antifouling mechanism of degradable and hydrolyzable polymers. (B) Structures of degradable and hydrolyzable polymers. Reproduced from ref (213) with permission. Copyright 2022 American Chemical Society.
Figure 44
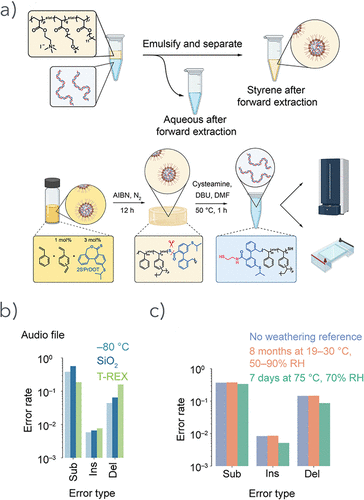
Figure 44. (a) Scheme for producing T-REX thermosets from polyplexes, reversible encapsulation, and subsequent characterization. (b) A comparative analysis of error rates in 210-bp dsDNA segments encoding digital data between samples stored in a frozen state without encapsulation and DNA recovered from both T-REX and silica-based encapsulated samples. (c) Comparison of error rates of T-REX-encapsulated samples containing 210-bp dsDNA encoding an image file subjected to real-time and accelerated weathering conditions. Reproduced from ref (221) with permission. Copyright 2024 American Chemical Society.
Figure 45

Figure 45. (a) During the DLW process, aliphatic polyester units are incorporated into the cross-linked network; after treatment with a nucleophile (Nu–), these units break down, degrading the microstructure. Cleavage of the ester bond by the nucleophile occurs between the carbonyl carbon and the oxygen (not shown for clarity). (b) Partial degradation (SEM images) of microdonut structures fabricated with MDO:PETA 90:10 (scale bar 20 μm). Only the final part degrades, leaving a bite mark. Reproduced from ref (222) with permission. Copyright 2022 Wiley-VCH.
Figure 46

Figure 46. (A) Preparation of degradable 3D objects by VAT photopolymerization and chain cleavage. (B) Example of a 3D product with and without cleavable copolymer additive. Reproduced from ref (223) with permission. Copyright 2022 Wiley-VCH. (C) Concept of self-destruct materials via the combination of thermolatent base and cleavable comonomers.
Figure 47

Figure 47. (A) Chemical structures of a 3D printing resin derived from α-lipoic acid building blocks. The resin components include n-butyl acrylate, a mixture of cross-linkers DIS-Lp2/TEG-Lp2, and the photoinitiator (BAPO). (B) Diagram illustrating self-healing, degradation, and recycling of the printed material using DIS-Lp2 as the cross-linker. Reproduced from ref (225) with permission. Copyright 2024 American Chemical Society.
Figure 48

Figure 48. Method enabling polymerization–depolymerization cycles of dynamic disulfide bonds, allowing for the formulation of 3D-printing resins from renewable sources that are suitable for closed-loop chemical recycling. (a) Chemical composition of the formulated resin. (b) An example of a complex 3D-printed part. (c) Photograph of 3D-printed parts in powder form. (d) Photograph of the resin recovered in 98% yield after depolymerizing a 3D-printed part. e) SEC of initial resin compared to recovered resin. Adapted from ref (81) with permission. Copyright 2024 Springer Nature.
Figure 49

Figure 49. a) Synthesis strategy for the design of gemcitabine-based degradable polymeric prodrugs via nitroxide-mediated polymerization initiated by a Gem-alkoxyamine initiator. Reproduced from ref (230) with permission. Copyright 2018 Royal Society of Chemistry. b) Design and preclinical development of (degradable) polyacrylamide (PAAm)-based prodrugs for the SC administration of the anticancer drug gemcitabine (Gem). Reproduced from ref (231) with permission. Copyright 2025 Royal Society of Chemistry.
Figure 50

Figure 50. (A) Synthesis of poly(N,N-dimethylaminoethyl methacrylate-co-methacrylic acid-co-5,6-benzo-2-methylene-1,3-dioxepane) poly(DMAEMA-co-MAA-co-BMDO) terpolymers by RAFT terpolymerization of DMAEMA, TBDMSMA, and BMDO with CPADB as a RAFT agent, followed by deprotection of TBDMSMA units. (234) (B) Schematic of the monolayer cryopreservation and post-thaw process. (C) Cell recovery 24 h post-thaw, relative to prefreezing, determined using Trypan blue exclusion test (left) and cell viability 24 h post thaw determined using Trypan blue exclusion test (right). One-way ANOVA with Tukey’s posthoc test. * = p < 0.05, ** = p < 0.001 considered as statistically significant different using a 95% confidence level, ns = not significant. Reproduced from ref (234) with permission. Copyright 2022 American Chemical Society.
Figure 51

Figure 51. (A) rROP of MDO and tBA yielding poly(MDO-co-tBA), followed by acid-mediated tert-butyl deprotection to obtain degradable poly(MDO-co-AA). (B) Overview of biodegradability, through initial hydrolysis of the main-chain esters into short oligomers, followed by complete biodegradation. Reproduced from ref (237) with permission. Copyright 2022 Elsevier.
Figure 52

Figure 52. (A) Synthesis of P(AAm-co-BMDO). (B) Evolution of the Mn with time during hydrolytic degradation in physiological conditions (PBS, pH 7.4, T = 37 °C) of (1) PAAm (P9), P(AAm-co-BMDO) copolymers with different BMDO contents (P10–P13 and P17) and (2) PLA and PLGA. (C) Evolution of the Mn with time during enzymatic degradation with lipases (Candida antartica, PBS, pH 7.4, T = 37 °C) of (1) PAAm (P9), P(AAm-co-BMDO) copolymers P13 and P17 and (2) PLA and PLGA. Reproduced from ref (239) with permission. Copyright 2022 Springer Nature.
Figure 53

Figure 53. Proposed reaction pathway for a 17O-labeled experiment for the hydrolysis of MDO with H217O at pH 2. Reproduced from ref (50) with permission. Copyright 2023 Wiley-VCH.
Figure 54
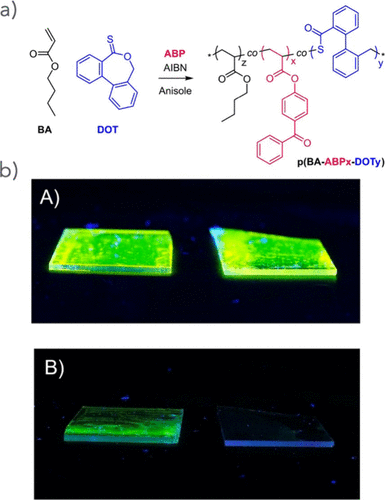
Figure 54. (a) Synthesis of UV cross-linkable degradable thioester-functional PSA. (c) Photos of dye-labeled photo-cross-linked copolymer films on glass substrates of the nondegradable control BA-ABP0.05-NBDA0.25 (left) and degradable BA-ABP0.05-DOT0.25-NBDA0.25 (right) A) before immersion and B) after immersion in 2 M n-propylamine in THF for 120 s, confirming visually the presence of insoluble residue for the control sample only. Reproduced from ref (244) with permission. Copyright 2023 Wiley-VCH.
Figure 55

Figure 55. (a) In situ rROP of CKA and comonomers to form a degradable and functional macromolecular chain by redox initiation benzoyl peroxide/N,N-dimethyl-p-toluidine (BPO/DMPT). (b) Tunable preparation of the adhesive called backbone-degradable robust adhesives (BDRAs) that achieve strong adhesion by forming a covalent interpenetrating network by in situ rROP and the synergy of intermolecular and chemical bonds. (c) Adhesion strength and setting time of BDRAs and the existing tissue adhesives for hard and soft tissues. (d) Bearing capacity of bonded fractured bovine bone using BDRAs. (e) Adhesion strength of BDRAs and commercial medical adhesives on different biological tissues, represented by flexural strength for bone and shear strength for pigskin. Data are presented as the means ± SDs, n = 3 independent samples per group. (f) Shear adhesion strength for low-surface-energy polymers adhered by a BDRA and commercial engineering adhesives. PP polypropylene, PE polyethylene, PTFE polytetrafluoroethylene. Data are presented as the means ± SDs, n = 3 independent samples. Reproduced from ref (251) with permission. Copyright 2023 Springer Nature.
Figure 56

Figure 56. (A) rROP of ethylene, vinyl acetate and thionolactone for the production of chemically degradable PE and EVA. (B) Thermogravimetric analyses of P(E-co-TCL). (C) SEC analyses of pristine and degraded (dashed lines) P(E-co-VAc-co-TCL). Adapted from ref (178) with permission. Copyright 2024 American Chemical Society.
Figure 57

Figure 57. a) Degradation of a PMMA-based copolymer via a specific TBAF triggering. b) SEC of pristine and degraded copolymer ([MMA]:[BMDO]:[SiOMMA] = 25:18:57, Mn = 31,100 g·mol–1, D = 1.54). c) Evolution of Mp with an increasing of amount of MMA in the copolymer. Reproduced from ref (257) with permission. Copyright 2024 American Chemical Society.
Figure 58

Figure 58. (a) Triblock copolymer synthesis and its stepwise photodegradation: a diblock copolymer consisting of a PDMA nondegradable block and photodegradable copolymer of the coumarin cycloadduct and DMA block is prepared by green light-initiated RAFT polymerization. Chain extension of this polymer with RAFT copolymerization of DMA and the cyclic monomer resulted from intramolecular [2 + 2] cycloaddition of styrylpyrene under blue light yields a triblock polymer with the copolymer of DMA and styrylpyrene cycloadduct as the third block. Under UVA, the styrylpyrene cycloadduct experiences [2 + 2] cycloreversion, leading to the fragmentation of the third block. Subsequent UVB irradiation initiates the degradation of the second block as the coumarin dimer in the polymer backbone undergoes [2 + 2] cycloreversion. Reproduced from ref (259) with permission. Copyright 2023 Wiley-VCH. (b) Synthesis of a polypeptide mimic and its SEC traces before (P1-tBu) and after (P1) deprotection. DOSY NMR of P1 at a 1 mg/mL concentration and CD spectra of P1 (0.2 mg/mL) at basic (pink) and acidic (purple) pH. The CD traces have not been smoothed. Reproduced from ref (260) with permission. Copyright 2024 Wiley-VCH.
Figure 59

Figure 59. a) Degree of hydrolysis of PCL, PMDOs, and PMe-MDO as a function of time. b) Biodegradation test results of PCL, PMDO-DB 10%, PMDO-DB 18% and PMe-MDO in river water. Mean and standard deviations (SDs) are calculated based on the biodegradation achieved in three replicate bottles for two biological replicate per inoculum. Reproduced from ref (263) with permission. Copyright 2023 Royal Society of Chemistry.
Figure 60

Figure 60. Proposed degradation pathways of a pDOT chain, where X = H and Me, and Y = OH and OTf for BF3·Et2O-initiated and MeOTf-initiated polymers, respectively. Reproduced from ref (265) with permission. Copyright 2024 Elsevier.
Figure 61

Figure 61. Evolution of the molar mass distribution of a P(S-co-POT) prepared at 80 °C and 5% POT before and after various degradation conditions. Reproduced from ref (69) with permission. Copyright 2023 American Chemical Society.
Figure 62

Figure 62. Temperature influence on the enzymatic degradation by proteinase K of P(NIPAM-co-MDO) hydrogels. Reproduced from ref (119) with permission. Copyright 2003 Wiley-VCH.
Figure 63

Figure 63. OECD 301 D Closed Bottle Test biodegradability (%) results of degradable P(MDO-co-AA) and nondegradable poly(AA) using secondary effluent from domestic wastewater treatment plant, as inoculum. Results shown are the average of the triplicates, Following the OECD Guideline for Ready Biodegradability, the test results were valid since reference compound achieves more than 60% biodegradability on Day 14. Reproduced from ref (237) with permission. Copyright 2022 Elsevier.
Figure 64

Figure 64. Various ecmhanisms of degradation for polyacrylate and polyacrylamide-based copolymers containing DOT units into the backbone. Reproduced from ref (267) with permission. Copyright 2022 American Chemical Society.
Figure 65

Figure 65. Evolution of the number-average molar mass, Mn, with time of different P(OEGA-co-MPDL) copolymers, PLA and PCL during the hydrolytic degradation in PBS (0.1 M, pH 7.4, 37 °C). Reproduced from ref (272) with permission. Copyright 2018 American Chemical Society.
Figure 66

Figure 66. a) Various degradation conditions for polymethacrylate derivatives containing SCM5–7 monomers. b) SEC traces of the PMMA-based copolymer containing 0.3% of SCM7 and 0.8% of SCM5 and its products after stepwise degradation. Adapted from ref (41) with permission. Copyright 2009 American Chemical Society.
Figure 67

Figure 67. Change of SEC-measured molar mass (normalized to intact species) versus time during the hydrolysis (in 8 mM NaOH in water–methanol) of three p(R-SCM-co-HPMAm) copolymers. Values are the fitted rates of degradation for each copolymer. Adapted from ref (275) with permission. Copyright 2025 American Chemical Society.
Figure 68
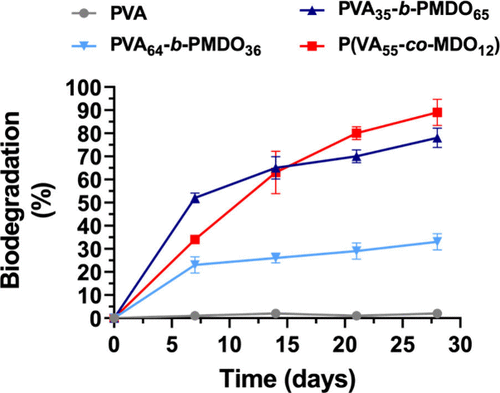
Figure 68. OECD 301 D Closed Bottle Test biodegradability (%) results of solid sample of nondegradable PVA (Mn = 5,100 g·mol–1), degradable P(VA-co-MDO) (88% VA units, Mn = 1,700 g·mol–1) and nanoparticles of degradable P(VA-b-MDO) (35% VA units, Mn = 7,300 g·mol–1) and degradable P(VA-b-MDO) (64% VA units, Mn = 12,000 g·mol–1) using secondary effluent from domestic wastewater treatment plant, as inoculum. The results shown are the average of the triplicates. Adapted from refs (199) and (200) with permission. Copyright 2023 Elsevier.
Figure 69

Figure 69. Enzymatic hydrolysis test results for CKA-NVP polymers. Adapted from ref (163) with permission. Copyright 2024 Elsevier.
Figure 70

Figure 70. a) Biodegradation curves of CKA-based polymers following OECD 301F (solid lines) and modified OECD 302B (dashed line) protocols. b) SEC analysis of poly(MTC-co-NVP) before and after modified OECD 302B biodegradation test. The assigned mass peak sets for species comprising one MTC unit from the spectrum (B) are listed in spectra (C–F), corresponding to oligomer structures (2)–(5). (G) Proposed biodegradation pathways, based on mass spectrometry analysis. Adapted from ref (163) with permission. Copyright 2024 Elsevier.
Figure 71

Figure 71. Evolution of the weight-average molar mass (Mw) during the hydrolytic degradation under accelerated conditions (0.05 wt % NaOH MeOH:THF 1:1 v/v) (a, b) or under physiological conditions (PBS, pH 7.4, 37 °C) (c, d) of P(MDO-co-BVE) and P(MTC-co-BVE) copolymers, and P(MDO-co-TEGVE) and P(MTC-co-TEGVE) copolymers. Adapted from ref (168) with permission. Copyright 2023 American Chemical Society.
Figure 72
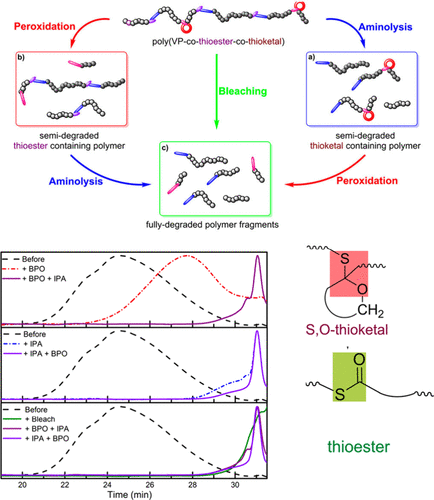
Figure 72. Schematic representation of the two-step (peroxidation/aminolysis) and one-step (bleach) degradation processes for P (vinyl pivalate-co-thionocaprolactone). Experimental SEC chromatograms before and after degradation: P(TCL0.68-co-VP0.32) with benzyl peroxide (BPO) and subsequent addition of N-isopropylamine (IPA); IPA and subsequent addition of BPO; and bleach in comparison to the two-step degradation. Adapted from ref (276) with permission. Copyright 2023 American Chemical Society.
Figure 73

Figure 73. a) Synthetic scheme illustrating the deconstruction and recycling of high-molar-mass PS. SEC traces for the deconstruction/recycling cycles of P(S-co-DOT). b) Application of the deconstruction/reconstruction strategy to an acrylic copolymer; SEC traces for the deconstruction/recycling cycles of P(nBA-co-DOT). Reproduced from ref (78) with permission. Copyright 2022 American Chemical Society.
Figure 74

Figure 74. A) Removal of label adhesive (αLA-ELp-nBA and nBA-AA) attached to recyclable plastic bottles. B) Model adhesive (ELp-nBA) with functional chain-ends produced after degradation can undergo repeated oxidative repolymerization and reductive degradation for closed-loop recycling, as evidenced by c) size-exclusion chromatography analysis. Reproduced from ref (130) with permission. Copyright 2023 American Chemical Society.
Figure 75

Figure 75. Closed-loop recycling of PS-PnBA multiblock copolymers containing disulfide linkages. (i) Mild oxidation (I2/pyridine) reconnects α,ω-dithiol PS and PnBA blocks into high-molar-mass copolymers; (ii) reductive treatment (TCEP) cleaves the disulfide bonds. Reproduced from ref (278) with permission. Copyright 2025 Wiley-VCH.
Figure 76
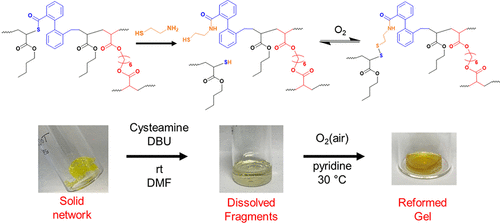
Figure 76. Degradation and regelation scheme for PBA-DOT networks prepared by RAFT polymerization. Reproduced from ref (217) with permission. Copyright 2023 Royal Society of Chemistry.
References
This article references 278 other publications.
- 1Braunecker, W.; Matyjaszewski, K. Controlled/Living Radical Polymerization: Features, Developments, and Perspectives. Prog. Polym. Sci. 2007, 32 (1), 93– 146, DOI: 10.1016/j.progpolymsci.2006.11.002There is no corresponding record for this reference.
- 2Corrigan, N.; Jung, K.; Moad, G.; Hawker, C.; Matyjaszewski, K.; Boyer, C. Reversible-Deactivation Radical Polymerization (Controlled/Living Radical Polymerization): From Discovery to Materials Design and Applications. Prog. Polym. Sci. 2020, 111, 101311, DOI: 10.1016/j.progpolymsci.2020.101311There is no corresponding record for this reference.
- 3Perrier, S. 50th Anniversary Perspective: RAFT Polymerization-A User Guide. Macromolecules 2017, 50 (19), 7433– 7447, DOI: 10.1021/acs.macromol.7b00767There is no corresponding record for this reference.
- 4Moad, G.; Rizzardo, E.; Thang, S. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58 (6), 379– 410, DOI: 10.1071/CH05072There is no corresponding record for this reference.
- 5Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-Mediated Polymerization. Prog. Polym. Sci. 2013, 38 (1), 63– 235, DOI: 10.1016/j.progpolymsci.2012.06.002There is no corresponding record for this reference.
- 6Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101 (9), 2921– 2990, DOI: 10.1021/cr940534gThere is no corresponding record for this reference.
- 7Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45 (10), 4015– 4039, DOI: 10.1021/ma3001719There is no corresponding record for this reference.
- 8Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-Catalyzed Living Radical Polymerization. Chem. Rev. 2001, 101 (12), 3689– 3745, DOI: 10.1021/cr9901182There is no corresponding record for this reference.
- 9Pan, X.; Tasdelen, M.; Laun, J.; Junkers, T.; Yagci, Y.; Matyjaszewski, K. Photomediated Controlled Radical Polymerization. Prog. Polym. Sci. 2016, 62, 73– 125, DOI: 10.1016/j.progpolymsci.2016.06.005There is no corresponding record for this reference.
- 10Chen, M.; Zhong, M.; Johnson, J. Light-Controlled Radical Polymerization: Mechanisms, Methods, and Applications. Chem. Rev. 2016, 116 (17), 10167– 10211, DOI: 10.1021/acs.chemrev.5b00671There is no corresponding record for this reference.
- 11Blasco, E.; Sims, M.; Goldmann, A.; Sumerlin, B.; Barner-Kowollik, C. 50th Anniversary Perspective: Polymer Functionalization. Macromolecules 2017, 50 (14), 5215– 5252, DOI: 10.1021/acs.macromol.7b00465There is no corresponding record for this reference.
- 12Iha, R.; Wooley, K.; Nyström, A.; Burke, D.; Kade, M.; Hawker, C. Applications of Orthogonal “Click” Chemistries in the Synthesis of Functional Soft Materials. Chem. Rev. 2009, 109 (11), 5620– 5686, DOI: 10.1021/cr900138tThere is no corresponding record for this reference.
- 13Günay, K.; Theato, P.; Klok, H. Standing on the Shoulders of Hermann Staudinger: Post-Polymerization Modification from Past to Present. J. Polym. Sci., Part A: Polym. Chem. 2013, 51 (1), 1– 28, DOI: 10.1002/pola.26333There is no corresponding record for this reference.
- 14Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and Chemical Recycling of Solid Plastic Waste. Waste Management 2017, 69, 24– 58, DOI: 10.1016/j.wasman.2017.07.044There is no corresponding record for this reference.
- 15Al-Salem, S.; Lettieri, P.; Baeyens, J. Recycling and Recovery Routes of Plastic Solid Waste (PSW): A Review. Waste Management 2009, 29 (10), 2625– 2643, DOI: 10.1016/j.wasman.2009.06.004There is no corresponding record for this reference.
- 16Geyer, R.; Jambeck, J. R.; Law, K. L. Production, Use, and Fate of All Plastics Ever Made. Sci. Adv. 2017, 3, e1700782 DOI: 10.1126/sciadv.1700782There is no corresponding record for this reference.
- 17Lefay, C.; Guillaneuf, Y. Recyclable/Degradable Materials via the Insertion of Labile/Cleavable Bonds Using a Comonomer Approach. Prog. Polym. Sci. 2023, 147, 101764, DOI: 10.1016/j.progpolymsci.2023.101764There is no corresponding record for this reference.
- 18Tardy, A.; Nicolas, J.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. Radical Ring-Opening Polymerization: Scope, Limitations, and Application to (Bio)Degradable Materials. Chem. Rev. 2017, 117 (3), 1319– 1406, DOI: 10.1021/acs.chemrev.6b00319There is no corresponding record for this reference.
- 19Colombani, D. Driving Forces in Free Radical Addition-Fragmentation Processes. Prog. Polym. Sci. 1999, 24 (3), 425– 480, DOI: 10.1016/S0079-6700(99)00005-2There is no corresponding record for this reference.
- 20Errede, L. A. The Chemistry of Xylylenes. X. Some Polymers and Telomers of Spiro-Di-o-Xylylene. J. Polym. Sci. 1961, 49, 253, DOI: 10.1002/pol.1961.1204915209There is no corresponding record for this reference.
- 21Van Volkenburgh, R.; Greenlee, K. W.; Derfer, J. M.; Boord, C. E. A Synthesis of Vinylcyclopropane. J. Am. Chem. Soc. 1949, 71 (11), 3595– 3597, DOI: 10.1021/ja01179a004There is no corresponding record for this reference.
- 22Takahashi, T.; Yamashita, I. 1,5-Polymerization of Vinylcyclopropane. Journal of Polymer Science Part B: Polymer Letters 1965, 3 (4), 251– 255, DOI: 10.1002/pol.1965.110030403There is no corresponding record for this reference.
- 23Endo, T.; Bailey, W. J. Radical Ring-Opening Polymerization of 3,9-Dimethylene-1,5,7,11-Tetraoxaspiro-[5,5] Undecane. Journal of Polymer Science: Polymer Letters Edition 1975, 13 (4), 193– 195, DOI: 10.1002/pol.1975.130130401There is no corresponding record for this reference.
- 24Endo, T.; Bailey, W. J. Synthesis and Radical Ring-Opening Polymerization of 2-Methylene-1,4,6-Trioxaspiro[4,4] Nonane. Journal of Polymer Science: Polymer Letters Edition 1980, 18 (1), 25– 27, DOI: 10.1002/pol.1980.130180106There is no corresponding record for this reference.
- 25Lalande, R.; Maillard, B.; Cazaux, M. Addition radicalaire du dioxolanne-1,3 a l’octene-1. Tetrahedron Lett. 1969, 10 (9), 745– 748, DOI: 10.1016/S0040-4039(01)87798-4There is no corresponding record for this reference.
- 26Bailey, W. J.; Ni, Z.; Wu, S.-R. Synthesis of Poly-r-Caprolactone via a Free Radical Mechanism. Free Radical Ring-Opening Polymerization of 2-Methylene- 1,3-Dioxepane. Journal of Polymer Science: Polymer Chemistry Edition 1982, 20, 3021– 3030, DOI: 10.1002/pol.1982.170201101There is no corresponding record for this reference.
- 27Cho, I.; Kim, J.-B. Exploratory Ring-Opening Polymerization. VIII. Radical Ring-Opening Polymerization of 2-Phenyl-3-Vinyloxirane: A C–C Bond Scission Polymerization of the Epoxide Ring. Journal of Polymer Science: Polymer Letters Edition 1983, 21 (6), 433– 436, DOI: 10.1002/pol.1983.130210605There is no corresponding record for this reference.
- 28Bailey, W. J. Free Radical Ring-Opening Polymerization. Polym. J. 1985, 17, 85, DOI: 10.1295/polymj.17.85There is no corresponding record for this reference.
- 29Cho, I.; Kim, S.-K.; Lee, M.-H. Exploratory Ring-Opening Polymerization. XIIL Ring-Opening Polymerization of 2-Vinyl Cyclic Sulfones. Journal of Polymer Science: Polymer Symposia 1986, 74 (1), 219– 226, DOI: 10.1002/polc.5070740119There is no corresponding record for this reference.
- 30Bailey, W. J.; Chou, J. L.; Feng, P.-Z.; Issari, B.; Kuruganti, V.; Zhou, L.-L. Recent Advances in Free-Radical Ring-Opening Polymerization. Journal of Macromolecular Science: Part A - Chemistry 1988, 25 (5–7), 781– 798, DOI: 10.1080/00222338808053398There is no corresponding record for this reference.
- 31Evans, R. A.; Moad, G.; Rizzardo, E.; Thang, S. H. New Free-Radical Ring-Opening Acrylate Monomers. Macromolecules 1994, 27 (26), 7935– 7937, DOI: 10.1021/ma00104a062There is no corresponding record for this reference.
- 32Evans, R. A.; Rizzardo, E. Free-Radical Ring-Opening Polymerization of Cyclic Allylic Sulfides. Macromolecules 1996, 29 (22), 6983– 6989, DOI: 10.1021/ma960573pThere is no corresponding record for this reference.
- 33Tang, H.; Tsarevsky, N. V. Lipoates as Building Blocks of Sulfur-Containing Branched Macromolecules. Polym. Chem. 2015, 6 (39), 6936– 6945, DOI: 10.1039/C5PY01005EThere is no corresponding record for this reference.
- 34Albanese, K. R.; Morris, P. T.; Read De Alaniz, J.; Bates, C. M.; Hawker, C. J. Controlled-Radical Polymerization of α-Lipoic Acid: A General Route to Degradable Vinyl Copolymers. J. Am. Chem. Soc. 2023, 145 (41), 22728– 22734, DOI: 10.1021/jacs.3c08248There is no corresponding record for this reference.
- 35Huang, H.; Sun, B.; Huang, Y.; Niu, J. Radical Cascade-Triggered Controlled Ring-Opening Polymerization of Macrocyclic Monomers. J. Am. Chem. Soc. 2018, 140 (33), 10402– 10406, DOI: 10.1021/jacs.8b05365There is no corresponding record for this reference.
- 36Bingham, N. M.; Roth, P. J. Degradable Vinyl Copolymers through Thiocarbonyl Addition-Ring-Opening (TARO) Polymerization. Chem. Commun. 2019, 55 (1), 55– 58, DOI: 10.1039/C8CC08287AThere is no corresponding record for this reference.
- 37Smith, R. A.; Fu, G. Y.; McAteer, O.; Xu, M. Z.; Gutekunst, W. R. Radical Approach to Thioester-Containing Polymers. J. Am. Chem. Soc. 2019, 141 (4), 1446– 1451, DOI: 10.1021/jacs.8b12154There is no corresponding record for this reference.
- 38Jackson, A. W. Reversible-Deactivation Radical Polymerization of Cyclic Ketene Acetals. Polym. Chem. 2020, 11 (21), 3525– 3545, DOI: 10.1039/D0PY00446DThere is no corresponding record for this reference.
- 39Agarwal, S. Chemistry, Chances and Limitations of the Radical Ring-Opening Polymerization of Cyclic Ketene Acetals for the Synthesis of Degradable Polyesters. Polym. Chem. 2010, 1, 953– 964, DOI: 10.1039/c0py00040jThere is no corresponding record for this reference.
- 40Deng, Y.; Mehner, F.; Gaitzsch, J. Current Standing on Radical Ring-Opening Polymerizations of Cyclic Ketene Acetals as Homopolymers and Copolymers with One Another. Macromol. Rapid Commun. 2023, 44, e2200941 DOI: 10.1002/marc.202200941There is no corresponding record for this reference.
- 41Paulusse, J. M. J.; Amir, R. J.; Evans, R. A.; Hawker, C. J. Free Radical Polymers with Tunable and Selective Bio- and Chemical Degradability. J. Am. Chem. Soc. 2009, 131 (28), 9805– 9812, DOI: 10.1021/ja903245pThere is no corresponding record for this reference.
- 42Zhang, S.; Wang, Y.; Huang, H.; Cao, D. A Strategy for Controlling the Polymerizations of Thiyl Radical Propagation by RAFT Agents. Angew. Chem. Int. Ed 2023, 62 (37), e202308524 DOI: 10.1002/anie.202308524There is no corresponding record for this reference.
- 43Hu, H.; Yi, P.; He, J.; Cao, D.; Huang, H. Controlling Thiyl Radical Polymerization via in Situ Desulfurization. Polym. Chem. 2025, 16 (2), 174– 180, DOI: 10.1039/D4PY01140FThere is no corresponding record for this reference.
- 44Wang, Y.; Du, J.; Huang, H. Reversible Thiyl Radical Addition-Fragmentation Chain Transfer Polymerization. Angew. Chem. Int. Ed. 2024, 63 (12). DOI: 10.1002/anie.202318898 .There is no corresponding record for this reference.
- 45Stockmayer, W. H.; Howard, R. O.; Clarke, J. T. Copolymerization of Vinyl Acetate with a Cyclic Disulfide. J. Am. Chem. Soc. 1953, 75 (7), 1756– 1757, DOI: 10.1021/ja01103a528There is no corresponding record for this reference.
- 46Tobolsky, A. V.; Baysal, B. The Reaction between Styrene and Ring Disulfides: Copolymerization Effected by the Chain Transfer Reaction. J. Am. Chem. Soc. 1953, 75 (7), 1757– 1757, DOI: 10.1021/ja01103a529There is no corresponding record for this reference.
- 47Nambu, Y.; Acar, M. H.; Suzuki, T.; Endo, T. Thermal and Photoinitiated Copolymerization of the Cyclic Disulfide Lipoamide with Styrene. Makromol. Chem. 1988, 189 (3), 495– 500, DOI: 10.1002/macp.1988.021890302There is no corresponding record for this reference.
- 48Suzuki, T.; Nambu, Y.; Endo, T. Radical Copolymerization of Lipoamide with Vinyl Monomers. Macromolecules 1990, 23 (6), 1579– 1582, DOI: 10.1021/ma00208a004There is no corresponding record for this reference.
- 49Albanese, K. R.; Read De Alaniz, J.; Hawker, C. J.; Bates, C. M. From Health Supplement to Versatile Monomer: Radical Ring-Opening Polymerization and Depolymerization of α-Lipoic Acid. Polymer 2024, 304, 127167, DOI: 10.1016/j.polymer.2024.127167There is no corresponding record for this reference.
- 50Mothe, S. R.; Chennamaneni, L. R.; Tan, J.; Lim, F. C. H.; Zhao, W.; Thoniyot, P. A Mechanistic Study on the Hydrolysis of Cyclic Ketene Acetal (CKA) and Proof of Concept of Polymerization in Water. Macro Chemistry & Physics 2023, 224 (19), 2300221, DOI: 10.1002/macp.202300221There is no corresponding record for this reference.
- 51McElvain, S. M.; Curry, M. J. Ketene Acetals. XIX. 2-Methylene-1,3-Dioxolanes and 1,3-Dioxanes. J. Am. Chem. Soc. 1948, 70 (11), 3781– 3786, DOI: 10.1021/ja01191a071There is no corresponding record for this reference.
- 52Tran, J.; Guégain, E.; Ibrahim, N.; Harrisson, S.; Nicolas, J. Efficient Synthesis of 2-Methylene-4-Phenyl-1,3-Dioxolane, a Cyclic Ketene Acetal for Controlling the NMP of Methyl Methacrylate and Conferring Tunable Degradability. Polym. Chem. 2016, 7 (26), 4427– 4435, DOI: 10.1039/C6PY00778CThere is no corresponding record for this reference.
- 53Folini, J.; Murad, W.; Mehner, F.; Meier, W.; Gaitzsch, J. Updating Radical Ring-Opening Polymerisation of Cyclic Ketene Acetals from Synthesis to Degradation. Eur. Polym. J. 2020, 134, 109851, DOI: 10.1016/j.eurpolymj.2020.109851There is no corresponding record for this reference.
- 54Folini, J.; Huang, C.-H.; Anderson, J. C.; Meier, W. P.; Gaitzsch, J. Novel Monomers in Radical Ring-Opening Polymerisation for Biodegradable and pH Responsive Nanoparticles. Polym. Chem. 2019, 10 (39), 5285– 5288, DOI: 10.1039/C9PY01103JThere is no corresponding record for this reference.
- 55Petasis, N. A.; Lu, S.-P. Methylenations of Heteroatom-Substituted Carbonyls with Dimethyl Titanocene. Tetrahedron Lett. 1995, 36 (14), 2393– 2396, DOI: 10.1016/0040-4039(95)00320-CThere is no corresponding record for this reference.
- 56Deng, Y.; Frezel, A.; Mehner, F.; Friedel, P.; Gaitzsch, J. Amine-Bearing Cyclic Ketene Acetals for pH-Responsive and Degradable Polyesters through Radical Ring-Opening Polymerisation. Polym. Chem. 2023, 14 (37), 4275– 4281, DOI: 10.1039/D3PY00929GThere is no corresponding record for this reference.
- 57Jiang, N.-C.; Zhou, Z.; Niu, J. Quantitative, Regiospecific, and Stereoselective Radical Ring-Opening Polymerization of Monosaccharide Cyclic Ketene Acetals. J. Am. Chem. Soc. 2024, 146 (8), 5056– 5062, DOI: 10.1021/jacs.3c14244There is no corresponding record for this reference.
- 58Ko, Y.-J.; Shim, S.-B.; Shin, J.-H. Facile Synthesis of 2- O -Iodoacetyl Protected Glycosyl Iodides: Useful Precursors of 1→2-Linked 1,2- Trans -Glycosides. Org. Lett. 2009, 11 (3), 609– 612, DOI: 10.1021/ol8026472There is no corresponding record for this reference.
- 59Sznaidman, M. L.; Johnson, S. C.; Crasto, C.; Hecht, S. M. Carbohydrate Cyclic Ketene Acetals. J. Org. Chem. 1995, 60 (13), 3942– 3943, DOI: 10.1021/jo00118a006There is no corresponding record for this reference.
- 60Hardy, C.; Levere, M. E.; Kociok-Köhn, G.; Buchard, A. Radical Ring Opening Polymerization of Cyclic Ketene Acetals Derived From D-Glucal. ACS Macro Lett. 2023, 12 (11), 1443– 1449, DOI: 10.1021/acsmacrolett.3c00397There is no corresponding record for this reference.
- 61Evans, R. A.; Rizzardo, E. Free Radical Ring-Opening Polymerization of Cyclic Allylic Sulfides: Liquid Monomers with Low Polymerization Volume Shrinkage. J. Polym. Sci. A Polym. Chem. 2001, 39 (1), 202– 215, DOI: 10.1002/1099-0518(20010101)39:1<202::AID-POLA230>3.0.CO;2-6There is no corresponding record for this reference.
- 62Chaumont, P.; Asgarzadeh, F.; Colombani, D.; Arotcarena, M.; Baudouin, A. Synthesis, Characterization and Hydrolysis of Poly[Styrene-Co-(6-Methylene-1,4-Oxathiepane-7-One)] and Poly[Styrene-Co-(6-Methylene-5-Methyl-1,4-Oxathiepane-7-One)]. Macromol. Chem. Phys. 1998, 199 (11), 2577– 2582, DOI: 10.1002/macp.1998.021991128There is no corresponding record for this reference.
- 63Phelan, M.; Aldabbagh, F.; Zetterlund, P. B.; Yamada, B. Mechanism and Kinetics of the Free Radical Ring-Opening Polymerization of Eight-Membered Cyclic Allylic Disulfide Monomers. Macromolecules 2005, 38 (6), 2143– 2147, DOI: 10.1021/ma047894iThere is no corresponding record for this reference.
- 64Sbordone, F.; Veskova, J.; Richardson, B.; Do, P.; Micallef, A.; Frisch, H. Embedding Peptides into Synthetic Polymers: Radical Ring-Opening Copolymerization of Cyclic Peptides. J. Am. Chem. Soc. 2023, 145 (11), 6221– 6229, DOI: 10.1021/jacs.2c12517There is no corresponding record for this reference.
- 65Do, P.; Sbordone, F.; Kalmer, H.; Sokolova, A.; Zhang, C.; Thai, L.; Golberg, D.; Chapman, R.; Poad, B.; Frisch, H. Main Chain Selective Polymer Degradation: Controlled by the Wavelength and Assembly. Chemical Science 2024, 15 (31), 12410– 12419, DOI: 10.1039/D4SC02172JThere is no corresponding record for this reference.
- 66Huang, H.; Wang, W.; Zhou, Z.; Sun, B.; An, M.; Haeffner, F.; Niu, J. Radical Ring-Closing/Ring-Opening Cascade Polymerization. J. Am. Chem. Soc. 2019, 141 (32), 12493– 12497, DOI: 10.1021/jacs.9b05568There is no corresponding record for this reference.
- 67Zhang, S.; Cao, C.; Jiang, S.; Huang, H. A General Strategy for Radical Ring-Opening Polymerization of Macrocyclic Allylic Sulfides. Macromolecules 2022, 55 (21), 9411– 9419, DOI: 10.1021/acs.macromol.2c01636There is no corresponding record for this reference.
- 68Meijs, G. F.; Rizzardo, E.; Le, T. P. T.; Chen, Y.-C. Influence of Thionoesters on the Degree of Polymerization of Styrene, Meth. Makromol. Chem. 1992, 193 (2), 369– 378, DOI: 10.1002/macp.1992.021930209There is no corresponding record for this reference.
- 69Luzel, B.; Gil, N.; Désirée, P.; Monot, J.; Bourissou, D.; Siri, D.; Gigmes, D.; Martin-Vaca, B.; Lefay, C.; Guillaneuf, Y. Development of an Efficient Thionolactone for Radical Ring-Opening Polymerization by a Combined Theoretical/Experimental Approach. J. Am. Chem. Soc. 2023, 145 (50), 27437– 27449, DOI: 10.1021/jacs.3c08610There is no corresponding record for this reference.
- 70Ko, K.; Lundberg, D.; Johnson, A.; Johnson, J. Mechanism-Guided Discovery of Cleavable Comonomers for Backbone Deconstructable Poly(Methyl Methacrylate). J. Am. Chem. Soc. 2024, 146 (13), 9142– 9154, DOI: 10.1021/jacs.3c14554There is no corresponding record for this reference.
- 71Plummer, C. M.; Gil, N.; Dufils, P. E.; Wilson, D. J.; Lefay, C.; Gigmes, D.; Guillaneuf, Y. Mechanistic Investigation of Epsilon-Thiono-Caprolactone Radical Polymerization: An Interesting Tool to Insert Weak Bonds into Poly(Vinyl Esters). Acs Applied Polymer Materials 2021, 3 (6), 3264– 3271, DOI: 10.1021/acsapm.1c00569There is no corresponding record for this reference.
- 72Ivanchenko, O.; Authesserre, U.; Coste, G.; Mazières, S.; Destarac, M.; Harrisson, S. ε-Thionocaprolactone: An Accessible Monomer for Preparation of Degradable Poly(Vinyl Esters) by Radical Ring-Opening Polymerization. Polym. Chem. 2021, 12 (13), 1931– 1938, DOI: 10.1039/D1PY00080BThere is no corresponding record for this reference.
- 73Kamiki, R.; Kubo, T.; Satoh, K. Addition-Fragmentation Ring-Opening Polymerization of Bio-Based Thiocarbonyl L-Lactide for Dual Degradable Vinyl Copolymers. Macromol. Rapid Commun. 2023, 44 (2), 2200537, DOI: 10.1002/marc.202200537There is no corresponding record for this reference.
- 74Ivanchenko, O.; Mazières, S.; Harrisson, S.; Destarac, M. Lactide-Derived Monomers for Radical Thiocarbonyl Addition Ring-Opening Copolymerisation. Polym. Chem. 2022, 13 (39), 5525– 5529, DOI: 10.1039/D2PY00893AThere is no corresponding record for this reference.
- 75Ivanchenko, O.; Mazières, S.; Mallet-Ladeira, S.; Harrisson, S.; Destarac, M. Radical Copolymerization of Thionoglycolide and Derivatives for Preparation of Degradable Vinyl Polymers. Macromolecules 2024, 57 (16), 8059– 8066, DOI: 10.1021/acs.macromol.4c01259There is no corresponding record for this reference.
- 76Brendel, J.; Schmidt, W.; Below, P. 2’-Substituted 1,1’-Biphenyl-2- Carboxamides, Processes for Their Preparation, Their Use as Medicaments, and Pharmaceutical Preparations Comprising Them. US 6,531,495 B1, March 11, 2003.There is no corresponding record for this reference.
- 77Johnson, J. A.; Cardoso Da Costa, L. C.; Prince, E. Cleavable Comonomer Strategy for Accelerating Removal of Gel Nail Polish. WO2024119140A1, June 6, 2024.There is no corresponding record for this reference.
- 78Kiel, G. R.; Lundberg, D. J.; Prince, E.; Husted, K. E. L.; Johnson, A. M.; Lensch, V.; Li, S.; Shieh, P.; Johnson, J. A. Cleavable Comonomers for Chemically Recyclable Polystyrene: A General Approach to Vinyl Polymer Circularity. J. Am. Chem. Soc. 2022, 144 (28), 12979– 12988, DOI: 10.1021/jacs.2c05374There is no corresponding record for this reference.
- 79Luzel, B.; Raggio, L.; Benharrous, E.; Monot, J.; Bourissou, D.; Siri, D.; Gigmes, D.; Lefay, C.; Martin-Vaca, B.; Guillaneuf, Y. Closer Look at the Substituent Effects on the Copolymerization of Thionolactones by Radical Ring-Opening Polymerization. Macromolecules 2025, 58 (9), 4627– 4635, DOI: 10.1021/acs.macromol.5c00448There is no corresponding record for this reference.
- 80Schomaker, J. M.; Travis, B. R.; Borhan, B. Direct Lactonization of Alkenols via Osmium Tetroxide-Mediated Oxidative Cleavage. Org. Lett. 2003, 5 (17), 3089– 3092, DOI: 10.1021/ol035057fThere is no corresponding record for this reference.
- 81Machado, T. O.; Stubbs, C. J.; Chiaradia, V.; Alraddadi, M. A.; Brandolese, A.; Worch, J. C.; Dove, A. P. A Renewably Sourced, Circular Photopolymer Resin for Additive Manufacturing. Nature 2024, 629 (8014), 1069– 1074, DOI: 10.1038/s41586-024-07399-9There is no corresponding record for this reference.
- 82Tardy, A.; Gil, N.; Plummer, C. M.; Siri, D.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. Polyesters by a Radical Pathway: Rationalization of the Cyclic Ketene Acetal Efficiency. Angew. Chem. Int. Ed 2020, 59 (34), 14517– 14526, DOI: 10.1002/anie.202005114There is no corresponding record for this reference.
- 83Ochiai, B.; Endo, T. Computational Evaluation of Radical Ring-Opening Polymerization. J. Pol. Sci.: Part A: Polym. Chem. 2007, 45, 2827– 2834, DOI: 10.1002/pola.22039There is no corresponding record for this reference.
- 84Reddy Mothe, S.; Tan, J. S. J.; Chennamaneni, L. R.; Aidil, F.; Su, Y.; Kang, H. C.; Lim, F. C. H.; Thoniyot, P. A Systematic Investigation of the Ring Size Effects on the Free Radical Ring-Opening Polymerization (rROP) of Cyclic Ketene Acetal (CKA) Using Both Experimental and Theoretical Approach. J. Polym. Sci. 2020, 58 (12), 1728– 1738, DOI: 10.1002/pol.20200210There is no corresponding record for this reference.
- 85Jin, S.; Gonsalves, K. E. A Study of the Mechanism of the Free-Radical Ring-Opening Polymerization of 2-Methylene-1,3-Dioxepane. Macromolecules 1997, 30 (10), 3104– 3106, DOI: 10.1021/ma961590hThere is no corresponding record for this reference.
- 86Agarwal, S.; Speyerer, C. Degradable Blends of Semi-Crystalline and Amorphous Branched Poly(Caprolactone): Effect of Microstructure on Blend Properties. Polymer 2010, 51 (5), 1024– 1032, DOI: 10.1016/j.polymer.2010.01.020There is no corresponding record for this reference.
- 87Tardy, A.; Honoré, J.-C.; Siri, D.; Nicolas, J.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. A Comprehensive Kinetic Study of the Conventional Free-Radical Polymerization of Seven-Membered Cyclic Ketene Acetals. Polym. Chem. 2017, 8 (34), 5139– 5147, DOI: 10.1039/C7PY00337DThere is no corresponding record for this reference.
- 88Tardy, A.; Delplace, V.; Siri, D.; Lefay, C.; Harrisson, S.; De Fatima Albergaria Pereira, B.; Charles, L.; Gigmes, D.; Nicolas, J.; Guillaneuf, Y. Scope and Limitations of the Nitroxide-Mediated Radical Ring-Opening Polymerization of Cyclic Ketene Acetals. Polym. Chem. 2013, 4 (17), 4776, DOI: 10.1039/c3py00719gThere is no corresponding record for this reference.
- 89Delplace, V.; Tardy, A.; Harrisson, S.; Mura, S.; Gigmes, D.; Guillaneuf, Y.; Nicolas, J. Degradable and Comb-Like PEG-Based Copolymers by Nitroxide-Mediated Radical Ring-Opening Polymerization. Biomacromolecules 2013, 14 (10), 3769– 3779, DOI: 10.1021/bm401157gThere is no corresponding record for this reference.
- 90Delplace, V.; Harrisson, S.; Tardy, A.; Gigmes, D.; Guillaneuf, Y.; Nicolas, J. Nitroxide-Mediated Radical Ring-Opening Copolymerization: Chain-End Investigation and Block Copolymer Synthesis. Macromol. Rapid Commun. 2014, 35 (4), 484– 491, DOI: 10.1002/marc.201300809There is no corresponding record for this reference.
- 91Delplace, V.; Guégain, E.; Harrisson, S.; Gigmes, D.; Guillaneuf, Y.; Nicolas, J. A Ring to Rule Them All: A Cyclic Ketene Acetal Comonomer Controls the Nitroxide-Mediated Polymerization of Methacrylates and Confers Tunable Degradability. Chem. Commun. 2015, 51 (64), 12847– 12850, DOI: 10.1039/C5CC04610FThere is no corresponding record for this reference.
- 92Albergaria Pereira, B. de F.; Tardy, A.; Monnier, V.; Guillaneuf, Y.; Gigmes, D.; Charles, L. Elucidation of a Side Reaction Occurring during Nitroxide-Mediated Polymerization of Cyclic Ketene Acetals by Tandem Mass Spectrometric End-Group Analysis of Aliphatic Polyesters. Rapid Commun. Mass Spectrom. 2015, 29 (23), 2302– 2308, DOI: 10.1002/rcm.7397There is no corresponding record for this reference.
- 93Jackson, A. W.; Chennamaneni, L. R.; Thoniyot, P. Main-Chain Degradable, pH-Responsive and Covalently Cross-Linked Nanoparticles via a One-Step RAFT-Based Radical Ring-Opening Terpolymerization. Eur. Polym. J. 2020, 122, 109391, DOI: 10.1016/j.eurpolymj.2019.109391There is no corresponding record for this reference.
- 94Jackson, A. W.; Chennamaneni, L. R.; Mothe, S. R.; Thoniyot, P. A General Strategy for Degradable Single-Chain Nanoparticles via Cross-Linker Mediated Chain Collapse of Radical Copolymers. Chem. Commun. 2020, 56 (68), 9838– 9841, DOI: 10.1039/D0CC03792CThere is no corresponding record for this reference.
- 95Wais, U.; Chennamaneni, L. R.; Thoniyot, P.; Zhang, H.; Jackson, A. W. Main-Chain Degradable Star Polymers Comprised of pH-Responsive Hyperbranched Cores and Thermoresponsive Polyethylene Glycol-Based Coronas. Polym. Chem. 2018, 9 (39), 4824– 4839, DOI: 10.1039/C8PY01113CThere is no corresponding record for this reference.
- 96Jiang, S.; Huang, H. Oxygen-Initiated Radical Ring-Opening Polymerization of Macrocyclic Allylic Sulfides under Ambient Conditions. Polymer 2024, 303, 127106, DOI: 10.1016/j.polymer.2024.127106There is no corresponding record for this reference.
- 97Spick, M. P.; Bingham, N. M.; Li, Y.; de Jesus, J.; Costa, C.; Bailey, M. J.; Roth, P. J. Fully Degradable Thioester-Functional Homo- and Alternating Copolymers Prepared through Thiocarbonyl Addition-Ring-Opening RAFT Radical Polymerization. Macromolecules 2020, 53 (2), 539– 547, DOI: 10.1021/acs.macromol.9b02497There is no corresponding record for this reference.
- 98Rix, M.; Higgs, S.; Dodd, E.; Coles, S.; Bingham, N. M.; Roth, P. J. Insertion of Degradable Thioester Linkages into Styrene and Methacrylate Polymers. ChemRxiv , 2022, DOI: 10.26434/chemrxiv-2022-cdt52 .There is no corresponding record for this reference.
- 99Kazmi, T.; Hepburn, K. S.; Nisa, Q.; Neogi, S.; Bingham, N. M.; Roth, P. J. Single-Unit Comonomer Insertion Initiates Radical Polymerization of Thionolactone to Give Chemically and Thermally Degradable Polythioesters. Macromolecules 2025, 58 (18), 9617– 9628, DOI: 10.1021/acs.macromol.5c01418There is no corresponding record for this reference.
- 100Prebihalo, E. A.; Luke, A. M.; Reddi, Y.; LaSalle, C. J.; Shah, V. M.; Cramer, C. J.; Reineke, T. M. Radical Ring-Opening Polymerization of Sustainably-Derived Thionoisochromanone. Chem. Sci. 2023, 14 (21), 5689– 5698, DOI: 10.1039/D2SC06040JThere is no corresponding record for this reference.
- 101Raeisi, M.; Tsarevsky, N. V. Radical Ring-opening Polymerization of Lipoates: Kinetic and Thermodynamic Aspects. J. Polym. Sci. 2021, 59 (8), 675– 684, DOI: 10.1002/pol.20200765There is no corresponding record for this reference.
- 102Lee, K.; Han, S.; Morris, P. T.; Bates, C. M.; Hawker, C. J.; Boyer, C. Controlled Synthesis of Lipoate Homopolymers via Reversible Addition-Fragmentation Chain Transfer Polymerization. J. Am. Chem. Soc. 2025, 147 (42), 38796– 38806, DOI: 10.1021/jacs.5c14377There is no corresponding record for this reference.
- 103Gigmes, D.; Van Steenberge, P. H. M.; Siri, D.; D’Hooge, D. R.; Guillaneuf, Y.; Lefay, C. Simulation of the Degradation of Cyclic Ketene Acetal and Vinyl-Based Copolymers Synthesized via a Radical Process: Influence of the Reactivity Ratios on the Degradability Properties. Macromol. Rapid Commun. 2018, 39 (19), 1800193, DOI: 10.1002/marc.201800193There is no corresponding record for this reference.
- 104Liausvia, F.; Rusli, W.; van Herk, A. Prediction of the Oligomer Distribution after Degradation of (Co)Polymers with Inserted Break Points. Macromol. Theory Simul. 2021, 30 (6), 2100038, DOI: 10.1002/mats.202100038There is no corresponding record for this reference.
- 105Lundberg, D. J.; Ko, K.; Kilgallon, L. J.; Johnson, J. A. Defining Reactivity-Deconstructability Relationships for Copolymerizations Involving Cleavable Comonomer Additives. ACS Macro Lett. 2024, 13 (5), 521– 527, DOI: 10.1021/acsmacrolett.4c00106There is no corresponding record for this reference.
- 106Callan, D. H.; Bates, C. M. Efficient Deterministic Modeling of Reversible Copolymerizations. Macromolecules 2025, 58 (19), 10289– 10300, DOI: 10.1021/acs.macromol.5c01421There is no corresponding record for this reference.
- 107Wen, M.; Wong, W.; Junkers, T. Synthesis of Biodegradable Vinyl Copolymers via Enforced Regular Sequence Distribution from Automated Radical Ring Opening Polymerisation. Chem. Sci. 2025, 16 (42), 19624– 19631, DOI: 10.1039/D5SC03738GThere is no corresponding record for this reference.
- 108Bailey, W. J.; Wu, S.-R.; Ni, Z. Free Radical Ring-Opening Polymerization of 4-n-Hexyl- and 4-n-Decyl-2-Methylene-1,3-Dioxolanes. Journal of Macromolecular Science: Part A - Chemistry 1982, 18 (6), 973– 986, DOI: 10.1080/00222338208077212There is no corresponding record for this reference.
- 109Bailey, W. J.; Endo, T.; Gapud, B.; Lin, Y.-N.; Ni, Z.; Pan, C.-Y.; Shaffer, S. E.; Wu, S.-R.; Yamazaki, N.; Yonezawa, K. Synthesis of Functionally-Terminated Oligomers by Free Radical Ring-Opening Polymerization. Journal of Macromolecular Science: Part A - Chemistry 1984, 21 (8–9), 979– 995, DOI: 10.1080/00222338408056586There is no corresponding record for this reference.
- 110Jackson, A. W.; Mothe, S. R.; Chennamaneni, L. R.; van Herk, A.; Thoniyot, P. Unraveling the History and Revisiting the Synthesis of Degradable Polystyrene Analogues via Radical Ring-Opening Copolymerization with Cyclic Ketene Acetals. Materials 2020, 13 (10), 2325, DOI: 10.3390/ma13102325There is no corresponding record for this reference.
- 111Gil, N.; Caron, B.; Siri, D.; Roche, J.; Hadiouch, S.; Khedaioui, D.; Ranque, S.; Cassagne, C.; Montarnal, D.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. Degradable Polystyrene via the Cleavable Comonomer Approach. Macromolecules 2022, 55 (15), 6680– 6694, DOI: 10.1021/acs.macromol.2c00651There is no corresponding record for this reference.
- 112Wickel, H.; Agarwal, S. Synthesis and Characterization of Copolymers of 5,6-Benzo-2-Methylene-1,3-Dioxepane and Styrene. Macromolecules 2003, 36 (16), 6152– 6159, DOI: 10.1021/ma034551wThere is no corresponding record for this reference.
- 113Hiracuri, Y.; Tokiwa, Y. Synthesis of Copolymers Composed of 2-Methylene-1,3,6-Trioxocane and Vinyl Monomers and Their Enzymatic Degradation. J. Polym. Sci., Part A: Polym. Chem. 1993, 31 (12), 3159– 3163, DOI: 10.1002/pola.1993.080311233There is no corresponding record for this reference.
- 114Rix, M. F. I.; Collins, K.; Higgs, S. J.; Dodd, E. M.; Coles, S. J.; Bingham, N. M.; Roth, P. J. Insertion of Degradable Thioester Linkages into Styrene and Methacrylate Polymers: Insights into the Reactivity of Thionolactones. Macromolecules 2023, 56 (23), 9787– 9795, DOI: 10.1021/acs.macromol.3c01811There is no corresponding record for this reference.
- 115Okayama, Y.; Morris, P.; Albanese, K.; Olsen, S.; Mori, A.; de Alaniz, J.; Bates, C.; Hawker, C. Enhanced Degradation of Vinyl Copolymers Based on Lipoic Acid. J. Polym. Sci. 2025, 63 (6), 1345– 1351, DOI: 10.1002/pol.20241085There is no corresponding record for this reference.
- 116Li, J.; Yang, H.; Jiang, Q.; Li, J.; Jiang, B.; Xue, X.; Komarneni, S.; Huang, W. A New Strategy to Synthesize Degradable Polystyrene of Ultra-High Molecular Weight. Chem. Commun. 2025, 61 (18), 3692– 3695, DOI: 10.1039/D4CC05166AThere is no corresponding record for this reference.
- 117Sun, L. F.; Zhuo, R. X.; Liu, Z. L. Synthesis and Enzymatic Degradation of 2-Methylene-1,3-Dioxepane and Methyl Acrylate Copolymers. J. Polym. Sci., Part A: Polym. Chem. 2003, 41 (18), 2898– 2904, DOI: 10.1002/pola.10868There is no corresponding record for this reference.
- 118Maji, S.; Zheng, M.; Agarwal, S. Functional Degradable Polymers via Radical Ring-Opening Polymerization and Click Chemistry. Macromol. Chem. Phys. 2011, 212 (23), 2573– 2582, DOI: 10.1002/macp.201100375There is no corresponding record for this reference.
- 119Sun, L.-F.; Zhuo, R.-X.; Liu, Z.-L. Studies on the Synthesis and Properties of Temperature Responsive and Biodegradable Hydrogels. Macromol. Biosci. 2003, 3 (12), 725– 728, DOI: 10.1002/mabi.200300012There is no corresponding record for this reference.
- 120Huang, J.; Gil, R.; Matyjaszewski, K. Synthesis and Characterization of Copolymers of 5,6-Benzo-2-Methylene-1,3-Dioxepane and n-Butyl Acrylate. Polymer 2005, 46 (25), 11698– 11706, DOI: 10.1016/j.polymer.2005.09.048There is no corresponding record for this reference.
- 121Yuan, J.-Y.; Pan, C.-Y. “Living” Free Radical Ring-Opening Copolymerization of 4,7-Dimethyl-2-Methylene-1,3-Dioxepane and Conventional Vinyl Monomers. Eur. Polym. J. 2002, 38 (10), 2069– 2076, DOI: 10.1016/S0014-3057(02)00085-XThere is no corresponding record for this reference.
- 122Lena, J.-B.; Van Herk, A. M. Toward Biodegradable Chain-Growth Polymers and Polymer Particles: Re-Evaluation of Reactivity Ratios in Copolymerization of Vinyl Monomers with Cyclic Ketene Acetal Using Nonlinear Regression with Proper Error Analysis. Ind. Eng. Chem. Res. 2019, 58 (46), 20923– 20931, DOI: 10.1021/acs.iecr.9b02375There is no corresponding record for this reference.
- 123Lena, J.-B.; Jackson, A. W.; Chennamaneni, L. R.; Wong, C. T.; Lim, F.; Andriani, Y.; Thoniyot, P.; Van Herk, A. M. Degradable Poly(Alkyl Acrylates) with Uniform Insertion of Ester Bonds, Comparing Batch and Semibatch Copolymerizations. Macromolecules 2020, 53 (10), 3994– 4011, DOI: 10.1021/acs.macromol.0c00207There is no corresponding record for this reference.
- 124Wang, W.; Zhou, Z.; Sathe, D.; Tang, X.; Moran, S.; Jin, J.; Haeffner, F.; Wang, J.; Niu, J. Degradable Vinyl Random Copolymers via Photocontrolled Radical Ring-Opening Cascade Copolymerization. Angew. Chem., Int. Ed. 2022, 61 (8), e202113302 DOI: 10.1002/anie.202113302There is no corresponding record for this reference.
- 125Luzel, B.; Efimov, I.; Edeleva, M.; Kouider, S.; Gil, N.; Montarnal, D.; Lefay, C.; Gigmes, D.; Van Steenberge, P. H. M.; D’hooge, D. R.; Guillaneuf, Y. The successful impossible radical ring-opening copolymerization of thionolactones and methacrylates via an auxiliary third comonomer. Nature Communications 2025, 1, 9468, DOI: 10.1038/s41467-025-64502-yThere is no corresponding record for this reference.
- 126Bingham, N. M.; Nisa, Q. un; Chua, S. H. L.; Fontugne, L.; Spick, M. P.; Roth, P. J. Thioester-Functional Polyacrylamides: Rapid Selective Backbone Degradation Triggers Solubility Switch Based on Aqueous Lower Critical Solution Temperature/Upper Critical Solution Temperature. ACS Appl. Polym. Mater. 2020, 2 (8), 3440– 3449, DOI: 10.1021/acsapm.0c00503There is no corresponding record for this reference.
- 127Un Nisa, Q.; Theobald, W.; Hepburn, K. S.; Riddlestone, I.; Bingham, N. M.; Kopeć, M.; Roth, P. J. Degradable Linear and Bottlebrush Thioester-Functional Copolymers through Atom-Transfer Radical Ring-Opening Copolymerization of a Thionolactone. Macromolecules 2022, 55 (17), 7392– 7400, DOI: 10.1021/acs.macromol.2c01317There is no corresponding record for this reference.
- 128Jazani, A. M.; Bernat, R.; Matyjaszewski, K. Visible Light-Induced Photo-Radical Ring-Opening Copolymerization of Thionolactone and Acrylates. Polymer 2024, 302, 127032, DOI: 10.1016/j.polymer.2024.127032There is no corresponding record for this reference.
- 129Morris, P. T.; Watanabe, K.; Albanese, K. R.; Kent, G. T.; Gupta, R.; Gerst, M.; Read De Alaniz, J.; Hawker, C. J.; Bates, C. M. Scalable Synthesis of Degradable Copolymers Containing α-Lipoic Acid via Miniemulsion Polymerization. J. Am. Chem. Soc. 2024, 146 (44), 30662– 30667, DOI: 10.1021/jacs.4c12438There is no corresponding record for this reference.
- 130Albanese, K. R.; Okayama, Y.; Morris, P. T.; Gerst, M.; Gupta, R.; Speros, J. C.; Hawker, C. J.; Choi, C.; De Alaniz, J. R.; Bates, C. M. Building Tunable Degradation into High-Performance Poly(Acrylate) Pressure-Sensitive Adhesives. ACS Macro Lett. 2023, 12 (6), 787– 793, DOI: 10.1021/acsmacrolett.3c00204There is no corresponding record for this reference.
- 131Albanese, K. R.; Morris, P. T.; Roehrich, B.; Read de Alaniz, J.; Hawker, C. J.; Bates, C. M. Selective Electrochemical Degradation of Bottlebrush Elastomers. J. Polym. Sci. 2024, 62 (18), 4326– 4331, DOI: 10.1002/pol.20240393There is no corresponding record for this reference.
- 132Giri, A.; Aquib, M.; Choudhury, A.; Kannaujiya, V. K.; Lim, J. L.; Gu, Z.; Lenardon, M. D.; Boyer, C. Lipoic Acid Based Redox-Responsive Degradable Antimicrobial Polymers. Macromol. Rapid Commun. 2025, 46 (17), e00224 DOI: 10.1002/marc.202500224There is no corresponding record for this reference.
- 133Choi, C.; Okayama, Y.; Morris, P. T.; Robinson, L. L.; Gerst, M.; Speros, J. C.; Hawker, C. J.; Read de Alaniz, J.; Bates, C. M. Digital Light Processing of Dynamic Bottlebrush Materials. Adv. Funct. Mater. 2022, 32 (25), 2200883, DOI: 10.1002/adfm.202200883There is no corresponding record for this reference.
- 134Lai, H.; Le Dot, M.; Chen, J.; Zhang, J.; Xiao, P. Biomass-Derived Photoresins for Digital Light Processing 3D Printing of Degradable Objects. ACS Sustainable Resour. Manage. 2025, 2 (5), 833– 841, DOI: 10.1021/acssusresmgt.5c00094There is no corresponding record for this reference.
- 135Pesenti, T.; Nicolas, J. 100th Anniversary of Macromolecular Science Viewpoint: Degradable Polymers from Radical Ring-Opening Polymerization: Latest Advances, New Directions, and Ongoing Challenges. ACS Macro Lett. 2020, 9 (12), 1812– 1835, DOI: 10.1021/acsmacrolett.0c00676There is no corresponding record for this reference.
- 136Roberts, G. E.; Coote, M. L.; Heuts, J. P. A.; Morris, L. M.; Davis, T. P. Radical Ring-Opening Copolymerization of 2-Methylene 1,3-Dioxepane and Methyl Methacrylate: Experiments Originally Designed To Probe the Origin of the Penultimate Unit Effect. Macromolecules 1999, 32 (5), 1332– 1340, DOI: 10.1021/ma9813587There is no corresponding record for this reference.
- 137Agarwal, S. Microstructural Characterisation and Properties Evaluation of Poly (Methyl Methacrylate-Co-Ester)s. Polym. J. 2007, 39 (2), 163– 174, DOI: 10.1295/polymj.PJ2006137There is no corresponding record for this reference.
- 138Zhang, Y.; Zheng, M.; Kissel, T.; Agarwal, S. Design and Biophysical Characterization of Bioresponsive Degradable Poly(Dimethylaminoethyl Methacrylate) Based Polymers for In Vitro DNA Transfection. Biomacromolecules 2012, 13 (2), 313– 322, DOI: 10.1021/bm2015174There is no corresponding record for this reference.
- 139Lutz, J.-F.; Andrieu, J.; Üzgün, S.; Rudolph, C.; Agarwal, S. Biocompatible, Thermoresponsive, and Biodegradable: Simple Preparation of “All-in-One” Biorelevant Polymers. Macromolecules 2007, 40 (24), 8540– 8543, DOI: 10.1021/ma7021474There is no corresponding record for this reference.
- 140Agarwal, S.; Ren, L.. Polycaprolactone-Based Novel Degradable Ionomers by Radical Ring-Opening Polymerization of 2-Methylene-1,3-Dioxepane. Macromolecules 2009, 42 (5), 1574– 1579, DOI: 10.1021/ma802615fThere is no corresponding record for this reference.
- 141Zhang, Y.; Aigner, A.; Agarwal, S. Degradable and Biocompatible Poly(N,N-Dimethylaminoethyl Methacrylate-Co-Caprolactone)s as DNA Transfection Agents. Macromol. Biosci. 2013, 13 (9), 1267– 1275, DOI: 10.1002/mabi.201300043There is no corresponding record for this reference.
- 142Jin, Q.; Maji, S.; Agarwal, S. Novel Amphiphilic, Biodegradable, Biocompatible, Cross-Linkable Copolymers: Synthesis, Characterization and Drug Delivery Applications. Polym. Chem. 2012, 3 (10), 2785, DOI: 10.1039/c2py20364bThere is no corresponding record for this reference.
- 143Zhang, Y.; Chu, D.; Zheng, M.; Kissel, T.; Agarwal, S. Biocompatible and Degradable Poly(2-Hydroxyethyl Methacrylate) Based Polymers for Biomedical Applications. Polym. Chem. 2012, 3 (10), 2752, DOI: 10.1039/c2py20403gThere is no corresponding record for this reference.
- 144Ko, J. H.; Terashima, T.; Sawamoto, M.; Maynard, H. D. Fluorous Comonomer Modulates the Reactivity of Cyclic Ketene Acetal and Degradation of Vinyl Polymers. Macromolecules 2017, 50 (23), 9222– 9232, DOI: 10.1021/acs.macromol.7b01973There is no corresponding record for this reference.
- 145Lai, H.; Ouchi, M. Backbone-Degradable Polymers via Radical Copolymerizations of Pentafluorophenyl Methacrylate with Cyclic Ketene Acetal: Pendant Modification and Efficient Degradation by Alternating-Rich Sequence. ACS Macro Lett. 2021, 10 (10), 1223– 1228, DOI: 10.1021/acsmacrolett.1c00513There is no corresponding record for this reference.
- 146Grabe, N.; Zhang, Y.; Agarwal, S. Degradable Elastomeric Block Copolymers Based on Polycaprolactone by Free-Radical Chemistry. Macromol. Chem. Phys. 2011, 212 (13), 1327– 1334, DOI: 10.1002/macp.201100031There is no corresponding record for this reference.
- 147Gardoni, G.; Zanoni, A.; González De San Román, E.; Moscatelli, D.; Gabirondo, E.; Leiza, J. R.; Barquero, A.; Sardon, H. Enhancing the Incorporation of 2-Methylen-1,3-Dioxepane (MDO) into Industrial Monomers by the Addition of Crotonate Comonomers. Polymer 2024, 307, 127298, DOI: 10.1016/j.polymer.2024.127298There is no corresponding record for this reference.
- 148Gao, Y.; Böhmer, V. I.; Zhou, D.; Zhao, T.; Wang, W.; Paulusse, J. M. J. Main-Chain Degradable Single-Chain Cyclized Polymers as Gene Delivery Vectors. J. Controlled Release 2016, 244, 375– 383, DOI: 10.1016/j.jconrel.2016.07.046There is no corresponding record for this reference.
- 149Ratcliffe, L. P. D.; Couchon, C.; Armes, S. P.; Paulusse, J. M. J. Inducing an Order-Order Morphological Transition via Chemical Degradation of Amphiphilic Diblock Copolymer Nano-Objects. Biomacromolecules 2016, 17 (6), 2277– 2283, DOI: 10.1021/acs.biomac.6b00540There is no corresponding record for this reference.
- 150Wang, W.; Rondon, B.; Wang, Z.; Wang, J.; Niu, J. Macrocyclic Allylic Sulfone as a Universal Comonomer in Organocatalyzed Photocontrolled Radical Copolymerization with Vinyl Monomers. Macromolecules 2023, 56 (5), 2052– 2061, DOI: 10.1021/acs.macromol.2c02025There is no corresponding record for this reference.
- 151Keyser, S. P.; Fairbanks, B. D.; Bahns, T.; Bowman, C. N. Dithiolane-Ene Copolymerization: Enabling Tunable, Dynamic, Dual-Cure Networks via Real-Time UV-Vis/FTIR Kinetics and Compositional Analysis. Macromolecules 2025, 58 (15), 8468– 8478, DOI: 10.1021/acs.macromol.5c00977There is no corresponding record for this reference.
- 152Agarwal, S.; Kumar, R.; Kissel, T.; Reul, R. Synthesis of Degradable Materials Based on Caprolactone and Vinyl Acetate Units Using Radical Chemistry. Polym. J. 2009, 41 (8), 650– 660, DOI: 10.1295/polymj.PJ2009091There is no corresponding record for this reference.
- 153Undin, J.; Illanes, T.; Finne-Wistrand, A.; Albertsson, A.-C. Random Introduction of Degradable Linkages into Functional Vinyl Polymers by Radical Ring-Opening Polymerization, Tailored for Soft Tissue Engineering. Polym. Chem. 2012, 3 (5), 1260, DOI: 10.1039/c2py20034aThere is no corresponding record for this reference.
- 154Bell, C. A.; Hedir, G. G.; O’Reilly, R. K.; Dove, A. P. Controlling the Synthesis of Degradable Vinyl Polymers by Xanthate-Mediated Polymerization. Polym. Chem. 2015, 6 (42), 7447– 7454, DOI: 10.1039/C5PY01156FThere is no corresponding record for this reference.
- 155Hedir, G. G.; Bell, C. A.; Ieong, N. S.; Chapman, E.; Collins, I. R.; O’Reilly, R. K.; Dove, A. P. Functional Degradable Polymers by Xanthate-Mediated Polymerization. Macromolecules 2014, 47 (9), 2847– 2852, DOI: 10.1021/ma500428eThere is no corresponding record for this reference.
- 156Bailey, W. J.; Wu, S.-R.; Ni, Z. Synthesis and Free Radical Ring-Opening Polymerization of 2-Methylene-4-Phenyl-1,3-Dioxolane. Makromol. Chem. 1982, 183 (8), 1913– 1920, DOI: 10.1002/macp.1982.021830811There is no corresponding record for this reference.
- 157Tardy, A.; Honoré, J.-C.; Tran, J.; Siri, D.; Delplace, V.; Bataille, I.; Letourneur, D.; Perrier, J.; Nicoletti, C.; Maresca, M.; Lefay, C.; Gigmes, D.; Nicolas, J.; Guillaneuf, Y. Radical Copolymerization of Vinyl Ethers and Cyclic Ketene Acetals as a Versatile Platform to Design Functional Polyesters. Angew. Chem., Int. Ed. 2017, 56 (52), 16515– 16520, DOI: 10.1002/anie.201707043There is no corresponding record for this reference.
- 158Hedir, G.; Stubbs, C.; Aston, P.; Dove, A. P.; Gibson, M. I. Synthesis of Degradable Poly(Vinyl Alcohol) by Radical Ring-Opening Copolymerization and Ice Recrystallization Inhibition Activity. ACS Macro Lett. 2017, 6 (12), 1404– 1408, DOI: 10.1021/acsmacrolett.7b00905There is no corresponding record for this reference.
- 159Hedir, G. G.; Bell, C. A.; O’Reilly, R. K.; Dove, A. P. Functional Degradable Polymers by Radical Ring-Opening Copolymerization of MDO and Vinyl Bromobutanoate: Synthesis, Degradability and Post-Polymerization Modification. Biomacromolecules 2015, 16 (7), 2049– 2058, DOI: 10.1021/acs.biomac.5b00476There is no corresponding record for this reference.
- 160Ding, D.; Pan, X.; Zhang, Z.; Li, N.; Zhu, J.; Zhu, X. A Degradable Copolymer of 2-Methylene-1,3-Dioxepane and Vinyl Acetate by Photo-Induced Cobalt-Mediated Radical Polymerization. Polym. Chem. 2016, 7 (33), 5258– 5264, DOI: 10.1039/C6PY01061JThere is no corresponding record for this reference.
- 161Odian, G. Principles of Polymerization; John Wiley & Sons, 2004.There is no corresponding record for this reference.
- 162Jin, S.; Gonsalves, K. E. Synthesis and Characterization of Functionalized Poly(ε-Caprolactone) Copolymers by Free-Radical Polymerization. Macromolecules 1998, 31 (4), 1010– 1015, DOI: 10.1021/ma9707289There is no corresponding record for this reference.
- 163Du, Y.; Ren, L.; Sloan, J.; Chong, S.; Lamprou, A.; Du, Y.; Coughlin, E. B. Comparative Study of N-Vinyl Pyrrolidone and Cyclic Ketene Acetal Copolymer Degradation under Alkaline, Enzymatic, or Wastewater Conditions. Polymer 2024, 309, 127444, DOI: 10.1016/j.polymer.2024.127444There is no corresponding record for this reference.
- 164Du, Y.; Du, Y.; Lazzari, S.; Reimers, T.; Konradi, R.; Holcombe, T. W.; Coughlin, E. B. Mechanistic Investigation of Cyclic Ketene Acetal Radical Ring-Opening Homo- and Co-Polymerization and Preparation of PEO Graft Copolymers with Tunable Composition. Polym. Chem. 2022, 13 (41), 5829– 5840, DOI: 10.1039/D2PY00986BThere is no corresponding record for this reference.
- 165Hiraguri, Y.; Tokiwa, Y. Synthesis of a Novel Water-Soluble Copolymer Composed of 2-Methylene-1,3,6-Trioxocane and N-Vinyl 2-Pyrrolidone and Its Enzymatic Degradation. Clean Prod Processes 2001, 3 (3), 303– 306, DOI: 10.1007/s100980100120There is no corresponding record for this reference.
- 166Choi, S.; Lee, K.; Kwon, S.; Kim, H. Preparation of Fine Particles of Poly(N-Vinyl-2-Pyrrolidone-Co-2-Methylene-1,3-Dioxepane) Using Supercritical Antisolvent. Journal of Supercritical Fluids 2006, 37 (3), 287– 291, DOI: 10.1016/j.supflu.2005.11.023There is no corresponding record for this reference.
- 167Pamungkas, M. S.; Ando, T.; Ajiro, H. Copolymer Synthesis of N -Vinylacetamide and 2-Methylene-1,3-Dioxepane Provides a New Degradable Polymer. J. Polym. Sci. 2025, 63 (6), 1306– 1318, DOI: 10.1002/pol.20240935There is no corresponding record for this reference.
- 168Pesenti, T.; Gillon, E.; Ishii, S.; Messaoudi, S.; Guillaneuf, Y.; Imberty, A.; Nicolas, J. Increasing the Hydrophilicity of Cyclic Ketene Acetals Improves the Hydrolytic Degradation of Vinyl Copolymers and the Interaction of Glycopolymer Nanoparticles with Lectins. Biomacromolecules 2023, 24 (2), 991– 1002, DOI: 10.1021/acs.biomac.2c01419There is no corresponding record for this reference.
- 169Pesenti, T.; Domingo-Lopez, D.; Gillon, E.; Ibrahim, N.; Messaoudi, S.; Imberty, A.; Nicolas, J. Degradable Glycopolyester-like Nanoparticles by Radical Ring-Opening Polymerization. Biomacromolecules 2022, 23 (9), 4015– 4028, DOI: 10.1021/acs.biomac.2c00851There is no corresponding record for this reference.
- 170Tran, J.; Pesenti, T.; Cressonnier, J.; Lefay, C.; Gigmes, D.; Guillaneuf, Y.; Nicolas, J. Degradable Copolymer Nanoparticles from Radical Ring-Opening Copolymerization between Cyclic Ketene Acetals and Vinyl Ethers. Biomacromolecules 2019, 20 (1), 305– 317, DOI: 10.1021/acs.biomac.8b01500There is no corresponding record for this reference.
- 171Ivanchenko, O.; Mazières, S.; Poli, R.; Harrisson, S.; Destarac, M. Ring Size-Reactivity Relationship in Radical Ring-Opening Copolymerisation of Thionolactones with Vinyl Pivalate. Polym. Chem. 2022, 13 (45), 6284– 6292, DOI: 10.1039/D2PY01153KThere is no corresponding record for this reference.
- 172Calderón-Díaz, A.; Boggiano, A. C.; Xiong, W.; Kaiser, N.; Gutekunst, W. R. Degradable N -Vinyl Copolymers through Radical Ring-Opening Polymerization of Cyclic Thionocarbamates. ACS Macro Lett. 2024, 13 (11), 1390– 1395, DOI: 10.1021/acsmacrolett.4c00550There is no corresponding record for this reference.
- 173Hepburn, K. S.; Zulkifli, N. S.; Delorme, C.; Kazmi, T.; Abu Bakar, R.; Roth, P. J. Thioester-Rich Degradable Copolymers from a Thionolactone and S-Vinyl and P-Vinyl Monomers. Polymer 2024, 311, 127485, DOI: 10.1016/j.polymer.2024.127485There is no corresponding record for this reference.
- 174Shi, Y.; Agarwal, S. Thermally Stable Optically Transparent Copolymers of 2-Methylene-1,3-Dioxepane and N-Phenyl Maleimide with Degradable Ester Linkages. e-Polym. 2015, 15 (4), 217– 226, DOI: 10.1515/epoly-2015-0096There is no corresponding record for this reference.
- 175Hill, M. R.; Guégain, E.; Tran, J.; Figg, C. A.; Turner, A. C.; Nicolas, J.; Sumerlin, B. S. Radical Ring-Opening Copolymerization of Cyclic Ketene Acetals and Maleimides Affords Homogeneous Incorporation of Degradable Units. ACS Macro Lett. 2017, 6 (10), 1071– 1077, DOI: 10.1021/acsmacrolett.7b00572There is no corresponding record for this reference.
- 176Hill, M. R.; Kubo, T.; Goodrich, S. L.; Figg, C. A.; Sumerlin, B. S. Alternating Radical Ring-Opening Polymerization of Cyclic Ketene Acetals: Access to Tunable and Functional Polyester Copolymers. Macromolecules 2018, 51 (14), 5079– 5084, DOI: 10.1021/acs.macromol.8b00889There is no corresponding record for this reference.
- 177Hall, H. K.; Padias, A. B. Bond Forming Initiation of “Charge-Transfer” Polymerizations and the Accompanying Cycloadditions. Acc. Chem. Res. 1997, 30 (8), 322– 329, DOI: 10.1021/ar950222fThere is no corresponding record for this reference.
- 178Vu, N. D.; Ivanchenko, O.; Baffie, F.; Guillaneuf, Y.; Gigmes, D.; Harrisson, S.; Monteil, V.; Destarac, M.; D’Agosto, F. Synthesis of Degradable Polyethylene and Poly(Ethylene- Co -Vinyl Acetate) by Radical Co- and Terpolymerization of Ethylene, ε-Thionocaprolactone, and Vinyl Acetate. Macromolecules 2024, 57 (9), 4516– 4523, DOI: 10.1021/acs.macromol.4c00085There is no corresponding record for this reference.
- 179Hiraguri, Y.; Katase, K.; Tokiwa, Y. Synthesis of Biodegradable Detergent Builder by Alternating Copolymerization of 2-Methylene-1,3,6-trioxocane and Maleic Anhydride. Journal of Macromolecular Science, Part A 2007, 44 (8), 893– 897, DOI: 10.1080/10601320701407979There is no corresponding record for this reference.
- 180Du, Y.; Yang, Y.; Deglmann, P.; Holcombe, T. W.; Du, Y.; Coughlin, E. B. The Competition between Radical Copolymerization and Charge Separation of Cyclic Ketene Acetals and Electron-deficient Olefins. J. Polym. Sci. 2024, 62 (16), 3716– 3729, DOI: 10.1002/pol.20230480There is no corresponding record for this reference.
- 181Lovell, P. A.; Schork, F. J. Fundamentals of Emulsion Polymerization. Biomacromolecules 2020, 21 (11), 4396– 4441, DOI: 10.1021/acs.biomac.0c00769There is no corresponding record for this reference.
- 182Schork, F. J. In Support of Commercial Applications of Miniemulsion Polymerization. Ind. Eng. Chem. Res. 2023, 62 (5), 2329– 2335, DOI: 10.1021/acs.iecr.2c01746There is no corresponding record for this reference.
- 183Asua, J. M. Miniemulsion Polymerization. In Encyclopedia of Polymeric Nanomaterials; Kobayashi, S., Müllen, K., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2015; pp 1267– 1275. DOI: 10.1007/978-3-642-29648-2_263 .There is no corresponding record for this reference.
- 184Siebert, J. M.; Baumann, D.; Zeller, A.; Mailänder, V.; Landfester, K. Synthesis of Polyester Nanoparticles in Miniemulsion Obtained by Radical Ring-Opening of BMDO and Their Potential as Biodegradable Drug Carriers. Macromol. Biosci. 2012, 12 (2), 165– 175, DOI: 10.1002/mabi.201100236There is no corresponding record for this reference.
- 185d’Ayala, G. G.; Malinconico, M.; Laurienzo, P.; Tardy, A.; Guillaneuf, Y.; Lansalot, M.; D’Agosto, F.; Charleux, B. RAFT/MADIX Copolymerization of Vinyl Acetate and 5,6-benzo-2-methylene-1,3-dioxepane. J. Polym. Sci., Part A: Polym. Chem. 2014, 52 (1), 104– 111, DOI: 10.1002/pola.26976There is no corresponding record for this reference.
- 186Wickel, H.; Agarwal, S.; Greiner, A. Homopolymers and Random Copolymers of 5,6-Benzo-2-Methylene-1,3-Dioxepane and Methyl Methacrylate: Structural Characterization Using 1D and 2D NMR. Macromolecules 2003, 36 (7), 2397– 2403, DOI: 10.1021/ma025983uThere is no corresponding record for this reference.
- 187Galanopoulo, P.; Sinniger, L.; Gil, N.; Gigmes, D.; Lefay, C.; Guillaneuf, Y.; Lages, M.; Nicolas, J.; D’Agosto, F.; Lansalot, M. Degradable Vinyl Polymer Particles by Radical Aqueous Emulsion Copolymerization of Methyl Methacrylate and 5,6-Benzo-2-Methylene-1,3-Dioxepane. Polym. Chem. 2023, 14 (11), 1224– 1231, DOI: 10.1039/D3PY00040KThere is no corresponding record for this reference.
- 188Carter, M. C. D.; Hejl, A.; Woodfin, S.; Einsla, B.; Janco, M.; DeFelippis, J.; Cooper, R. J.; Even, R. C. Backbone-Degradable Vinyl Acetate Latex: Coatings for Single-Use Paper Products. ACS Macro Lett. 2021, 10 (5), 591– 597, DOI: 10.1021/acsmacrolett.1c00172There is no corresponding record for this reference.
- 189Carter, M. C. D.; DeFelippis, J.; Even, R. C.; Hejl, A. Aqueous Dispersion of Copolymer Particles of Vinyl Acetate and a Cyclic Ketene Acetal Monomer. US 11,111,328 B2, July 9, 2021.There is no corresponding record for this reference.
- 190Kordes, B. R.; Ascherl, L.; Rüdinger, C.; Melchin, T.; Agarwal, S. Competition between Hydrolysis and Radical Ring-Opening Polymerization of MDO in Water. Who Makes the Race?. Macromolecules 2023, 56 (3), 1033– 1044, DOI: 10.1021/acs.macromol.2c01653There is no corresponding record for this reference.
- 191Carter, M. C. D.; Hejl, A.; Janco, M.; DeFelippis, J.; Yang, P.; Gallagher, M.; Liang, Y. Emulsion Polymerization of 2-Methylene-1,3-Dioxepane and Vinyl Acetate: Process Analysis and Characterization. Macromolecules 2023, 56 (15), 5718– 5729, DOI: 10.1021/acs.macromol.3c00571There is no corresponding record for this reference.
- 192Wenzel, F.; Aguirre, M.; Leiza, J. R. Emulsion Copolymerization of 2-Methylene-1,3-Dioxepane (MDO) and Acrylate Monomers: Incorporation vs Hydrolysis. Polymer 2024, 307, 127285, DOI: 10.1016/j.polymer.2024.127285There is no corresponding record for this reference.
- 193Movafagh, M.; Meek, K. M.; Dubé, M. A. Butyl Acrylate/2-Methylene-1,3-Dioxepane/Vinyl Acetate Emulsion Terpolymerization: Incorporating Backbone Degradable Linkages into Adhesive Applications. ChemSusChem 2025, 18 (9), e202402478 DOI: 10.1002/cssc.202402478There is no corresponding record for this reference.
- 194Mothe, S. R.; Zhao, W.; Van Herk, A. M.; Thoniyot, P. Backbone-Degradable Acrylate Latex: Toward Overcoming Hydrolysis Limitations of Cyclic Ketene Acetal Monomers. Macromolecules 2024, 57 (6), 2937– 2949, DOI: 10.1021/acs.macromol.3c02474There is no corresponding record for this reference.
- 195Zhu, C.; Nicolas, J. Towards Nanoparticles with Site-Specific Degradability by Ring-Opening Copolymerization Induced Self-Assembly in Organic Medium. Polym. Chem. 2021, 12 (4), 594– 607, DOI: 10.1039/D0PY01425GThere is no corresponding record for this reference.
- 196Guégain, E.; Zhu, C.; Giovanardi, E.; Nicolas, J. Radical Ring-Opening Copolymerization-Induced Self-Assembly (rROPISA). Macromolecules 2019, 52 (10), 3612– 3624, DOI: 10.1021/acs.macromol.9b00161There is no corresponding record for this reference.
- 197D’Agosto, F.; Rieger, J.; Lansalot, M. RAFT-Mediated Polymerization-Induced Self-Assembly. Angew. Chem. Int. Ed 2020, 59 (22), 8368– 8392, DOI: 10.1002/anie.201911758There is no corresponding record for this reference.
- 198Zhu, C.; Denis, S.; Nicolas, J. A Simple Route to Aqueous Suspensions of Degradable Copolymer Nanoparticles Based on Radical Ring-Opening Polymerization-Induced Self-Assembly (rROPISA). Chem. Mater. 2022, 34 (4), 1875– 1888, DOI: 10.1021/acs.chemmater.1c04151There is no corresponding record for this reference.
- 199Mothe, S. R.; Kanaujia, P.; Oh, A. B. Y.; Ang, P.; Thoniyot, P. PVA Free-Radical Diblock Copolymer for Nanoencapsulation with Enhanced Biodegradability in the Environment. Eur. Polym. J. 2023, 196, 112287, DOI: 10.1016/j.eurpolymj.2023.112287There is no corresponding record for this reference.
- 200Mothe, S. R.; Ang, P.; Lau, H. H.; Oh, A. B. Y.; Thoniyot, P. Investigating the Potential of Degradable Poly(Vinyl Acetate) Copolymer Microparticles for Encapsulation and in Vitro Release Studies. Next Materials 2023, 1 (4), 100055, DOI: 10.1016/j.nxmate.2023.100055There is no corresponding record for this reference.
- 201Galanopoulo, P.; Gil, N.; Gigmes, D.; Lefay, C.; Guillaneuf, Y.; Lages, M.; Nicolas, J.; Lansalot, M.; D’Agosto, F. One-Step Synthesis of Degradable Vinylic Polymer-Based Latexes via Aqueous Radical Emulsion Polymerization. Angew. Chem., Int. Ed. 2022, 61 (15), e202117498 DOI: 10.1002/anie.202117498There is no corresponding record for this reference.
- 202Galanopoulo, P.; Gil, N.; Gigmes, D.; Lefay, C.; Guillaneuf, Y.; Lages, M.; Nicolas, J.; d’Agosto, F.; Lansalot, M. RAFT-Mediated Emulsion Polymerization-Induced Self-Assembly for the Synthesis of Core-Degradable Waterborne Particles. Angew. Chem., Int. Ed. 2023, 62 (16), e202302093 DOI: 10.1002/anie.202302093There is no corresponding record for this reference.
- 203Georgiou, P. G.; Neal, T. J.; Newell, M. A.; Hepburn, K. S.; Tyler, J. J. S.; Farmer, M. A. H.; Chohan, P.; Roth, P. J.; Nicolas, J.; Armes, S. P. Degradable Diblock Copolymer Vesicles via Radical Ring-Opening Polymerization-Induced Self-Assembly in Aqueous Media. J. Am. Chem. Soc. 2025, 147 (23), 19817– 19828, DOI: 10.1021/jacs.5c03744There is no corresponding record for this reference.
- 204Lages, M.; Gil, N.; Galanopoulo, P.; Mougin, J.; Lefay, C.; Guillaneuf, Y.; Lansalot, M.; D’Agosto, F.; Nicolas, J. Degradable Vinyl Copolymer Nanoparticles/Latexes by Aqueous Nitroxide-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2022, 55 (21), 9790– 9801, DOI: 10.1021/acs.macromol.2c01734There is no corresponding record for this reference.
- 205Lages, M.; Gil, N.; Galanopoulo, P.; Mougin, J.; Lefay, C.; Guillaneuf, Y.; Lansalot, M.; D’Agosto, F.; Nicolas, J. Degradable Latexes by Nitroxide-Mediated Aqueous Seeded Emulsion Copolymerization Using a Thionolactone. Macromolecules 2023, 56 (19), 7973– 7983, DOI: 10.1021/acs.macromol.3c01478There is no corresponding record for this reference.
- 206Fuji, K.; Kitayama, Y.; Harada, A. Miniemulsion Ring-Opening Radical Polymerization with Dibenzo[c, e]Oxepan-5-Thione for Degradable Polymer Particles. ACS Appl. Polym. Mater. 2025, 7 (5), 3349– 3357, DOI: 10.1021/acsapm.5c00088There is no corresponding record for this reference.
- 207Fadil, Y.; Fan, D.; Thompson, S. W.; Zetterlund, P. B. Synthesis of Degradable Vinyl Copolymers Based on Lipoic Acid via Ab Initio Emulsion Polymerization. ACS Sustainable Chem. Eng. 2025, 13 (38), 16117– 16126, DOI: 10.1021/acssuschemeng.5c06894There is no corresponding record for this reference.
- 208Riachi, C.; Schüwer, N.; Klok, H.-A. Degradable Polymer Brushes Prepared via Surface-Initiated Controlled Radical Polymerization. Macromolecules 2009, 42 (21), 8076– 8081, DOI: 10.1021/ma901537xThere is no corresponding record for this reference.
- 209Xie, F.; Deng, X.; Kratzer, D.; Cheng, K. C. K.; Friedmann, C.; Qi, S.; Solorio, L.; Lahann, J. Backbone-Degradable Polymers Prepared by Chemical Vapor Deposition. Angew. Chem. 2017, 129 (1), 209– 213, DOI: 10.1002/ange.201609307There is no corresponding record for this reference.
- 210Dai, G.; Xie, Q.; Chen, S.; Ma, C.; Zhang, G. Biodegradable Poly(Ester)-Poly(Methyl Methacrylate) Copolymer for Marine Anti-Biofouling. Prog. Org. Coat. 2018, 124, 55– 60, DOI: 10.1016/j.porgcoat.2018.08.003There is no corresponding record for this reference.
- 211Xie, Q.; Ma, C.; Zhang, G.; Bressy, C. Poly(Ester)-Poly(Silyl Methacrylate) Copolymers: Synthesis and Hydrolytic Degradation Kinetics. Polym. Chem. 2018, 9 (12), 1448– 1454, DOI: 10.1039/C8PY00052BThere is no corresponding record for this reference.
- 212Dai, G.; Xie, Q.; Ai, X.; Ma, C.; Zhang, G. Self-Generating and Self-Renewing Zwitterionic Polymer Surfaces for Marine Anti-Biofouling. ACS Appl. Mater. Interfaces 2019, 11 (44), 41750– 41757, DOI: 10.1021/acsami.9b16775There is no corresponding record for this reference.
- 213Pan, J.; Ai, X.; Ma, C.; Zhang, G. Degradable Vinyl Polymers for Combating Marine Biofouling. Acc. Chem. Res. 2022, 55 (11), 1586– 1598, DOI: 10.1021/acs.accounts.2c00187There is no corresponding record for this reference.
- 214Ai, X.; Pan, J.; Xie, Q.; Ma, C.; Zhang, G. UV-Curable Hyperbranched Poly(Ester- Co -Vinyl) by Radical Ring-Opening Copolymerization for Antifouling Coatings. Polym. Chem. 2021, 12 (31), 4524– 4531, DOI: 10.1039/D1PY00810BThere is no corresponding record for this reference.
- 215Irvine, G.; Dawson, F.; George, A.; Kopec, M. Are Polymer Gels Synthesized by Free Radical Polymerization with Cleavable Crosslinkers Really Degradable?. Eur. Polym. J. 2024, 213, 113089, DOI: 10.1016/j.eurpolymj.2024.113089There is no corresponding record for this reference.
- 216Elliss, H.; Dawson, F.; Nisa, Q. un; Bingham, N. M.; Roth, P. J.; Kopeć, M. Fully Degradable Polyacrylate Networks from Conventional Radical Polymerization Enabled by Thionolactone Addition. Macromolecules 2022, 55 (15), 6695– 6702, DOI: 10.1021/acs.macromol.2c01140There is no corresponding record for this reference.
- 217Dawson, F.; Kazmi, T.; Roth, P.; Kopec, M. Strands vs. Crosslinks: Topology-Dependent Degradation and Regelation of Polyacrylate Networks Synthesised by RAFT Polymerisation. Polym. Chem. 2023, 14 (47), 5166– 5177, DOI: 10.1039/D3PY01008BThere is no corresponding record for this reference.
- 218Dawson, F.; Irvine, G.; Kopeć, M. Lipoic Acid/Ethyl Lipoate as Cleavable Comonomers for Synthesis of Degradable Polymer Networks. Polym. Chem. 2025, 16 (22), 2659– 2669, DOI: 10.1039/D5PY00379BThere is no corresponding record for this reference.
- 219Choi, C.; Self, J. L.; Okayama, Y.; Levi, A. E.; Gerst, M.; Speros, J. C.; Hawker, C. J.; Read De Alaniz, J.; Bates, C. M. Light-Mediated Synthesis and Reprocessing of Dynamic Bottlebrush Elastomers under Ambient Conditions. J. Am. Chem. Soc. 2021, 143 (26), 9866– 9871, DOI: 10.1021/jacs.1c03686There is no corresponding record for this reference.
- 220Maw, M.; Dashtimoghadam, E.; Keith, A. N.; Morgan, B. J.; Tanas, A. K.; Nikitina, E.; Ivanov, D. A.; Vatankhah-Varnosfaderani, M.; Dobrynin, A. V.; Sheiko, S. S. Sticky Architecture: Encoding Pressure Sensitive Adhesion in Polymer Networks. ACS Cent. Sci. 2023, 9 (2), 197– 205, DOI: 10.1021/acscentsci.2c01407There is no corresponding record for this reference.
- 221Prince, E.; Cheng, H.; Banal, J.; Johnson, J. Reversible Nucleic Acid Storage in Deconstructable Glassy Polymer Networks. J. Am. Chem. Soc. 2024, 146 (25), 17066– 17074, DOI: 10.1021/jacs.4c01925There is no corresponding record for this reference.
- 222Carlotti, M.; Tricinci, O.; Mattoli, V. Novel, High-Resolution, Subtractive Photoresist Formulations for 3D Direct Laser Writing Based on Cyclic Ketene Acetals. Advanced Materials Technologies 2022, 7 (9), 2101590, DOI: 10.1002/admt.202101590There is no corresponding record for this reference.
- 223Gil, N.; Thomas, C.; Mhanna, R.; Mauriello, J.; Maury, R.; Leuschel, B.; Malval, J.-P.; Clément, J.-L.; Gigmes, D.; Lefay, C.; Soppera, O.; Guillaneuf, Y. Thionolactone as a Resin Additive to Prepare (Bio)Degradable 3D Objects via VAT Photopolymerization. Angew. Chem., Int. Ed. 2022, 61 (18), e202117700 DOI: 10.1002/anie.202117700There is no corresponding record for this reference.
- 224Kouider, S.; Buchon, L.; Choulot, L.; Bratasanu, J.; Désirée, P.; Mauriello, J.; Maarek, E.; Clément, J.-L.; Montarnal, D.; Soppera, O.; Gigmes, D.; Lefay, C.; Guillaneuf, Y. Self-Destruct Thermosets: A Combination of the Comonomer Approach and Thermolatent-Base Compounds. ChemRxiv , 2025, DOI: 10.26434/chemrxiv-2025-z051f .There is no corresponding record for this reference.
- 225Han, S.; Bobrin, V. A.; Michelas, M.; Hawker, C. J.; Boyer, C. Sustainable and Recyclable Acrylate Resins for Liquid-Crystal Display 3D Printing Based on Lipoic Acid. ACS Macro Lett. 2024, 13 (11), 1495– 1502, DOI: 10.1021/acsmacrolett.4c00600There is no corresponding record for this reference.
- 226Biying, A. O.; Mothe, S. R.; Jackson, A. W.; Kanaujia, P.; Thoniyot, P. Effects of Polymer Microstructure Introduced by Radical Ring-Opening Polymerization on Nanoencapsulation and Controlled Release. Polymer 2024, 307, 127284, DOI: 10.1016/j.polymer.2024.127284There is no corresponding record for this reference.
- 227Balaji, A.; Prior, A. R.; O’Reilly, R. K.; Dove, A. P.; Thurecht, K. J.; Bell, C. A. Biodegradable mPEG- b -Poly(MDO- Co -Vinyl Esters) Block Copolymers as a Viable Nanocarrier Platform with Tuneable Disassembly. Polym. Chem. 2024, 15 (12), 1152– 1165, DOI: 10.1039/D4PY00049HThere is no corresponding record for this reference.
- 228Ahmed, S. E.; Fletcher, N. L.; Prior, A. R.; Huda, P.; Bell, C. A.; Thurecht, K. J. Development of Targeted Micelles and Polymersomes Prepared from Degradable RAFT-Based Diblock Copolymers and Their Potential Role as Nanocarriers for Chemotherapeutics. Polym. Chem. 2022, 13 (27), 4004– 4017, DOI: 10.1039/D2PY00257DThere is no corresponding record for this reference.
- 229Deng, Y.; Debbeler, J.; Traeger, A.; Gaitzsch, J. Degradable Nanocarriers Capable of Gene Delivery Derived from pH-Responsive Polyester: RROP Copolymerization Between Cyclic Ketene Acetals. Macromol. Rapid Commun. 2025, e00722 DOI: 10.1002/marc.202500722There is no corresponding record for this reference.
- 230Guégain, E.; Tran, J.; Deguettes, Q.; Nicolas, J. Degradable Polymer Prodrugs with Adjustable Activity from Drug-Initiated Radical Ring-Opening Copolymerization. Chem. Sci. 2018, 9 (43), 8291– 8306, DOI: 10.1039/C8SC02256AThere is no corresponding record for this reference.
- 231Guerassimoff, L.; Cao, J.; Auguste, M.; Bossion, A.; Zhu, C.; Le, D.; Cailleau, C.; Ismail, S. M.; Mercier-Nomé, F.; Nicolas, J. Degradable Water-Soluble Polymer Prodrugs for Subcutaneous Delivery of Irritant Anticancer Drugs. Chem. Sci. 2025, 16 (31), 14323– 14341, DOI: 10.1039/D5SC02967HThere is no corresponding record for this reference.
- 232Zhu, C.; Beauseroy, H.; Mougin, J.; Lages, M.; Nicolas, J. In Situ Synthesis of Degradable Polymer Prodrug Nanoparticles. Chem. Sci. 2025, 16 (6), 2619– 2633, DOI: 10.1039/D4SC07746FThere is no corresponding record for this reference.
- 233Lau, U. Y.; Pelegri-O’Day, E. M.; Maynard, H. D. Synthesis and Biological Evaluation of a Degradable Trehalose Glycopolymer Prepared by RAFT Polymerization. Macromol. Rapid Commun. 2018, 39 (5), 1700652, DOI: 10.1002/marc.201700652There is no corresponding record for this reference.
- 234Pesenti, T.; Zhu, C.; Gonzalez-Martinez, N.; Tomás, R. M. F.; Gibson, M. I.; Nicolas, J. Degradable Polyampholytes from Radical Ring-Opening Copolymerization Enhance Cellular Cryopreservation. ACS Macro Lett. 2022, 11 (7), 889– 894, DOI: 10.1021/acsmacrolett.2c00298There is no corresponding record for this reference.
- 235Barber Nunez, B.; Guillaneuf, Y.; Grande, D.; Carbonnier, B.; Le Droumaguet, B. Degradable Biporous Polymeric Networks Based on 2-Methylene-1,3-Dioxepane: Towards Hierarchically Structured Materials Meant for Biomedical Applications. Polymer 2024, 308, 127332, DOI: 10.1016/j.polymer.2024.127332There is no corresponding record for this reference.
- 236Barber Nunez, B.; Carbonnier, B.; Grande, D.; Le Droumaguet, B. Biporous Polycaprolactone-like Networks from 2-Methylene-1,3-Dioxepane: A Versatile Platform towards Functional and Reactive Polyesters. React. Funct. Polym. 2025, 214, 106284, DOI: 10.1016/j.reactfunctpolym.2025.106284There is no corresponding record for this reference.
- 237Jackson, A. W.; Mothe, S. R.; Ang, P.; Chennamaneni, L. R.; Herk, A. M. V.; Thoniyot, P. Backbone Degradable Poly(Acrylic Acid) Analogue via Radical Ring-Opening Copolymerization and Enhanced Biodegradability. Chemosphere 2022, 293, 133487, DOI: 10.1016/j.chemosphere.2021.133487There is no corresponding record for this reference.
- 238Däbritz, S. B.; Neubauer, K.; Kropf, C.; Agarwal, S. Evaluating Polymerization Methods and Deprotection Strategies for Making Water Soluble Poly(Acrylic Acid) with Hydrolyzable Breaking Points. Macro Chemistry & Physics 2025, 226 (11), 2500080, DOI: 10.1002/macp.202500080There is no corresponding record for this reference.
- 239Bossion, A.; Zhu, C.; Guerassimoff, L.; Mougin, J.; Nicolas, J. Vinyl Copolymers with Faster Hydrolytic Degradation than Aliphatic Polyesters and Tunable Upper Critical Solution Temperatures. Nat. Commun. 2022, 13 (1), 2873, DOI: 10.1038/s41467-022-30220-yThere is no corresponding record for this reference.
- 240Kertsomboon, T.; Agarwal, S.; Chirachanchai, S. UCST-Type Copolymer through the Combination of Water-Soluble Polyacrylamide and Polycaprolactone-Like Polyester. Macromol. Rapid Commun. 2020, 41 (21), 2000243, DOI: 10.1002/marc.202000243There is no corresponding record for this reference.
- 241Sugihara, S.; Endo, A.; Raje, K.; Matsumoto, A.; Fujita, S.; Maeda, Y. Design and Synthesis of Thermoresponsive Degradable Copolymers: Integrating Hydroxy-Functional Vinyl Ethers with Cyclic Ketene Acetals. Polym. Chem. 2025, 16 (26), 3059– 3069, DOI: 10.1039/D5PY00398AThere is no corresponding record for this reference.
- 242Neal, T. J.; Nicolas, J. Aqueous Degradability of Water-Soluble, Thioester-Containing Polyacrylamides with UCST-Type Behaviour in Salt Solutions Obtained by rROP. Chem. Commun. 2024, 60 (96), 14260– 14263, DOI: 10.1039/D4CC04718DThere is no corresponding record for this reference.
- 243Levkovsky, I. O.; Trachsel, L.; Murata, H.; Matyjaszewski, K. Versatile and Controlled Synthesis of Degradable, Water-Soluble Bottlebrush Polymers with Poly(Disulfide) Backbones Derived from α-Lipoic Acid. ACS Macro Lett. 2025, 14 (2), 207– 213, DOI: 10.1021/acsmacrolett.4c00839There is no corresponding record for this reference.
- 244Abu Bakar, R.; Hepburn, K. S.; Keddie, J. L.; Roth, P. J. Degradable, Ultraviolet-Crosslinked Pressure-Sensitive Adhesives Made from Thioester-Functional Acrylate Copolymers. Angew. Chem. Int. Ed 2023, 62 (34), e202307009 DOI: 10.1002/anie.202307009There is no corresponding record for this reference.
- 245Kohsaka, Y.; Naganuma, K. Main-Chain Scission of Acrylic Polymers via Intrachain Transesterification and Their Application in UV-Curable Dismountable Adhesives. ACS Polym. Au 2025, 5 (5), 481– 487, DOI: 10.1021/acspolymersau.5c00060There is no corresponding record for this reference.
- 246Pal, S.; Shin, J.; DeFrates, K.; Arslan, M.; Dale, K.; Chen, H.; Ramirez, D.; Messersmith, P. B. Recyclable Surgical, Consumer, and Industrial Adhesives of Poly(α-Lipoic Acid). Science 2024, 385 (6711), 877– 883, DOI: 10.1126/science.ado6292There is no corresponding record for this reference.
- 247Pal, S.; Salzman, E. E.; Ramirez, D.; Chen, H.; Perez, C. A.; Dale, K.; Ghosh, S. K.; Lin, L.; Messersmith, P. B. Versatile Solid-State Medical Superglue Precursors of α-Lipoic Acid. J. Am. Chem. Soc. 2025, 147 (16), 13377– 13384, DOI: 10.1021/jacs.4c18448There is no corresponding record for this reference.
- 248Shi, Y.; Zhou, P.; Jérôme, V.; Freitag, R.; Agarwal, S. Enzymatically Degradable Polyester-Based Adhesives. ACS Biomater. Sci. Eng. 2015, 1 (10), 971– 977, DOI: 10.1021/acsbiomaterials.5b00217There is no corresponding record for this reference.
- 249Yang, Y.; Shi, K.; Yu, K.; Xing, F.; Lai, H.; Zhou, Y.; Xiao, P. Degradable Hydrogel Adhesives with Enhanced Tissue Adhesion, Superior Self-Healing, Cytocompatibility, and Antibacterial Property. Adv. Healthcare Mater. 2022, 11 (4), 2101504, DOI: 10.1002/adhm.202101504There is no corresponding record for this reference.
- 250Ding, J.; Han, Y.; Wu, J.; Chen, D.; Xiong, C.; Huang, D.; Xiong, Z. Degradable Underwater Adhesive Utilizing Free Radical Ring-Opening Polymerization. ACS Appl. Polym. Mater. 2024, 6 (10), 5980– 5987, DOI: 10.1021/acsapm.4c00666There is no corresponding record for this reference.
- 251Yang, R.; Zhang, X.; Chen, B.; Yan, Q.; Yin, J.; Luan, S. Tunable Backbone-Degradable Robust Tissue Adhesives via in Situ Radical Ring-Opening Polymerization. Nat. Commun. 2023, 14 (1), 6063, DOI: 10.1038/s41467-023-41610-1There is no corresponding record for this reference.
- 252Bailey, W. J.; Gapud, B. Synthesis of Biodegradable Polyethylene. In Polymer Stabilization and Degradation; ACS Symposium Series; American Chemical Society: Washington, D.C., 1985; Vol. 280, pp 423– 431. DOI: 10.1021/bk-1985-0280 .There is no corresponding record for this reference.
- 253Wu, B.; Lenz, R. W. Synthesis, Characterization, and Hydrolytic Degradation of Copolymers of 2-Methylene-1,3-Dioxepane with Ethylene and with Styrene. Journal of Polymers and the Environment 1998, 6 (1), 23– 29, DOI: 10.1023/A:1022874411834There is no corresponding record for this reference.
- 254Zeng, T.; You, W.; Chen, G.; Nie, X.; Zhang, Z.; Xia, L.; Hong, C.; Chen, C.; You, Y. Degradable PE-Based Copolymer with Controlled Ester Structure Incorporation by Cobalt-Mediated Radical Copolymerization under Mild Condition. iScience 2020, 23 (3), 100904, DOI: 10.1016/j.isci.2020.100904There is no corresponding record for this reference.
- 255Wolpers, A.; Bergerbit, C.; Ebeling, B.; D’Agosto, F.; Monteil, V. Aromatic Xanthates and Dithiocarbamates for the Polymerization of Ethylene through Reversible Addition-Fragmentation Chain Transfer (RAFT). Angew. Chem. Int. Ed 2019, 58 (40), 14295– 14302, DOI: 10.1002/anie.201905629There is no corresponding record for this reference.
- 256Zeng, T.-Y.; Xia, L.; Zhang, Z.; Hong, C.-Y.; You, Y.-Z. Dithiocarbamate-Mediated Controlled Copolymerization of Ethylene with Cyclic Ketene Acetals towards Polyethylene-Based Degradable Copolymers. Polym. Chem. 2021, 12 (2), 165– 171, DOI: 10.1039/D0PY00200CThere is no corresponding record for this reference.
- 257Kohsaka, Y.; Toyama, K.; Kawauchi, M.; Naganuma, K. Fast and Selective Main-Chain Scission of Vinyl Polymers Using the Domino Reaction in the Alternating Sequence for Transesterification. ACS Macro Lett. 2024, 13 (8), 1016– 1021, DOI: 10.1021/acsmacrolett.4c00295There is no corresponding record for this reference.
- 258Yang, Y.; Xing, F.; Zhou, Y.; Xiao, P. Hydrolysis/Photolysis Dual-Stimuli-Responsive Backbone-Degradable Copolymers Featuring Cyclic Ketene Acetal and Ortho-Nitrobenzyl Pendants. Eur. Polym. J. 2022, 181, 111659, DOI: 10.1016/j.eurpolymj.2022.111659There is no corresponding record for this reference.
- 259Do, P. T.; Poad, B. L. J.; Frisch, H. Programming Photodegradability into Vinylic Polymers via Radical Ring-Opening Polymerization. Angew. Chem. Int. Ed 2023, 62 (6), e202213511 DOI: 10.1002/anie.202213511There is no corresponding record for this reference.
- 260Sbordone, F.; Micallef, A.; Frisch, H. pH-Controlled Reversible Folding of Copolymers via Formation of Β-sheet Secondary Structures. Angew. Chem. Int. Ed 2024, 63 (10), e202319839 DOI: 10.1002/anie.202319839There is no corresponding record for this reference.
- 261Woodruff, M. A.; Hutmacher, D. W. The Return of a Forgotten Polymer─Polycaprolactone in the 21st Century. Prog. Polym. Sci. 2010, 35 (10), 1217– 1256, DOI: 10.1016/j.progpolymsci.2010.04.002There is no corresponding record for this reference.
- 262Ikada, Y.; Tsuji, H. Biodegradable Polyesters for Medical and Ecological Applications. Macromol. Rapid Commun. 2000, 21 (3), 117– 132, DOI: 10.1002/(SICI)1521-3927(20000201)21:3<117::AID-MARC117>3.0.CO;2-XThere is no corresponding record for this reference.
- 263Mousa, M.; Jonsson, M.; Wilson, O.; Geerts, R.; Bergenudd, H.; Bengtsson, C.; Larsson Kron, A.; Malmström, E. Branched Polyesters from Radical Ring-Opening Polymerization of Cyclic Ketene Acetals: Synthesis, Chemical Hydrolysis and Biodegradation. Polym. Chem. 2023, 14 (47), 5154– 5165, DOI: 10.1039/D3PY00630AThere is no corresponding record for this reference.
- 264Hiracuri, Y.; Tokiwa, Y. Synthesis of Copolymers Composed of 2-methylene-1,3,6-trioxocane and Vinyl Monomers and Their Enzymatic Degradation. J. Polym. Sci. A Polym. Chem. 1993, 31 (12), 3159– 3163, DOI: 10.1002/pola.1993.080311233There is no corresponding record for this reference.
- 265Neogi, S.; un Nisa, Q.; Abu Bakar, R.; Bingham, N. M.; Roth, P. J. Ambient Cationic Ring-Opening Polymerization of Dibenzo[c,e]Oxepine-5 (7H)-Thione (DOT): Thermal and Nucleophile-Initiated Depolymerization. Eur. Polym. J. 2024, 219, 113390, DOI: 10.1016/j.eurpolymj.2024.113390There is no corresponding record for this reference.
- 266Truong, T. H. A.; Mothe, S. R.; Min, J. L.; Tan, H. M.; Jackson, A. W.; Nguyen, D. T.; Ye, D. K. J.; Kanaujia, P.; Thoniyot, P.; Dang, T. T. Immuno-Modulatory Effects of Microparticles Formulated from Degradable Polystyrene Analogue. Macromol. Biosci. 2022, 22 (7), 2100472, DOI: 10.1002/mabi.202100472There is no corresponding record for this reference.
- 267Bingham, N. M.; Nisa, Q. U.; Gupta, P.; Young, N. P.; Velliou, E.; Roth, P. J. Biocompatibility and Physiological Thiolytic Degradability of Radically Made Thioester-Functional Copolymers: Opportunities for Drug Release. Biomacromolecules 2022, 23 (5), 2031– 2039, DOI: 10.1021/acs.biomac.2c00039There is no corresponding record for this reference.
- 268Hiraguri, Y.; Tokiwa, Y. Synthesis of Thermosensitive Polymers Having Enzymatic Degradability. Clean Techn Environ. Policy 2002, 4 (2), 122– 124, DOI: 10.1007/s10098-002-0140-4There is no corresponding record for this reference.
- 269Ren, L.; Agarwal, S. Synthesis, Characterization, and Properties Evaluation of Poly[(N-isopropylacrylamide)-co-ester]s. Macromol. Chem. Phys. 2007, 208 (3), 245– 253, DOI: 10.1002/macp.200600484There is no corresponding record for this reference.
- 270Siegwart, D. J.; Bencherif, S. A.; Srinivasan, A.; Hollinger, J. O.; Matyjaszewski, K. Synthesis, Characterization, and in Vitro Cell Culture Viability of Degradable Poly(N -isopropylacrylamide- Co −5,6-benzo-2-methylene-1,3-dioxepane)-based Polymers and Crosslinked Gels. J. Biomedical Materials Res. 2008, 87A (2), 345– 358, DOI: 10.1002/jbm.a.31708There is no corresponding record for this reference.
- 271Barbon, S. M.; Carter, M. C. D.; Yin, L.; Whaley, C. M.; Albright, V. C.; Tecklenburg, R. E. Synthesis and Biodegradation Studies of Low-Dispersity Poly(Acrylic Acid). Macromol. Rapid Commun. 2022, 43 (13), 2100773, DOI: 10.1002/marc.202100773There is no corresponding record for this reference.
- 272Guégain, E.; Michel, J.-P.; Boissenot, T.; Nicolas, J. Tunable Degradation of Copolymers Prepared by Nitroxide-Mediated Radical Ring-Opening Polymerization and Point-by-Point Comparison with Traditional Polyesters. Macromolecules 2018, 51 (3), 724– 736, DOI: 10.1021/acs.macromol.7b02655There is no corresponding record for this reference.
- 273Volz, H. K.-H.; Agarwal, S. High Tg Poly(Ester- Co -Vinyl)s with Compositional Homogeneity by Radical Ring-Opening Polymerization. ACS Appl. Polym. Mater. 2023, 5 (10), 8754– 8763, DOI: 10.1021/acsapm.3c01970There is no corresponding record for this reference.
- 274Luzel, B.; Efimov, I.; Edeleva, M.; Kouider, S.; Gil, N.; Montarnal, D.; Lefay, C.; Gigmes, D.; Van Steenberge, P. H. M.; D’hooge, D. R.; Guillaneuf, Y. The Successful Impossible Radical Ring-Opening Copolymerization of Thionolactones and Methacrylates via an Auxiliary Third Comonomer. Nat. Commun. 2025, 16 (1), 9468, DOI: 10.1038/s41467-025-64502-yThere is no corresponding record for this reference.
- 275Bingham, N. M.; Collins, K. E.; Walsh, J.; Williams, A.; Roth, P. J. Bespoke Degradable Polymers: Modifying Cyclic Allyl Sulfide Lactones to Tune Polymer Degradation Rates and Lower Critical Solution Temperatures. Macromolecules 2025, 58 (1), 516– 525, DOI: 10.1021/acs.macromol.4c02681There is no corresponding record for this reference.
- 276Ivanchenko, O.; Mazières, S.; Harrisson, S.; Destarac, M. On-Demand Degradation of Thioester/Thioketal Functions in Vinyl Pivalate-Derived Copolymers with Thionolactones. Macromolecules 2023, 56 (11), 4163– 4171, DOI: 10.1021/acs.macromol.3c00312There is no corresponding record for this reference.
- 277Rahimi, A.; García, J. M. Chemical Recycling of Waste Plastics for New Materials Production. Nat. Rev. Chem. 2017, 1 (6), 0046, DOI: 10.1038/s41570-017-0046There is no corresponding record for this reference.
- 278Czuczola, M.; Hossain, M. S.; Shannon, D. P.; Morris, P. T.; Getty, P. T.; Bates, C. M.; Read de Alaniz, J.; Hawker, C. J. Telechelic Dithiol Copolymers as Tunable Building Blocks for Synthesizing Multiblock Materials. J. Polym. Sci. 2025, 63 (3), 759– 765, DOI: 10.1002/pol.20240876There is no corresponding record for this reference.