

About the Cover:
Perspectives
Thermodynamic Modeling of Mixed Solvent Electrolyte Solutions: Challenges and Practical Guide
Saheb Maghsoodloo *- ,
Tri Dat Ngo *- ,
Jean-Charles de Hemptinne *- ,
Edouard Moine - ,
Shu Wang - ,
Bjorn Maribo-Mogensen - ,
Emrah Altuntepe - ,
Salvador Asensio-Delgado - ,
Stephanie Peper - ,
Andrés González de Castilla - ,
Pascal Ferrari - ,
Ellen Steimers - , and
Susanna Kuitunen

This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
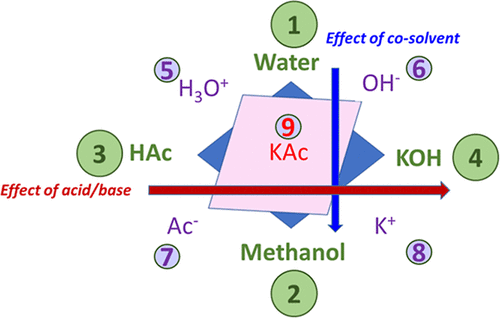
Electrolyte systems are characterized by their (i) strong nonideal behavior requiring advanced thermodynamic models, (ii) reactive components necessitating knowledge of formation properties, and (iii) multiphase nature, which demands robust reactive algorithms. These complexities are crucial in industrial applications, leading to the creation of the Joint Industry Project (JIP) EleTher, which aims to promote further academic exploration and provide recommendations for parametrizing such systems. To better understand interactions in electrolyte systems, the study focuses on quaternary systems comprising water, an acid, a base, and a cosolvent. This approach isolates pH effects from the dielectric constant influences. Due to limited data, only ternary subsystems were analyzed. The study evaluated the accuracy of three approaches: full dissociation (FD), no dissociation (ND), and partial dissociation (PD). While FD and ND approaches performed adequately for vapor–liquid equilibrium, they failed when speciation became significant. The PD approach, though more accurate, revealed challenges with parameter optimization as multiple local minima resulted in varying species distributions. A hybrid approach combining FD, ND, and PD models is recommended for achieving the most physically meaningful results. The study utilized the eNRTL method CPA equation of state within in-house tools (Carnot and ATOUT) to conduct the analysis.
Chemical Databases as a Promising Tool for Obtaining the Vaporization Characteristics of Organic Compounds
Dmitrii N. Bolmatenkov *- ,
Ilyas I. Nizamov - ,
Boris N. Solomonov - , and
Mikhail I. Yagofarov
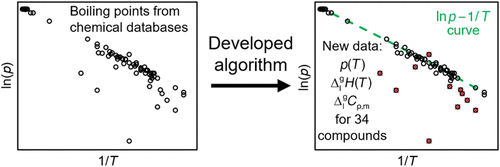
Chemical databases, such as Reaxys and SciFinder, contain extensive physicochemical data on organic compounds, including normal and reduced pressure boiling points. Recently, we proposed an approach for extracting the vaporization enthalpies and vapor pressures from these data. Although the raw values from the databases are typically obtained by relatively simple, nonspecialized equipment, the errors in the vaporization enthalpies and vapor pressures derived according to this procedure were estimated at the level of 2 kJ mol–1 and 20%, respectively. In this work, we discuss how this approach can complement classical experimental methods for determination of the vaporization characteristics. We used it to obtain saturated vapor pressures and vaporization enthalpies of 34 poorly studied aromatic compounds between 298 K and their normal boiling point. We focused on substances whose vaporization characteristics were not reported before, were available at a single temperature, or deviated significantly from expected values. Where possible, a comparison with the existing literature data and estimation methods was performed.
Thermophysical and Thermochemical Properties
Molecular Interactions of Ketorolac Tromethamine with Protic Ionic Liquids: Combined Volumetric and Spectroscopic Studies
Khajuria Deepika Amirchand - and
Vickramjeet Singh *
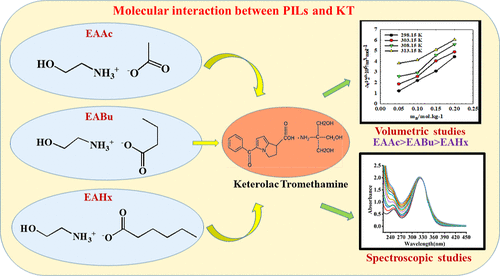
The density and speed of sound of ketorolac tromethamine (KT) in aqueous protic ionic liquid (PIL) solutions were systematically measured over a range of concentrations and temperatures between 298.15 to 313.15 K at atmospheric pressure. These experimental measurements enabled the determination of apparent molar volume (V2,ϕ) and apparent isentropic compressibility (Ks,2,ϕ), which were further used to calculate the partial molar volume (V2°) and partial molar isentropic compressibility (Ks,2°) at infinite dilution through appropriate data-fitting techniques. Thus, the estimated thermodynamic parameters are of utmost importance concerning the solvation phenomenon and interactions at the molecular level between the solvents and drug molecules, regarding the structural modifications caused by PILs. Investigations using UV–visible spectroscopy showed hyperchromic shifts with increased PIL concentrations, indicating marked changes in electronic transitions related to specific solvation effects. Increasing the anionic chain length of PILs also exhibited an effect; its influence was critical for modifying drug solubility and stability, and such studies would lead to novel insights in designing PIL-based pharmaceutical formulations. This highlights the potential of PILs in modulating physicochemical properties through controlled solvation interactions to act as novel excipients to improve drug delivery and bioavailability.
Boosting CO2 Hydrate Formation in Saline Systems with Cyclopentane Hydrate Seeds and Tetrahydrofuran: Experiments and Implications for Ocean Carbon Sequestration
Jun Fu - ,
Chun-Gang Xu *- ,
Yun-Hao Li - ,
Xiao-Sen Li *- ,
Yi Wang - , and
Zhao-Yang Chen
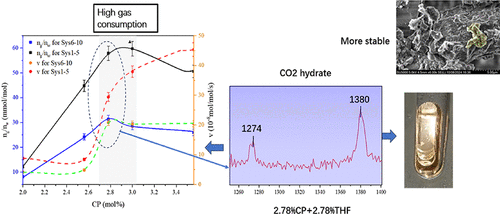
Hydrate-based ocean carbon sequestration is a highly effective approach to carbon capture and storage (CCS). The key to its success lies in the efficient formation of CO2 hydrates in seawater and their stability. This research integrated macroscopic experiments using CP (cyclopentane) hydrate crystal seeds and THF (tetrahydrofuran) as a combined promoter with microscopic analyses via Raman spectroscopy and scanning electron microscopy (SEM). The goal was to thoroughly examine how different concentrations of the solid–liquid promoters affect CO2 hydrate formation in the saltwater system, ultimately identifying the most effective concentration mix. Additionally, we studied how CO2 hydrates from various systems settle and disperse over time under simulated marine conditions. The results show that the combination of 2.78% CP and 2.78% THF is the most effective, leading to a CO2 gas consumption of 23.28 mmol in the saltwater system, which is 33% higher than in the pure water system, and the average formation rate improves by 40%. CO2 hydrates formed in the saltwater system also show a significant increase in density under a pressure of 5.50 MPa and exhibit superior stability in simulated marine sedimentation experiments. In contrast, CO2 hydrates formed in pure water are more prone to decomposition, which hinders their long-term storage.
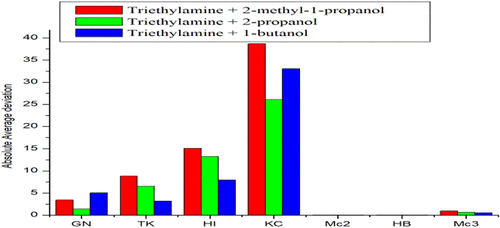
Thermodynamic Properties and Viscosity Modeling for Binary Liquid Mixtures of Triethylamine with 2-Methyl-1-propanol, 2-Propanol, and 1-Butanol at Different Temperatures
Krishan Kumar - ,
Likhish Dhingra - , and
Gyan Prakash Dubey *
The present discussion embodies the studies on binary mixtures containing triethylamine with 2-methyl-1-propanol, 2-propanol, and 1-butanol. For this purpose, the density and speed of sound for pure liquids and their binary mixtures were measured within the temperature range 293.15–313.15 K, while viscosity was measured from 298.15 to 308.15 K. Various excess and deviation parameters have been calculated using the measured properties. The calculated parameters reveal the formation of strong intermolecular interactions upon mixture formation. Excess and deviation parameters were fitted to the Redlich–Kister polynomial. The correlation ability of viscosity-related models has also been tested for the studied binary mixtures.

Supramolecular Interactions of a Biguanide-Type Antidiabetic Drug and d-(+)-Glucose in Aqueous Solution: Exploring the Role of Temperature and Concentration
Mohammad Hossain - and
Md. Abu Bin Hasan Susan *
In this work, volumetric, acoustic, viscometric, photon correlation, and near-infrared spectroscopic studies of a biguanide-type antidiabetic drug, metformin hydrochloride (M·HCl), have been reported for binary solutions with water and ternary solutions with d-(+)-glucose in aqueous media. The volumetric and acoustic studies show a kosmotropic nature of M·HCl in aqueous systems (from 0.1534 × 10–3 mol·kg–1 to 60.9483 × 10–3 mol·kg–1). Structure breaking progresses with increasing temperature (from 290.0 to 330.0 K). Strong interactions between M·HCl and water are revealed by viscometric analysis, and compared to the transition state, the ground state shows more prominent interactions. Again, the studies unveil that water structure breaking occurs with the addition of d-(+)-glucose molecules for ternary systems (up to 10.0 × 10–3 mol·kg–1). Self-association of glucose occurs with an increasing concentration of d-(+)-glucose. Water molecules surround glucose molecules more, and the kosmotropic nature of M·HCl is observed (from 10.0 × 10–3 mol·kg–1 to 20.0 × 10–3 mol·kg–1). Photon correlation and near-infrared spectroscopy studies also yielded the same result. Structure making progresses with rising temperature (from 290.0 to 315.0 K), but a rise to a greater extent (from 315.0 to 330.0 K) leads to the breaking of the water structure.
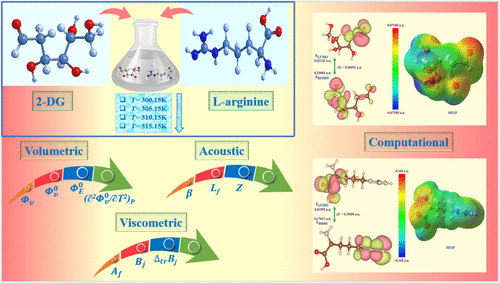
Thermophysical, Spectral, and Computational Analysis of Molecular Interactions between the Anti-COVID-19 Drug 2-DG and l-Arginine in an Aqueous Environment
Sunaina Sharma - ,
Parveen Kumar - ,
Palak Ahir - ,
Vishal Thakur - ,
Palak Verma - ,
Inesh Kumar - , and
Sunil Kumar *
The interactions between drugs and biologically active compounds have attracted significant attention due to their importance in the pharmacological and biological fields. Thus, current research is undertaken to investigate the molecular interaction between the COVID-19 drug 2-deoxy-d-glucose (2-DG) and l-arginine in an aqueous medium using experimental and computational methods. Experimental studies include the measurements of the thermophysical properties for 2-DG (0.001–0.010) mol·kg–1 in water and aqueous l-arginine (0.01) mol·kg–1 solution at four equidistant temperatures (T = 300.15–315.15 K) under barometric pressure (P = 101.3 kPa). The experimentally determined densities (ρ) and sound velocities (u) were used to evaluate the various thermoacoustic parameters. Moreover, the activation energy (Eη) and activation parameters, Δμ10#, Δμ20#, ΔH20#, and ΔS20# have also been evaluated by using transition state theory. The temperature and concentration dependencies of the evaluated parameters were estimated in terms of the structural behavior of 2-DG and its molecular interactions prevailing in an aqueous solution of l-arginine. The UV-spectral and DFT studies have been carried out to support thermophysical outcomes. The experimental results could be helpful in predicting the pharmacokinetic and pharmacodynamic action of the drug in the human body.
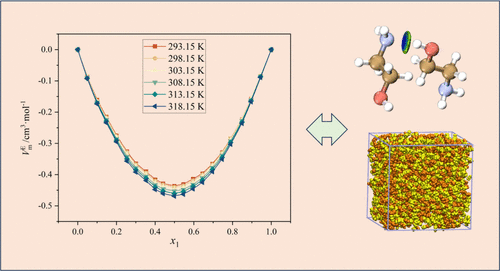
Probing the Interactions of Calcium Lactate with Glycine and l-Leucine in Water Using Volumetric Properties
Vickramjeet Singh *- ,
Arpita Padhan - , and
Sanjay Chandaka
The hydration behavior of glycine and L-leucine was examined in aqueous solutions of calcium lactate. The experiments were accomplished at atmospheric pressure across a temperature range from 293.15 to 318.15 K, emphasizing the nature of solute–solvent interactions. The volumetric properties were estimated by employing experimentally measured density and speed of sound results. The interactions between leucine or glycine and calcium lactate were analyzed by using the values of apparent molar volume, apparent molar isentropic compressibility, infinite dilution partial molar volume, and volume of transfer. The volumetric transfer parameters were determined and analyzed using the cosphere overlap model. Among all of the interactions present in the system, hydrophilic interactions were found to be the most dominant. These interactions were more pronounced in the case of glycine compared to leucine. The positive contribution to the volume may be attributed to the interaction between calcium ions and the carbonyl or amino groups of the amino acids. Additionally, the hydrophobic lactate group of calcium lactate may engage in hydrophobic interactions with the nonpolar regions of the amino acids in aqueous solution. However, the hydrophilic interactions significantly outweigh the hydrophobic contributions.
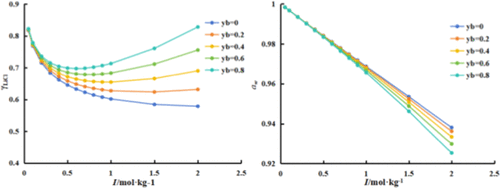
Mean Activity Coefficients and Phase Equilibria of the LiCl–KCl–H2O Ternary System at 273.15 K
Yang-Dian-Dian Wang - ,
Shi-Hua Sang *- ,
Kuangyi Zhu - , and
Ling-Xuan Wang
The mean activity coefficients of KCl (γ±KCl) in LiCl–KCl–H2O mixed salt solutions were investigated at 273.15 K by the cell potential method, and the cell potential value of a KCl–H2O solution, the standard potential value E0, and electrode response slope κ were obtained. The results indicated the ion selective electrodes have a good Nernst response. The mean activity coefficients of KCl (γ±KCl) in LiCl–KCl–H2O mixed salt solutions were determined, the total ionic strength I ranged from 0.0500 to 2.0000 mol·kg–1, and the ionic strength fractions yb of LiCl were 0.8, 0.6, 0.4, 0.2, and 0.0. The LiCl single salt parameters of the Pitzer model β(0), β(1), CΦ, and Pitzer mixed ion interaction parameters θK,Li, ψK+,Li+,Cl– for the ternary system at 273.15 K were fitted by the Pitzer model. The related thermodynamic properties, including the mean activity coefficients of LiCl (γ±LiCl), water activity (aw), osmotic coefficients (Φ), and excess Gibbs free energy (GE) were calculated. Moreover, the phase equilibrium predictions were in good accordance with the experimental values, which means that the fitted parameters have good applicability.
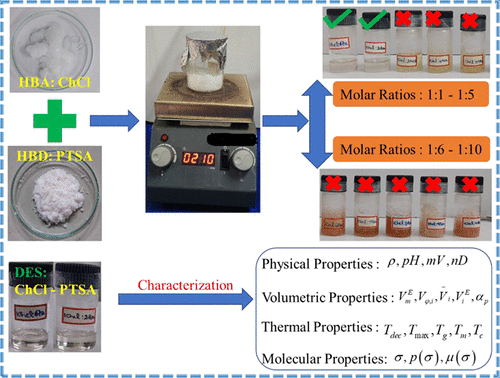
p-Toluene Sulfonic Acid-Based Deep Eutectic Solvents: Preparation and Characterization of Their Physical Properties and Thermal Stability
Gayathri Mahavishnu *- and
Sathish Kumar Kannaiyan
In this work, p-toluene sulfonic acid (PTSA)-based deep eutectic solvents (DESs) were prepared at different mole ratios (DESs: 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, and 1:10) by mixing of choline chloride (ChCl) and PTSA. Among them, 1:1 (DES1) and 1:2 (DES2) molar ratios show a clear and homogeneous liquid, even after 24 h. The density of DES1 and DES2 was measured at temperatures from 298.15 to 343.15 K and 101.305 kPa. The refractive index (nD) of DES1 and DES2 was measured at 298.15 K and 101.305 kPa. The density of DES1 + water and DES2 + water mixtures over the whole mole fractions was measured at temperatures from 298.15 to 343.15 K and 101.305 kPa. The refractive index of DES1 and DES2 with water over the whole mole fractions was measured at 298.15 K and 101.305 kPa. From the measured density data, the excess molar volume (VmE), apparent molar volume (Vφi), partial molar volume ((V¯l)), excess partial molar volume (V¯iE), and coefficient of isobaric expansion (αP) at constant pressure were calculated. The formation of DES was confirmed through FTIR spectra, 1H NMR spectra, and 13C spectra. Further, the thermal stability and thermal behavior of all the prepared DESs were determined using TG-DSC thermogram.
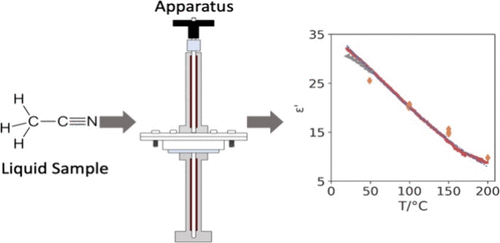
Novel Reactor for In Situ Dielectric Constant Measurements of Fluids at High Temperature and Pressure
San Lin Htun - and
Jillian L. Goldfarb *
The dielectric constant measures the influence of an electric field on a material. As it probes solute–solvent interactions within a fluid, it could shed light on reactions such as the hydrothermal conversion of biomass to biofuels. However, such measurements are difficult to obtain due to the low temperature and electrical conductivity operating limits of conventional dielectric constant apparati. An apparatus was designed to measure the dielectric constant of fluids up to 220 °C consisting of a cylindrical resonant cavity that operates in TM010 mode at 1 GHz. The liquid sample is contained in a bespoke quartz tube reactor with an internal thermocouple and a pressure transducer. The sample temperature is manipulated with a dual zone heating unit. The system was validated with pure subcritical water between 20 to 220 °C and was employed to in situ detect changes in the dielectric constant of five organic solvents up to 200 °C, beyond values currently available in the literature. The maximum deviation from reference data is approximately 5% for water. The maximum deviation extends to 20% with less polar organic solvents when compared to values using single-Debye models in high-temperature regions.
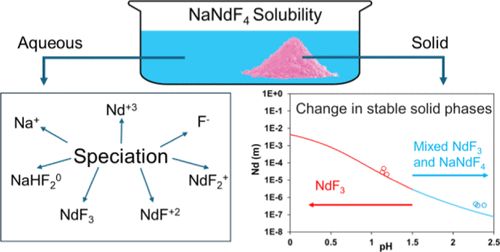
Nd–Na–F– H2O System: Solubility Measurements at 23 °C and Thermodynamic Modeling
Paul Antonick - ,
Jiangping Liu - ,
Gaurav Das *- ,
Malgorzata M. Lencka - ,
Richard E. Riman - ,
Alexandra Navrotsky - , and
Andrzej Anderko
A thermodynamic model has been developed for calculating solubilities and speciation in aqueous Nd–Na–F–H2O systems. The model combines experimental data from the literature for NdF3 and NaF in aqueous environments with new measurements of the solubility of both NdF3 and NaNdF4 as a function of pH at room temperature. For NaNdF4, the solubility data complement the recently reported solid state property data, thus establishing a comprehensive database for this hitherto poorly characterized compound. The solubilities of NdF3 and NaNdF4 have been measured through the dissolution method in HCl (0.1, 0.01 mol·kg–1 H2O) solutions at target pH of ∼1 and ∼2 over an equilibration period of up to 12 weeks at room temperature. The composition of the solution phase was determined through Inductively Coupled Plasma–Optical Emission Spectrometry. X-ray diffraction was performed to determine the remaining equilibrated solid phases. For modeling, the Mixed-Solvent Electrolyte framework has been used, which was previously parametrized and validated for various rare earth systems. The model combines a comprehensive speciation treatment in the aqueous phase with solid-state thermodynamics to reproduce the solubility of solids at varying processes conditions, thus providing a foundation for designing novel processes.

Elucidating the Solvation Dynamics of Polyethylene Glycols 200 and 400 in the Presence of Disodium EDTA: A Physicochemical and Spectroscopic Approach
Manisha Lamba - ,
Nabaparna Chakraborty - , and
Kailash Chandra Juglan *
An effective and nondestructive method for examining the complex molecular interactions and structural alterations taking place in liquid systems involves the use of ultrasonic and volumetric measurements. Ultrasonic techniques were used in this investigation because of their remarkable sensitivity to intermolecular forces, solute–solvent interactions, and microstructural changes. This study examines the physicochemical properties, including density (ρ) and sound speed (c), for various compositions of Polyethylene Glycols (PEG 200/400) in water and mixed aqueous disodium ethylenediaminetetraacetic acid (EDTA) solvent media across four temperatures ranging from 288.15 to 318.15 K under ambient pressure (0.1 MPa). The collected empirical data were used to calculate various volumetric and acoustic parameters, including apparent molar parameters (Vϕ, Kϕ,s), partial molar parameters (Vϕ0, Kϕ,s0), transfer parameters (Vϕ,tr0, Kϕ,s,tr0), isobaric thermal expansion (α), and pair and triplet interaction coefficients (VAB, KAB, VABB, KABB). The temperature derivative (∂Vϕ0∂T) was used to explore the solute’s structure-making ability. The quality of taste was determined from the apparent specific volume (vϕ) at (288.15 to 318.15) K. The cosphere overlap model was applied to understand the intermolecular interactions within the mixture. FT-IR spectra of the aqueous solution were recorded with spectral analysis indicating strong intermolecular interactions.
Density, Speed of Sound, and Refractive Index of the Binary Solvent System n-Propanol + Monoethanolamine and Their Intermolecular Interactions
Jingxiao Zhang - ,
Mengru Xie - ,
Ye Wang - ,
Xuanyi Ni - ,
Yongqi Su - ,
Tongtong Yang - ,
Zixin Qi - , and
Lilei Zhang *
n-Propanol and monoethanolamine are widely used organic solvents and reaction media in the chemical industry. This work reports rigorous thermodynamic data, including density, speed of sound, and refractive index for the binary n-propanol + monoethanolamine system at 0.1 MPa and temperatures ranging from 293.15 to 318.15 K. Excess properties, such as excess molar volume, excess speed of sound, excess isentropic compressibility, and excess refractive index, were calculated to elucidate the system’s thermodynamic behavior and the nature of intermolecular interactions. Molecular dynamics simulations and quantum mechanical calculations combined with 1H NMR and IR spectra reveal that complex hydrogen bonding between the hydroxyl group of n-propanol and the amino/hydroxyl groups of monoethanolamine, coupled with van der Waals forces, drives significant nonideal behavior in the mixture. This study provides reliable thermodynamic data and theoretical insights for optimizing solvent formulations, chemical processes, and design strategies involving n-propanol and monoethanolamine, thereby facilitating technological advancements in related fields.
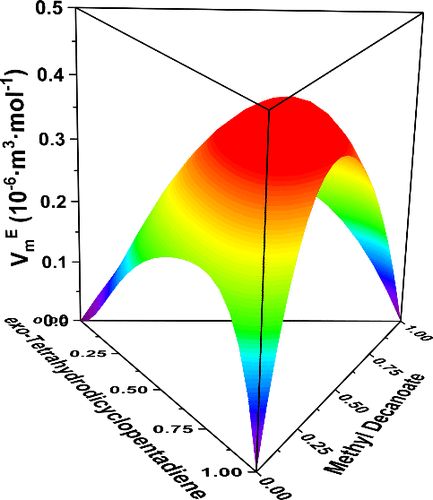
Densities and Viscosities for the Ternary Mixtures of exo-Tetrahydrodicyclopentadiene (1) + Methyl Decanoate (2) + 1-Pentanol (3) and Corresponding Binaries at T = (293.15 to 333.15) K and p = 0.1 MPa
Yunfei Wei - ,
Pengfei Jiang - ,
Ji Mi - ,
Jingdi Zhu - ,
Yitong Dai *- ,
Yongsheng Guo - , and
Wenjun Fang *
It has been proven that biofuels could enhance the performance of endothermic hydrocarbon fuels (EHFs) such as reducing harmful emissions and adjusting physical properties. In order to understand the properties of EHFs containing biofuels, the densities (ρ) and viscosities (η) of the ternary system exo-tetrahydrodicyclopentadiene (1) + methyl decanoate (2) + 1-pentanol (3) and the corresponding binary systems were investigated at T = (293.15 to 333.15) K and p = 0.1 kPa. The excess molar volume (VmE) and viscosity deviation (Δη) of the binary mixtures were calculated and fitted into the Redlich–Kister equation. The VmE and Δη of the ternary system were calculated and fitted to four different semiempirical equations (Singh, Cibulka, Nagata–Tamura, and Redlich–Kister). The results show that all VmE values of binary systems and ternary system are positive while all Δη values are negative. This work can provide reliability data for the compatibility of biofuels and EHFs.
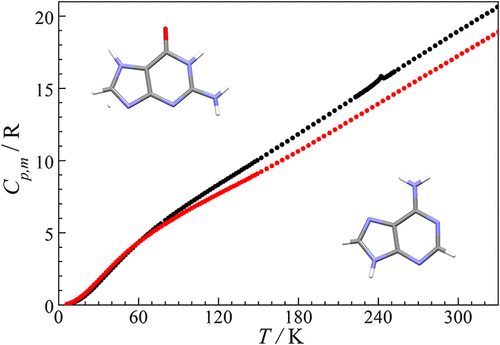
Low-Temperature Thermodynamic Properties of Purine Bases: Adenine and Guanine
Michael A. Bespyatov *
The work presents precision data on the heat capacity of adenine (C5H5N5; IUPAC name: 9H-purin-6-amine; CAS Number: 73–24–5) and guanine (C5H5N5O; IUPAC name: 2-amino-1,9-dihydro-6H-purin-6-one; CAS Number: 73–40–5) in the range 6–330 K obtained by adiabatic calorimetry. The data were used to calculate the thermodynamic functions (entropy, enthalpy increment, and Gibbs reduced energy) between 0 and 330 K. For the first time, a λ-type anomaly was revealed in the functional behavior of the heat capacity of guanine in the range of 225–250 K, which indicates the presence of a second-order phase transition in this temperature range. The anomalous component was segregated from the experimental data. Approximation of the anomalous component was done to find its thermodynamic parameters (entropy and enthalpy increment) and the critical temperature of phase transition: Ttr = 242.7 ± 0.1 K, ΔSan = 0.042 ± 0.004 J·mol–1K–1, ΔHan = 10.2 ± 1.0 J·mol–1.
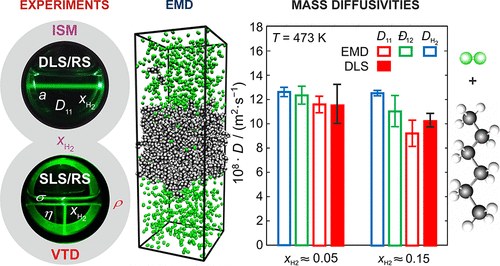
Thermophysical Properties of n-Hexane under the Influence of Dissolved Hydrogen by Experiments and Equilibrium Molecular Dynamics Simulations
Paul Damp - ,
Yongzhen Sun - ,
Chathura J. Kankanamge - ,
Julius H. Jander - ,
Michael H. Rausch - ,
Tobias Klein - ,
Thomas M. Koller *- , and
Andreas P. Fröba
This study demonstrates the applicability of different experimental techniques in combination with equilibrium molecular dynamics (EMD) simulations for investigating the influence of hydrogen (H2) on various thermophysical properties of n-hexane at temperatures T from (298 to 473) K and pressures p from (0.1 to 20) MPa at or close to vapor-liquid equilibrium. The amount fraction of H2 in the liquid phase, xH2, and the liquid density ρ are determined via the isochoric saturation method and vibrating-tube densimetry. Polarization-difference Raman spectroscopy (PDRS) serves to characterize xH2 during dynamic light scattering (DLS) experiments allowing access to the thermal diffusivity a and the Fick diffusion coefficient D11 in the liquid phase as well as surface light scattering (SLS) experiments used to determine the liquid dynamic viscosity η and the vapor-liquid interfacial tension σ. With increasing p and xH2, the measurement results for a and D11 are not significantly affected, except for states approaching the critical point. The experimental data for ρ, η, and σ show decreasing values with increasing xH2. The EMD simulations predict the influence of H2 on D11, η, σ, and ρ well, and allow to discuss the behavior of D11 and σ in connection with the fluid structure.
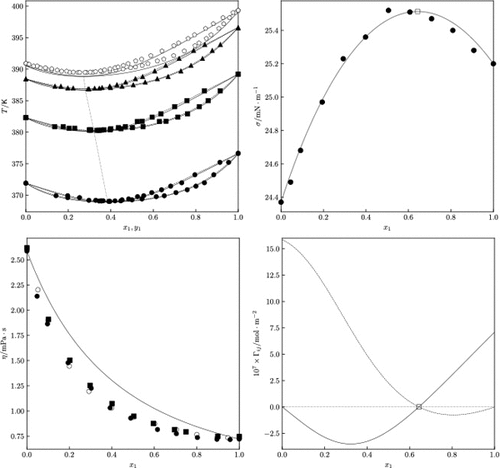
Experimental Determination and Theoretical Modeling of Diethyl Carbonate +1-Butanol Binary Mixture
Marcela Cartes - ,
Melissa Morales - ,
Juan M. Uceda - , and
Andrés Mejía *
This work reports the vapor–liquid equilibria (VLE), liquid viscosity, and surface tension of a potential biofuel mixture formed by diethyl carbonate and 1-butanol. VLE measurements are performed at 50.00, 75.00, and 94.00 kPa, revealing a minimum-boiling azeotrope. The measured VLE data passed the point–point Fredenlund test and are well-correlated using the ϕ−γ approach, where the Wilson model displays the most accurate model for the liquid phase. This model is also incorporated into the Peng–Robinson Stryjek-Vera EoS using the MHV mixing rule to predict VLE data and extend their application over a broad pressure range. The liquid dynamic viscosity is measured at 298.15 K, exhibiting a negative deviation from linear behavior, decreasing as the mole fraction of diethyl carbonate increases. This behavior is predicted by the proposed extension of the scaling viscosity theory based on the EoSs used. The surface tension at 298.15 K showed a positive deviation and exhibited a maximum anisotropy point. The latter results are successfully modeled using the Chunxi model coupled to the reported Wilson model, allowing for the calculation of the relative Gibbs adsorption. The findings provide valuable information to advance the validation of this mixture as a potential biofuel or bio-oxygenate additive for fuels.

Liquid Viscosity and Interfacial Tension of Binary Mixtures of n-Pentane, n-Decane, n-Hexadecane, or Squalane with Dissolved Carbon Dioxide or Propane by Surface Light Scattering
Fabian Lorig - ,
Ziwen Zhai - ,
Chathura J. Kankanamge - ,
Pedro M. Gonzalez - ,
Markus Richter - ,
Thomas M. Koller - ,
Tobias Klein *- , and
Andreas P. Fröba
The present study investigates the influence of dissolved gases on the liquid viscosity ηL and interfacial tension σ of linear and branched alkanes under saturated conditions by using surface light scattering (SLS). Five binary mixtures consisting of n-pentane, n-decane, n-hexadecane, or 2,6,10,15,19,23-hexamethyltetracosane (squalane) with dissolved propane or carbon dioxide are studied at temperatures T between 255.7 and 423.15 K, pressures p up to 7.8 MPa, and solute amount fractions up to 0.79. Using SLS, ηL and σ were determined with an average expanded experimental uncertainty (coverage factor k = 2) of 2.0%. Polarization-difference Raman spectroscopy was simultaneously applied to SLS experiments to determine the liquid-phase composition. The influence of the dissolved gas is discussed by comparing the thermophysical properties of the mixtures to those of the pure solvents. It could be observed that the molecular characteristics of the solute have a minimal influence on ηL of the mixture, which is primarily determined by the solvent’s characteristics. In contrast, the molecular characteristics of the solvent and solute strongly influence σ. Overall, the results of this study contribute to expanding the database for ηL and σ for binary mixtures, which can be considered surrogate mixtures for refrigeration oil–refrigerant systems.

Refractive Index and Related Properties for the Binary Mixture (1,4-Dioxane + Chloroform) at T = (295.15, 298.15, 301.15, 304.15, 307.15, 310.15, and 313.15) K
Taoufik Kouissi *- ,
Adel Toumi - , and
Moncef Bouanz
For the binary mixture (1,4-dioxane (1) + chloroform (2)), the refractive index (nD) was measured at temperatures T = (295.15, 298.15, 301.15, 304.15, 307.15, 310.15, and 313.15) K and at atmospheric pressure over the whole composition range. The molar refraction (Rm), reduced molar free volumes (Vm/Rm), molecular radius (r), internal pressure(Pint), and their excess properties were calculated from experimental data. These values were then analyzed to determine the type and nature of specific intermolecular interactions among the components. The refractive indices of the binary mixture were calculated using nine different mixing rules (Gladstone–Dale, Arago–Biot, Weiner, Heller, Lorentz–Lorentz, Eykmen, Eyring–John, Oster, and Newton), and the results were compared with experimental measurements at each temperature. The nature of the molecular interactions was explored by analyzing the thermodynamic properties, specifically by fitting the excess parameters to the Redlich–Kister polynomial equation to derive the corresponding coefficients and standard errors.
Vapor-Liquid Equilibria and Supercritical Fluid Equilibria
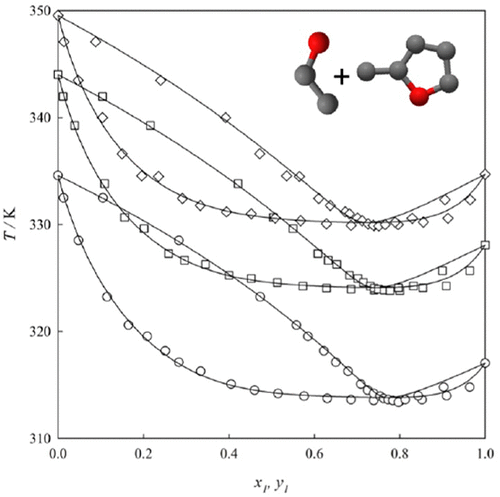
Experimental Determination of Selected Thermophysical Properties of the 2-Methylfuran + Ethanol Binary Mixture
Ariel Hernandez - ,
Melissa Morales - ,
Marcela Cartes - , and
Andrés Mejía *
Vapor–liquid equilibrium (VLE), liquid mass density, liquid dynamic viscosity, and surface tension have been measured over the entire mole fraction range for a new potential biofuel formed by a 2-methylfuran + ethanol binary mixture. The VLEs are measured at 50.01, 75.01, and 94.01 kPa, and over a temperature range of 313 to 350 K. The other mixture properties have been measured at 298.15 K and 101.33 kPa. According to the VLE results, this binary mixture displays a minimum-boiling azeotropy under the explored conditions, and the data are well-correlated by classical activity coefficient models, with the Wilson model showing lower deviations. The liquid mass density increases with the mole fraction of 2-methylfuran, and the sign of the excess volumes changes from negative to positive as the concentration of 2-methylfuran increases. This behavior is correlated to two parameters of the Redlich–Kister expansion. The liquid dynamic viscosity decreases as the mole fraction of 2-methylfuran increases, and this relationship is correlated by two parameters using the Redlich–Kister expansion. The surface tension exhibits unusual behavior, where it increases as the mixture becomes more volatile, showing a positive deviation from linear behavior. The surface tension determinations are well-correlated using the four-parameter Myers–Scott polynomial.
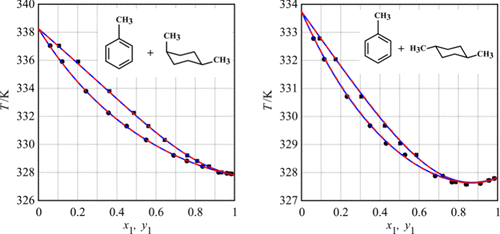
Experimental Determination of Isobaric Vapor–Liquid Equilibria for the Binary Systems Containing Toluene and Isomers of 1,4-Dimethylcyclohexane at 15, 25, and 40 kPa
Tomáš Sommer *- ,
Vojtěch Strnad - ,
Martin Zapletal - ,
Jana Vostrá - ,
Jiří Zbytovský - , and
Jiří Trejbal

This publication is Open Access under the license indicated. Learn More
Toluene is a valuable chemical used in various industrial applications, making its purification a critical process. Removing aliphatic hydrocarbons, such as dimethylcyclohexanes, is one of the main challenges in producing high-purity toluene; however, the thermodynamic properties of mixtures containing dimethylcyclohexanes and toluene remain largely undocumented in the literature. Therefore, this work focuses on studying the equilibrium behavior of two of its isomers in mixtures with toluene. The vapor–liquid equilibria of binary mixtures of cis- and trans-1,4-dimethylcyclohexane with toluene were measured at 15, 25, and 40 kPa across the full range of molar fractions. The thermodynamic consistency of the obtained data was verified using the Van Ness method modified by Fredenslund, as well as the Van Ness point-to-point test and the Herington test. The behavior of the systems was described by using the NRTL and UNIQUAC models. For the mixture of toluene with trans-1,4-dimethylcyclohexane, experimental data and both models confirm the existence of a minimum-boiling azeotrope; however, the mixture of toluene with cis-1,4-dimethylcyclohexane does not exhibit azeotropic behavior. The resulting models will be helpful in process simulations, which is crucial in the toluene production process.
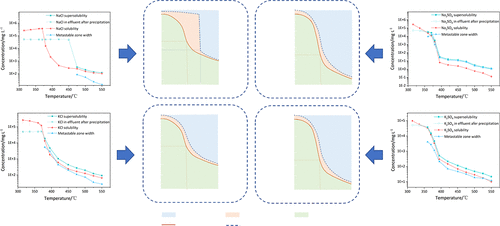
Dissolution Characteristics of Inorganic Salts in Sub/supercritical Water: Type 1 and Type 2 Salts
Yishu Zhang *- ,
Dong Han - ,
Yuanwang Duan - , and
Shuzhong Wang *
Supercritical water’s unique properties enable organic decomposition and inorganic synthesis, but salt precipitation risks clogging. This study investigates the solubility, supersolubility, and metastable zone width (MSZW) of type 1 and type 2 salts using salt bed dissolution (cooling) and precipitation (warming) methods. At supercritical conditions (25 ± 0.1 MPa), type 1 salts show a moderate solubility decline (1–2 orders of magnitude, to ∼10–1 to 10° mmol·kg–1), while Type 2 salts exhibit a drastic drop (3–4 orders, to ∼10–3 to 10–2 mmol·kg–1). Type 1a salts display a wide metastable zone between the critical point and three-phase equilibrium, whereas type 1b salts show stable MSZW. Type 2a and 2b salts exhibit minimal dissolution differences. Most salts (except type 1a) have a wider MSZW under transcritical conditions but are narrower under supercritical conditions. These findings clarify salt-specific precipitation behaviors, aiding in clogging prediction and mitigation strategies for supercritical water processes.

Biobased Solvents as Entrainers for Extractive Distillation in Isobutyl Acetate–Isobutanol Separation: Experimental Investigation, Toxicity Evaluation, and Mechanism Analysis
Jun Li *- ,
Renting Li - ,
Jing Yang - ,
Sen Li - ,
Zhenyu Zhang - ,
Haigang Liu - ,
Zhanhua Ma - , and
Lanyi Sun *
In the industrial synthesis of isobutyl acetate (IbAC) from isobutyl alcohol (IbOH) and acetic acid, excess IbOH forms an azeotrope with IbAC. Extractive distillation is widely used for azeotrope separation, yet conventional solvents are often toxic and volatile. Biobased solvents, being biodegradable and ecofriendly, offer a promising alternative. Screening with the conductor-like screening model for segment activity coefficient identified cinene and α-pinene as effective entrainers to break the IbOH–IbAC azeotrope. Ternary vapor–liquid equilibrium experiments confirmed that both solvents, when added at 20 mol %, eliminated the azeotrope. The nonrandom two-liquid model accurately correlated the experimental data. However, α-pinene formed a new azeotrope with IbOH at an atmospheric pressure. Toxicity assessment showed cinene has lower mammalian toxicity but higher aquatic toxicity than those of conventional solvents. Quantum chemical calculations revealed that stronger van der Waals interactions between cinene and IbAC reduce the activity coefficient of IbAC, facilitating separation.
Liquid-Liquid Equilibria and Vapor-Liquid-Liquid Equilibria
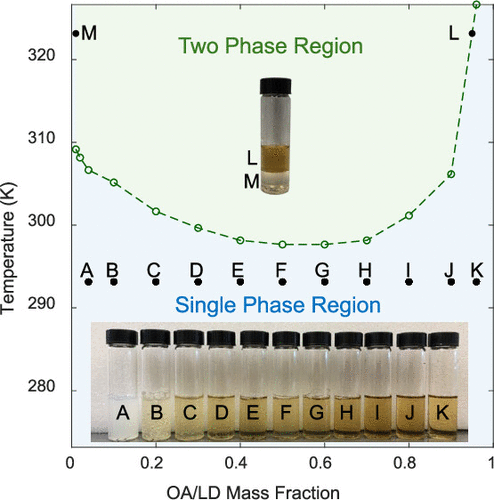
Lower Critical Solution Temperature Phase Behavior and Water Activity of a Ternary Mixture of Oleic Acid, Lidocaine, and Water
Jordan D. Kocher - ,
Ahmed Mahfouz - ,
Hunter T. Bell - ,
Joshua M. Rinehart - , and
Akanksha K. Menon *
This publication is Open Access under the license indicated. Learn More
Mixtures that possess a lower critical solution temperature (LCST) phase behavior form a homogeneous single phase at temperatures below the LCST and separate into two liquid phases (water-rich, WR, and water-scarce, WS) above the LCST. This unique thermoresponsive phase behavior can be leveraged in various thermodynamic cycles, which are used for applications such as desalination and dehumidification. In addition to their phase diagram, the performance of aqueous LCST mixtures is dictated by their water activity (i.e., the chemical potential of water in the mixture). Recently, a ternary mixture of oleic acid (OA), lidocaine (LD), and water was shown to possess an LCST of ∼298.15 K, but the phase diagram over the full range of concentrations and water activity has not been reported. In this work, we experimentally characterize the phase diagram (liquid–liquid equilibrium, LLE), water activity (vapor–liquid equilibrium, VLE), chemical potential of water, and osmotic pressure of OA/LD/H2O under conditions that are relevant to the aforementioned applications. Our results suggest that OA/LD/H2O can outperform other LCST mixtures (e.g., ionic liquids) given its broad phase diagram, low LCST, and purity of the two phases after separation.
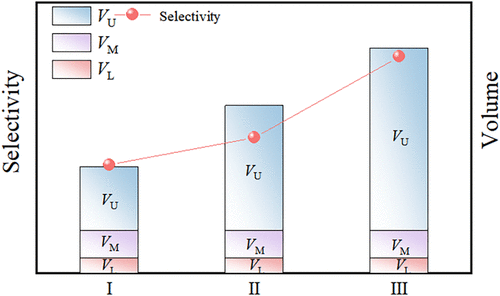
Multistage Fractionation of Polycyclic Aromatic Hydrocarbon Isomers Using a Deep Eutectic Solvent-Based Aqueous Three-Phase System
Jinwan Xu - ,
Jinhuan Fu - ,
Hao Xin - ,
Shaoyan Wang - , and
Xiuhong Wu *
This study demonstrates an innovative multiple-stage separation strategy for anthracene and phenanthrene by using a deep eutectic solvent (DES)-based aqueous three-phase system (ATrPS). The system, composed of choline chloride–PEG400–glycerol DES (1:1:1 molar ratio) and K2HPO4, achieved good separation efficiency through three key mechanisms: DES-mediated polarity adjustment of phase environments, salt-induced phase boundary modification, and multistage fractionation amplification. The optimal system (70.0 wt % DES + 17.5 wt % K2HPO4) showed superior performance with an initial selectivity of 1.21 ± 0.05, which increased to 2.44 ± 0.05 after three extraction stages, representing a 2.22-fold enhancement over aqueous two-phase systems composed of 55.0 wt % DES and 17.5 wt % K2HPO4. The DES’s tunable physicochemical properties, coupled with K2HPO4’s salting-out effect, created an ideal environment for isomer discrimination. This green separation platform offers significant advantages for challenging separations in pharmaceutical and petrochemical applications.
Phase Behavior of Green Aqueous Biphasic Systems Based on Ethyl Lactate and Choline Salts
Marion Engole - ,
Kridsada Aunnankat - ,
Patricia Thornley - ,
Worapon Kiatkittipong - ,
Prakorn Ramakul - ,
Robert Evans - , and
Vesna Najdanovic-Visak *
This publication is Open Access under the license indicated. Learn More
Aqueous biphasic systems (ABS) have recently emerged as an economic and sustainable solution for the separation and isolation of biomolecules. Ethyl lactate (EL) is an attractive phase-forming component, as it is a biorenewable, biodegradable, and nontoxic solvent. In this study, cloud points and tie-line data for ethyl lactate (EL)-based aqueous biphasic systems (ABS) with four choline salts─choline bicarbonate (ChHCO3), choline chloride (ChCl), choline bitartrate (ChBitar), and choline dihydrogen citrate (ChH2Cit)─were experimentally determined at 298.2 and 328.2 K. For both temperatures, three models were used to fit the data: the three-parameter Merchuk’s equation, a two-parameter correlation, and the effective excluded volume. The molecular-level interactions and dynamic behavior within the ABS systems were investigated using diffusion nuclear magnetic resonance. EL–ChH2Cit showed the most significant changes in diffusion coefficients and water shifts, indicating increased viscosity and altered water structuring. In contrast, ChCl effects were primarily viscosity-driven, ChBitar exhibited complex, nonlinear trends suggestive of solvation or aggregation phenomena, whereas ChHCO3 uniquely displayed peak splitting, pointing to multiple EL environments. This work provides novel insights into the design of green solvent systems and contributes to the development of alternatives to hazardous organic solvents, with potential applications across biotechnology, pharmaceuticals, and the green chemistry industries.
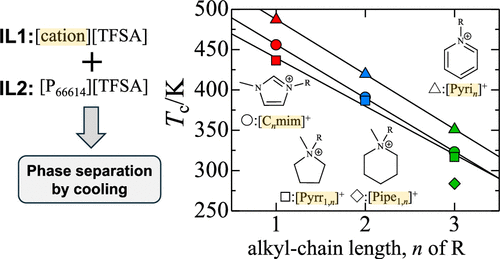
Phase Behavior of Imidazolium-, Pyrrolidinium-, Pyridinium-, and Piperidinium-Based Ionic Liquid + Phosphonium-Based Ionic Liquid Binary Mixtures with a Common Anion
Takuya Shimomura *- ,
Taiga Murayama - , and
Masaki Kurebayashi
Phase separation temperatures of mixtures of four different ionic liquids (ILs) with trihexyltetradecylphosphonium bis(trifluoromethanesulfonyl)amide ([P66614][TFSA]) have been measured in order to elucidate the effects of cation structures on the liquid–liquid phase behavior of IL + IL mixtures with a common anion. 1-Alkyl-3-methylimidazolium ([Cnmim]+), 1-methyl-1-alkylpyrrolidinium ([Pyrr1,n]+), 1-alkylpyridinium ([Pyrin]+), and 1-alkyl-1-methylpiperidinium ([Pipe1,n]+) (n = 1, 2, and 3) were used for four different ILs as cations. [TFSA]− was selected as a common anion. The IL + IL mixtures with [TFSA]− caused phase separation by cooling. The phase separation temperatures of the mixtures decreased with an increase in the alkyl-chain lengths n of cations. The upper critical solution temperatures Tc for the mixtures linearly decreased with increasing n. This suggests that the liquid–liquid phase behavior of the IL + IL mixtures with [TFSA]− is affected by the hydrophobicity of ILs. However, the Tc values for the mixtures are higher in the cation order of [Pyrin]+ > [Cnmim]+ > [Pyrr1,n]+ > [Pipe1,n]+. This tendency is inconsistent with the hydrophobicity of the ILs. These results suggest that the liquid–liquid phase behavior of IL + IL mixtures with a common anion is affected not only by the hydrophobicity of ILs but also by cation–anion interactions in the mixtures.
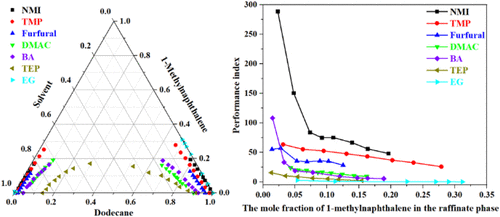
Experimental Determination and Correlation of Liquid–Liquid Equilibrium Data for Ternary Systems of Dodecane + 1-Methylnaphthalene + Solvent
Chong Yang *- ,
Yuyao Xie - ,
Shimin Xu - ,
Yuwei Luo - ,
Huaiyu Zhu - , and
Hui Wang
Liquid–liquid equilibrium (LLE) data for the ternary system of dodecane + 1-methylnaphthalene + solvent were experimentally determined at 298.2 K and 101.3 kPa using N-methylimidazole, trimethyl phosphate, furfural, N,N-dimethylacetamide, benzyl alcohol, triethyl phosphate, and ethylene glycol as extractants. Using the obtained LLE data, the distribution constant, separation factor, and performance index were calculated to evaluate the performance of these extractants in extracting and separating 1-methylnaphthalene from dodecane. The reliability of the LLE data was examined using the Hand and Othmer–Tobias equations, with the correlation coefficients of the fitted equations exceeding 0.96. Furthermore, the nonrandom two-liquid (NRTL) and universal quasichemical (UNIQUAC) thermodynamic models were used to correlate the LLE data and obtain binary interaction parameters. The root-mean-square deviations between the experimental and calculated results were 0.15–0.97 for NRTL and 0.13–1.05 for UNIQUAC. The derived binary interaction parameters were validated using GMcal_TieLinesLL, a specialized software package for LLE calculations. These findings highlight that both the NRTL and UNIQUAC thermodynamic models can accurately describe the liquid–liquid phase equilibrium behavior of ternary systems, providing a theoretical basis for the industrial application of catalytic cracking diesel extraction and aromatics removal technology.

Measurement and Correlation of Liquid–Liquid Equilibrium Data for Extracting Acetic Acid from Water Using a Mixed Solvent Containing n-Hexane + Isopropyl Acetate + Methyl Ethyl Ketone
Shiyue Li - ,
Xiaojing Hou *- ,
Yuchen He *- ,
Pengfei Xie - , and
Kejun Wu
The mixed-solvent extraction–distillation method is widely employed for industrial acetic acid (HAc) recovery, and the water content in the extract critically determines the energy required for downstream distillation. Consequently, obtaining reliable liquid–liquid equilibrium (LLE) data for a mixed-solvent/HAc/water system is essential to optimize process energy consumption. This study focused on determining comprehensive LLE data, assessing predictive modeling, and analyzing process performance. Experiments were carried out at 298.15 K and 101.33 kPa for two ternary systems, n-hexane (HEX) + isopropyl acetate (IPAC) + H2O and methyl ethyl ketone (MEK) + IPAC + H2O, and for a HEX + MEK + HAc + IPAC + H2O quinary system over the temperature range of 298.15–318.15 K. Both NRTL and UNIQUAC thermodynamic models yielded RMSDs below 0.015 for all systems. Detailed analysis of the solvent composition, feed concentration, and phase ratio effects showed significant impacts on the water content in the organic phase and extraction efficiency. Further Aspen simulations validated the accuracy of NRTL model parameters with all deviations in the predicted water content and extraction efficiency limited to 0.0073 and 8.84%, respectively. These results provide accurate LLE data and actionable guidelines for optimizing HAc recovery processes in the industry.
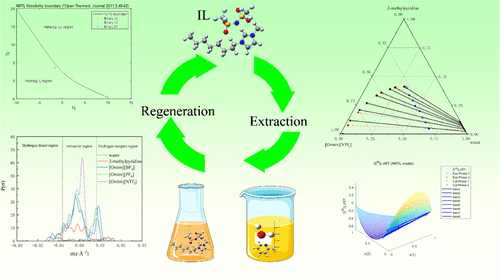
Liquid–Liquid Equilibrium Behavior for Ternary Mixtures Water and 2-Methylpyridine with Hydrophobic Imidazolium-Based Ionic Liquids
Jun Gao - ,
Ying Qin - ,
Dongmei Xu *- ,
Lianzheng Zhang - ,
Yixin Ma - , and
Yinglong Wang
To recover 2-methylpyridine from aqueous solution using an extraction process, three hydrophobic imidazolium-based ionic liquids (ILs), 1-octyl-3-methylimidazolium tetrafluoroborate ([Omim][BF4]), 1-octyl-3-methylimidazolium hexafluorophosphate ([Omim][PF6]), and 1-octyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([Omim][NTf2]), were selected as the extractants. The liquid–liquid equilibrium (LLE) investigation was conducted for ternary mixtures (water + 2-methylpyridine + hydrophobic imidazolium-based ionic liquids) at a temperature of 298.15 K and pressure of 101.3 kPa. Distribution coefficient and selectivity computed with the ascertained LLE tie-line data were employed to evaluate the separation performance of the ILs. The results indicated that [Omim][NTf2] exhibited superior extraction efficiency followed by [Omim][PF6] and [Omim][BF4]. The tie-line data was fitted by the NRTL model with the most root-mean-square deviation value of 0.9995%. Additionally, the intermolecular interactions between the ILs and 2-methylpyridine were explored by quantum chemical calculations. The findings demonstrate that the hydrophobic ILs exhibit significant potential as sustainable extractants in separating 2-methylpyridine from an aqueous solution.

Evaluation of Four Inexpensive Extractants for Pyridine Separation from n-Heptane at 303.2 K: Liquid–Liquid Equilibrium and Thermodynamic Modeling
Jingli Sun - ,
Xuqiang Li - ,
Lu Chen - ,
Yujie Zhen - ,
Siyu Zheng - ,
Shaolong Dong - , and
Yingmin Yu *
In order to reduce the impact of nitrogen-containing compounds in oil products on the environment, this paper selected n-heptane as the oil product model and pyridine as the nitrogen-containing compound. Four inexpensive extractants (furfuryl alcohol, 1,4-butanediol, N,N-dimethylacetamide, and dimethyl sulfoxide) were selected as the extractants to separate pyridine from the oil model at 303.2 K, and the liquid–liquid equilibrium (LLE) data were measured. The partition coefficients (D) and separation coefficients (S) of the extractants were calculated and used to evaluate the extractants. Based on the LLE data, correlations were performed using NRTL and UNIQUAC models, and the interaction parameters of the corresponding optimization models were obtained. The root-mean-square deviation (RMSD) values of the NRTL and UNIQUAC models were 0.98% and 1.09%, respectively. The GUI-MATLAB tool was selected to evaluate the accuracy of the binary interaction parameter regression by a Gibbs energy topology analysis. To probe the underlying mechanism of the extraction process between the extractants and pyridine, the σ-profile was used to analyze the interaction patterns between the extractants and pyridine. Given their high extraction efficiency, the studied extractants can be considered as potential extractants for extractive denitrification processes.
Solid-Solid Equilibria and Solid-Fluid Equilibria
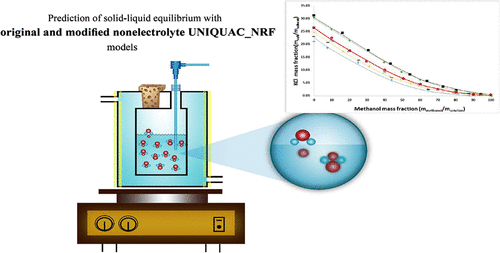
Solubility of KCl, NaCl, Na2SO4, and K2SO4 in Water–Methanol: Experimental and Modeling
Ali Hosseini Eghbal - and
Seyed Hossein Mazloumi *
To measure the solubility of KCl, NaCl, Na2SO4, and K2SO4 through a methodical-analytical approach, a simple and accurate apparatus is applied. The solubility of these salt types is measured in H2O and CH3OH at 278.15 and 288.15 K. The nonelectrolyte UNIQUAC-NRF and modified nonelectrolyte UNIQUAC-NRF models are developed to correlate the salt solubility data based on the excess Gibbs free energy. In these models, the composition of short-range interaction and long-range contribution is represented through the local composition model and a Pitzer–Debye–Hückel equation, respectively. Each one of these two models has two binary adjustable parameters, which are obtained by fitting the experimental solubility data of the water–methanol–salt mixtures at 278.15 and 288.15 K. The adjustment of the parameters is essential in predicting the solubility of the salt types for H2O–CH3OH–salt mixtures at electrolyte concentrations up to the saturation point and temperatures within 283.15 to 328.15 K. The findings here indicate that these two models can accurately describe salt solubility in water–methanol–salt mixtures, only through the binary adjustable parameters.
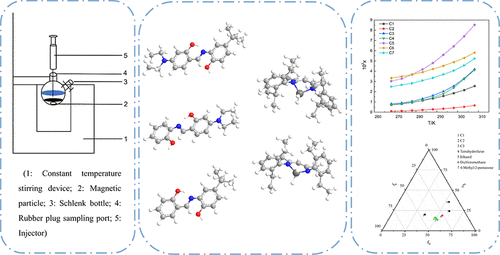
Solid–Liquid Phase Equilibrium Characteristics and Thermodynamic Analysis of Asymmetric Schiff Base Ligands and Their Organic Phenol-Aluminum Compounds
Qifeng Li - ,
Xiaoli Ma *- ,
Wenliang Yan - ,
Ziyuan Pang - ,
Congjian Ni - ,
Yiwen Chen - , and
Zhi Yang *
In this work, a static analysis method was used to measure the solubility of nine compounds: C1 and C2 in three solvents, C3 in four solvents, and C4–C7 in four pure solvents, with temperatures ranging from 268.15 to 328.15 K (measurements in p-xylene were performed over the temperature interval of 288.15 to 328.15 K, whereas for other systems, the experimental temperature range was maintained between 268.15 and 308.15 K). Experimental results demonstrate enhanced solubility for all compounds at selected temperatures. Solubility data were correlated using seven thermodynamic models: Yaws, polynomial, van’t Hoff, λh, Wilson, NRTL, and UNIQUAC, yielding superior fits characterized by an average absolute relative deviation (ARD) below 5% and root-mean-square deviation (RMSD) under 0.2%. Among these, the polynomial model exhibited optimal performance for empirical correlations, whereas the NRTL model provided the best fit among activity coefficient models. Solvent-dependent solubility variations were further elucidated through Hansen solubility parameters. Thermodynamic calculations confirmed dissolution to be endothermic and entropy-driven in most systems. The measured solubility and fusion enthalpy data establish fundamental references for optimizing homogeneous catalysis and crystallization processes of organic aluminum compounds.
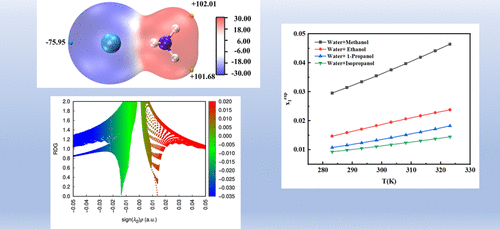
Ammonium Chloride Solubility Measurement, Molecular Simulation, and Correlation in Binary Solvents from 283.15 to 323.15 K
Mingting Yuan - ,
Sheng Liu - ,
Yimin Jia - ,
Yingchen Wang - ,
Jiaqi Luo - ,
Qiutong Zhang - ,
Wenhao Yan - , and
Qiushuo Yu *
The solubility of ammonium chloride in water + methanol/ethanol/1-propanol/isopropanol binary solvent systems was determined by the gravimetric method at atmospheric pressure. The temperature was varied from 283.15 to 323.15 K. Within this temperature range, solubility increased with temperature. The mole fraction solubility in the binary solvent systems followed this order: water + methanol > water + ethanol > water + 1-propanol > water + isopropanol. Molecular electrostatic potential analysis and reduced density gradient (RDG) plot analysis were used to study the intermolecular interactions. The solubility data were correlated using the modified Apelblat equation, the van’t Hoff equation, the λh equation, and the Jouyban–Acree model. Understanding these solubility behaviors is essential for designing crystallization and purification processes for ammonium chloride.
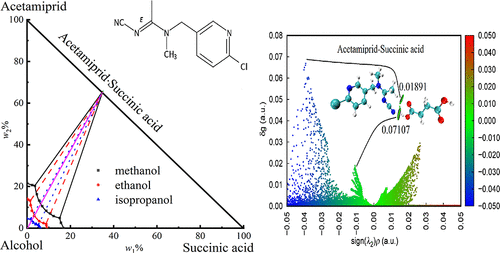
Cocrystal Phase Diagrams, Cocrystalline Region Discovery, and DFT Study for Acetamiprid + Succinic Acid/l-Tartaric Acid + Methanol/Ethanol/Isopropanol Systems
Shuai Huang - ,
Mingke Lei - ,
Yahui Xu - ,
Lijuan Guo - ,
Chuanzhong Yang - ,
Xinding Yao - , and
Hongkun Zhao *
Cocrystal phase diagrams are useful for making cocrystal preparations and determining the starting composition point for the cooling/solution cocrystallization process. Herein, we used the isothermal saturation technique to determine the mutual solubilities of acetamiprid (ACE) + succinic acid (SU) + methanol/ethanol/isopropanol and acetamiprid (ACE) + l-tartaric acid (l-Tar) + methanol/ethanol/isopropanol systems at 298.15 K. Six cocrystal phase diagrams were constructed through the Schreinemaker wet residue method, and the cocrystal crystalline region of ACE with SU/l-Tar (ACE·SU/l-Tar, 1:1, mole ratio) was identified. The stability level of the ACE·l-Tar cocrystal is higher compared to that of the ACE·SU cocrystal. The mutual solubilities along the crystalline curves of ACE·SU/l-Tar cocrystals were correlated using the solution complex technique. Relative standard error has a maximum value of 5.52%. The hydrogen bonding and vdW forces are viewed as positive weak interactions when creating ACE·SU/l-Tar cocrystals. This assessment is based on an independent gradient model based on Hirshfeld partitions (IGMH), a 2D fingerprint plot, DFT analysis, and examination of the Hirshfeld surface. The cocrystal phase diagrams and their modeling can provide a powerful approach to designing cocrystal screening and to formulating solutions with cocrystal components where crystallization does not occur.

Determination and Correlation of Solubility of Benzotrifuroxan in 12 Pure Solvents at Temperatures Ranging from 293.15 to 333.15 K
Sha Bai - ,
Yunlu Li *- ,
Shangbiao Feng - ,
Guanchao Lan - ,
Le Song - ,
Lizhen Chen - , and
Jianlong Wang *
Benzotrifuroxan (BTF), as a green hydrogen-free explosive, has a critical impact on its application performance due to its purity. However, there is a lack of systematic research on its solubility in the literature. This study used laser dynamics to systematically measure the solubility data of BTF in 12 pure solvents (methanol, ethanol, n-propanol, n-butanol, chloroform, carbon tetrachloride, polar solvents: acetonitrile, acetone, ethyl acetate, formic acid, acetic acid, and propionic acid) within the temperature range of 293.15–333.15 K for the first time. To establish a reliable solubility prediction model, the Apelblat equation, Yaws model, Van’t Hoff equation, NRTL model, and Wilson model were used to fit and analyze the experimental data. The results indicate that the Yaws equation has the highest goodness of fit (R2 > 0.99) and can accurately correlate the dependence of BTF solubility on temperature and solvent properties. This study not only fills the gap in the basic physical property data of BTF, but also provides a theoretical basis for solvent screening and process optimization in its industrial refining process, further promoting the application of BTF in the field of energetic materials.
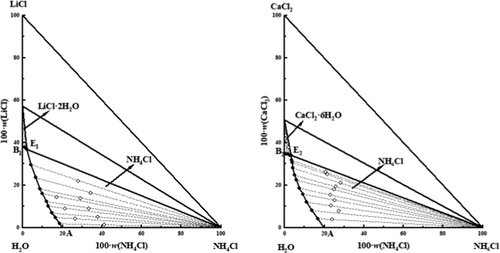
Solid–Liquid Phase Equilibria in the Ternary Systems NH4Cl–LiCl–H2O and NH4Cl–CaCl2–H2O at 258.15 K
Lin-Xuan Cui - ,
Shi-Hua Sang *- ,
Guang Tan - ,
Yu-Qiu Cen - , and
Ling-Xuan Wang
Based on the characteristics of oilfield waters in the Nanyi Mountain area of western Qaidam basin at subzero temperature, the solid–liquid phase equilibria of ternary systems NH4Cl–LiCl–H2O and NH4Cl–CaCl2–H2O at 258.15 K were investigated by using an isothermal solution equilibrium method. The phase diagrams of ternary systems NH4Cl–LiCl–H2O and NH4Cl–CaCl2–H2O at 258.15 K were plotted, respectively. The results show that the equilibrium phase diagram of the ternary system NH4Cl–LiCl–H2O at 258.15 K has one invariant point, two univariate curves, and two solid-phase crystallization zones (NH4Cl and LiCl·2H2O), belonging to hydrate type I. In the equilibrium phase diagram of the ternary system NH4Cl–CaCl2–H2O at 258.15 K, there are one invariant point, two univariate curves, and two solid-phase crystallization zones (NH4Cl and CaCl2·6H2O). In addition, the unreported Pitzer parameters were obtained based on the solubility data of the ternary systems. Using the Pitzer model, the solubilities of salts in the ternary systems NH4Cl–LiCl–H2O and NH4Cl–CaCl2–H2O at 258.15 K were modeled in detail. The modeling solubilities are in great agreement with the experimental results, indicating that the Pitzer model can be well applied to study the thermodynamic phase equilibria at subzero temperature.
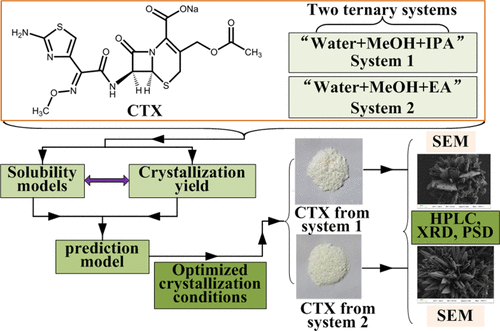
Solubility Modeling of Cefotaxime Sodium in Two Ternary Systems and Its Application in Optimizing Solvent Amount for the Crystallization Process
Xiaohua Jing *- ,
Tiancun Wang - ,
Cui Gan - ,
Yuhan Zhao - ,
Xiangyu Zhang - , and
Mengyao Li
In this study, the solubility of cefotaxime sodium (CTX) in two new ternary systems of “water+methanol(MeOH)+isopropanol(IPA)” and “water+MeOH+ethyl acetate(EA)” at various temperatures was determined and represented using the CNIBS/Redlich–Kister model. In the first ternary system, the mean relative deviation percentages (MRDs%) were 2.6, 6.6, 6.1, and 6.2% for the CTX solubility at temperatures of 278.2, 283.2, 288.2, and 293.2 K, respectively. The MRDs% were 5.9, 7.2, 7.8, and 8.9% for the CTX solubility in the second system. It was concluded that the model used adequately predicted the CTX solubility in two new ternary systems. In addition, the apparent thermodynamic properties of the dissolution process indicated that the CTX dissolution process is endothermic and enthalpy-driven. Furthermore, the solvent amounts for crystallization processes in two ternary systems were optimized based on the obtained solubility model, and the overall MRDs% between the experimental and predicted yields in two systems were 4.0 and 6.7% at 278.2 K. The solvent amounts of the crystallization process were effectively optimized based on the predictions of reliable solubility models. Finally, the solid powders of CTX from two optimized crystallization systems were analyzed by powder X-ray diffraction (XRD), particle size detector (PSD), and scanning electron microscopy (SEM).
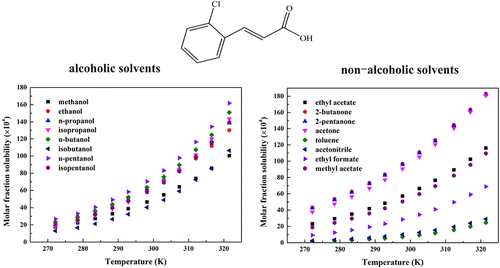
Determination of 2-Chlorocinnamic Acid Solubility in 16 Solvents: Model Correlation, Molecular Simulation and Solvent Effects Analysis
Bin Ou - ,
Xiaofang Li - ,
Lemei Huang - ,
Yujiang Ke - ,
Chaohui Che - ,
Xiaobing Liu - ,
Yajun Li - , and
Kui Wu *
The solubility of 2-chlorocinnamic acid in 16 pure organic solvents, including alcohols, esters, ketones, toluene, and acetonitrile, was determined by the gravimetric method at 272.15–321.55 K under 101.3 kPa. Results showed that solubility of 2-chlorocinnamic acid increased with temperature in all solvents. To correlate the experimental data, five thermodynamic models, i.e., the modified Apelblat, Buchowski–Ksiazaczak λh, NRTL, Wilson, and Yaws equations, were employed. Model accuracy was evaluated using average relative deviation (ARD) and root-mean-square deviation (RMSD), and all models provided satisfactory correlations. In addition, electrostatic potential energy surface analysis was carried out to preliminarily assess possible solute–solvent interactions, while density functional theory (DFT) calculations were used to further examine molecular interactions during dissolution. The Kamlet–Abboud–Taft linear solvation energy relationship (KAT-LSER) model was applied to analyze solvent effects. Thermodynamic functions, including enthalpy, entropy, and Gibbs free energy of mixing, were calculated using the Wilson model. The results revealed that the dissolution of 2-chlorocinnamic acid in the studied solvents is an endothermic, entropy-driven, and spontaneous process, indicating favorable solute–solvent interactions and enhanced solubility at higher temperatures.
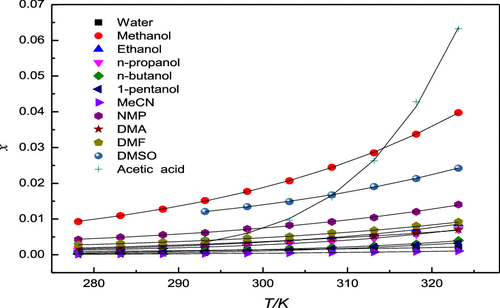
Solid–Liquid Phase Equilibrium Solubility and Model Correlation of Difenidol Hydrochloride in 12 Neat Solvents from (278.15 to 323.15) K
Yu Bi - ,
Xiao chen Ye - ,
Jingwen Zhang - ,
Ziyin Wang - ,
Zenan Gong - ,
Yanmin Shen *- , and
Wenju Liu *
In the study, the experiment solubility of difenidol hydrochloride was determined in 12 neat solvents [water, methanol, ethanol, n-propanol, n-butanol, 1-pentanol, N-methyl-2-pyrrolidone(NMP), N,N-dimethylformamide(DMF), N,N-dimethylacetamide (DMA), dimethyl sulfoxide (DMSO), acetonitrile (MeCN), and acetic acid] at a temperature range from 278.15 to 323.15 K by a gravimetric method under 0.1 MPa. Results indicated that the experimental mole fraction solubility (10x) order of difenidol hydrochloride in 12 neat solvents at the temperature 298.15 K was: methanol(0.1771)>DMSO(0.1343)>NMP(0.0725)>acetic acid (0.0601)>DMF(0.0467)>DMA(0.0357)>ethanol (0.0336)>n-propanol (0.0255)>n-butanol (0.0137)≈water (0.0135)>1-pentanol (0.0098)>MeCN (0.0041), and solubility increased with the increase in temperature. Three models, including Modified Apelblat model, Van’ t Hoff model, and Yaws model, were applied to correlate the experimental data and analyze the solubility data of difenidol hydrochloride. Based on the principle of similar compatibility, the law of solubility and miscibility of difenidol hydrochloride in selected solvents was discussed according to Hansen solubility parameters (HSP) and solvent effect for predicting solubility behavior.
Comments
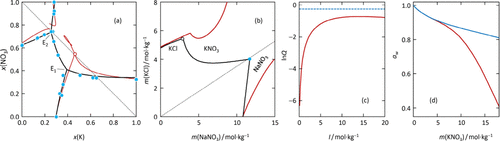
Comment on “A Consistent Set of Pitzer Interaction Parameters for NO3– Ions in the Senary Aqueous Oceanic System at 25 °C: The Zdanovskii Approach” by A. G. Muñoz (J. Chem. Eng. Data2025, 70, 4–18)
Michael Steiger *
This publication is Open Access under the license indicated. Learn More
In a recent article ( J. Chem. Eng. Data 2025, 70, 4−18), Muñoz presents parameters of a conventional Pitzer ion interaction model. The author claims that the model is accurate and consistent. However, this brief comment demonstrates that the model of Muñoz is not thermodynamically consistent and does not allow for accurate predictions of solubilities.
Mastheads
Issue Editorial Masthead
This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More