


ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Practically Significant Method Comparison Protocols for Machine Learning in Small Molecule Drug DiscoveryClick to copy article linkArticle link copied!
- Jeremy R. AshJeremy R. AshJohnson & Johnson Innovative Medicine, Spring House, Pennsylvania 19477, United StatesMore by Jeremy R. Ash
- Cas Wognum*Cas Wognum*Email: [email protected]Valence Laboratories, Montréal, Québec H2S 3G6, CanadaRecursion Pharmaceuticals, Salt Lake City, Utah 84101, United StatesMore by Cas Wognum
- Raquel Rodríguez-Pérez
- Matteo AldeghiMatteo AldeghiBayer Research and Innovation Center, Cambridge, Massachusetts 02142, United StatesMore by Matteo Aldeghi
- Alan C. ChengAlan C. ChengMerck & Co., Inc., South San Francisco, California 94080, United StatesMore by Alan C. Cheng
- Djork-Arné Clevert
- Ola EngkvistOla EngkvistDepartment of Computer Science and Engineering, Chalmers University of Technology & University of Gothenburg, Gothenburg, Mölndal 412 58, SwedenMolecular AI, Discovery Sciences AstraZeneca R&D, Gothenburg, Mölndal 431 83, SwedenMore by Ola Engkvist
- Cheng FangCheng FangBlueprint Medicines Corporation, Cambridge, Massachusetts 02139, United StatesMore by Cheng Fang
- Daniel J. PriceDaniel J. PriceNimbus Therapeutics, Boston, Massachusetts 02210, United StatesMore by Daniel J. Price
- Jacqueline M. Hughes-OliverJacqueline M. Hughes-OliverDepartment of Statistics, North Carolina State University, Raleigh, North Carolina 27607, United StatesMore by Jacqueline M. Hughes-Oliver
- W. Patrick WaltersW. Patrick WaltersRelay Therapeutics, Cambridge, Massachusetts 02139, United StatesMore by W. Patrick Walters
Abstract
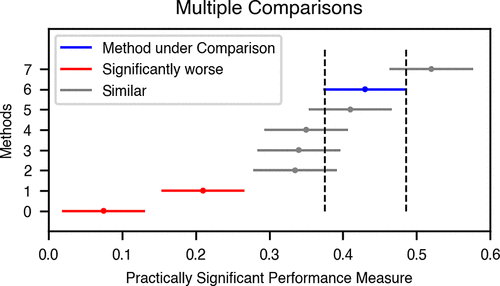
Machine Learning (ML) methods that relate molecular structure to properties are frequently proposed as in silico surrogates for expensive or time-consuming experiments. In small molecule drug discovery, such methods inform high-stakes decisions like compound synthesis and in vivo studies. This application lies at the intersection of multiple scientific disciplines. When comparing new ML methods to baseline or state-of-the-art approaches, statistically rigorous method comparison protocols and domain-appropriate performance metrics are essential to ensure replicability and ultimately the adoption of ML in small molecule drug discovery. This paper proposes a set of guidelines to incentivize rigorous and domain-appropriate techniques for method comparison tailored to small molecule property modeling. These guidelines, accompanied by annotated examples using open-source software tools, lay a foundation for robust ML benchmarking and thus the development of more impactful methods.
Cited By
This article is cited by 23 publications.
- Woruo Chen, Yao Tian, Nian Liao, Youchao Deng, Dejun Jiang, Dongsheng Cao. TabPFN Opens New Avenues for Small-Data Tabular Learning in Drug Discovery. Journal of Chemical Information and Modeling 2026, Article ASAP.
- Karmen Čondić-Jurkić, Irfan Alibay, Woody Sherman, Mallory R. Tollefson, W. Patrick Walters, Zachary Baker, Lillian T. Chong, Jennifer N. Wei, Jeffrey Gray, Brian D. Weitzner, Daniel G. A. Smith, Julia Koehler Leman, Chris Bahl, David L. Mobley. The Open Molecular Software Foundation (OMSF) and the Growing Role of Open Source Software in Molecular Modeling. Journal of Chemical Information and Modeling 2026, 66
(6)
, 2967-2984. https://doi.org/10.1021/acs.jcim.5c03137
- Manal A. Nael, Laxman M. Alakonda, Khaled M. Elokely. Defining the Data set Defines the QSAR Claim. Journal of Chemical Information and Modeling 2026, 66
(6)
, 2951-2954. https://doi.org/10.1021/acs.jcim.6c00514
- Martin Adamczewski, Britta Nisius, Nina Kausch-Busies, Niklas Tötsch. Combining High-Throughput Screening and In Silico Modeling to Derisk Novel Agrochemicals for Androgen Receptor Binding. Chemical Research in Toxicology 2026, Article ASAP.
- Cole Baker, Francis A. Acquah, Lakshmi G. Chivukula, Liping Wu, Laurence Philippe-Venec, Mostafa Abedi, Yujun Tao, Daniel Ramírez, Matthew D. McCoy, Brandon Moore, Jennifer O. Asher. PEGASUS: Unlocking Polarity in Cell-Permeable Cyclic Peptides Using AI Models Built on Massively Parallel Biological Assays. Journal of Medicinal Chemistry 2026, 69
(5)
, 5175-5198. https://doi.org/10.1021/acs.jmedchem.5c01836
- Raquel Parrondo-Pizarro, Jessica Lanini, Raquel Rodríguez-Pérez. Uncertainty Quantification in Molecular Machine Learning for Property Predictions under Data Shifts. Journal of Chemical Information and Modeling 2026, 66
(2)
, 923-935. https://doi.org/10.1021/acs.jcim.5c02381
- Mohamed Kouider Amar, Mohamed Hentabli, Nabil Touzout, Ilhem Bouaziz, Soufiane Rahal, Maamar Laidi, Abdeltif Amrane, Salah Hanini, Jie Zhang, Muhammad Farhan Saeed, Aftab Jamal. Interpretable Yield Prediction of Supercritical CO2 Extraction from Various Essential Oil Sources Using Optimized Machine Learning and PCA-Based Descriptors. Journal of Chemical Information and Modeling 2026, 66
(1)
, 194-215. https://doi.org/10.1021/acs.jcim.5c02171
- Jie Li, Santeri Aikonen, Nicolás M. Morato, Bo Hao, Zhicai Shi, Iulia I. Strambeanu, R. Graham Cooks. Catalyst-Free C–N Coupling under Ambient Conditions via High-Throughput Microdroplet Reactions. The Journal of Organic Chemistry 2025, 90
(51)
, 18172-18180. https://doi.org/10.1021/acs.joc.5c02022
- Bola Khalil, Kajetan Schweighofer, Natalia Dyubankova, Gerard J. P. van Westen, Herman van Vlijmen. Combining Bayesian and Evidential Uncertainty Quantification for Improved Bioactivity Modeling. Journal of Chemical Information and Modeling 2025, 65
(24)
, 13057-13069. https://doi.org/10.1021/acs.jcim.5c01597
- Yaëlle Fischer, Thibaud Southiratn, Dhoha Triki, Ruel Cedeno. Deep Learning vs Classical Methods in Potency and ADME Prediction: Insights from a Computational Blind Challenge. Journal of Chemical Information and Modeling 2025, 65
(24)
, 13115-13131. https://doi.org/10.1021/acs.jcim.5c01982
- Rajarshi Guha. Paths to cheminformatics: Q&A with Rajarshi Guha. Journal of Cheminformatics 2026, 18
(1)
https://doi.org/10.1186/s13321-025-01133-x
- Tiantao Liu, Jiangcheng Xu, Xinke Zhan, Shaolong Lin, Shirley W. I. Siu. Enzyformer: a two-stage pretrained model for enzymatic retrosynthesis. Journal of Cheminformatics 2026, 18
(1)
https://doi.org/10.1186/s13321-026-01164-y
- Zhenyong Cheng, Dinghao Liu, Yuanpeng Fu, Kewei Sheng, Yan Xing, Yanling Qiao, Shangxuan Cai, Jubo Wang, Peng Xu, Bin Di, Jun Liao. MSIGN: A deep learning framework based on multi-scale interaction graph neural networks for predicting binding of synthetic cannabinoids to receptors. Digital Discovery 2026, 5
(3)
, 1351-1362. https://doi.org/10.1039/D5DD00317B
- Jibai Li, Xintong Qu, Wenlong Zhang, Shifa Zhong. Collision-free morgan fingerprints: a principled approach to enhance machine learning performance and interpretability in chemistry. Journal of Cheminformatics 2026, 5 https://doi.org/10.1186/s13321-026-01170-0
- Petra Čechová, Petra Kührová, Martin Šrejber, Mariana Valério, Mariia Borbuliak, Paulo C. T. Souza, Michal Otyepka, Markéta Paloncýová. Computational Microscopy of Lipid Confined Systems: Challenges and Opportunities. Small Structures 2026, 7
(3)
https://doi.org/10.1002/sstr.202500697
- Amit Gangwal, Antonio Lavecchia. IMPACT
Framework: Establishing Global Standards for Artificial Intelligence Implementation, Methodology, and Translation in Drug Discovery. WIREs Computational Molecular Science 2026, 16
(2)
https://doi.org/10.1002/wcms.70072
- Simon D Rihm, Aleksandar Kondinski, Markus Kraft. Product design, synthesis, and lab automation with The World Avatar. Current Opinion in Chemical Engineering 2026, 51 , 101203. https://doi.org/10.1016/j.coche.2025.101203
- Rafael F. Lameiro, Luiz F. Barbosa, Evelin R. Cardoso, Beatriz Siqueira Ho, Felipe Cardoso Prado Martins, Bruna C. de Melo, Fabiana Rosini, Anwar Shamim, Priscila M. Souza, Wellington Falcão de Souza, Carlos A. Montanari. Machine Learning‐Guided Repositioning of a SARS‐CoV‐2‐Targeting Molecular Series as Cruzain Inhibitors. ChemMedChem 2026, 21
(2)
https://doi.org/10.1002/cmdc.202500630
- Peer Schliephacke, Daniel Kuhn, Lukas Friedrich. Improving Absorption, Distribution, Metabolism, and Excretion Property Predictions by Integrating Public and Proprietary Data. ChemMedChem 2026, 21
(1)
https://doi.org/10.1002/cmdc.202500713
- Oleg V. Tinkov, Pavel E. Gurevich, Sergei A. Nikolenko, Shamil D. Kadyrov, Natalya S. Bogatyreva, Veniamin Y. Grigorev, Dmitry N. Ivankov, Marina A. Pak. KRASAVA—An Expert System for Virtual Screening of KRAS G12D Inhibitors. International Journal of Molecular Sciences 2026, 27
(1)
, 120. https://doi.org/10.3390/ijms27010120
- Daniyar Mazitov, Timur Gimadiev, Assima Poyezzhayeva, Valentina Afonina, Timur Madzhidov. Conditional Variational AutoEncoder to Predict Suitable Conditions for Hydrogenation Reactions. Molecules 2026, 31
(1)
, 75. https://doi.org/10.3390/molecules31010075
- Raquel Parrondo-Pizarro, Luca Menestrina, Ricard Garcia-Serna, Adrià Fernández-Torras, Jordi Mestres. Enhancing molecular property prediction through data integration and consistency assessment. Journal of Cheminformatics 2025, 17
(1)
https://doi.org/10.1186/s13321-025-01103-3
- Kiarash Farajzadehahary, Shaghayegh Hamzehlou, Nicholas Ballard. Adding machine learning to the polymer reaction engineering toolbox. Progress in Polymer Science 2025, 170 , 102029. https://doi.org/10.1016/j.progpolymsci.2025.102029

Article Views are the COUNTER-compliant sum of full text article downloads since November 2008 (both PDF and HTML) across all institutions and individuals. These metrics are regularly updated to reflect usage leading up to the last few days.
Citations are the number of other articles citing this article, calculated by Crossref and updated daily. Find more information about Crossref citation counts.
The Altmetric Attention Score is a quantitative measure of the attention that a research article has received online. Clicking on the donut icon will load a page at altmetric.com with additional details about the score and the social media presence for the given article. Find more information on the Altmetric Attention Score and how the score is calculated.