

About the Cover:
Reviews

Advances and Challenges in Machine Learning for RNA-Small Molecule Interaction Modeling: Review
Tingting Sun *- ,
Wentao Xia - ,
Jiasai Shu - ,
Chunjiang Sang - ,
Mei Feng - , and
Xiaojun Xu *
RNA plays a pivotal role in biological processes such as gene expression regulation and protein synthesis. Targeting RNA with small molecules offers a novel therapeutic strategy for various diseases by directly modulating these processes. However, the structural diversity and complexity of RNA pose significant challenges for experimentally characterizing RNA-small molecule interactions. Recently, machine learning-based approaches have emerged as powerful tools for modeling RNA-small molecule interactions, enabling accurate prediction of binding sites, poses, preferences, and affinities. This review provides a comprehensive overview of state-of-the-art machine learning algorithms designed for RNA-small molecule interaction modeling, focusing on their applications in predicting binding characteristics and their underlying mechanisms. We also highlight the limitations of current methods and systematically discuss the challenges that remain to be addressed. By advancing these computational approaches, the ultimate goal is to enable the rational design of RNA-targeted small molecule drugs with high specificity and efficacy, paving the way for novel therapeutic interventions.
Perspective: Vibronic Coupling Potentials for Trajectory-Based Excited-State Dynamics
Sandra Gómez *- ,
Patricia Vindel-Zandbergen *- ,
Dilara Farkhutdinova - , and
Leticia González *

This publication is Open Access under the license indicated. Learn More
This Perspective reviews the use of vibronic coupling (VC) potentials in trajectory-based excited-state dynamics simulations. Originally developed to provide simplified yet physically grounded representations of nonadiabatic interactions, VC models─particularly their linear version (LVC)─have facilitated extensive investigations of photophysical and photochemical processes, in both molecular and condensed-phase systems. By effectively capturing the coupling between electronic and vibrational motions, VC models enable efficient dynamical simulations, making it feasible to investigate larger and more complex systems, for longer time scales or relying on potential energy surfaces calculated with high levels of theory. These models provide valuable insights into energy and charge transfer mechanisms following photoexcitation, shedding light on excited-state lifetimes and intricate relaxation pathways. Here, we discuss their integration with three trajectory-based computational families of methods: surface hopping, variational multiconfigurational Gaussian, and exact-factorization-derived approaches. We showcase how VC models have helped uncovering key mechanistic insights, including state-specific intersystem crossing pathways and vibrational mode selectivity. As the field progresses, VC-based approaches are expected to be increasingly combined with machine learning, anharmonic corrections, and hybrid LVC/MM frameworks, broadening their applicability to complex, flexible, and solvated environments. We highlight the advantages of VC-based potentials for trajectory-based simulations, emphasizing their computational efficiency and usefulness for benchmarking and exploring photophysical processes in molecular systems.
Dynamics

Variational Quantum Simulation of Open Quantum Dynamics via Non-Markovian Stochastic Schrödinger Equation with Complex Frequency Modes
Yukai Guo - and
Xing Gao *
Simulating non-Markovian open quantum dynamics is crucial for understanding complex quantum systems, yet it poses significant challenges for standard quantum hardware. These challenges stem from the non-Hermitian nature of such dynamics, which results in nonunitary evolution, as well as constraints imposed by limited quantum resources. To address this, we propose a hybrid quantum–classical algorithm designed for simulating dissipative dynamics in systems coupled to non-Markovian environments. Our approach includes formulating a non-Markovian Stochastic Schrödinger equation with complex frequency modes (cNMSSE) where the non-Markovianity is characterized by the mode excitation. We then employ variational quantum simulation to capture the nonunitary evolution within the cNMSSE framework, leading to a substantial reduction in qubit requirements. To demonstrate our approach, we investigated dissiaptive dynamics in the spin-boson model (SBM) and excitation energy transfer processes in both a prototype dimer system and the biologically relevant Fenna–Matthews–Olson (FMO) complex.

FLAMES─Fast, Low-Storage, Accurate, and Memory-Efficient Adaptive Sampling─Approach to Resolve Spatially Dependent Dynamics of Molecular Liquids
Guang Chen - ,
Suresh Narayanan - ,
Gregory Brian Stephenson - ,
Michael J. Servis - , and
Subramanian K. R. S. Sankaranarayanan *
Many critical phenomena in soft matter occur at large length scales, necessitating the resolution of their structure and dynamics at low wavenumbers. However, resolving wavenumber-dependent dynamics computationally via molecular dynamics simulations presents significant challenges, as these phenomena span several orders of magnitude in both time and length scales, resulting in high computational costs and memory demands. This work highlights the computational and memory challenges associated with analyzing molecular trajectories in reciprocal space and demonstrates a method to address them. We introduce FLAMES─Fast, Low-storage, Accurate, and Memory-Efficient adaptive Sampling, which is a direct method for calculation of structure factors, allowing us to select only the required number of wavevectors for binning. We also use wavenumber-dependent time steps to extract dynamics. Our FLAMES approach effectively mitigates computational and memory/storage bottlenecks. We demonstrate the method using simulations of a model system, liquid octane, at various temperatures. Comparisons with experimental data and real space computation show that the FLAMES technique achieves high accuracy in resolving temperature- and spatially dependent dynamics while being significantly more computationally efficient and requiring less memory and storage than methods based on a uniform wavevector grid and fixed temporal spacing.

Computationally Efficient DFT-Based Sampling of Ion Diffusion in Crystalline Solids
Hannes Gustafsson - ,
Fabian Schwarz - ,
Thijs Smolders - ,
Senja Barthel - , and
Amber Mace *
This publication is Open Access under the license indicated. Learn More
We present a method for large-scale DFT-based screening of ion diffusion in crystalline solids. This is accomplished by extending the Ionic TuTraSt method to sample the potential energy surface by using single-point DFT calculations. To drastically reduce the number of single-point DFT calculations, symmetry, interpolation, and exclusion of high-energy regions are employed. This approach is tested on a large data set of solid-state Li-ion conductors, for which the interpolation and high-energy exclusion are optimized to balance computational efficiency and accuracy of the obtained diffusion properties. Furthermore, the developed workflow is validated by comparison with ab initio molecular dynamics (AIMD) simulations on a set of known Li-ion superconducting materials.
Roadmap to CCSD(T)-Quality Machine-Learned Potentials for Condensed Phase Simulations
Eric D. Boittier - ,
Silvan Käser - , and
Markus Meuwly *
Accurate, yet computationally efficient energy functions are essential for state-of-the-art molecular dynamics (MD) studies of condensed phase systems. Here, a generic workflow based on a combination of machine-learning-based and empirical representations of intra- and intermolecular interactions is presented. The total energy is decomposed into internal contributions and electrostatic and van der Waals interactions between monomers. The monomer potential energy surface is described using a neural network, whereas for the electrostatics the flexible minimally distributed charge model is employed. Remaining contributions between reference energies from electronic structure calculations and the model are fitted to standard Lennard-Jones (12-6) terms. For water as a topical example, reference energies for the monomers are determined from CCSD(T)-F12 calculations, whereas for an ensemble of cluster structures containing [2, 60] and [2, 4] monomers DFT and CCSD(T) energies, respectively, were used to adjust the van der Waals contributions. Based on the bulk liquid density and heat of vaporization, the best-performing set of LJ(12-6) parameters was selected and a wide range of condensed phase properties were determined and compared with experiment. MD simulations on the multiple-nanosecond time scale were carried out for water boxes containing 2000 to 8000 monomers, depending on the property considered. The performance of such a generic ML-inspired parametrization scheme is very promising and future improvements and extensions are discussed, also in view of recent advances for water in particular in the literature.
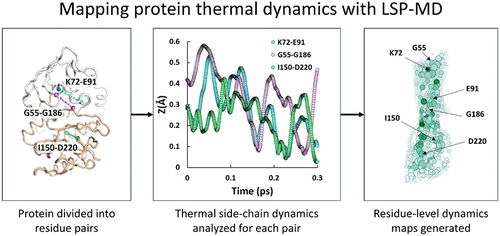
LSP-MD: A Fast Computational Method to Study Allostery Driven by Thermal Vibrations
Alexandr P. Kornev *
Conformational entropy associated with thermal vibrations plays fundamental roles in protein function, from ligand binding and catalysis to allosteric regulation. Cooper and Dryden first proposed entropy-driven allostery as an example of these effects. However, measuring the underlying thermal motions remains technically challenging. Here, we introduce LSP-MD, a computational method that builds on the Local Spatial Pattern (LSP) alignment to track side-chain stability in molecular dynamics (MD) simulations. LSP-MD uses graph-based Protein Residue Networks (PRNs) with edge weights derived from fast, local geometric fluctuations. Applied to protein kinase A (PKA), the method captures picosecond-time scale vibrations with amplitudes in the 0–2 Å range and frequencies below 100 cm–1─within the range implicated in entropy-mediated signaling. Centrality measures derived from LSP-MD networks remain stable across different simulation lengths, vector definitions, and force fields, confirming robustness. Importantly, LSP-MD reproduces key findings from traditional LSP analysis, while offering a clearer physical basis and greater computational efficiency. This method opens new opportunities for exploring entropy-driven allosteric behavior in diverse macromolecular systems.

ILVES: Accurate and Efficient Bond Length and Angle Constraints in Molecular Dynamics
Lorién López-Villellas *- ,
Carl Christian Kjelgaard Mikkelsen - ,
Juan José Galano-Frutos - ,
Santiago Marco-Sola - ,
Jesús Alastruey-Benedé - ,
Pablo Ibáñez - ,
Pablo Echenique - ,
Miquel Moretó - ,
Maria Cristina De Rosa - , and
Pablo García-Risueño *
This publication is Open Access under the license indicated. Learn More
All-atom, force field-based molecular dynamics simulations are essential tools in computational chemistry, enabling the prediction and analysis of biomolecular systems with atomic-level resolution. However, as system sizes and simulation time scales increase, so does the associated computational cost. To extend simulated time using the same resources, a common strategy is to constrain the fastest degrees of freedom, such as bond lengths, allowing for larger integration time steps without compromising accuracy. The de facto state-of-the-art algorithms for this purpose─SHAKE, LINCS, and P-LINCS─are integrated into most molecular dynamics packages and widely adopted across the field. Despite their impact, these methods exhibit limitations: all converge slowly when high numerical accuracy is required, and the LINCS and P-LINCS algorithms cannot handle general angular constraints, limiting further increases in time step. In this article, we introduce ILVES, a family of parallel algorithms that converge so rapidly that it is now practical to solve bond length and associated angular constraint equations as accurately as the hardware will allow. We have integrated ILVES into Gromacs, and our analysis demonstrates that it is superior to the state-of-the-art when constraining bond lengths. Due to its better convergence properties, we also show that if the time step is increased up to 3.5 fs by enforcing angular constraints, ILVES enables a 1.65× increase in simulated time using the same computational resources and wall-clock time, an outcome unattainable with current methods. This advance can significantly reduce the computational cost of most all-atom molecular dynamics simulations while improving their accuracy and extending access to larger systems and longer time scales.
Quantum Electronic Structure
Pruned-ADAPT-VQE: Compacting Molecular Ansätze by Removing Irrelevant Operators
Nonia Vaquero-Sabater - ,
Abel Carreras *- , and
David Casanova *
This publication is Open Access under the license indicated. Learn More
The adaptive derivative-assembled problem-tailored variational quantum eigensolver (ADAPT-VQE) is one of the most widely used algorithms for electronic structure calculations in quantum computers. It adaptively selects operators based on their gradient, constructing ansätze that continuously evolve to match the energy landscape, helping avoid local traps and barren plateaus. However, this flexibility in reoptimization can lead to the inclusion of redundant or inefficient operators that have almost zero parameter value, barely contributing to the ansatz. We identify three phenomena responsible for the appearance of these operators: poor operator selection, operator reordering, and fading operators. In this work, we propose an automated, cost-free refinement method that removes unnecessary operators from the ansatz without disrupting convergence. Our approach evaluates each operator after ADAPT-VQE optimization by using a function that considers both its parameter value and position in the ansatz, striking a balance between eliminating low-coefficient operators while preserving the natural reduction of coefficients as the ansatz grows. Additionally, a dynamic threshold based on the parameters of recent operators enables efficient convergence. We apply this method to several molecular systems and find that it reduces ansatz size and accelerates convergence, particularly in cases with flat energy landscapes. The refinement process incurs, at most, a small additional computational cost and consistently improves or maintains ADAPT-VQE performance.
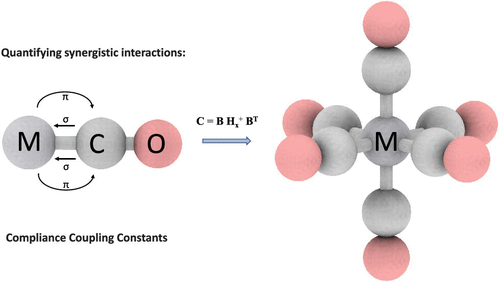
Effective Computation of Coupling Force Constants: Metal Carbonyls as a Test Case
Henrik Borgman - ,
Somi Vasisth - , and
Jörg Grunenberg *
This publication is Open Access under the license indicated. Learn More
An automated protocol enabling the efficient computation of unique potential coupling constants is presented. Several modern density functional (DFT) methods are tested against coupled cluster theory (CCSD(T)) in order to evaluate their quality in producing reliable compliance matrix off-diagonal elements. While force coupling constants could serve as descriptors of electron delocalization in general, we tested the ability of coupling compliance constants as descriptors of the Dewar–Chatt–Duncanson model in VCO–, CrCO, MnCO+, FeCO2+, NiCO, CuCO+, FeCO+ and the isoelectronic hexacarbonyls of the 3d and 5d series from Ti to Co, and Hf to Ir, respectively. A robust semiautomated algorithm including the computation of all compliance coupling constants as inverse covariant second derivatives is implemented in our open source version of the COMPLIANCE code.
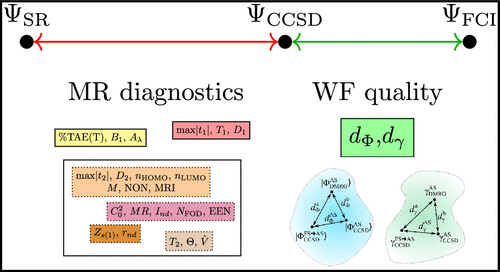
Assessing the Reliability of Truncated Coupled Cluster Wave Function: Estimating the Distance from the Exact Solution
Ádám Ganyecz *- ,
Zsolt Benedek - ,
Klára Petrov - ,
Gergely Barcza - ,
András Olasz - ,
Miklós A. Werner - , and
Örs Legeza
This publication is Open Access under the license indicated. Learn More
A new approach is proposed to assess the reliability of the truncated wave function methods by estimating the deviation from the full configuration interaction (FCI) wave function. While typical multireference diagnostics compare some derived property of the solution with the ideal picture of a single determinant, we try to answer a more practical question: how far is the solution from the exact one. Using the density matrix renormalization group (DMRG) method to provide an approximate FCI solution for the self-consistently determined relevant active space, we compare the low-level CI expansions and one-body reduced density matrixes to determine the distance of the two solutions (d̃Φ, d̃γ). We demonstrate the applicability of the approach for the CCSD method by benchmarking on the W4–17 data set, as well as on transition-metal-containing species. We also show that the presented moderate-cost, purely wave function-based metric is truly unique in the sense that it does not correlate with any popular multireference measures. We also explored the usage of CCSD natural orbitals (d̃γ,NO) and its effect on the active space size and the metric. The proposed diagnostic can also be applied to other wave function approximations, and it has the potential to provide a quality measure for post-Hartree–Fock procedures in general.
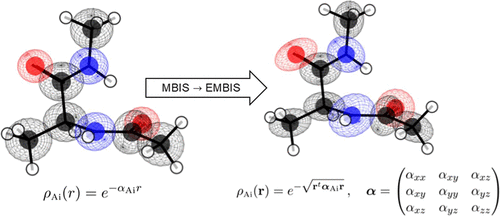
Minimal Basis Iterative Stockholder Decomposition with Ellipsoidal Atoms
Anker M. H. Nielsen - and
Frank Jensen *
The minimal basis iterative Stockholder (MBIS) decomposition of molecular electron densities into atomic contributions is extended from spherical to ellipsoidal atomic basins. Despite the more flexible parametrization, the derived atomic multipole moments do not systematically improve the reproduction of molecular multipole moments and electrostatic potentials relative to a decomposition into spherical atomic densities. The decomposition can be constrained to exactly reproduce molecular multipole moments, in the present work extended up to hexadecapole moments, and this slightly improves the ability to reproduce the electrostatic potential. A byproduct of the ellipsoidal decomposition is a set of atomic parameters that describe the anisotropic decay of the electron density with distance from the nucleus, and this may be useful in developing anisotropic atomic parameters for use in force fields as well as for defining anisotropic atomic densities for use in quantum crystallography.
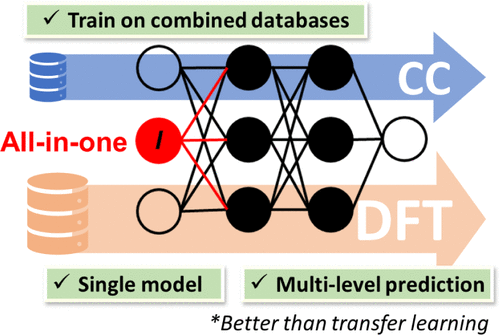
One to Rule Them All: A Universal Interatomic Potential Learning across Quantum Chemical Levels
Yuxinxin Chen - and
Pavlo O. Dral *
With the development of universal machine learning interatomic potentials, a rapidly growing number of chemical space data sets have appeared. One of the biggest challenges is that these data sets are mostly generated at different quantum chemical (QC) levels. However, a general framework that is scalable to learning across both the chemical space and quantum chemical levels remains unmet. In this work, we propose an all-in-one approach that enables simultaneous learning on an arbitrary number of QC levels from various data sets, presenting a more general and easier-to-use alternative to transfer learning. We showcase the superiority of our all-in-one strategy by creating OMNI-P1─the first-ever universal interatomic potential capable of simultaneously learning and making predictions at different QC levels. The generalization capability of the universal model OMNI-P1 for organic molecules is comparable to semiempirical GFN2-xTB and common density functional theory (DFT) methods with a double-ζ basis set, while its speed is orders of magnitude faster. Due to its unique ability to make predictions at different levels, a single model trained with our approach provides a straightforward way to also generate correction terms. This can be used in Δ-learning models without the need to train a dedicated correction model. We utilized this capability of OMNI-P1 to correct the DFT ωB97X-D4 level to obtain the Ω-ωB97X-D4 method with superior accuracy.
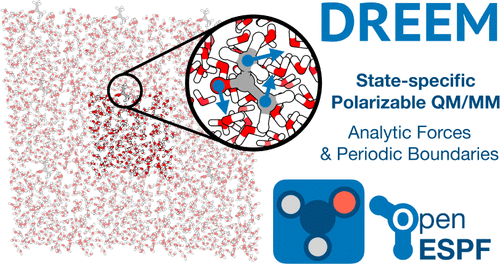
Analytic Gradients and Periodic Boundary Conditions for Direct Reaction Field Polarizable QM/MM with Electrostatic Potential Fitting
Thomas P. Fay *- ,
Miquel Huix-Rotllant - , and
Nicolas Ferré
Our recently developed Direct Reaction field with ESPF Embedding Model (DREEM) method offers an efficient and physically rigorous framework for incorporating polarizable molecular mechanics (MM) environments into quantum mechanics/molecular mechanics (QM/MM) simulations. By coupling the QM and MM regions through the instantaneous MM electrostatic polarization response to QM charge density fluctuations, DREEM enables consistent treatment of ground and excited electronic states, capturing electronic state-specific polarization and dispersion effects absent in conventional mean-field or linear response approaches. The use of the electrostatic potential fitting (ESPF) approximation method to describe charge density fluctuations significantly improves the computational efficiency compared to the integral-exact direct reaction field. In this work, we present two methodological advancements to extend the applicability of DREEM to realistic condensed-phase simulations: first, the development of efficient analytic energy gradients, enabling geometry optimization, transition state searches, and molecular dynamics; and second, a formulation of periodic boundary conditions (PBC) compatible with the DREEM framework. These capabilities are implemented in the open-source OpenESPF code, interfacing PySCF and OpenMM for high-performance QM and MM calculations. We demonstrate that the resulting implementation enables practical simulations of excited-state optical properties in periodic polarizable environments, where we calculate the fluorescence spectrum of acetone in water, including quantum vibronic and non-Condon effects. This paves the way for predictive modeling of photochemical reactivity and spectroscopy in complex systems where environment polarization is important.
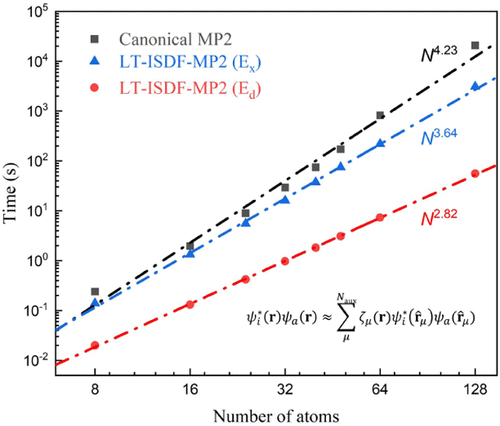
Low-Rank Approximations for Accurate and Efficient Plane-Wave Second-Order Møller–Plesset Perturbation Theory
Zhaolong Luo - ,
Xinming Qin *- ,
Wei Hu *- , and
Jinlong Yang *
The second-order Møller–Plesset perturbation (MP2) theory is a post-Hartree–Fock method widely used to describe weak correlation energies in solids and molecules, but its high computational cost scales as O(N5). Herein, we present an accurate and efficient implementation of MP2 within the plane-wave (PW) basis set for both periodic and molecular systems, which incorporates the interpolative separable density fitting (ISDF) decomposition and the Laplace transformation (LT) of the energy denominator. These innovations avoid the direct construction of electron repulsion integrals (ERIs) and reduce the computational complexity of MP2 from O(N5) to O(N4). The key idea for reducing the scaling is to exploit the numerical redundancy of occupied-virtual molecular orbital pairs on the real-space grid in the plane-wave basis set, which enables ERIs to be factorized into lower-rank quantities. This leads to further cost reductions in both the direct and exchange terms of the MP2 correlation energy. For a bulk silicon system consisting of 128 atoms, the LT-ISDF-MP2 method demonstrates a 13.5-fold speedup in total computation time compared to the standard approach. Using this plane-wave LT-ISDF-MP2 method, we simulate the π–π stacking interaction in the 1,3-butadiene dimer, successfully capturing the dispersion interaction and reproducing the self-assembled configuration.

Qubit-Efficient Quantum Chemistry with the ADAPT Variational Quantum Eigensolver and Double Unitary Downfolding
Harjeet Singh - ,
Luke W. Bertels *- ,
Daniel Claudino - ,
Sophia E. Economou - ,
Edwin Barnes - ,
Nicholas P. Bauman - ,
Karol Kowalski - , and
Nicholas J. Mayhall *
In this work, we combine the recently developed double unitary coupled cluster (DUCC) theory with the adaptive, problem-tailored variational quantum eigensolver (ADAPT-VQE) to explore the accuracy of unitary downfolded Hamiltonians for quantum simulation of chemistry. We benchmark the ability of DUCC effective Hamiltonians to recover dynamical correlation energy outside of an active space. We consider the effects of strong correlation, commutator truncation, higher-body terms, and approximate external amplitudes on the accuracy of these effective Hamiltonians. When combining these DUCC Hamiltonians with ADAPT-VQE, we observe similar convergence of the ground state as compared with bare active space Hamiltonians, demonstrating that DUCC Hamiltonians provide increased accuracy without increasing the load on the quantum processor.
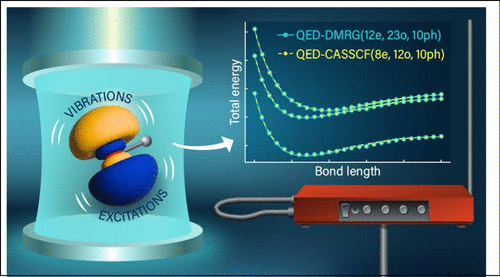
Modeling Strong Light-Matter Coupling in Correlated Systems: State-Averaged Cavity Quantum Electrodynamics Complete Active Space Self-Consistent Field Theory
Nam Vu *- ,
Kenny Ampoh - ,
Mikuláš Matoušek - ,
Libor Veis - ,
Niranjan Govind - , and
Jonathan J. Foley IV*
This publication is Open Access under the license indicated. Learn More
The description of strongly correlated systems interacting with quantized cavity modes poses significant theoretical challenges due to the combinatorial scaling of the electronic and photonic degrees of freedom. Recent advances addressing this complexity include cavity quantum electrodynamics (QED) generalizations of complete active space configuration interaction and density matrix renormalization group methods. In this work, we introduce a QED extension of state-averaged complete active space self-consistent field theory, which incorporates cavity-induced correlations through a second-order orbital optimization framework with robust convergence properties. The method is implemented using both photon number state and coherent state representations, with the latter showing robust origin invariance in the energies, regardless of the completeness of the photonic Fock space. The implementation enables symmetry-free orbital relaxations to account for photon-mediated symmetry breaking in polaritonic systems. Numerical validation on lithium hydride, hydroxide anion, and magnesium hydride cation demonstrates that this method achieves significantly improved accuracy in modeling ground-state and polariton potential energy surfaces compared with QED-CASCI in a fixed orbital basis. In these studies, we reach sub kcal/mol accuracy in potential energy surface in much smaller active spaces than are required for QED-CASCI. This advancement provides a more robust approach for studying cavity-altered chemical landscapes for ground and excited strongly coupled systems.

A Reduced Cost Two-Component Relativistic Equation-of-Motion Coupled Cluster Method for Ionization Potential
Somesh Chamoli - ,
Malaya K. Nayak - , and
Achintya Kumar Dutta *
We report an efficient implementation of the ionization potential (IP) variant of the equation-of-motion coupled cluster (IP-EOM-CC) method based on the exact two-component atomic mean field (X2CAMF) framework, utilizing Cholesky decomposition (CD) and frozen natural spinors (FNS). The CD approximation significantly reduces memory demands, whereas the FNS approximation lowers the number of floating-point operations. Together, these techniques make the method computationally efficient for accurate relativistic IP-EOM-CC calculations of molecules containing heavy elements. The calculated IP values are almost identical to those obtained by the four-component relativistic IP-EOM-CC method. Benchmark studies show good agreement with experimental ionization energies and photoelectron spectra, demonstrating the method’s reliability. The practical applicability of the approach is demonstrated by IP calculations on the medium-sized [I(H2O)12]− complex, with 1698 virtual spinors.

Slimmer Geminals For Accurate F12 Electronic Structure Models
Samuel R. Powell - ,
Kshitijkumar A. Surjuse - ,
Bimal Gaudel - , and
Edward F. Valeev *
This publication is Open Access under the license indicated. Learn More
The Slater-type F12 geminal length scales originally tuned for the second-order Mo̷ller-Plesset F12 method are too large for higher-order F12 methods formulated using the SP (diagonal fixed-coefficient spin-adapted) F12 ansatz. The new geminal parameters reported herein reduce the basis set incompleteness errors (BSIEs) of absolute coupled-cluster singles and doubles F12 correlation energies by a significant─and increase with the cardinal number of the basis─margin. The effect of geminal reoptimization is especially pronounced for the cc-pVXZ-F12 basis sets (specifically designed for use with F12 methods) relative to their conventional aug-cc-pVXZ counterparts. The BSIEs of relative energies are less affected, but substantial reductions can be obtained, especially for atomization energies and ionization potentials with the cc-pVXZ-F12 basis sets. The new geminal parameters are therefore recommended for all applications of high-order F12 methods, such as coupled-cluster F12 methods and transcorrelated F12 methods.
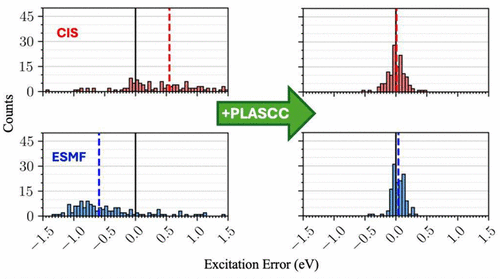
Aufbau Suppressed Coupled Cluster As a Post-Linear-Response Method
Trine Kay Quady - ,
Harrison Tuckman - , and
Eric Neuscamman *
We investigate the ability of Aufbau suppressed coupled cluster theory to act as a post-linear-response correction to widely used linear response methods for electronically excited states. We find that the theory is highly resilient to shortcomings in the underlying linear response method, with final results from less accurate starting points nearly as good as those from the best starting points. This pattern is especially stark in charge transfer states, where the approach converts starting points with multi-eV errors into post-linear-response results with errors on the order of 0.1 eV. These findings highlight the ability of Aufbau suppressed coupled cluster to perform its own orbital relaxations and raise the question of whether initializing it with an orbital relaxed reference is worth the trouble.
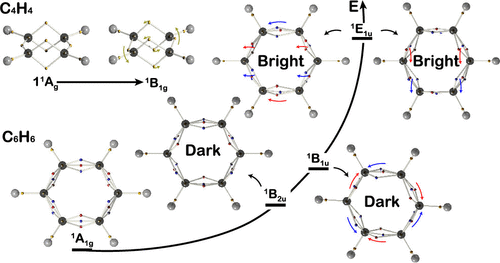
Visualizing Electronic Vibrations on the Wave Function Tiles of the Low-Lying Singlet Excited States of Benzene
Hui Zhang - ,
Terry J. Frankcombe - ,
Timothy W. Schmidt - ,
Wei Ren *- , and
Yu Liu *
The representation of the electronic structure of benzene is important for understanding the properties of planar and monocyclic organic carbon compounds. Resonant Kekulé and conjugated structures based on localized and delocalized electronic theories, respectively, can be used to depict the ground state of benzene; however, depictions of its electrons vibrating in the excited states remain to be clarified. This paper presents a novel algorithm for exploring the three lowest lying vertically singlet excited states of benzene, focusing on the electronic excitations between occupied π and unoccupied π* orbitals. We show that electronic vibrations between neighboring carbon nuclei on the wave function tile of benzene undergo excitation in the π → π* transition. Furthermore, we reveal that electronic vibrations from the ground state can explain the optical dark or bright properties of the relevant excited states, as well as transition dipole moments (TDMs) calculated from the centroid of electron densities. Moreover, our method shows the potential intramolecular change of the molecular structures in the bright excited states. This study provides new insights into the singlet excited states of benzene and validates the algorithm as a useful tool for introducing the high-dimensional wave function to the general chemical community.
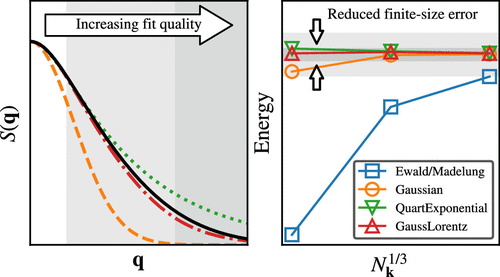
Optimized Auxiliary Functions for Robust Mitigation of Finite-Size Errors in Periodic Hybrid Density Functional Theory
Stephen Jon Quiton - ,
Juan D. F. Pottecher - ,
Xin Xing - ,
Martin Head-Gordon *- , and
Lin Lin *
When calculating properties of periodic systems at the thermodynamic limit (TDL), the dominant source of finite size error (FSE) arises from the long-range Coulomb interaction, and can manifest as a slowly converging quadrature error when approximating an integral in the reciprocal space by a finite sum. The singularity subtraction (SS) method offers a systematic approach for reducing this quadrature error and thus the FSE. In this work, we first investigate the performance of the SS method in the simplest setting, aiming at reducing the FSE in exact exchange calculations by subtracting the Coulomb contribution with a single, adjustable Gaussian auxiliary function. We demonstrate that a simple fitting method can robustly estimate the optimal Gaussian width and leads to rapid convergence toward the TDL. Furthermore, we suggest new forms of the auxiliary function, whose optimal parameters could also be determined through least-squares fitting. For a range of semiconductors and insulators, the proposed auxiliary functions achieve robust, millihartree-level accuracy in hybrid density functional theory calculations, including cases with sparse k-meshes and large basis sets.

A Computational Study of the Vibrational and Rotational g-Factors of the Diatomic Molecules LiH, LiF, CO, CS, SiO, and SiS
Anna Thorn Ekstrøm - and
Stephan P. A. Sauer *
The purpose of this article is to present theoretical values for the vibrational and rotational g-factors of several diatomic molecules. The calculations were carried out at the multiconfigurational self-consistent field (MCSCF) level of theory. To determine the most reliable method and basis set for these calculations, the Hartree–Fock (HF) and density functional theory (DFT) approaches were also considered. Different DFT functionals, including B3LYP, BHandHLYP, PBE0, B3PW91, and KT3, have been employed. Furthermore, different active spaces were evaluated to optimize MCSCF. To establish the accuracy of the methods, the computed rotational g-factors were compared to experimental values. The benchmark study of CO and CS shows that the MCSCF method provides the most reliable results and that the aug-cc-pCV5Z basis set is the most sufficient. The aug-cc-pCVQZ basis set for Li and aug-cc-pV5Z basis set for H gave the best results for LiH. The active spaces tested for CO and CS do not yet converge toward the experimental values when more determinants were included. However, if the g-factors are vibrationally averaged, the computed values are seen to move toward the experimental value.
Reaction Mechanisms

Reactive Active Learning: An Efficient Approach for Training Machine Learning Interatomic Potentials for Reacting Systems
Siddarth K. Achar - ,
Priyanka B. Shukla - ,
Chinmay V. Mhatre - ,
Leonardo Bernasconi - ,
Caitlyn Y. Vinger - , and
J. Karl Johnson *
This publication is Open Access under the license indicated. Learn More
Discovering chemical reaction pathways using quantum mechanics is impractical for many systems of practical interest because of unfavorable scaling and computational cost. While machine learning interatomic potentials (MLIPs) trained on quantum mechanical data offer a promising alternative, they face challenges for reactive systems due to the need for extensive sampling of the potential energy surface in regions that are far from equilibrium geometries. Unfortunately, traditional MLIP training protocols are not designed for comprehensive reaction exploration. We present a reactive active learning (RAL) framework that is designed to efficiently train MLIPs to achieve near-quantum mechanical accuracy for reactive systems for situations where one may not have prior knowledge of the possible transition states, reaction pathways, or even the potential products. Our method combines automated reaction exploration, uncertainty-driven active learning, and transition state sampling to build accurate potentials. We demonstrate RAL’s effectiveness across three different systems: uncatalyzed ammonia synthesis (gas-phase), methanimine hydrolysis (solution phase), and methane activation on titanium carbide surfaces (heterogeneous). The resulting MLIPs accurately predict reaction barriers and transition states. For catalysis, we show that RAL-trained MLIPs identify Ti2C as the most active methane activation surface (90% decomposition at 1000 K) through C-vacancy mediated mechanisms. The framework’s ability to simulate large systems (∼900 atoms) over nanosecond time scales provides previously inaccessible insights into surface poisoning and reaction networks. We show that reactive exploration is essential for adequately capturing the potential energy surface, with chemical and configurational sampling working synergistically to improve model accuracy. Our results establish general guidelines for training robust reactive potentials and open new possibilities for computational discovery of catalysts and reaction mechanisms.
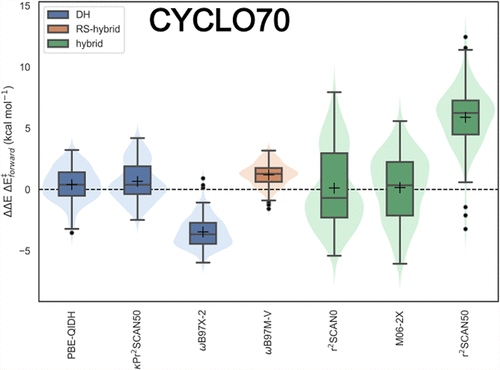
CYCLO70: A New Challenging Pericyclic Benchmarking Set for Kinetics and Thermochemistry Evaluation
Javier E. Alfonso-Ramos - ,
Carlo Adamo - ,
Éric Brémond *- , and
Thijs Stuyver *
Here, a new challenging benchmarking data set for cycloaddition reactions, CYCLO70, is presented and analyzed. CYCLO70 has been generated with the specific aim of being representative of the most challenging regions of the chemical reaction space surrounding Diels–Alder, dipolar cycloadditions, and (sigmatropic) rearrangement reactions with the help of an active learning approach. Testing 93 different functionals, spanning from spin-local density approximation to the most recent double-hybrid functionals, we observe that the errors on CYCLO70 are significantly bigger than those on the cycloaddition subset of BH9, the most popular benchmarking data set for this reaction class. Furthermore, we observe that the range-separated hybrid ωB97M-V is the best performing functional to model barrier heights and reaction energies, with a deviation closest to the desirable “chemical accuracy”; among the double hybrids, PBE-QIDH performs best, and among the fixed-range hybrids, M06–2X and r2SCAN0 emerge as the most balanced in terms of simultaneously reproducing both properties. Next, we perform a principal component analysis on the errors across the data set and demonstrate not only that the errors across different functional approximations correlate to a significant extent (the first two components explain 98% of the variance), but we also observe that functionals belonging to the same rung of Jacob’s ladder cluster together in the constructed two-dimensional plot. These results were further validated on a set of Diels–Alder reactions relevant to self-healing polymer design, reinforcing the practical relevance of CYCLO70.
Molecular Mechanics
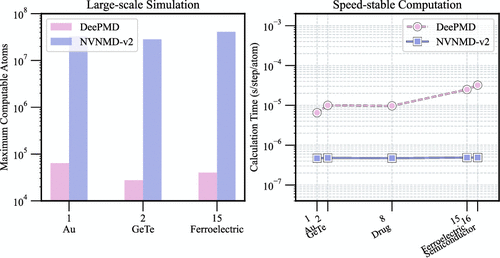
NVNMD-v2: Scalable and Accurate Deep Learning Molecular Dynamics Model Based on Non-Von Neumann Architectures
Xiaoyun Yu - ,
Guang Yang - ,
Zhuoying Zhao - ,
Junhua Li - ,
Xinyu Xiao - ,
Xin Zhang - ,
Jie Liu - , and
Pinghui Mo *
Molecular dynamics (MD) simulations have emerged as a transformative computational microscope for probing atomic interactions spanning catalysis, energy storage, biotechnology, and beyond. However, existing machine-learning MD (MLMD) frameworks face a trilemma in balancing accuracy, scalability, and energy efficiency, particularly in compositionally complex systems like high-entropy alloys and multiferroic perovskites. Here, we introduce NVNMD-v2, a co-designed algorithm-hardware architecture that integrates a generalized deep neural-network potential (GDNNP) within a processing-in-memory (PIM) accelerator. Building on the foundation of NVNMD-v1, which was limited to four-element systems, NVNMD-v2 employs optimized type-embedding descriptors to support multielement systems with up to 32 species, eliminating species-dependent parameter scaling. Deployed on a single FPGA, NVNMD-v2 maintains DFT-level accuracy while achieving a flat per-atom computational cost (∼10–7 s/step/atom), enabling simulations of system up to 20 million atoms─a 103 × scale-up over DeePMD on an NVIDIA V100 GPU, with ∼120 × energy reduction. These advances unlock quantum-accurate MD for multielement materials, from semiconductor heterostructures to biomolecular assemblies, bridging the gap between atomic fidelity and industrial-scale simulations.
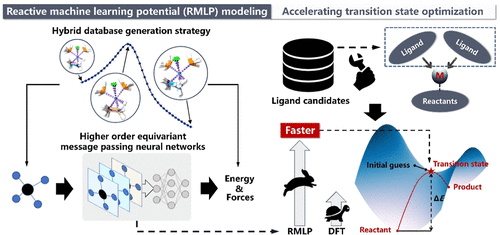
Accelerating Transition State Search and Ligand Screening for Organometallic Catalysis with Reactive Machine Learning Potential
Kun Tang - ,
Yujing Zhao - ,
Lei Zhang - ,
Jian Du - ,
Qingwei Meng - , and
Qilei Liu *
Organometallic catalysis lies at the heart of numerous industrial processes that produce bulk and fine chemicals. The search for transition states and screening for organic ligands are vital in designing highly active organometallic catalysts with efficient reaction kinetics. However, identifying accurate transition states necessitates computationally intensive quantum chemistry calculations. In this work, a reactive machine learning potential (RMLP) model is developed to accelerate transition state optimizations and ligand screening for organometallic catalysis based on an automated transition state database construction method and a higher-order equivariant message passing neural network. In case studies involving the ethylene hydrogenation reaction catalyzed by organometallic catalysts, RMLP rapidly predicts potential energy surfaces along intrinsic reaction coordinate paths, achieving speeds nearly 3 orders of magnitude faster than those of rigorous quantum chemistry calculations. Meanwhile, it maintains comparable accuracy with a root-mean-square deviation of 0.307 Å for transition state geometries and a mean absolute error of 0.871 kJ·mol–1 for reaction barriers on the external test set, significantly outperforming semiempirical quantum chemistry methods. Our RMLP model offers an effective alternative to both rigorous and semiempirical quantum chemistry approaches for rapid and precise transition state optimizations, facilitating high-throughput screening of advanced organometallic catalyst ligands.

Simulating Metal Complex Formation and Ligand Exchange: Unraveling the Interplay between Entropy, Kinetics, and Mechanisms on the Chelate Effect
Luca Sagresti - ,
Luca Benedetti - ,
Kenneth M. Merz Jr.- , and
Giuseppe Brancato *
This publication is Open Access under the license indicated. Learn More
Metal coordination is ubiquitous in Nature and central in many applications, ranging from nanotechnology to catalysis and environmental chemistry. Complex formation results from the subtle interplay between different thermodynamic, kinetic, and mechanistic contributions, which remain largely elusive to standard experimental methodologies and challenging for typical modeling approaches. Here, considering some prototypical metal complexes between Cd(II) and Ni(II) with various amine ligands, we present a comprehensive atomistic-level description of their chemical equilibrium, complex formation, and ligand exchange dynamics in aqueous solution, providing an excellent agreement with available association constants and formation rates spanning several orders of magnitude. This is achieved through an effective molecular simulation approach that combines finely tuned interatomic potentials with state-of-the-art enhanced sampling and kinetics techniques. Worthy of note, the nature of the chelate effect, a fundamental concept in coordination chemistry, is fully unravelled through the comparative analysis of the ligand binding reactions of monodentate and bidentate ligands in octahedral complexes. Results provide a complete picture illustrating all the concurrent contributions to this phenomenon, such as entropy, dissociation rates, and ligand binding mechanisms, in some cases contradicting previously held beliefs. This study represents a step forward for the in silico design and applications of coordination complex systems.
Spectroscopy and Excited States
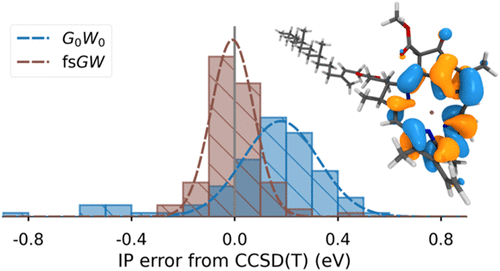
Self-Consistent GW via Conservation of Spectral Moments
Oliver J. Backhouse - ,
Marcus K. Allen - ,
Charles J. C. Scott - , and
George H. Booth *
This publication is Open Access under the license indicated. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
We expand on a recently introduced alternate framework for GW simulation of charged excitations [Scott, C. J. C. J. Chem. Phys. 2023, 158, 124102], based around the conservation of directly computed spectral moments of the GW self-energy. Featuring a number of desirable formal properties over other implementations, we also detail efficiency improvements and a parallelism strategy, resulting in an implementation with a demonstrably similar scaling to an established Hartree–Fock code, with only an order of magnitude increase in cost. We also detail the applicability of a range of self-consistent GW variants within this framework, including a scheme for full self-consistency of all dynamical variables, while avoiding the Matsubara axis or analytic continuation, allowing formal convergence at zero temperature. By investigating a range of self-consistency protocols over the GW100 molecular test set, we find a little-explored self-consistent variant based around a simpler coupled chemical potential and Fock matrix optimization to be the most accurate self-consistent GW approach. Additionally, we validate recently observed evidence that Tamm–Dancoff-based screening approximations within GW lead to higher accuracy than traditional random phase approximation screening over these molecular test cases. Finally, we consider the Chlorophyll A molecule, finding agreement with experiment within the experimental uncertainty, and a description of the full-frequency spectrum of charged excitations.

Computation of Exchange Couplings by Means of an Exchange-Dedicated Perturbation Theory
Michael Franz - ,
Frank Neese *- , and
Sabine Richert *
This publication is Open Access under the license indicated. Learn More
The accurate computation of high-spin/low-spin gaps remains a challenging task in computational chemistry, with significant implications for both theoretical studies and experimental applications. In this work, we present an exchange-dedicated perturbation theory (EDPT2) that allows an efficient calculation of exchange couplings in magnetic systems. Our approach builds on a previously developed second-order perturbative scheme based on de Loth’s formalism but refines the treatment of singlet wave functions by explicitly incorporating ionic determinants in the zeroth-order description. The EDPT2 method is derived from a two-electron-two-center model and can be applied to multispin systems using minimal CAS-generated orbitals. A key advantage of EDPT2 lies in its computational efficiency, with a scaling of N4, where N is the number of basis functions. Benchmark calculations on diverse test systems demonstrate that EDPT2 achieves high-spin/low-spin gaps with accuracy comparable to the commonly used FIC-NEVPT2 method. Beyond its efficiency, EDPT2 provides valuable information on the mechanisms that govern magnetic exchange. The method allows for a detailed decomposition of second-order contributions, facilitating the identification of dominant exchange pathways. This is exemplified on two bis(nitronyl nitroxide) biradicals, where dynamic spin polarization emerges as the key exchange mechanism. Furthermore, using the example of a trisnitroxide triradical, we demonstrate how the insights from EDPT2 can be used to prepare selective multireference CI approaches. A combined DDCI1 approach with EDPT2-derived corrections is shown to successfully reproduce the experimental doublet-quartet gap.

Efficient, Hierarchical, and Object-Oriented Electronic Structure Interfaces for Direct Nonadiabatic Dynamics Simulations
Sascha Mausenberger - ,
Severin Polonius - ,
Sebastian Mai *- , and
Leticia González *
This publication is Open Access under the license indicated. Learn More
We present a novel, flexible framework for electronic structure interfaces designed for nonadiabatic dynamics simulations, implemented in Python 3 using concepts of object-oriented programming. This framework streamlines the development of new interfaces by providing a reusable and extendable code base. It supports the computation of energies, gradients, various couplings─like spin–orbit couplings, nonadiabatic couplings, and transition dipole moments─and other properties for an arbitrary number of states with any multiplicities and charges. A key innovation within this framework is the introduction of hybrid interfaces, which can use other interfaces in a general hierarchical manner. Hybrid interfaces are capable of using one or more child interfaces to implement multiscale approaches, such as quantum mechanics/molecular mechanics where different child interfaces are assigned to different regions of a system. The concept of hybrid interfaces can be extended through nesting, where hybrid parent interfaces use hybrid child interfaces to easily setup complex workflows without the need for additional coding. We demonstrate the versatility of hybrid interfaces with two examples: one at the method level and one at the workflow level. The first example showcases the numerical differentiation of wave function overlaps, implemented as a hybrid interface and used to optimize a minimum-energy conical intersection with numerical nonadiabatic couplings. The second example presents an adaptive learning workflow, where nested hybrid interfaces are used to iteratively refine a machine learning model. This work lays the groundwork for more modular, flexible, and scalable software design in excited-state dynamics.

Excited-State Wave Functions and Energies Predicted by Machine Learning Based on Graph Neural Networks
Xiang-Yang Liu *- ,
Dongyi Xiao - ,
Wei-Hai Fang - , and
Ganglong Cui *
Accurate and efficient simulation of photoinduced dynamics in materials remains a significant challenge due to the computational cost of excited-state electronic structure calculations and the necessity to account for excitonic effects. In this work, we present a machine learning (ML)-accelerated approach to nonadiabatic molecular dynamics simulations that incorporates excitonic effects by predicting excited-state wave functions via configuration interaction coefficients and excitation energies using a graph neural network (GNN) architecture. The GNN model leverages molecular orbital information from ground-state calculations to construct input graphs, enabling efficient and accurate prediction of relevant excited-state wave functions and energies required for ab initio-based fewest-switches surface hopping simulations. Benchmarking on a zinc phthalocyanine-fullerene (ZnPc-C60) donor–acceptor system reveals that these ML-predicted properties agree closely with those obtained from linear-response time-dependent density functional theory calculations while boosting the computational efficiency significantly. The ML-accelerated simulations reproduce excited-state dynamics with high fidelity, demonstrating the methodological capability to study complex photodynamical processes in large systems. This work provides a general and scalable framework for efficient excited-state dynamics simulations in materials where excitonic effects play a vital role.
Condensed Matter, Interfaces, and Materials
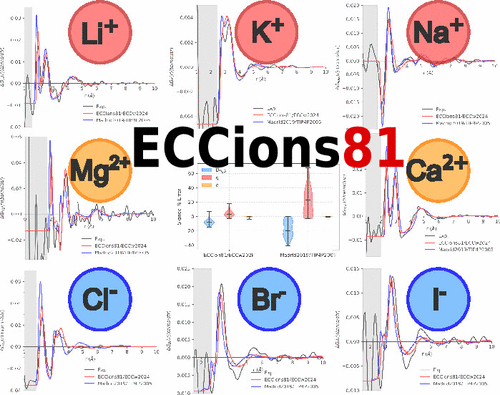
Charge Scaling Force Field for Biologically Relevant Ions Utilizing a Global Optimization Method
Shujie Fan - ,
Philip E. Mason - ,
Victor Cruces Chamorro - ,
Brennon L. Shanks - ,
Hector Martinez-Seara *- , and
Pavel Jungwirth *
This publication is Open Access under the license indicated. Learn More
Charge scaling, also denoted as the electronic continuum correction, has proven to be an efficient method for effectively including electronic polarization in force field molecular dynamics simulations without additional computational costs. However, scaling charges in existing force fields, fitted at least in part to experimental data, lead to inconsistencies, such as overscaling. We have, therefore, recently developed a four-site water model consistent with charge scaling, i.e., possessing the correct low-frequency dielectric constant of 45. Here, we build on top of this water model to develop charge-scaled models of biologically relevant Li+, Na+, K+, Ca2+, and Mg2+ cations as well as Cl–, Br–, and I– anions, employing machine learning to streamline and speed up the parametrization process. On the one hand, we show that the present model outperforms the best existing charge scaled model of aqueous ions. On the other hand, the present work points to a future need for consistently and simultaneously improving the water and ion models within the electronic continuum correction framework.
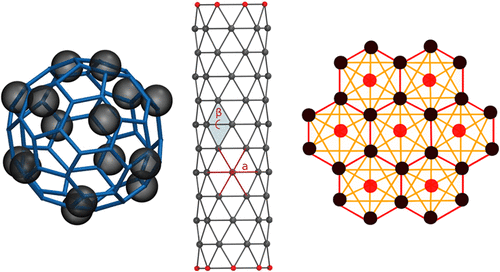
Martini 3 Coarse-Grained Models for Carbon Nanomaterials
Roshan Shrestha - ,
Riccardo Alessandri - ,
Martin Vögele - ,
Cecile Hilpert - ,
Paulo C. T. Souza - ,
Siewert J. Marrink - , and
Luca Monticelli *
The Martini model is a coarse-grained force field allowing simulations of biomolecular systems as well as a range of materials including different types of nanomaterials of technological interest. Recently, a new version of the force field (version 3) has been released that includes new parameters for lipids, proteins, carbohydrates, and a number of small molecules, but not yet carbon nanomaterials. Here, we present new Martini models for three major types of carbon nanomaterials: fullerene, carbon nanotubes, and graphene. The new models were parametrized within the Martini 3 framework, and reproduce semiquantitatively a range of properties for each material. In particular, the model of fullerene yields excellent solid-state properties and good properties in solution, including correct trends in partitioning between different solvents and realistic translocation across lipid membranes. The models of carbon nanotubes reproduce the atomistic behavior of nanotube porins spanning lipid bilayers. The model of graphene reproduces structural and elastic properties, as well as trends in experimental adsorption enthalpies of organic molecules. All new models can be used in large-scale simulations to study the interaction with the wide variety of molecules already available in the Martini 3 force field, including biomolecular and synthetic systems.
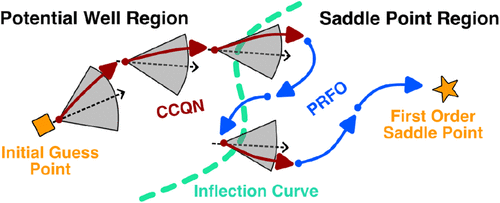
Cone-Shaped Constrained Quasi-Newton Method: Efficient and Robust Single-Ended Transition State Optimization Algorithm
Yinkai Wu - and
Haifeng Wang *
Efficient transition state location is a central challenge in heterogeneous catalysis. While single-ended methods are more efficient than double-ended methods, their convergence is often highly sensitive to the quality of the initial guess. Here, we propose a Cone-shaped Constrained Quasi-Newton (CCQN) method, which introduces a cone-shaped constraint to restrict the search direction, thereby effectively guiding the system from potential well regions toward saddle regions. After crossing the inflection curve, the optimization switches to the partitioned rational function optimization algorithm for further refinement. This curvature partitioned optimization strategy reduces the sensitivity to the quality of the initial guess while maintaining the efficiency of single-ended methods. Across 150 transition state optimization tasks with varying initial guess qualities, CCQN achieves an overall success rate of 93.3%, requiring only approximately 50 energy-force evaluations on average. The method exhibits strong robustness and convergence efficiency, offering a new tool for high-throughput transition state searches and mechanistic studies of complex catalytic reactions.

Hamiltonian Grid-Based QM/MM Method with Mean-Field Embedding for Simulating Arbitrary Slab Geometries
Hiroshi Nakano *- and
Hisao Nakamura
The quantum mechanics/molecular mechanics (QM/MM) method is a powerful approach for investigating solid surfaces in contact with various types of media, since it allows for flexible modeling of complex interfaces while maintaining an all-atom representation. The mean-field QM/MM method is an average reaction field model within the QM/MM framework. The method addresses the challenges associated with the statistical sampling of interfacial atomic configurations of a medium and enables efficient calculation of free energies. In this study, we propose a grid-based mean-field QM/MM method that leverages the particle-mesh approach in fractional coordinates, enabling simulations for arbitrary slab models in parallelepiped simulation cells. The charges of the MM atoms are assigned to nearby grid points using a Cn class assignment function with n ≥ 1. The QM-MM electrostatic forces acting on atoms are analytically derived from the total energy using the derivatives of the assignment function. The method is thus rigorously grounded in a fully Hamiltonian formalism, ensuring energy conservation, correct interfacial distribution, and reliable dynamics of the medium atoms sampled from long-time simulations. Furthermore, we demonstrate the feasibility of numerically rigorous free energy calculations through the use of analytical free energy gradients.
Polymers and Biopolymers
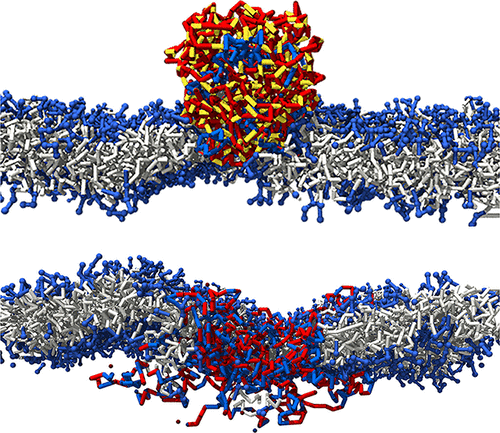
Soft Nanoparticle-Induced Permeability of Lipid Membranes: Interplay of Sequence and Hydrophobicity
Yiyang Zhang - ,
Xinyu Hu - ,
Wenfei Li - ,
Yachong Guo *- ,
Marco Werner *- , and
Wei Wang *
Single-chain nanoparticles (SCNPs) represent a class of folded macromolecules that mimic biologically derived structures through the covalent cross-linking of polymer backbones. This study explores the interactions between SCNPs and lipid bilayers, focusing on the modulation of the membrane permeability and lipid dynamics. By employing coarse-grained Monte Carlo simulations, we investigate the behavior of diblock, triblock, and random copolymers of varying hydrophobicities and cross-linking densities in contact with lipid membranes. Our results reveal that the structural properties and sequence of SCNPs significantly influence their adsorption behavior and the resulting membrane permeability. Specifically, SCNPs can embed within bilayers, forming transient nanopores that facilitate solvent and ion transport, contingent upon the balance of hydrophobicity and cross-linking. We elucidate the mechanism by which SCNPs affect lipid flip-flop rates and bilayer stability, providing insights into their potential applications in targeted drug delivery and biomimetic material design.
Biomolecular Systems
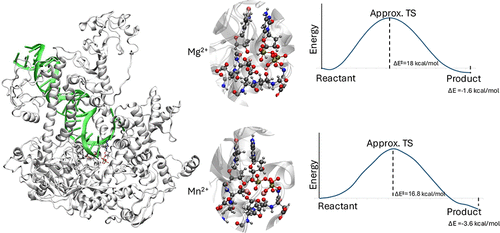
Comparison of Magnesium and Manganese Ions on the Structural and Catalytic Properties of Human DNA Polymerase Gamma
Arkanil Roy - and
G. Andrés Cisneros *
DNA polymerases are essential enzymes responsible for accurate genome replication and repair, with divalent metal cofactors playing a crucial role in their catalytic function. Polymerase γ (Pol γ) is the primary DNA polymerase in mitochondria, ensuring the faithful replication of mitochondrial DNA. The choice of metal cofactor, typically magnesium (Mg2+) or manganese (Mn2+), influences its structural stability, enzymatic activity, and fidelity. In this study, we employed molecular dynamics (MD) simulations and hybrid quantum mechanics/molecular mechanics (QM/MM) calculations to investigate how Mg2+ and Mn2+ affect the flexibility, active site stabilization, and catalytic efficiency of Pol γ. Intermolecular interaction analysis of individual residues is consistent with experimental mutagenesis reports and highlights the importance of specific residues, many of which are evolutionarily conserved, and some are involved in pathogenic mutations. It is also observed that Mn2+ enhances catalytic efficiency, exhibiting higher exoergicity (−3.65 kcal mol–1 vs −1.61 kcal mol–1 for Mg2+) and a lower activation barrier. Intermolecular interaction analysis reveals that Mn2+ provides larger stabilization of the transition state and product complex, favoring reaction progression. Investigation of the effects of the electric field in the active site suggests that the O3′ atom on the DNA primer base experiences larger polarization in the system with Mn2+ ions when compared to Mg2+, with dipole directions consistent with the catalytic reaction progress. Our findings highlight a trade-off between structural stability and catalytic efficiency, providing insights into the role of metal ions in mitochondrial polymerase function and their implications for mutagenesis and mitochondrial disorders.
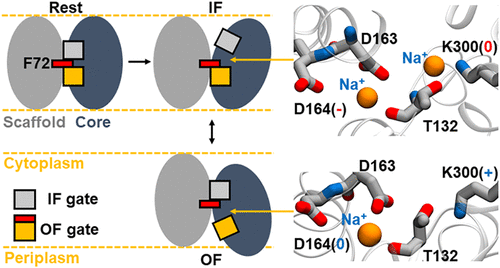
Molecular Mechanism of Na+/H+ Antiporting in NhaA
Tengyu Xie - ,
Jiahao He - ,
Shuo Sun - ,
Yulei Chen - ,
Jing Huang - , and
Yandong Huang *
Sodium–proton antiporter NhaA of Escherichia coli is a paradigm to investigate the mechanistic basis of the fundamental Na+/H+ exchange in cells. However, all existing crystal structures of NhaA are inward-facing (IF), and the putative outward-facing (OF) structures are still unsolved by experiment, limiting a complete understanding of the transport cycle in which Lys300 plays a key role in both structural stability and transport function. Here, we report a set of atomistic molecular dynamics (MD) simulations that start from the structure predicted by an artificial intelligence model that generates function-relevant alternative conformations. It is found that NhaA rapidly relaxes into either the IF or the OF conformation. Furthermore, the neutralization of Lys300 allows the binding of two sodium ions, a configuration that is associated with enhanced conformational sampling. Based on these observations, we propose a sodium-coupled mechanism of Na+/H+ antiporting.
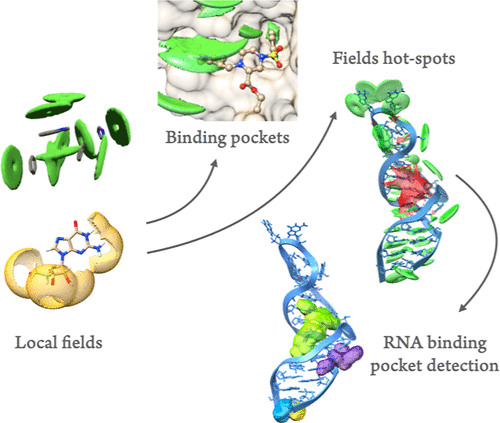
Statistical Molecular Interaction Fields: A Fast and Informative Tool for Characterizing RNA and Protein-Binding Pockets
Diego Barquero Morera - ,
Giovanni Mattiotti - ,
Alexandar Kocev - ,
Amshuman Rousselot - ,
Louis Meuret - ,
Lucas Rouaud - ,
Hubert Santuz - ,
Marc Baaden *- ,
Antoine Taly *- , and
Samuela Pasquali *
Developing a physical understanding of the interactions between a macromolecular target and its ligands is a crucial step in structure-based drug design. Although many tools exist to characterize protein-binding pockets in silico, this is not yet the case for RNA, which has been recognized only recently as a suitable target for small ligands. Molecular Interaction Fields (MIFs) are useful tools to characterize the interactions of a given binding pocket. However, classical MIFs heavily rely on the use of probes, which makes their calculations accurate but very specific to the binding partners in question. We develop here a simple version of MIF, that we call Statistical Molecular Interaction Fields (SMIFs), based on functional forms inspired by coarse-grained models and parametrized based on PDB structures and previous statistical analysis of the main form of interactions typical of macromolecules, namely, hydrogen bonding, stacking, and hydrophobic interactions. We show that these fields, despite their simplicity, are very informative and, overall, in agreement with pharmacophoric models. Thanks to a carefully optimized code, our calculations are fast and can be performed in bulk on a large set of binding pockets or even on a full macromolecule. As shown in a few representative examples, the latter possibility opens the way to the analysis of systems as large as 20000 to 80000 atoms in relation to the surrounding environment, i.e., a lipidic membrane, a small ligand, or another macromolecular partner, allowing for a detailed visualization of the possible interactions. The complete software and its documentation are available here: https://smiffer.mol3d.tech/
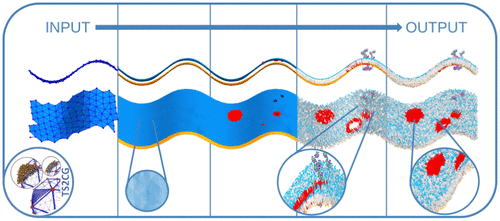
TS2CG as a Membrane Builder
Fabian Schuhmann - ,
Jan A. Stevens - ,
Neda Rahmani - ,
Isabell Lindahl - ,
Chelsea M. Brown - ,
Christopher Brasnett - ,
Dimitrios Anastasiou - ,
Adrià Bravo Vidal - ,
Beatrice Geiger - ,
Siewert J. Marrink *- , and
Weria Pezeshkian *
This publication is Open Access under the license indicated. Learn More
Molecular dynamics (MD) simulations excel at capturing biological processes at the molecular scale but rely on a well-defined initial structure. As MD simulations now extend to whole-cell-level modeling, new tools are needed to efficiently build initial structures. Here, we introduce TS2CG version 2, designed to construct coarse-grained membrane structures with any desired shape and lateral organization. This version enables precise placement of lipids and proteins based on curvature preference, facilitating the creation of large, near-equilibrium membranes. Additional features include controlled pore generation and the placement of specific lipids at membrane edges for stabilization. Moreover, a Python interface allows users to extend functionality while maintaining the high performance of the C++ core. To demonstrate its capabilities, we showcase challenging simulations, including a Möbius strip membrane, a vesicle with lipid domains as continental plates (Martini globe), and entire mitochondrial membranes exhibiting lipid heterogeneity due to curvature, along with a comprehensive set of tutorials.

Molecular Dynamics of the Intrinsically Disordered Protein COR15A─A Force Field Validation on Structure and Dynamics
Tobias Rindfleisch - ,
Ricky Nencini - ,
O. H. Samuli Ollila - ,
Dirk Walther - ,
Markus S. Miettinen *- , and
Anja Thalhammer *
This publication is Open Access under the license indicated. Learn More
Intrinsically disordered proteins (IDPs) pose a challenge for structural characterization, as experimental methods lack the subnanometer/subnanosecond resolution to capture their dynamic conformational ensembles. Molecular dynamics (MD) simulations can, in principle, provide this information, but for the simulation of IDPs, dedicated protein and water force fields are needed, as traditional MD models for folded proteins prove inadequate for IDPs. Substantial effort was invested to develop IDP-specific force fields, but their performance in describing IDPs that undergo conformational changes─such as those induced by molecular partner binding or changes in solution environment─remains underexplored. In this study, we investigated the ability of 20 MD models to accurately simulate structural and dynamic aspects of COR15A, an IDP just on the verge of folding, with a particular focus on their ability to capture subtle structural differences. We employ a two-step approach: (i) validation of short 200 ns simulations against small-angle X-ray scattering (SAXS) data and (ii) detailed evaluation of the six best-performing MD models through extended 1.2 μs MD simulations against nuclear magnetic resonance (NMR) data, including a single-point mutant with slightly increased helicity. Only DES-amber and ff99SBws capture helicity differences between wild-type and mutant, but ff99SBws overestimates helicity. Notably, only DES-amber adequately reproduces the COR15A dynamics, as assessed by NMR relaxation times at two different magnetic field strengths. Among the tested force fields, DES-amber emerges as the best MD model for the simulation of COR15A. Its application provides insights into its dynamic conformational landscape, albeit not perfectly reproducing all experimental data. Our study highlights the need for rigorous force field validation for IDPs and identifies remaining discrepancies in need of further force-field development.

Investigating the Impact of Cholesterol on GP41-Driven HIV Membrane Fusion
Jatin Soni - and
Taraknath Mandal *
Membrane fusion is a critical step in HIV infection, allowing the virus envelope to merge with the host cell membrane and deliver genetic material. Cholesterol is believed to play a pivotal role in HIV fusion; however, the specific mechanism by which it facilitates membrane fusion remains poorly understood. Our molecular dynamics simulation study elucidates the dual role of cholesterol: it promotes GP41 aggregation by reducing the dimerization free energy and enhances the strengths of the protein-induced membrane curvatures. The free energy calculations reveal that by enhancing membrane curvatures, cholesterol significantly reduces the energy barriers and stabilizes the fusion intermediates. These findings highlight cholesterol’s essential role in facilitating HIV entry and offer insights for potential antiviral strategies.
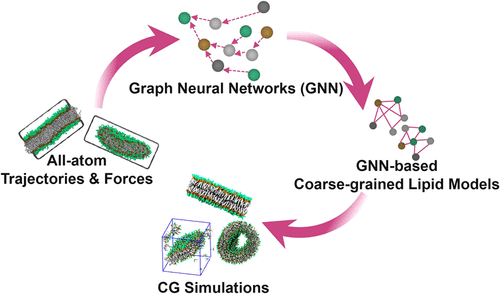
Development of Coarse-Grained Lipid Force Fields Based on a Graph Neural Network
Zhenyu Liao - ,
Ting Si - ,
Tairan Wang - ,
Ji-Jung Kai - ,
Christophe Chipot - , and
Jun Fan *
This publication is Open Access under the license indicated. Learn More
Coarse-grained (CG) lipid models enable efficient simulations of large-scale membrane events. However, achieving both speed and atomic-level accuracy remains challenging. Graph neural networks (GNNs) trained on all-atom (AA) simulations can serve as CG force fields, which have demonstrated success in CG simulations of proteins. Herein, we built data sets of AA simulations of DOPC, DOPS, and mixed DOPC/DOPS lipid bilayers and developed the first GNN-based CG lipid models based on the TorchMD-GN architecture. The CG lipid models reproduce the structural correlations of the AA simulations, accelerate the lipid dynamics by 9.4 times, and exhibit some degree of temperature transferability. Moreover, we demonstrate that training CG models on lipid bicelles enhances the performance of models in the lipid self-assembly and vesicle simulations. Our findings indicate that GNN-based CG lipid force fields show promise as a powerful approach for large-scale membrane simulations.
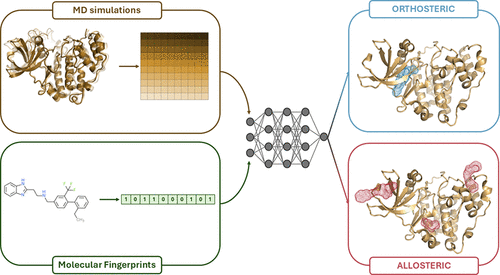
MOLECULE: Molecular-dynamics and Optimized deep Learning for Entropy-regularized Classification and Uncertainty-aware Ligand Evaluation
Ivan Cucchi - ,
Elena Frasnetti - ,
Francesco Frigerio - ,
Fabrizio Cinquini - ,
Silvia Pavoni - ,
Luca F. Pavarino *- , and
Giorgio Colombo *
This publication is Open Access under the license indicated. Learn More
Machine learning (ML) and deep learning (DL) methodologies have significantly advanced drug discovery and design in several aspects. Additionally, the integration of structure-based data has proven to successfully support and improve the models’ predictions. Indeed, we previously demonstrated that combining molecular dynamics (MD)-derived descriptors with ML models allows to effectively classify kinase ligands as allosteric or orthosteric. Extending this approach, we curated a wide and diverse kinase data set (comprising 280 experimentally resolved structures) to train and evaluate a new dual-modal deep neural network classifier, which is tailored to process separately and efficiently the dynamical and structural data to predict the mode of action of a compound. The developed model demonstrated robust classification performance, effective uncertainty handling, and underscored the critical importance of incorporating protein dynamics data. Remarkably, our method maintained high performance even with imputed dynamics data, enabling rapid compound screening and prioritization, without the need for extensive MD simulations.
Structure Prediction

Graph Learning-Based Scoring of RNA–Protein Complex Structures
Zheng Jiang - ,
Ye Zhang - ,
Guipu Yang - , and
Rong Liu *
Development of suitable scoring functions is essential for the prediction of RNA–protein complex structures. Conventional statistical potential-based scoring functions suffered from deficiencies in handling conformational flexibility. The recent application of convolutional neural network (CNN) to this field has shown the potential to address the problem. Compared to CNN, however, the graph deep learning generally exhibited better performance for biomolecule-related structural and functional prediction tasks. Herein, we propose EGARPS+, a novel attempt to apply the graph learning theory to evaluate RNA–protein complex structures. This algorithm comprised the intermolecular and intramolecular modules, which were established on the equivariant graph neural networks and specifically designed attention mechanisms. Additionally, we adopted previously unexplored sequence, structural and interaction features to fully represent interface regions. Our algorithm consistently outperformed the CNN-based approach and traditional statistical potentials on both bound and unbound data sets. The proposed model excelled in processing complexes with larger conformational changes, smaller interface sizes, and lower structural similarities. EGARPS+ could also improve the de novo RNA–protein complex prediction by RoseTTAFoldNA and AlphaFold3. Finally, interpretability analyses underscored the importance of conserved motifs and hydrogen bonding in RNA–protein interactions.
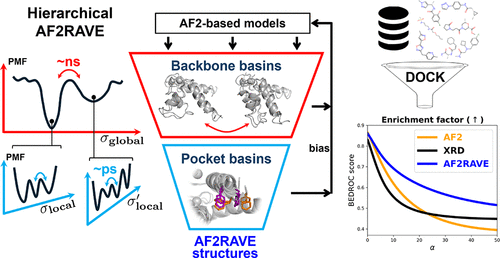
Hierarchical AF2RAVE for Multiconformation Virtual Screening Targeting S100 Ca2+-Binding Proteins
Xinyu Gu - ,
Venkata Sai Sreyas Adury - ,
Akashnathan Aranganathan - ,
Xinhao Zhuang - ,
Kristen M. Varney - ,
David J. Weber - , and
Pratyush Tiwary *
Protein function is driven by transitions between metastable conformations, many of which are not conserved across homologues, offering opportunities for selective drug design. Accurately modeling both backbone and side chain metastability, and generating structures suitable for rigid docking in high-throughput virtual screening, is thus desirable yet challenging. Here, we present a hierarchical AF2RAVE pipeline that integrates AlphaFold2 with machine learning-based enhanced sampling to systematically explore the free energy landscape and metastability of protein systems, particularly at both backbone and side chain levels. Applied to the calcium-binding S100 protein family, this approach enables the generation of diverse holo-like conformations, starting from sequence. Retrospective docking and enrichment testing with a new Ca2+-S100B inhibitor data set demonstrates that AF2RAVE-generated structures outperform standard AlphaFold2 and even outperform experimentally resolved X-ray structures in enrichment testing. Our results highlight the potential of AF2RAVE for high-throughput virtual screening and selective inhibitor discovery, particularly for challenging targets such as the Ca2+-S100 family.
Mastheads
Issue Editorial Masthead

This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More