

About the Cover:
Reviews
Consistent and Generalizable Effective Model Hamiltonian Framework for Studying Nonadiabatic Dynamics in the Condensed Phase
Zengkui Liu - ,
Hao Zeng - , and
Xiang Sun *

This publication is Open Access under the license indicated. Learn More
Simulating nonadiabatic dynamics in complex, condensed-phase systems presents a formidable computational challenge, demanding the development of effective model Hamiltonians that capture the essential physics of electronic and nuclear interactions. This Review charts the evolution of such models, from the foundational two-state spin-boson model to multistate Frenkel exciton models, highlighting the limitations of traditional approaches, particularly the isolated bath assumption, which neglects crucial environmental correlations. We focus on the recently developed multistate harmonic (MSH) model, a general and consistent framework for mapping information from all-atom simulations onto an effective Hamiltonian. The MSH model overcomes the shortcomings of previous models by systematically satisfying all pairwise reorganization energy constraints for a multistate system. This is achieved through a novel extension of the nuclear coordinate space, which provides a physically grounded and geometrically intuitive representation of a globally shared, correlated bath. We detail the construction of the MSH Hamiltonian and its equivalent multistate reaction coordinate (MRC) representation and discuss its applications in conjunction with various dynamical methods, including perturbative quantum master equations and semiclassical nonadiabatic dynamics approaches, as well as rate constant and time-dependent rate. The MSH/MRC models not only provide a robust platform for predictive simulations of charge and energy transfer in the condensed phase but also serve as an invaluable tool for benchmarking the accuracy of approximate quantum dynamics methods.
Dynamics
Molecular Models of Symmetry-Protected Quantum Batteries: Electronic Structure and Exciton Dynamics
Harold Mena - ,
Zohreh Khodadad - ,
Tao Zeng *- , and
Gabriel Hanna *
Quantum batteries, which store energy in long-lived excited states, have been theoretically predicted to possess several advantages over conventional classical batteries. While quantum battery research has been predominantly theoretical, preliminary experimental demonstrations have emerged. To advance the field beyond abstract theoretical models, we propose in this work molecular models that bring quantum batteries closer to physical realization. These models consist of anthracene-based chromophores in a hexagonal arrangement. Time-dependent density functional theory calculations confirm the existence of the previously predicted dark states in these models and yield monomeric excitation energies and couplings between the monomeric excited states, which are needed for parametrizing the Frenkel exciton Hamiltonians. Following the parametrizations, exciton dynamics simulations are carried out for all models starting from their respective dark states, under both symmetry-preserving (storage) and symmetry-breaking (discharge) conditions. Both the magnitude and sign of the on-site energy gaps are found to influence the exciton discharge rates, exhibiting a turnover as this gap is varied from large negative to large positive values. Notably, the rate exhibits a maximum when the energy gap is negative, and the turnover behavior is asymmetric about this point, with higher rates for negative gaps. Marcus theory provides a qualitative framework for explaining the trends in the simulated exciton discharge rates as both the sign and magnitude of the energy gap are varied. Overall, this work establishes a computational approach for designing molecular models of quantum batteries, sheds light on the nature of the dark states for exciton storage, and establishes design principles for controlling exciton transfer rates.
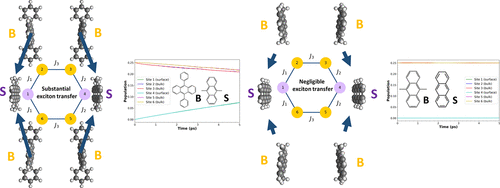
Ab Initio Nonadiabatic Molecular Dynamics in Weakly Coupled Nanosystems
Wei Li *- ,
Pingzhi Zhang - ,
Deyang Kong - ,
Lingjun Zhou - ,
Elizabeth Stippell - ,
David Beljonne - , and
Oleg V. Prezhdo *
Nanoscale systems often contain weakly coupled components, as exemplified by layered materials. Time-domain atomistic modeling of excited state processes in such systems with nonadiabatic (NA) molecular dynamics (MD) runs into severe challenges due to the divergence of the NA coupling. At the same time, standard NAMD methods work well within each component. We develop an efficient ab initio NAMD methodology using a mixed diabatic-adiabatic representation (dNAMD), implement decoherence-induced surface hopping (DISH) within the dNAMD framework, and demonstrate its utility with long-range charge transfer in 2D perovskites taking place on nano- to microsecond time scales. The dNAMD method bypasses the trivial state crossing issue of traditional NAMD by using a diabatization technique to derive diabatic electronic coupling integrals between weakly coupled components, while employing adiabatic representation within each component. We demonstrate the approach by application to 2D perovskites, which are promising materials for optoelectronic applications, but show limited efficiencies because of the insulating nature of organic spacer cations and slow interlayer charge transport. The interlayer charge transfer time scales predicted by DISH-dNAMD are consistent with experimental data and Marcus rate constants. The simulations show that phenethylammonium spacers enhance inorganic lattice rigidity via strong hydrogen bonding and π–π stacking interactions, and reduce electron-vibrational coupling while increasing interlayer spacing and charge localization. These effects significantly reduce the electronic couplings, yielding charge transfer rates that are 1–2 orders of magnitude lower than those for the more structurally flexible butylammonium spacers. The DISH-dNAMD simulations highlight the critical role of the spacer rigidity in the interlayer charge transport of 2D perovskites. The developed dNAMD framework provides an efficient and versatile tool for simulating and elucidating excited state dynamics in weakly coupled nanoscale and condensed phase systems at the atomistic level and in the time domain as it occurs in nature and experiments, advancing the design of next-generation optoelectronic devices.
Statistical Mechanics
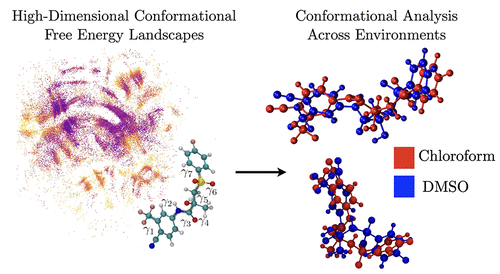
Sampling High-Dimensional Conformational Free Energy Landscapes of Active Pharmaceutical Ingredients
Alexandre Ferreira - ,
Rui Guo - ,
Ivan Marziano - , and
Matteo Salvalaglio *
This publication is Open Access under the license indicated. Learn More
We present a gridless framework for computing high-dimensional conformational free energy surfaces (FES) of flexible molecules using enhanced sampling trajectories. By combining concurrent well-tempered metadynamics with Density Peaks Advanced (DPA) clustering, our approach bypasses the dimensionality limitations of conventional grid-based FES reconstruction. Free energies are assigned on a per-configuration basis via local density estimation and Zwanzig reweighting, allowing for a direct, resolution-independent mapping of the conformational ensemble. Conformers are identified as density peaks in torsional angle space, and convergence is assessed via systematic consistency metrics. We validate this approach by reproducing the paradigmatic FES of alanine dipeptide and extend it to explore molecules with 4-, 7-, and 11-dimensional torsional angle spaces. As a key application, we investigate the solvent-dependent conformational preferences of bicalutamide in vacuum, chloroform, and DMSO. The predicted global minima reflect the known solvent-induced conformational shift between open and closed forms, in agreement with NMR and crystallographic data. These results demonstrate that our workflow provides a scalable route to high-dimensional conformational free energy landscapes, with direct relevance for polymorphism, solvation, and drug design.
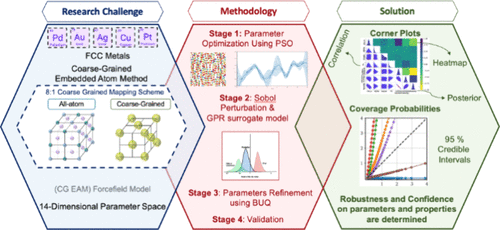
Development and Bayesian Uncertainty Quantification of Coarse-Grained Models of Metals Based on Embedded Atom Method Potentials
Abhishek T. Sose - ,
Troy Gustke - ,
Karteek K. Bejagam - ,
Fangxi Wang - ,
Aditya Savara *- , and
Sanket A. Deshmukh *
This publication is Open Access under the license indicated. Learn More
Coarse-grained (CG) molecular dynamics (MD) simulations have emerged as a powerful and cost-effective approach for modeling materials by simplifying atomic structures into CG beads. However, accurately parametrizing interatomic potential models (force fields, FFs) that can reliably reproduce material properties and quantifying the uncertainties associated with both the model parameters and their predictions remains a major challenge. In this study, we developed coarse-grained embedded atom method (CG EAM) potentials to model interatomic interactions in face-centered cubic (FCC) metals, including palladium (Pd), gold (Au), silver (Ag), copper (Cu), and platinum (Pt). The CG EAM potentials combine the physical interpretability of a traditional EAM with the computational efficiency of coarse-graining. We first employed a Particle Swarm Optimization (PSO) framework integrated with CG MD simulations to explore a 14-dimensional parameter space and identify CG EAM parameters that reproduce key physical, mechanical, and thermodynamic properties, such as cohesive energy, lattice constants, and elastic moduli. These parameters were subsequently refined using a Bayesian uncertainty quantification (BUQ) approach, which allowed the systematic assessment of uncertainties in both the FF parameters and the predicted properties. For all five metals, this framework yielded robust parameter ranges within which the predicted properties generally remained within their 95% confidence intervals. Overall, this integrated parameter optimization and BUQ approach provides an effective strategy for developing accurate and reliable interatomic potentials while offering a generalizable framework for designing both hard and soft materials with targeted properties.
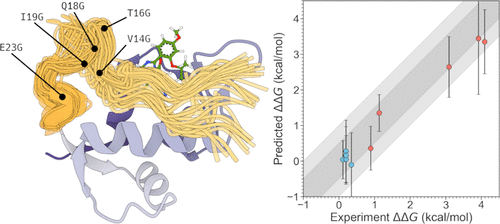
Accurate Prediction of Drug Resistance for Intrinsically Disordered Protein Regions
Audrius Kalpokas - ,
Mark Mackey - , and
Julien Michel *
Relative alchemical binding free energy calculations can be used to predict the effect of amino acid mutations on ligand binding affinities. However, these protocols are not well established for proteins containing intrinsically disordered regions (IDRs). In this work, we focus on the development of robust protein-free energy perturbation (FEP) protocols to reproduce experimental binding affinities that have been measured for a panel of mutants of the protein MDM2 against two ligands, AM-7209 and Nutlin-3a. We focus on mutations that occur in the N-terminal IDR lid of MDM2, which is known to undergo ligand-dependent folding upon binding. We systematically assess the effectiveness of both equilibrium and nonequilibrium alchemical protocols in reproducing these experimental binding affinities, in particular for mutations with slowly varying degrees of freedom. We show that the equilibrium protocol outperforms the nonequilibrium protocol in the precision of the free energy estimates obtained. In addition, we demonstrate the effect of the protein force field and the water model used to simulate the highly flexible IDR region. Overall, our findings demonstrate an accurate FEP protocol capable of reproducing these trends and further show the applicability of FEP protocols for elucidating the mutational effects on ligand binding affinity in highly dynamic intrinsically disordered protein regions.
Quantum Electronic Structure

The Grand Canonical General-Purpose Reactivity Indicator: A Conceptual DFT Approach to Predict Molecular Reactivity and Experimental Electrophilicity and Nucleophilicity Scales
Yoshio Barrera - ,
Tomás Rocha-Rinza - ,
Florian F. Mulks - ,
Paul W. Ayers - , and
James S. M. Anderson *
This publication is Open Access under the license indicated. Learn More
The Grand Canonical General-Purpose Reactivity Indicator (GC-GPRI) is introduced as a tool for predicting reactivity and for discerning the relative electrophilicity and nucleophilicity of electrophiles and nucleophiles, respectively. The GC-GPRI is derived within the zero-temperature grand canonical ensemble conceptual density-functional theory (CDFT) framework using a perturbative approach. In this model, the electrophile–nucleophile interaction energy is modeled by perturbations in the chemical and external potentials of the isolated species. The GC-GPRI accurately identifies the most reactive hard and soft atoms in complex molecules with multiple reactive sites. This model also reproduces experimental electrophilicity and nucleophilicity scales for 21 electrophiles and 20 nucleophiles, with R2 correlations of 0.98 and 0.94, respectively.
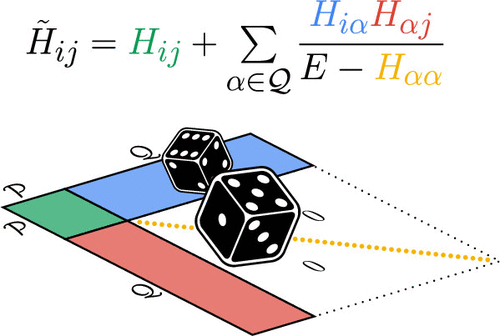
Stochastic-SplitGAS: A Quantum Monte Carlo Multi-Reference Perturbation Theory Based on the Imaginary-Time Evolution of Effective Hamiltonians
Luca Bonfirraro *- ,
Oskar Weser - ,
Maru Song - , and
Giovanni Li Manni *
This publication is Open Access under the license indicated. Learn More
Accurately modeling the electronic structure of systems with many unpaired electrons remains a major challenge in quantum chemistry. Qualitatively correct electronic structures generally require large active space multireference wave functions, while dynamic correlation effects beyond the active space are crucial for quantitatively accurate descriptions of magnetic, catalytic and optical properties of such systems. Here, we present an uncontracted multireference perturbation theory based on the FCIQMC imaginary-time evolution of effective Hamiltonians, built upon the generalized active space concept and Löwdin’s partitioning technique. The configurational interaction space is split into a reference space, consisting of the most important configurations, and a perturber space, containing the more numerous configurations responsible for dynamic correlation effects. The generalized active space algorithm allows the flexible partitioning of the configurational space. Löwdin’s partitioning technique is then used to construct an effective Hamiltonian which is stochastically solved. This strategy allows us to apply perturbative corrections on large active space reference wave functions, without requiring high-order reduced density matrices, which have been found the bottleneck in other perturbation theory strategies. The capabilities of the resulting method, called Stochastic-SplitGAS, are demonstrated on the triplet-quintet spin gap of an Fe(II)-porphyrin model system and the spin ladder of a [Fe(III)2S2]2– complex.
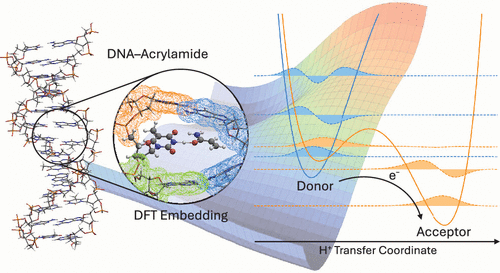
Large-Scale Modeling of Proton-Coupled Electron Transfer Based on Block-Localized Kohn–Sham Orbitals
Lukas Lampe - ,
Takeshi Yanai *- , and
Johannes Neugebauer *
The calculation of rate constants for proton-coupled electron transfer (PCET) reactions is a challenging task in quantum chemistry. This task involves identifying the mechanism of the process, that can take place either adiabatically or nonadiabatically, and calculating the necessary quantities, such as vibronic couplings, according to the mechanism. Due to different electronic configurations involved, it almost becomes inevitable to use wave function-based multireference methods. However, the accurate prediction of rate constants for large molecular systems is limited by the high computational cost of common choices such as complete active space self-consistent field (CASSCF). Since PCET reactions occur in a wide range of biological processes, the development of alternatives with large scalability is of particular interest. A promising alternative is the multistate density-functional theory method based on block-localized Kohn–Sham (BLKS) orbitals. This gives access to the diabatic donor and acceptor states, adiabatic ground and first excited states, and the electronic coupling. In this work, different operators for the construction of BLKS orbitals are considered. A comparison with CASSCF and N-electron valence state second-order perturbation theory shows that accurate vibronic couplings can be obtained using a non-Hermitian operator. As the method relies on a fragmentation of the system, spectator fragments can be explicitly included with a convenient computational cost. This is demonstrated by the example of a DNA–acrylamide complex.

Thermal Weight Determination and Interstate Coupling in State-Averaged ADAPT-VQE
Harper R. Grimsley *- and
Francesco A. Evangelista *
This publication is Open Access under the license indicated. Learn More
Characterizing electronic thermal states at low temperatures is an important but challenging task in quantum chemistry and condensed matter physics, making it a prime candidate for a useful application in quantum computing. One of the most successful methods for state preparation on quantum computers is the Adaptive, Problem-Tailored (ADAPT) Variational Quantum Eigensolver (VQE), which has recently been generalized to treat excited states within a state-averaged framework as well as Gibbs states. In this work, we introduce Helmholtz-Optimized Thermal (HOT) ADAPT-VQE, an ancilla-free strategy for preparing Gibbs states that directly minimizes the Helmholtz free energy by targeting the dominant eigenstates of the thermal ensemble. We demonstrate the usefulness of HOT-ADAPT-VQE by predicting the free energy of two model systems with strongly correlated ground states: (1) the Fe2+ cation in a magnetic field and (2) a [Cu2O7]10– fragment of the Mott insulator La2CuO4. Our results demonstrate that HOT-ADAPT-VQE significantly improves upon Gibbs-state estimates from multistate variants of ADAPT-VQE, often with substantially shallower quantum circuits, making it a promising candidate for thermal-state calculations.
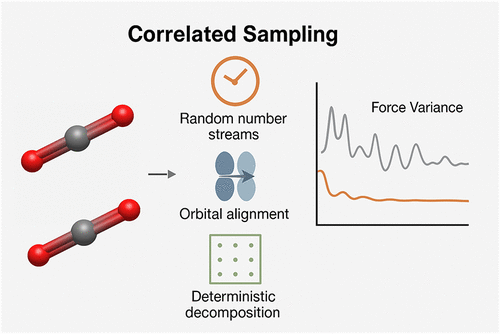
Molecular Properties in Quantum-Classical Auxiliary-Field Quantum Monte Carlo: Correlated Sampling with Application to Accurate Nuclear Forces
Joshua J. Goings *- ,
Kyujin Shin *- ,
Seunghyo Noh - ,
Woomin Kyoung - ,
Donghwi Kim - ,
Jihye Baek - ,
Martin Roetteler - ,
Evgeny Epifanovsky - , and
Luning Zhao
We extend correlated sampling to quantum-classical auxiliary-field quantum Monte Carlo (QC-AFQMC), enabling accurate nuclear force evaluation in strongly correlated systems. Computing forces via finite differences typically incurs prohibitive statistical noise in stochastic methods. We suppress this noise by maximizing correlation between geometries through synchronized random streams, orbital alignment, deterministic integral decomposition, and consistent classical shadow measurements. Crucially, a single shadow ensemble defined at the reference geometry suffices for all displaced structures, eliminating additional quantum measurements. This approach substantially reduces force variance while preserving accuracy. We validate the method on hydrogen chains across varying correlation regimes and demonstrate accurate forces for N2 dissociation and stretched H4 in strongly correlated regions where restricted coupled cluster methods fail qualitatively. Application to the MEA-CO2 carbon capture reaction, integrating quantum information metrics for active space selection and matchgate shadows for overlap estimation, demonstrates that QC-AFQMC delivers accurate forces for complex reaction pathways in strongly correlated systems where conventional methods are unreliable or prohibitively expensive.
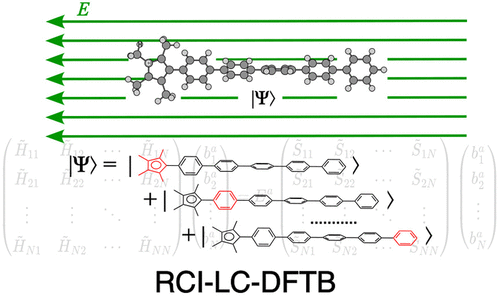
A Restriction-Based Configuration Interaction Approach Based on LC-DFTB: An Efficient Method for Field-Induced Charge Transfer in Molecular Systems
Ji Huang - ,
Tim Kowalczyk - ,
Yoshio Nishimoto - , and
Daisuke Yokogawa *
Investigating electron transfer behavior under external electric fields in molecular electronics is crucial for understanding the function of each component and for improving molecular design. Notably, the one-electron transfer is inevitable in molecular wires and switches, for which traditional density functional theory (DFT) and long-range corrected self-consistent-charge density functional tight binding (LC-DFTB) meet significant challenges. Inspired by previous studies on constrained configuration interaction schemes, we present restriction-based configuration interaction (RCI) LC-DFTB, a novel extension of LC-DFTB to deliver an accurate description of one-electron transfer under external electric fields. This approach retains the low cost of LC-DFTB while accurately capturing charge-resonance, localization versus delocalization, and field-induced response in large, structurally complex systems. We demonstrate its performance on a benzene assembly and a polyfluorene, showing that RCI-LC-DFTB efficiently describes the effects of molecular conformation and applied bias on electron localization and transfer. Our method thus provides a robust tool for the design of molecular electronic and organic photovoltaic materials.

Gold-Standard Chemical Database 137 (GSCDB137): A Diverse Set of Accurate Energy Differences for Assessing and Developing Density Functionals
Jiashu Liang - and
Martin Head-Gordon *
This publication is Open Access under the license indicated. Learn More
We present GSCDB137, a rigorously curated benchmark library of 137 data sets (8377 entries) covering main-group and transition-metal reaction energies and barrier heights, (intra- and intermolecular) noncovalent interactions, dipole moments, polarizabilities, electric-field response energies, and vibrational frequencies. Legacy data from GMTKN55 and MGCDB84 have been updated to today’s best reference values; redundant or low-quality points were removed, and many new, property-focused sets were added. Testing 29 popular density functional approximations (DFAs) confirms the expected Jacob’s-ladder hierarchy overall but also reveals notable exceptions: functional performance for frequencies and electric-field properties correlates poorly with that for other ground-state energetics. ωB97M-V and ωB97X-V are the most balanced hybrid meta-GGA and hybrid GGA, respectively; B97M-V and revPBE-D4 lead the meta-GGA and GGA classes. Double hybrids lower mean errors by about 30% versus their hybrid analogues but demand careful frozen-core, basis set, and spin contamination treatment. GSCDB137 offers a comprehensive, openly documented platform for rigorous validation of DFA and universal machine learning potentials, and training of the next generation of exchange-correlation functionals.
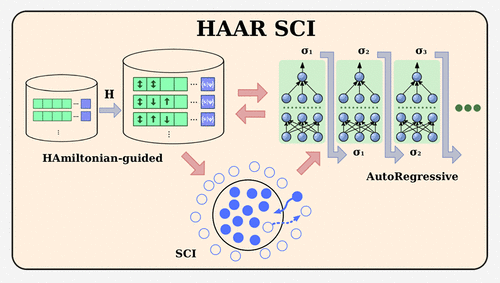
Hamiltonian-Guided Autoregressive Selected-Configuration Interaction Achieves Chemical Accuracy in Strongly Correlated Systems
Hao Zhang - ,
Xiongzhi Zeng *- ,
Zhenyu Li *- , and
Yi Zhou *
Strongly correlated molecules remain out of reach for most electronic-structure solvers because the exact wave function spans a determinant space that grows exponentially with the number of orbitals. State-of-the-art selected-CI (e.g., CIPSI and HCI) and ML-CI methods mitigate search by exploiting Hamiltonian sparsity and learned prescreening. Nonetheless, in strongly multireference regimes, the excitation-based candidate lists can grow rapidly, which may increase memory and screening costs. We present the Hamiltonian-guided autoregressive selected-configuration interaction (HAAR-SCI), a learn–sample–compress workflow that runs on a single GPU. A gated Transformer samples determinants autoregressively; Gumbel Top-K noise encourages exploration, and GPU min-heap kernels keep only configurations with the largest Hamiltonian couplings. The network is retrained after each expansion, and iterations stop when successive energies differ by ≤1 mHa. Across an 18-molecule benchmark set reaching 116 spin–orbitals, HAAR-SCI attains a mean absolute error of 0.51 mHa while using much fewer determinants than heat-bath CI. It traces the entire N2 dissociation curve within 0.67 mHa and achieves HCI accuracy for the 40-spin-orbital [Fe2S2(SCH3)4]2– cluster with a determinant reduction of 72%, demonstrating its power on systems considered intractable for conventional selected-CI solvers. A final probability-mass pruning compresses typical wave functions by a further 10–50×, retaining <0.01% of the Hilbert space yet still capturing >99.9% of the correlation energy. HAAR-SCI thus offers a compact and truly scalable route to chemical-accuracy quantum chemistry on commodity hardware.
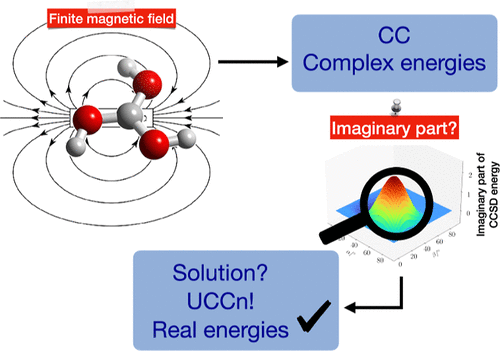
Unitary Coupled-Cluster Theory for the Treatment of Molecules in Strong Magnetic Fields
Laura Grazioli *- ,
Marios-Petros Kitsaras *- , and
Stella Stopkowicz *
This publication is Open Access under the license indicated. Learn More
In coupled-cluster (CC) theory, unphysical complex energies may arise in the presence of strong magnetic fields, near conical intersections, or in systems exhibiting complex Abelian point group symmetries. This issue originates from the non-Hermitian nature of the CC energy expression. A promising solution is provided by unitary coupled-cluster (UCC) theory, which retains the advantages of an exponential parametrization while ensuring real-valued energy eigenvalues. In this work, we present an implementation of finite-field second-order (ff-UCC2) and third-order (ff-UCC3) UCC theory. We assess the performance of these truncation levels in comparison to conventional finite-field CC methods, using the methylidyne ion, water, and boric acid.
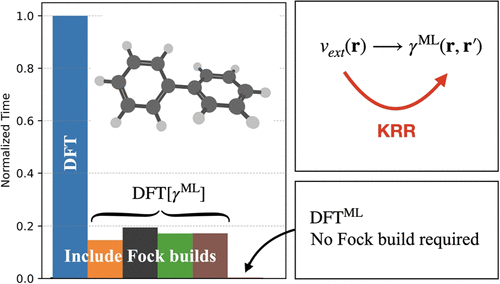
Learning the One-Electron Reduced Density Matrix at SCF Convergence Thresholds
Bhaskar Rana - ,
Nicolas Viot - ,
Jessica A. Martinez B - ,
Xuecheng Shao - ,
Pablo Ramos - , and
Michele Pavanello *
This publication is Open Access under the license indicated. Learn More
Machine learning of the one-electron reduced density matrix (1-RDM) provides a computationally efficient surrogate to conventional electronic structure methods. In this work, we train models that map the electron−nuclear interaction potential to the 1-RDM with such an accuracy that predicted 1-RDMs deviate from fully converged ones by no more than a standard self-consistent field (SCF) threshold. Through targeted model optimization strategies, we demonstrate that training set sizes substantially smaller than those required in our previous work [Shao, X. Nat. Commun. 14, 6281 (2023)] are sufficient to reach this accuracy. Furthermore, we introduce a force-correction algorithm that enables stable ab initio molecular dynamics powered by the machine learned 1-RDMs, extending the applicability of the surrogate electronic structure methods to molecules as large as biphenyl.
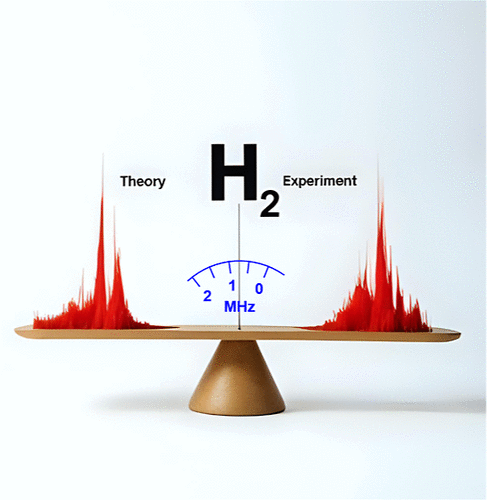
From First-Principles to Quantum Electrodynamics: Pushing the Limits of Theory with the Hydrogen Molecule
Krzysztof Pachucki *- and
Jacek Komasa *

This publication is free to access through this site. Learn More

ACS Editors' Choice® is a collection designed to feature scientific articles of broad public interest. Read the latest articles
Modern spectroscopic techniques enable the determination of the spacing between rovibrational levels of H2 with a relative accuracy of approximately 10–11. At this extreme level of precision, subtle quantum electrodynamic (QED) effects, such as electron self-interaction and vacuum polarization, are probed. A theoretical model aiming to achieve similar accuracy must precisely describe not only these relatively small QED effects but also the more significant contributions related to electron correlation, coupling between electronic and nuclear motions, and relativistic effects. Although the hydrogen molecule exhibits most of the phenomena found in larger molecules, it is simple enough to meet the requirements mentioned above. In this article, we report on enhancements to the current capabilities of quantum mechanical calculations for the hydrogen molecule. We present a method based on exponential functions that fully captures electron correlation or, more broadly, interparticle correlation, enabling a comprehensive description of effects related to nuclear motion. Specifically, we solve the four-particle Schrödinger equation without invoking commonly used approximations such as the one-electron or the Born–Oppenheimer approximation. The only source of nonrelativistic energy error comes from the finite size of the basis set. The explicitly correlated nonadiabatic wave function used here is then employed to determine the relativistic and QED effects. As a result, the dissociation energy for the lowest rovibrational levels in the electronic ground state of H2 has been obtained with a relative accuracy of 7 × 10–10, while the frequencies of intervals between these levels have been determined with sub-MHz accuracy, corresponding to a relative accuracy of 3 × 10–9. In consequence, the discrepancies between the highest precision measurements and earlier theoretical predictions have been resolved.
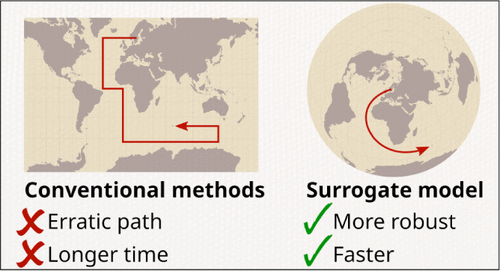
Accelerating SCF Orbital Optimization with S-GEK/RVO: Efficient Subspace Compression and Robust Convergence
Ignacio Fdez. Galván *- ,
Daniel Weßling - , and
Roland Lindh *
This publication is Open Access under the license indicated. Learn More
We present enhancements to the S-GEK/RVO method for self-consistent field (SCF) orbital optimization, aimed at improving computational efficiency and robustness. Building on a gradient-enhanced Kriging surrogate model and restricted-variance optimization, we introduce three key modifications: (i) a cost-effective subspace expansion using r-GDIIS or BFGS displacement predictions, (ii) a systematic undershoot mitigation strategy in flat energy regions, and (iii) rigorous coordinate and gradient transformations consistent with the exponential parametrization of orbital rotations. Benchmarking across an extensive set of molecular systems─including organic molecules, radicals, and transition-metal complexes─demonstrates that the new S-GEK/RVO variants consistently outperform the default (in OpenMolcas) r-GDIIS method in iteration count, convergence reliability, and wall time. These improvements make S-GEK/RVO a competitive alternative for SCF optimization and suggest broader applicability to other orbital optimization and localization problems.
Reaction Mechanisms
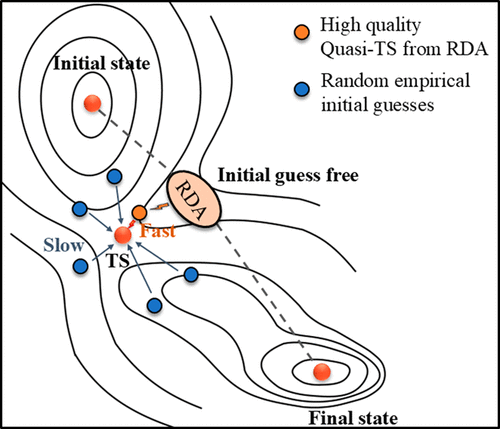
Fast-Tracking Transition-State Localization via Reaction Directional Analysis
Peipei Zhang - ,
Chenxi Guo *- , and
P. Hu *
Transition-state localization is critical for elucidating chemical reaction mechanisms but remains one of the most computationally demanding challenges in theoretical chemistry. Here, we introduce a novel method, reaction directional analysis-dimer (RDA-D), which integrates reaction directional analysis (RDA) with the dimer method to achieve efficient and reliable transition state searching. Reaction directional analysis generates high-quality quasi-transition-state structures directly from only the initial and final state geometries, combining dynamic interpolation, structural optimization, and directional analysis. These quasi-transition-state structures then serve as starting points for refinement via the dimer method. Benchmark tests on a diverse set of gas-phase and catalytic reactions on surfaces demonstrate that RDA-D is, on average, 5.83 times faster than the Nudged Elastic Band (NEB) method in CPU time and reduces the number of gradient evaluations by a factor of 4.74. Moreover, reaction directional analysis eliminates the need for predefined reaction coordinates or chemically intuitive initial guesses, providing a robust, scalable, and automation-friendly framework for transition-state localization.
Molecular Mechanics
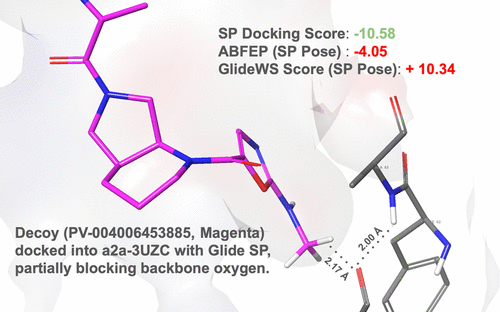
Glide WS: Methodology and Initial Assessment of Performance for Docking Accuracy and Virtual Screening
Richard A. Friesner *- ,
Robert B. Murphy - ,
Yuqi Zhang - ,
Yeyue Xiong - ,
Pierre A. Devlaminck - ,
Ivan Tubert-Brohman - , and
Steven V. Jerome *
Powered by dramatic advances in computer hardware, the advent of ultralarge make-on-demand virtual libraries, and a shift in small-molecule discovery toward more challenging targets with limited known actives, there has been a growing interest in the development of performant virtual screening methods that can reliably deliver novel hits. We report on a new method called Glide WS, that builds on our earlier efforts (WScore) to introduce an explicit representation of water structure and dynamics to an empirical scoring function suitable for high-throughput docking. This scoring function has been carefully tuned using absolute binding free energy perturbation calculations (ABFEP). Compared with Glide SP, Glide WS offers significant gains in the two primary tasks for molecular docking in drug discovery, pose prediction and virtual screening enrichment. For docking accuracy, Glide WS achieves a self-docking accuracy of 92% on a diverse set of 1477 protein ligand complexes as compared to 85% for Glide SP, using a criterion of 2.5 Å. We also demonstrate significantly improved virtual screening enrichment using a diverse data set covering of 38 targets together with three different computationally generated libraries of decoys, combined with standard known ChEMBL actives. We focus on ligands ranked in the top few percent of the database (the subset that is relevant to practical virtual screening efforts) and demonstrate that, along with improved enrichment of ChEMBL actives, Glide WS achieves a remarkable reduction in the number of poorly scoring decoys (as calibrated by ABFEP calculations), across a high percentage of targets, as compared to Glide SP. These results suggest that considerably higher hit rates will be observed, as compared to conventional rigid receptor docking, in practical virtual screening applications.

A Universal Augmentation Framework for Long-Range Electrostatics in Machine Learning Interatomic Potentials
Dongjin Kim - ,
Xiaoyu Wang - ,
Santiago Vargas - ,
Peichen Zhong - ,
Daniel S. King - ,
Theo Jaffrelot Inizan - , and
Bingqing Cheng *
Most current machine learning interatomic potentials (MLIPs) rely on short-range approximations, without explicit treatment of long-range electrostatics. To address this, we recently developed the Latent Ewald Summation (LES) method, which infers electrostatic interactions, polarization, and Born effective charges (BECs), just by learning from energy and force training data. Here, we present LES as a standalone library, compatible with any short-range MLIP, and demonstrate its integration with methods such as MACE, NequIP, Allegro, CACE, CHGNet, and UMA. We benchmark LES-enhanced models on distinct systems, including bulk water, polar dipeptides, and gold dimer adsorption on defective substrates, and show that LES not only captures correct electrostatics but also improves accuracy. Additionally, we scale LES to large and chemically diverse data by training MACELES-OFF on the SPICE set containing molecules and clusters, making a universal MLIP with electrostatics for organic systems, including biomolecules. MACELES-OFF is more accurate than its short-range counterpart (MACE-OFF) trained on the same data set, predicts dipoles and BECs reliably, and has better descriptions of bulk liquids. By enabling efficient long-range electrostatics without directly training on electrical properties, LES paves the way for electrostatic foundation MLIPs.
Spectroscopy and Excited States
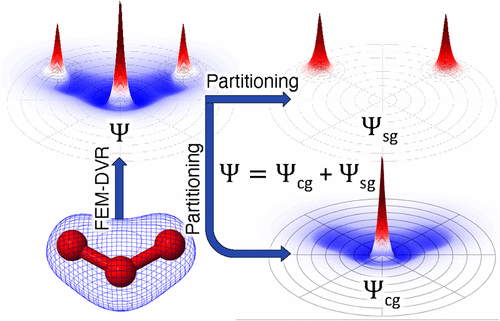
Overset-Grid Method with Smooth Orbital Partitioning for Molecular Scattering Calculations
Yuchen Liu - ,
Jan Dvořák - ,
Loren Greenman - ,
C. William McCurdy *- , and
Robert R. Lucchese *
To solve molecular photoionization and electron scattering problems, we use an overset-grid representation of electronic continuum functions, which has an extended central spherical grid that overlaps small spherical grids (subgrids) centered on each atom of a polyatomic molecule. Here, we present an improved algorithm that smoothly partitions the total wave function between the central grid and the atomic subgrids. The smooth partitioning allows one to use approximately one-fourth the number of partial waves on the central grid compared to our previous implementation with switching functions. The resulting numerical method for treating electron scattering and photoionization of polyatomic molecules combines the accuracy and flexibility of pure numerical grid representations with the rapid convergence of hybrid combinations of atom-centered basis-set expansions and grid methods. The overset-grid representation is implemented using the complex Kohn variational principle for scattering and photoionization amplitudes. The faster convergence with respect to the number of central grid partial waves is demonstrated and accuracy is verified by comparisons with the previous implementation and with far more computationally demanding single-center numerical expansions in electron-molecule scattering and photoionization calculations on the neon dimer (Ne2) system, carbon tetrafluoride (CF4) molecule, and the pyridine (C5H5N) molecule in the static-exchange approximation.
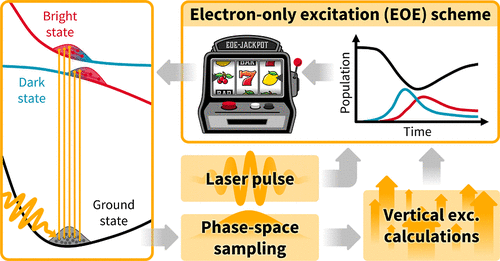
An Excitation Strategy for the Initial Condition Generation for Surface Hopping Trajectories Using Electron-Only Dynamics Including Explicit Laser Pulses
Lorenz Grünewald - ,
Laurens van Dam - , and
Sebastian Mai *
This publication is Open Access under the license indicated. Learn More
Nonadiabatic dynamics simulations, e.g., via trajectory surface hopping, are nowadays used regularly to describe various photoinduced phenomena in molecules. For a number of reasons, in the setup of such simulations, the actual photoexcitation process is often described by rather crude and approximate excitation schemes (e.g., vertical excitation by an implicit delta pulse), and more attention is directed to the ensuing dynamics simulations. However, several studies have implied the importance of properly considering the spatial, temporal, and spectral details of the exciting laser pulse. Here, we suggest the “electron-only explicit” (“EOE”) excitation scheme for setting up trajectory surface hopping initial conditions based on an explicit laser pulse at little computational cost. The scheme is based on solving the time-dependent electronic Schrödinger equation including the explicit influence of a laser pulse within the frozen-nuclei approximation. The obtained time-dependent, coherently excited, electronic populations are then used to stochastically select the initial electronic states in a postprocessing step. Here, the electronic populations are renormalized such that one excites a reasonable fraction of the initial condition even when operating well within the weak-field regime. The new scheme is made freely available as part of the SHARC 4 dynamics package. We illustrate and validate the new excitation scheme by means of several simulations of sodium iodide and 6-cyanobenzquinuclidine excited with laser pulses of different energy and pulse duration, comparing to quantum dynamics results.

Improved TDDFT Excitation Energies with an Accurate Kohn–Sham Potential: Reassessment of the Double-Excitation Character of the Low Lying 21Ag Excited State of s-Trans-1,3-Butadiene
Evert Jan Baerends *- ,
Mario Amati *- , and
Sonia Stoia
Extensive benchmarks and reviews of time-dependent density functional theory (TDDFT) have been published, covering at least 50 functionals. Here, we do not use one of the (meta)GGA or hybrid functionals but highlight the particular TDDFT method that does not use the Kohn–Sham (KS) potential of some exchange-correlation functional, but uses the exact Kohn–Sham potential or a close approximation to it in the SCF calculations. Such KS-potential-based TDDFT results have been proven to yield excellent results. For routine application, it is required that a computationally simple approximation (as a density functional) to the exact KS potential is available. In this paper, we benchmark TDDFT calculations with a recently developed model KS potential and compare them to advanced quantum chemical methods and experimental data. The target systems here are medium-sized molecules. These TDDFT calculations based on good KS potentials prove to be competitive in accuracy with sophisticated ab initio methods. An advantage is the possibility to use large basis sets (also for large molecules), enabling a description of valence excitations and Rydberg (or mixed valence-Rydberg) excitations on the same footing. This is necessary for high accuracy; a sophisticated but expensive method that cannot handle large basis sets cannot achieve high accuracy. An advantage of the use of a (close to) exact Kohn–Sham potential is the realistic nature (shape and energy) of both the occupied and the virtual KS orbitals, affording an interpretation of excitations in terms of one or a few single orbital-to-orbital transitions. This obviates the need for extensive optimization of the virtual orbitals for the purpose of interpreting excitations. To highlight the importance of the realistic nature of the KS virtuals, including the Rydberg orbitals, we discuss and reassess the nature of the 21 Ag state of s-trans-1,3-butadiene, which has been widely considered as a prototype excitation with large double excitation character. Charge-transfer and true double excitations cannot be handled by the simple ALDA kernel used here.
Condensed Matter, Interfaces, and Materials

A Unified Langevin Framework for Bosonic and Fermionic Dissipation in Nonadiabatic Electrochemical Proton Transfer
Elvis F. Arguelles *- and
Osamu Sugino
We present an influence functional path integral framework for treating the coupled dynamics of solvated proton and electron transfer within a nonequilibrium open system. This method formulates a generalized Langevin equation describing dynamics in systems where proton is simultaneously coupled to fermionic (metal electrons) and bosonic (solvent phonons) reservoirs. It accounts for multiple dissipative channels without relying on phenomenological assumptions. With this scheme, we capture the relaxation of oscillations associated with large quantum zero-point fluctuations when protons are trapped in a harmonic potential. When the proton’s translational motion is slow, the dynamics become effectively Markovian. In this regime, dissipation to the electronic reservoir is characterized by a position-dependent electronic friction. Using an effective electronic model Hamiltonian, we demonstrate that electronic friction introduces a sharp, localized resistance when the proton level crosses the Fermi level, effectively delaying the reaction. In contrast, solvent friction arising from assumed Caldeira–Leggett-type coupling, exerts a uniform, position-independent drag. Both mechanisms contribute comparable amounts to the overall energy dissipation. This framework offers a computationally efficient route to simulate complex electrochemical environments involving multiple dissipative baths.
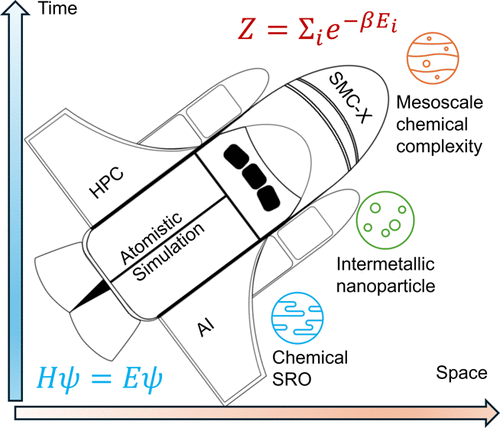
SMC-X: A Distributed, Scalable Monte Carlo Simulation Method for Chemically Complex Alloys
Xianglin Liu *- ,
Kai Yang - ,
Fanli Zhou - , and
Pengxiang Xu
To predict the complex chemical evolution in multicomponent alloys, it is highly desirable to have accurate atomistic simulation methods capable of reaching sufficiently large spatial and temporal scales. In this work, we advance the recently proposed SMC-X method through distributed computation on either GPUs or CPUs, pushing both spatial and temporal scales of atomistic simulation of chemically complex alloys to previously inaccessible scales. This includes a record-breaking 128-billion-atom HEA system extending to the micrometer regime in space, and a 1-billion-atom HEA evolved over more than three million Monte Carlo swap steps, approaching the minute regime in time. We show that such large-scale simulations are essential for bridging the gap between experimental observations and theoretical predictions of the nanoprecipitate sizes in HEAs, based on analysis using the Lifshitz–Slyozov-Wagner (LSW) theory for diffusion-controlled coarsening. This work demonstrates the great potential of SMC-X for simulation-driven exploration of the chemical complexity in high-entropy materials at large spatial and temporal scales.
Biomolecular Systems
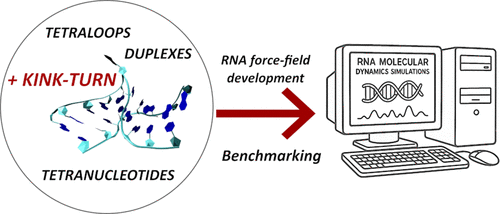
The Kink-Turn 7 Motif: An Additional Test for RNA Force Field Performance
Toon Lemmens - ,
Vojtěch Mlýnský - ,
Jiří Šponer - ,
Martin Pykal - ,
Pavel Banáš - ,
Michal Otyepka - , and
Miroslav Krepl *
This publication is Open Access under the license indicated. Learn More
The kink-turn is a recurrent RNA structural motif that induces a sharp bend (kink) in the A-form RNA helix. It is defined by key structural features, including consecutive sheared AG base pairs, an A-minor interaction, and multiple base–sugar interactions. An accurate representation of these densely packed noncanonical interactions by molecular dynamics simulations poses a significant challenge for contemporary force fields (FFs). Here, we present extended simulations of the ribosomal kink-turn 7 (Kt-7) from H.m., the so-called “consensual” kink-turn, using a broad spectrum of pair-additive and polarizable RNA FFs. None of the tested FFs manage to flawlessly describe all of the structural features of the Kt-7 although several FFs provide rather acceptable results and should not cause problems in simulations of larger RNAs containing a kink-turn. On aggregate, the widely used OL3 (ff99bsc0χOL3) and polarizable AMOEBA FFs achieve the best performance for this motif. Interestingly, some more recently parametrized FF variants struggle to describe the Kt-7’s tertiary A-minor interaction – a ubiquitous tertiary contact in RNA. This raises some concerns about the broader applicability of these FFs and suggests that they may be overfitted to small model systems, such as RNA tetranucleotides. In some cases, irreversible unkinking of the entire kink-turn motif can also be observed. The kink-turn motif is highly sensitive to variations in RNA FFs, and we strongly recommend its inclusion in training and benchmarking data sets as an important regression test to improve the robustness and accuracy of RNA FF parametrization.
Structure Prediction
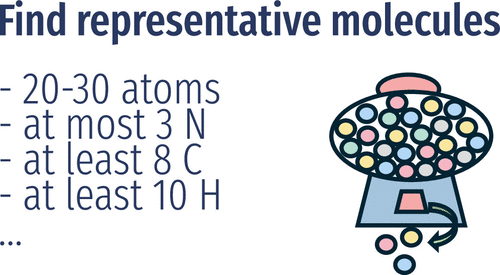
Representative Random Sampling of Chemical Space
Diego J. Monterrubio-Chanca - and
Guido Falk von Rudorff *
This publication is Open Access under the license indicated. Learn More
An overwhelming majority of molecules remain unexplored. This is mostly due to the sheer number of them, which prohibits any enumeration of chemical space, the set of all such molecules. In practice, only subsets of chemical space are considered, but those subsets exhibit substantial bias, prohibiting the data-driven characterization of chemical space itself. In this work, we provide a method to produce unbiased representative random samples of the chemical space without enumeration of constituent molecules and to estimate the number of molecules in any custom chemical space. The approach is applicable to molecules that can be represented as graphs and runs efficiently even for molecules of 30 atoms. We use it to estimate the representativeness of current databases with respect to their underlying chemical space and establish a necessary criterion for a lower bound of database sizes to be representative of an underlying chemical space.
Mastheads
Issue Editorial Masthead
This publication is free to access through this site. Learn More
Issue Publication Information
This publication is free to access through this site. Learn More